

Abstract
The composition of the cellular proteome is commonly thought to strictly adhere to the genetic code. However, accumulating evidence indicates that cells also regulate the synthesis of mutant protein molecules that deviate from the genetic code. Production of mutant proteins varies in amounts and specificity and generally occurs when cells are stressed or undergo environmental adaptation. The deliberate synthesis of protein mutants suggests that some of these proteins can be useful in cellular stress response and adaptation. This review describes the occurrence, the translation mechanisms, and the functional hypotheses on regulated synthesis of mutant proteins.
Keywords: Adaptive translation: synthesis of mutant proteins that benefit the cell, mistranslation, stress response, tRNA misacylation, ribosome misdecoding
Introduction
A central pillar of molecular biology is the accuracy of information transfer from DNA to proteins. It is commonly believed that the genetic code should be obeyed at all times, and any derivation of this rule reflects the imperfection of the translation process. The translation fidelity is commonly cited as between 10-5 to 10-3 per codon, depending on the measurement method and the codon context (27, 35, 36, 43, 61, 79). These error frequencies are typically interpreted as the tolerance threshold of the translation machinery. Less accurate translation would result in the synthesis of proteins that deviate from the genetic code.
The translation fidelity is maintained at two steps: the accuracy of tRNA aminoacylation and the ribosome matching the mRNA codon with the tRNA anticodon (Fig. 1A). tRNA aminoacylation or charging is performed by aminoacyl-tRNA synthetases (aaRS); there is typically one aaRS for each amino acid in the cell. Each aaRS selects its cognate tRNAs among all tRNAs in the cell and chemically attaches its cognate amino acid to the 3′ end of the cognate tRNA. In general, tRNA synthetases are highly accurate: the fidelity of aminoacylation is typically better than 10-4 when measured in vitro using purified tRNA synthetases (43). The ribosome matching mRNA codon with the correct tRNA anticodon involves Watson-Crick base pairing of the first and second codon nucleotide, and either Watson-Crick or wobble base pairing of the third codon nucleotide. The highly accurate matching involves many quality control steps and is typically on the order of 10-4 when measured in vitro using purified components (61, 79). For both aminoacylation and codon-anticodon matching, a common theme has emerged that fine tuning of every step of the process is important to ensure high fidelity of translation.
Fig. 1. Processes in protein synthesis that deviate from the genetic code.
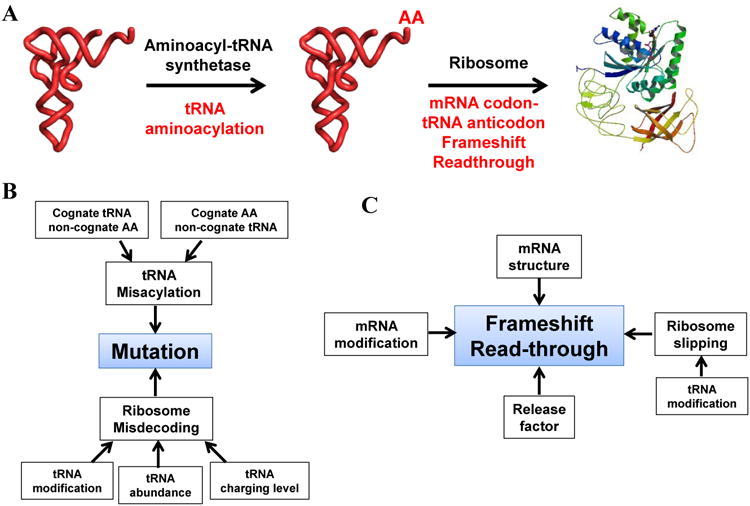
(A) Two steps in translation where translational fidelity is controlled (tRNA charging and ribosome decoding). AA: amino acid. (B) Mechanisms in making mutant proteins. (C) Mechanisms in making proteins through frameshift or stop codon readthrough.
An important consideration of translation fidelity is when a fine-tuned translational process may no longer be available in the cell. As early as the advent of two-dimensional gel electrophoresis in the 1970s, it was observed that under nutritional or environmental stress, cells often produce proteins that seem to deviate from those programmed by the genetic code (54). Starving E. coli for the amino acid asparagines (Asn) leads to readily detectable levels of proteins that contain non-Asn substitutions such as lysine (Lys, (56)). This result was interpreted as Asn starvation decreasing the amount of charged tRNAAsn (which reads AAC/AAU codons) so that the near-cognate tRNALys (which reads AAG/AAA codons) can read the Asn codons to make Asn-to-Lys mutant proteins. Mutant protein synthesis under an imbalance of charged tRNAAsn/tRNALys shows that there are potential advantages in making mutant proteins, which may be active in response to cell stress, over making no protein all. It is however not known whether any of the Lys-to-Asn mutant proteins serves a function distinct from the wild-type protein. A recent example of conditional dependence of synthesizing mutant proteins in mammalian cells shows that high level antibody production in Hamster cells leads to significant levels of Asn-to-Ser substituted antibody proteins as detected by mass spectrometry (74). This mutant protein production seems to be derived from insufficient supply of Asn in the growth medium: Asn supplement drastically reduces the amount of such mutant proteins.
Underappreciated until recently, cells and organisms have a high threshold of tolerance of decreased translational fidelity when one central component in translation is genetically mutated either in isolated mutant strains or occurring naturally, or expressed at inappropriate amounts. For example, the Ala734-to-Glu mutation in the mammalian Alanyl-tRNA synthetase (AlaRS) significantly increases the frequency of AlaRS charging of tRNAAla with serine or glycine (39). This high level of decreased translational fidelity is not lethal for homozygous mice bearing this genetic mutation, however. The known significant damage of these mice occurs in cerebellar Purkinje cells in the brain which is linked in some way to the accumulation of protein aggregates in these cells. This result also means though that a similar level of synthesis of mutant proteins does not induce comparable damage in many tissues and cell types. In E. coli, the expression of editing-deficient AspRS leads to significant generation of Asn-mischarged tRNAAsp which is then used in translation to generate Asp-to-Asn mutant proteins (62). Examining a reporter gene that is not under selection pressure shows that up to 10% of this substitution is tolerated in actively growing E. coli. In S. cerevisiae and E. coli, the expression of an editing-deficient PheRS leads to significant generation of Tyr-mischarged tRNAPhe (60). However, substitution of the wild-type with the inaccurate PheRS in E. coli or in the cytosol of S. cerevisiae has little effect on growth in rich media. The inaccurate PheRS cannot replace the wild-type PheRS in yeast mitochondria, indicating that different cellular compartment has distinct tolerance to translation accuracy.
The Candida branch of yeast contains a tRNA that is charged with serine but reads the CUG codon which codes for leucine in all other organisms (49, 64). Accordingly, all Candida species containing this tRNASer(CAG) gene have reprogrammed their genomes where CUG codes for serine. In Candida albicans, this tRNASer(CAG) is charged with serine at 95-97% and with leucine at 3-5%, and this charging heterogeneity is reflected by their incorporation into the Candida proteome (19). Interestingly, the tRNASer(CAG) from C. albicans can be expressed in a different yeast, Saccharomyces cerevisiae without causing lethality; it even benefits S. cerevisiae in responding to stress (63). In S. cerevisiae, tRNASer(CAG) is a de facto mutator-tRNA, it is still charged with Ser but competes with the endogenous tRNALeu(CAG) for decoding all CUG codons, resulting in the production of Leu-to-Ser mutant proteins. Yeast cells with low-fidelity proteomes do not fare as well as the wild-type cells under steady-state growth conditions. However, these cells do markedly better than the wild-type cells when challenged with chemical stressors. Making a lower fidelity proteome under steady-state growth elevates the production of stress response proteins such as hsp-protein chaperones before these cells are challenged with a stressor, thereby increasing their tolerance to stress through induction of cross-protection mechanisms.
These results indicate that cells can not only produce mutant proteins beyond the commonly cited threshold of translational fidelity, but also tolerate the persistent presence of such proteins. In the case of tRNASer(CAG) in S. cerevisiae, the expression of this foreign tRNA even confers a benefit in stress response. Two questions immediately come to mind: Can cells naturally regulate translation of mutant proteins and also benefit from this process? And if yes, how? Affirmative answers to these questions would indicate that regulated synthesis of mutant proteins can serve a heretofore much under-appreciated role in cellular stress response and adaptation. This review will discuss known cases that support the hypothesis of regulated translation of mutant proteins serving beneficial functions and provide my personal perspectives on future challenges of the field.
How cells regulate translation of mutant proteins: tRNA aminoacylation
Translation fidelity can be changed in many ways through errors in tRNA aminoacylation or ribosome codon-anticodon matching, occurrences in ribosome frameshifting or readthrough, and cellular variations of protein factors involved in translation (Fig. 1B, 1C).
There are two major ways to decrease fidelity in tRNA aminoacylation by AARS. The first involves the attachment of non-cognate amino acid to cognate tRNA. This type of error arises because the chemical structures of some amino acids are similar and differ by as little as a methyl group. One extensively studied example of this type is the mischarging of Val to tRNAIle by IleRS (22, 25, 53). The amino acids Ile and Val differ by a single methyl-group. The smaller Val can fit in the active site of IleRS and is mischarged at ∼1% level as Ile. More than 99% of the mischarged Val-tRNAIle is subsequently eliminated by the IleRS because only the smaller Val can fit in the active site of IleRS's editing domain. The editing efficiency of AARS in vivo can vary in several ways. Several AARSs in Mycoplasma parasites have natural mutations in their editing domains resulting in the inactivation of their editing activities (42). Proteomic mass spectrometry shows that the cellular proteomes in these bacterial parasites contain numerous mutations due to tRNA mischarging derived from these editing-defective AARSs. The generation of statistical proteomes in these bacteria may have a useful purpose that remains to be determined. The E. coli ThrRS has a conserved Cys-residue in its editing domain. This Cys-residue is sensitive to oxidation in vitro upon exposure to H2O2 and in vivo upon increasing intracellular concentration of reactive oxygen species (44). The Cys-oxidized ThrRS is defective in editing, thus generating a significant amount of serine mischarged tRNAThr which is used in translation in cells. Hence, the editing activity of ThrRS can be regulated during oxidative stress. Increased mischarging of tRNAThr results in Ser mis-incorporation, thereby producing misfolded proteins or generating potential new sites of phosphorylation. Such mutant proteins may be used in signaling pathways (44). Presumably, the wild-type ThrRS editing activity can be restored once the cells are no longer under oxidative stress.
The second way to decrease tRNA charging fidelity involves the attachment of cognate amino acid to non-cognate tRNA. This type of error arises because in cells, cognate tRNAs for each AARS are present together with all non-cognate tRNAs for the 19 other AARSs. Although the cognate AARS recognizes the identity element for their cognate tRNAs for positive identification, it must also avoid aminoacylation of non-cognate tRNAs. It was recognized in 1988 that individual tRNA concentrations and their cognate synthetases need to be properly balanced to avoid mischarging in vivo (71). In E. coli, overexpression of GlnRS leads to increased mischarging of a non-cognate tRNA with Gln; this mischarging is suppressed upon overexpression of tRNAGln at the same time. The Candida albicans tRNASer(CAG) reads CUG codons; it is mostly charged with serine, but also with 3-5% leucine in vivo depending on the cellular conditions (19). Since both Ser-tRNASer(CAG) and Leu-tRNASer(CAG) are used in translation, the proteome of C. albicans contains many proteins with either Ser or Leu at all CUG-codons. Expression of a foreign tRNALeu(CAG) in C. albicans results in up to 28% CUGmiscoding, and yet, C. albicans growth measured in the laboratory is not significantly impaired. Increasing CUG-miscoding using the foreign tRNALeu(CAG) leads to significantly larger diversity in colony morphologies, indicating that a more diverse proteome generated through ambiguous decoding can lead to phenotypic diversity (47). The C. elegans genome contains a tRNAGly and a tRNAIle gene with an additional stem-loop in the variable arm (7). tRNAs with a stem-loop in the variable arm (type II tRNA) are generally restricted to tRNALeu and tRNASer in eukaryotes, and all other tRNAGly and tRNAIle genes in C. elegans have a standard, short variable loop. The type II-tRNAGly and tRNAIle are expressed in C. elegans; but in vitro, they are charged with Leu and decode the corresponding Gly and Ile codons in translation (24). Thus, C. elegans and a few other worm species seem to contain mutator-tRNAs that read codons in different ways as dictated by the genetic code.
A recent discovery shows that mammalian cells contain many non-methionyl-tRNAs mischarged with methionine at levels that are highly regulated ((52); Fig. 2). The baseline misacylation for methionine in unstressed cells, i.e. the fraction of Met attached to nonmethionyl tRNA, is ∼1%, or at least 100-fold higher than expected for commonly cited fidelity for AARSs (<0.01% or 10-4). Remarkably, following infection of cells with several drastically different viruses, Met misacylation rises to up to ∼10%. Active virus infection is not required for this effect, but rather, misacylation is induced by Toll-like receptor (TLR) ligands, including inactivated virions, poly-IC, CpG oligonucleotides, and lipopolysaccharides. Induced misacylation above basal levels can also be triggered by exposure to oxidative chemicals such as H2O2 or arsenite. Induction of misacylation is inhibited in the presence of a small molecule inhibitor for a broad family of human NOX proteins which are NADPH oxidases capable of rapid production of reactive oxygen species (ROS). These results indicate that ROS is the cellular trigger of tRNA misacylation with Met. Met misacylation is selective for certain tRNA families and does not correlate with their abundance in cells. tRNA misacylation seems to be restricted to Met, as no misacylation was detected for amino acids Cys, Ile, Phe, Tyr or Val. The Met-misacylated tRNAs are used in translation as analyzed by global charged tRNA turnover kinetics, 1D and 2D gel electrophoresis and mass spectrometry of proteins.
Fig. 2. tRNA misacylation with Methionine.
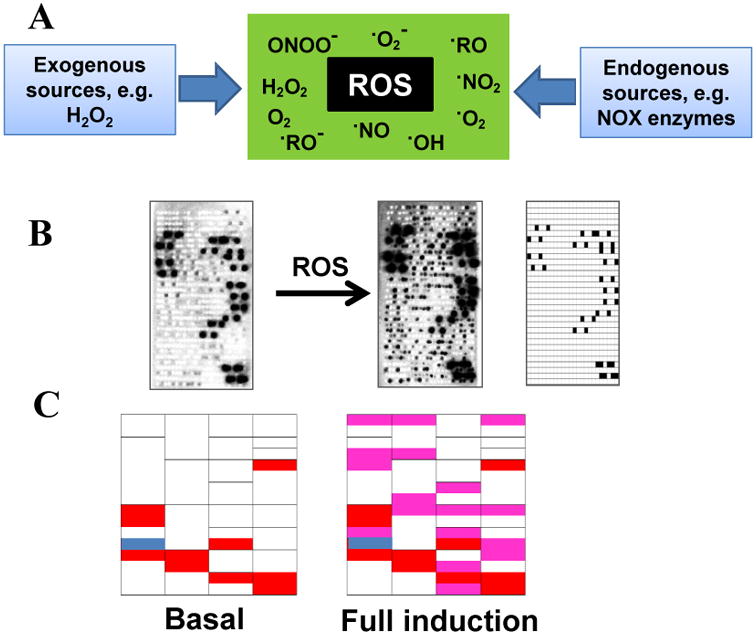
(A) tRNA misacylation with Met in mammalian cells is regulated by reactive oxygen species (ROS) which can be generated from both exogenous and endogenous sources. (B) HeLa cells exhibit a basal level of misacylation which increases further by ∼10-fold upon full induction. tRNA misacylation is detected by microarrays of total charged tRNA from cells pulse-labeled with 35S-Met (52). Array probes for tRNAMets are shown as black dots in the array layout. (C) In the codon tables, codons read by misacylated tRNAs under uninduced conditions are shown in red and under induced conditions are in red and pink. Met codon is in blue.
Additional studies show that the Met-misacylation is derived from the activity of methionyl-tRNA synthetase (MetRS). Recombinant E. coli MetRS misacylates two E. coli tRNA species in vitro (31). Further, E. coli MetRS mutants have been found that either increase or decrease the level of misacylation in vitro, suggesting misacylation by MetRS is a tunable activity of this enzyme. Met misacylation is also present in S. cerevisiae, and the recombinant yeast tRNA synthetase complex made of one subunit each of MetRS, GluRS and Arc1p protein misacylates numerous yeast tRNA species in vitro (75).
Are all misacylated tRNAs used in translation in vivo? A major mechanism known to selectively deliver correctly charged tRNAs into the A-site of ribosome is their selective binding by the bacterial elongation factor EF-Tu, also known as the EF-Tu filter (9, 15, 38). EF-Tu has separate binding affinities for the tRNA body and the 3′ attached amino acid. The free energy of both binding affinities adds up equally for correctly charged tRNAs, whereas incorrectly charged tRNA can bind too strongly, too weakly or similarly to correctly charged tRNAs. In vitro, the E. coli EF-Tu does not deliver strongly or weakly bound tRNAs to the ribosome. The extent of this discrimination remains to be determined in vivo where some mischarged tRNAs are shown to be used for protein synthesis at appreciable frequencies as analyzed by reporter gene activities or mass spectrometry of reporter proteins. Whether the eukaryotic homologue of EF-Tu, EF-1α, discriminates mischarged tRNA is unclear. At least some misacylated tRNAs seem to be used for protein synthesis in yeast and in mammalian cells (e.g. (52, 75)).
How cells regulate translation of mutant proteins: ribosome decoding
Altering translational fidelity on the ribosome can occur in several ways. One way is mismatching an mRNA codon with a tRNA anticodon, causing an amino acid substitution. Codon-anticodon mismatch can result through the imbalance of available, charged cognate tRNAs either through improper balance of tRNA abundance or selectively decreased tRNA charging levels in specific tRNAs. For example, charging levels of certain tRNAs can drop dramatically during amino acid starvation of the cell (12, 68, 78). The decreased availability of cognate, charged tRNAs increases the frequency of A-site entry of near-cognate tRNAs whose charging is not perturbed under these conditions.
Alterations in translational fidelity can also depend on the extent of specific tRNA modifications. Oxidative and methylation stresses in yeast induce highly dynamic variations of tRNA modification (5). For example, 5-methyl-C (m5C) modification in total yeast tRNA can increase or decrease depending on the type and severity of stress. This dynamic variation is biologically important as the m5C-modification enzyme knockout yeast strain is less tolerant to certain stresses. The m5C34 wobble modification in tRNALeu(CAA) selectively enhances translation of the UUG codons which are over-represented in numerous yeast proteins that are translated in response to oxidative stress (6). Another dynamic yeast modification involves methylation of the hypermodified U34 wobble in tRNAArg(UCU) and tRNAGlu(UUC) (2). This modification is inducible upon DNA damage and is necessary to maintain translation fidelity at codons read by the hypermodified tRNAArg(UCU) (57).
Hypomodification in specific tRNAs can also increase ribosomal frameshifting and stop codon readthrough. A well-characterized example is the N1-methyl-G37 (m1G) modification in several bacterial tRNAs which occurs at the immediate 3′ residue to the anticodon (3, 4). m1G37 modification reduces frameshifting frequencies for tRNAs that read codons with C in their first position (e.g. tRNAPro), because this modification prevents incorrect pairing with a C in the third position of the preceding codon. m1G modification is essential in bacteria; its complete loss is lethal to the cell.
How cells regulate translation of mutant proteins: mRNA and translation factors
The presence of certain mRNA structures or alteration of specific translation factors can also increase the frequency of ribosomal frameshifting and stop codon readthrough. Ribosome frameshifting or stop codon readthough is essential for the life cycle of mammalian retroviruses (16, 18). The structural genes of retroviruses (gag proteins) are linked with the catalytic genes (pol proteins) in a single mRNA transcript. To balance the amount of gag versus pol proteins, the pol proteins are produced at frequencies of ∼5-10% to gag proteins through ribosome frameshifting or stop codon readthrough as directed by specific mRNA structures in the viral mRNA.
Another way of altering translational fidelity is by reducing the amount of available protein factors through protein aggregation such as prion formation (23, 66, 72, 73). In yeast, prion formation is an inducible process where specific, soluble proteins are incorporated into aggregated states. One prion protein is Sup35 which is a release factor required for translational termination. Under specific conditions the N-terminal domain of Sup35 allows this protein to switch into an alternative conformation, leading to an aggregated prion-like state. Sup35 prion formation reduces the amount of available release factors which leads to stop codon readthrough at numerous genes. This process can be beneficial in yeast response to certain stresses, presumably because some of the new protein C-terminal extensions could be specifically useful in stress response. Another yeast prion protein is the tRNA N6- isopentenyl-A37 (i6A37) modification enzyme (70). i6A37 may control translation fidelity of decoding and the frequency of frameshifting using a mechanism similar to the m1G37 modification in bacterial tRNAs.
How cells regulate translation of mutant proteins: to be determined
Significant, dynamic fluctuations in the frequency of frameshifting and stop codon readthrough have been observed in the bacterium Bacillus subtilis using a GFP reporter and an antibiotic resistance gene (46). The frequency of frameshifting and stop codon readthrough depends on cellular conditions such as growth temperature, cell density or the presence of antibiotics. Although the mechanism of regulating translational fidelity in this case remains to be determined, it may be derived from dynamic variations in specific tRNA modifications, the availability of active release factors or something else.
Mistranslation dependent protein aggregation is a major cause of cell toxicity upon chromium stress (26). These protein aggregates are derived from newly synthesized proteins during chromium exposure. For example, the frequency of stop codon readthrough significantly increases, although this increase only occurs under aerobic conditions. The mechanism for altering translational fidelity upon chromium exposure remains to be elucidated; it may be derived from altered activity of release factors or dynamic variations in tRNA modifications.
Significant, genome-wide changes in frameshifting and stop codon readthrough have also been observed in yeast upon oxidative stress using the ribosome profiling technique (17). Ribosome profiling is a recently developed high-throughput method that determines the actively translating mRNAs at single codon resolution (28, 29). Frameshifts can be identified when ribosome density shifts its periodicity from the annotated coding sequences and/or drops off at an alternate stop codon. Stop codon readthrough can be identified when significant ribosome density is present past the annotated stop codon and drops off at a downstream stop codon. Although the mechanism of regulating translational fidelity in oxidative stress in yeast remains to be determined, it may again be derived from dynamic variations in specific tRNA modifications, the availability of active release factors or something else.
A potential mechanism in regulating the synthesis of mutant proteins involves mRNA modification with pseudo-uridine (Ψ, (33)). The first nucleotide of all three stop codons is Uridine. Modification of U to Ψ in all three stop codons leads to high levels of stop codon readthrough, and each Ψ-modified stop codon specifically codes for two amino acids. Computational analysis suggests that Ψ-modified sense codons can be recoded; for example, the standard UUU codes for Phe, but ΨUU may be read as Cys/Tyr by the ribosome (55). Ψ is a major modification in tRNA, rRNA and spliceosomal RNA (8). Ψ may also occur in mRNA, although the location and the frequency of Ψ modification in mRNA remain to be determined.
How cells benefit from making their own mutant proteins
Proteins produced through the mechanisms described above generally are not uniform molecules. They are either protein families that share a large degree of similarity in case of mutations, or groups of diverse protein species that share common domains in case of frameshift and stop codon readthrough. A common assumption for proteins generated through strictly obeying the genetic code is that each of these protein molecules is intended to perform useful function for the cell. In contrast, it is highly unlikely that every mutant protein produced by the cell is functional; in fact, some mutant proteins can be harmful to the cell either by nonspecific mechanisms such as aggregate formation, or possibly by dominant-negative functions. Nevertheless, since cells can produce appreciable amounts of mutant proteins in a regulated manner, can some of these mutant proteins serve beneficial functions for the cell? Several distinct fates for the mutant proteins can be envisioned, ranging from harmful to beneficial (Fig. 3):
Neutral: many mutant proteins fold correctly and are treated as regular proteins in cells. For example, tRNA misacylation with Met results in the generation of protein mutants that contain at most one Met substitution of non-Met residues in an average sized protein (52). Among others, these mutant proteins substitute a charged amino acid on the surface (e.g. Lys, Asp, Glu), or an internal hydrophobic amino acid (e.g. Leu, Val) with Met. Since the chemical property of the Met side chain is both hydrophobic and polar, many Met-substituted proteins likely fold like the wild-type protein.
Harmful through aggregation: mutant proteins with amino acid substitution of distinct chemical properties can lead to conformational change and aggregation. For example, AlaRS mutant that loses its editing activity misacylates tRNAAla with Ser, resulting in substituting Ala, a small hydrophobic amino acid with Ser, a polar amino acid. Over time, some of these mutant proteins accumulate in aggregates which are linked to damage of specific neurons (39).
Unfolded protein response: mutations leading to misfolded proteins are one of many mechanisms that trigger cellular response to unfolded proteins. This is a commonly discussed benefit of deliberate or increased production of mutant proteins (13, 21, 50, 59, 65). Since the mutant proteins from one single mRNA are not confined to one single sequence, there is a good chance that some of these proteins would misfold and thereby enhance the cellular unfolded protein response.
Protein fragments as ligands: A possible benefit of making mutant proteins is that some mutants misfold and can therefore be rapidly recognized by the cellular protein degradation machinery which degrades the full-length protein to generate protein fragments. For example, in mammalian cells rapid degradation of newly synthesized, misfolded proteins by the proteosome can immediately produce peptides that are loaded onto Major Histocompatibility Complexes (MHC) for antigen presentation. This deficient ribosome product (DRIP) mechanism enables constant survey of cellular translation status by immune cells (77). It is also conceivable that some protein fragments produced through degradation immediately upon synthesis could become ligands for other cellular components in e.g. signal transduction.
Mutant proteins that benefit directly: A few specific mutant protein molecules could directly benefit the cells because of the actual sequence of mutation. For example, the yeast prion state has substantially increased frequency of frameshift and readthrough. The prion state enhances the synthesis of antizyme by modulating frameshift required for its production (51). Antizyme is a negative regulator of polyamine biosynthesis, and the prion-induced translational control of antizyme significantly affects the cellular content of polyamines.
Fig. 3. Mutant proteins affect cells in many ways.

Perhaps the least explored and least understood of these is when mutant proteins benefit cells directly because of their specific sequences. This process is defined as adaptive translation in this review because these specific proteins could help cells adapt to stresses or new environments.
We have proposed a beneficial hypothesis for Met-substituted mutant proteins directly benefiting cells in response to oxidative stress (Fig. 4). This hypothesis is based on the unique property of Met which can be oxidized by ROS, but also reduced by peptidyl-Met sulfoxide reductases in cells (Fig. 4A, (41, 45, 48)). Many enzymes contain active site residues such as Trp, His or Cys that can be oxidized by ROS. ROS diffuses like water and can oxidize an active site residue resulting in permanent inactivation of the enzyme. To avoid this from occurring often, some proteins contain genetically encoded, strategically located Met residues that readily react with ROS before it gets into the active site (Fig. 4B). This process is reversible since the oxidized Met residues can be reduced by the MSRa/b proteins.
Fig. 4. A functional hypothesis on Met-substituted mutants being useful in oxidative stress response.
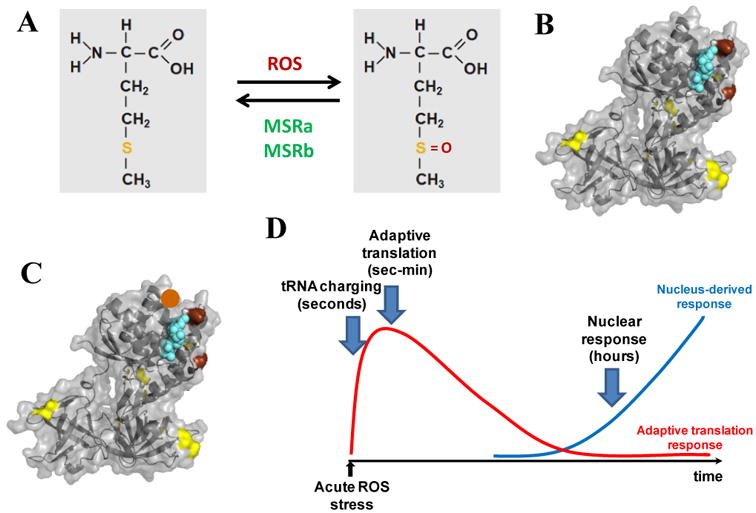
(A) Met-residues in proteins can undergo reversible oxidation by ROS and reduction by MSRa/b enzymes. (B) The E. coli EF-Tu (PDB 1efc) has two strategically positioned Met-residues (shown in brown) that protect the active site (bound GDP shown in cyan) from ROS oxidation. Other Met-residues in EF-Tu are shown in yellow. (C) A member in the Met-substituted protein library can get an extra Met-residue located at the right place (e.g. the position indicated by the brown ball) that offers extra protection. This particular mutant molecule should be more resistant against ROS inactivation. (D) tRNA misacylation-based translation may help buy time before the full engagement of the nuclear response.
For each protein coding gene, high fidelity translation produces identical protein molecules; in contrast, the use of Met-misacylated tRNAs in translation produces a library of protein molecules. Individual molecules in this library are highly similar to each other and most can remain biologically active, but they differ in having on average one extra Met residue distributed throughout the protein molecule (52). Within this library, some mutant molecules could be better protected against oxidation than the wild-type molecule, because the extra Met residue in these mutants can be strategically localized to augment protection. For example, this extra Met residue may be placed on the opposite side of the active site groove relative to the genetically encoded Met residues (Fig. 4C). In this way, cells can ensure that some molecules derived from the same gene needed to respond to a wide and distinct range of oxidative stresses are more resistant to oxidative inactivation. By enabling some individual protein molecules to remain active under all conditions, this protein library approach produced at low translational fidelity may enable better cellular adaptation to stress compared to the uniform protein approach produced at high translational fidelity.
Another important issue in stress response is time (Fig. 4D). When response is present at time zero, cells activate signal transduction pathways that send signals to the nucleus in order to synthesize new mRNAs and then new proteins to respond to stress. This nuclear-response to stress works very well; but it generally takes some time for these stress response proteins to take over. In mammalian cells, the nuclear response takes on the order of hours to be fully active, so cells must respond by other means first to buy time. The mechanism of tRNA misacylation with Met is well suited for this purpose. Charged tRNAs turn over on the second time scale in mammalian cells (30, 76). Thousands of human genes undergo translational regulation, so that many mRNAs are already present in the cytoplasm prior to the exposure of the stressor. These mRNAs can be translated within seconds or minutes to produce Met-substituted mutant protein libraries. Each library contains a few individual protein molecules with additional, strategically located Met-residues. These individual protein mutants are therefore better protected and should remain active longer than the wild-type proteins. These mutants need only remain active long enough for the nuclear response to arrive.
To quickly regulate tRNA misacylation with Met, we hypothesize that the cellular MetRS exists in at least two forms that differ in post-translational modifications. The hypomodified form aminoacylates at high fidelity and this form dominates in unstressed cells. Upon oxidative stress, some MetRS molecules become modified at specific residues, and the modified form aminoacylates at low fidelity. An important aspect of this hypothesis is reversible regulation: the modification that generates the low fidelity form of MetRS can be reversed when the stress has been dealt with. Potentially reversible modifications include phosphorylation/dephosphorylation carried out by protein kinases/phosphatases, or Cys/Met oxidation/reduction. Since the low fidelity form is supposed to increase upon elevated ROS levels which can directly oxidize Cys or Met residues, no enzymes may be needed to generate the low fidelity form of MetRS. The reduction of Cys can be carried out by cellular small molecule reductants or thioredoxin proteins, and the reduction of Met can be carried out by the MSRa/b proteins.
Future perspectives and challenges
Translational errors have been generally categorized as mistranslation in the past. The term “mistranslation” however implies that translational errors are solely mistakes to be avoided. As described here and in previous reviews (14, 50, 59, 77), sufficient evidence exists that point to useful applications for some of the mistranslated proteins. I tentatively use the term “adaptive translation” to describe mistranslation events that generate mutant proteins that play functional roles because of their specific mutated sequences (Fig. 3). Adaptive translation seems to be prevalent and inducible under stress, suggesting that a primary function of adaptive translation is for cellular stress response.
Once an adaptive translation process has been identified, several questions need to be addressed to fully evaluate the functional significance and the regulatory mechanisms. Using the tRNA misacylation with Met as an example, I describe below four questions that represent major challenges for the field.
-
What is the regulatory mechanism? tRNA misacylation with Met is carried out by methionyl-tRNA synthetases. However, cellular conditions have been found where the extent of misacylation can be anywhere between 10% to below detection by the microarray method, <0.025% (unpublished results), indicating that the fidelity of MetRS in cells is highly regulatable. One model of this regulation is through post-translational modification of MetRS. The mammalian MetRS is known to be phosphorylated and Cys-oxidized (37, 40, 58), any one of these modifications may alter the tRNA charging fidelity of this enzyme. Another model is through association/dissociation of MetRS with other cellular components. The mammalian MetRS is a subunit of an eleven protein complex, but it can also dissociate from this complex in cells (34, 37). This 11-protein complex is also known to associate with polysomes (10, 32, 69), and this polysome association is dependent on active translation of the mRNA in cells (10).
Identification of the regulatory mechanism will enable the construction of MetRS mutants that still perform its essential, housekeeping function of charging tRNAMet, but can no longer misacylate non-Met-tRNAs. These MetRS mutants will be crucial in the studies of phenotypic responses that benefit the organism.
What is the fitness benefit? Once a mutant MetRS is identified that cannot misacylate tRNAs in response to stress, bacterial or yeast strains or mammalian cell lines can be constructed where this mutant replaces the wild-type MetRS gene. These strains will allow for testing the physiological response of tRNA misacylation with Met. For example, our functional hypothesis (Fig. 4C) predicts that cells that can no longer misacylate tRNA should have a longer delay in early response to and will take longer to recover from oxidative stress.
What is the selectivity of the adaptive-translated proteome? Although we expect that many misacylated tRNA with Met are used by the ribosome, not all misacylated tRNAs will be used equally as the correctly charged tRNAs in protein synthesis. Some misacylated tRNAs with Met may not be efficiently delivered to the A-site by the elongator factor. Most studies on translational errors on the ribosome so far investigated ribosome proofreading upon codonanticodon mismatch. Misacylated tRNAs however can form perfect matches in codon-anticodon pairing. The ribosome may reject certain misacylated tRNAs because the amino acid and tRNA mismatch may not allow for efficient peptide bond formation, although how extensive this occurs remains to be determined. Understanding the ribosome's usage of misacylated tRNAs will lead to new decoding rules for protein synthesis that do not strictly adhere to the genetic codes.
What are the specific protein molecules that use adaptive translation to confer benefit?Adaptive translation using misacylated tRNAs with Met can generate protein libraries from many genes. Clearly, not all protein molecules in these libraries are useful for the cell. A functional hierarchy likely exists where protein libraries from a few genes confer a great deal of benefit, whereas libraries from many genes are neutral in functional significance. Furthermore, only a fraction of all members within each beneficial library would be useful for stress response. These particular members may have the extra Met-residue located at strategic positions to enhance their survivability under oxidative stress. Hence, the actual sequences of these mutant molecules matter. Identification of the functionally beneficial libraries and the specific mutant sequences within these libraries that truly matter will be necessary to understand the significance and the physiological response derived from adaptive translation.
Concluding remarks
For a long time, translational errors have been commonly thought to be avoided at all times. Recent results indicate that cells actively regulate translational errors, potentially for beneficial purposes such as in the response to stress. Among the two other processes in the central dogma of molecular biology, decreasing the fidelity of replication and transcription has already been shown to be beneficial in certain circumstances. For example, somatic hypermutation reduces the fidelity of DNA replication by over 1,000-fold and enables B-cells to generate diverse libraries of receptors and antibodies (11, 20). The ∼100-fold lower fidelity of retroviral reverse transcriptase enables the generation of diverse population of retroviruses, some of which can better resist cellular and pharmacological attacks (1, 67). It should not be surprising that cells could have also evolved to use regulated translational errors for stress response and adaptation (14, 50, 59, 77). I am confident that adaptive translation will be found to be more wide spread than currently known, and I eagerly anticipate that many more discoveries will be made in the near future.
Central issues.
Protein synthesis errors occur frequently, especially during stress.
Cells can tolerate a high level of protein synthesis errors than commonly assumed.
Cells actively regulate protein synthesis errors.
Mechanisms of altering protein synthesis errors include variations in tRNA misacylation, ribosome misdecoding, activity of release factors and others to be determined.
Some mistranslated protein molecules may benefit cells in stress response and in adaptation.
A potential benefit for mistranslated proteins is in response to oxidative stress, e.g. in response to changes in reactive oxygen species in cells.
Future issues.
There are four key questions to be addressed to fully establish the biology of adaptive translation.
What are the regulatory mechanisms?
What are the fitness benefits for the cell and for the organism?
What are the selectivities of the adaptive-translated proteome?
What are the specific protein molecules that use adaptive translation to confer benefit?
Acknowledgments
Research in this area in the Pan laboratory is supported by a NIH Director's Pioneer Award (DP1GM105386). I thank Drs. Allan Drummond, Jonathan Yewdell, Marc Parisien, Ana Cristina Gomes, Michael Schwartz and other members of my laboratory for stimulating discussions and insightful comments on the manuscript.
Abbreviations
- AARS
aminoacyl-tRNA synthetase
- MetRS
methionyl-tRNA synthetase
- ROS
reactive oxygen species
References
- 1.Angarano G, Monno L. Genotype and phenotype resistance: an overview. J Biol Regul Homeost Agents. 2000;14:11–4. [PubMed] [Google Scholar]
- 2.Begley U, Dyavaiah M, Patil A, Rooney JP, DiRenzo D, et al. Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol Cell. 2007;28:860–70. doi: 10.1016/j.molcel.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Björk GR. Biosynthesis and function of modified nucleosides. In: Söll D, RajBhandary U, editors. tRNA: structure, biosynthesis, and function. Washington, D.C.: ASM press; 1995. pp. 165–206. [Google Scholar]
- 4.Bjork GR, Wikstrom PM, Bystrom AS. Prevention of translational frameshifting by the modified nucleoside 1-methylguanosine. Science. 1989;244:986–9. doi: 10.1126/science.2471265. [DOI] [PubMed] [Google Scholar]
- 5.Chan CT, Dyavaiah M, DeMott MS, Taghizadeh K, Dedon PC, Begley TJ. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet. 2010;6:e1001247. doi: 10.1371/journal.pgen.1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan CT, Pang YL, Deng W, Babu IR, Dyavaiah M, et al. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat Commun. 2012;3:937. doi: 10.1038/ncomms1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan PP, Lowe TM. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009;37:D93–7. doi: 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Charette M, Gray MW. Pseudouridine in RNA: what, where, how, and why. IUBMB Life. 2000;49:341–51. doi: 10.1080/152165400410182. [DOI] [PubMed] [Google Scholar]
- 9.Dale T, Uhlenbeck OC. Amino acid specificity in translation. Trends Biochem Sci. 2005;30:659–65. doi: 10.1016/j.tibs.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 10.David A, Netzer N, Strader MB, Das SR, Chen CY, et al. RNA binding targets aminoacyltRNA synthetases to translating ribosomes. J Biol Chem. 2011;286:20688–700. doi: 10.1074/jbc.M110.209452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diaz M, Flajnik MF. Evolution of somatic hypermutation and gene conversion in adaptive immunity. Immunol Rev. 1998;162:13–24. doi: 10.1111/j.1600-065x.1998.tb01425.x. [DOI] [PubMed] [Google Scholar]
- 12.Dittmar KA, Sorensen MA, Elf J, Ehrenberg M, Pan T. Selective charging of tRNA isoacceptors induced by amino-acid starvation. EMBO Rep. 2005;6:151–7. doi: 10.1038/sj.embor.7400341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Drummond DA, Wilke CO. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell. 2008;134:341–52. doi: 10.1016/j.cell.2008.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drummond DA, Wilke CO. The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet. 2009;10:715–24. doi: 10.1038/nrg2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fahlman RP, Dale T, Uhlenbeck OC. Uniform binding of aminoacylated transfer RNAs to the ribosomal A and P sites. Mol Cell. 2004;16:799–805. doi: 10.1016/j.molcel.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 16.Farabaugh PJ. Programmed translational frameshifting. Annu Rev Genet. 1996;30:507–28. doi: 10.1146/annurev.genet.30.1.507. [DOI] [PubMed] [Google Scholar]
- 17.Gerashchenko MV, Lobanov AV, Gladyshev VN. Genome-wide ribosome profiling reveals complex translational regulation in response to oxidative stress. Proc Natl Acad Sci U S A. 2012;109:17394–9. doi: 10.1073/pnas.1120799109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goff SP. Genetic reprogramming by retroviruses: enhanced suppression of translational termination. Cell Cycle. 2004;3:123–5. [PubMed] [Google Scholar]
- 19.Gomes AC, Miranda I, Silva RM, Moura GR, Thomas B, et al. A genetic code alteration generates a proteome of high diversity in the human pathogen Candida albicans. Genome Biol. 2007;8:R206. doi: 10.1186/gb-2007-8-10-r206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu Rev Biochem. 2002;71:17–50. doi: 10.1146/annurev.biochem.71.083101.124707. [DOI] [PubMed] [Google Scholar]
- 21.Guo M, Schimmel P. Structural analyses clarify the complex control of mistranslation by tRNA synthetases. Curr Opin Struct Biol. 2011;22:119–26. doi: 10.1016/j.sbi.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hale SP, Auld DS, Schmidt E, Schimmel P. Discrete determinants in transfer RNA for editing and aminoacylation. Science. 1997;276:1250–2. doi: 10.1126/science.276.5316.1250. [DOI] [PubMed] [Google Scholar]
- 23.Halfmann R, Lindquist S. Epigenetics in the extreme: prions and the inheritance of environmentally acquired traits. Science. 2010;330:629–32. doi: 10.1126/science.1191081. [DOI] [PubMed] [Google Scholar]
- 24.Hamashima K, Fujishima K, Masuda T, Sugahara J, Tomita M, Kanai A. Nematode-specific tRNAs that decode an alternative genetic code for leucine. Nucleic Acids Res. 2012;40:3653–62. doi: 10.1093/nar/gkr1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hendrickson TL, Nomanbhoy TK, de Crecy-Lagard V, Fukai S, Nureki O, et al. Mutational separation of two pathways for editing by a class I tRNA synthetase. Mol Cell. 2002;9:353–62. doi: 10.1016/s1097-2765(02)00449-5. [DOI] [PubMed] [Google Scholar]
- 26.Holland S, Lodwig E, Sideri T, Reader T, Clarke I, et al. Application of the comprehensive set of heterozygous yeast deletion mutants to elucidate the molecular basis of cellular chromium toxicity. Genome Biol. 2007;8:R268. doi: 10.1186/gb-2007-8-12-r268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ibba M, Soll D. Quality control mechanisms during translation. Science. 1999;286:1893–7. doi: 10.1126/science.286.5446.1893. [DOI] [PubMed] [Google Scholar]
- 28.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–23. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jakubowski H, Goldman E. Quantities of individual aminoacyl-tRNA families and their turnover in Escherichia coli. J Bacteriol. 1984;158:769–76. doi: 10.1128/jb.158.3.769-776.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones TE, Alexander RW, Pan T. Misacylation of specific nonmethionyl tRNAs by a bacterial methionyl-tRNA synthetase. Proc Natl Acad Sci U S A. 2011;108:6933–8. doi: 10.1073/pnas.1019033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaminska M, Havrylenko S, Decottignies P, Le Marechal P, Negrutskii B, Mirande M. Dynamic Organization of Aminoacyl-tRNA Synthetase Complexes in the Cytoplasm of Human Cells. J Biol Chem. 2009;284:13746–54. doi: 10.1074/jbc.M900480200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karijolich J, Yu YT. Converting nonsense codons into sense codons by targeted pseudouridylation. Nature. 2011;474:395–8. doi: 10.1038/nature10165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ko YG, Kang YS, Kim EK, Park SG, Kim S. Nucleolar localization of human methionyltRNA synthetase and its role in ribosomal RNA synthesis. J Cell Biol. 2000;149:567–74. doi: 10.1083/jcb.149.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kramer EB, Farabaugh PJ. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA. 2007;13:87–96. doi: 10.1261/rna.294907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kramer EB, Vallabhaneni H, Mayer LM, Farabaugh PJ. A comprehensive analysis of translational missense errors in the yeast Saccharomyces cerevisiae. RNA. 2010;16:1797–808. doi: 10.1261/rna.2201210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwon NH, Kang T, Lee JY, Kim HH, Kim HR, et al. Dual role of methionyl-tRNA synthetase in the regulation of translation and tumor suppressor activity of aminoacyl-tRNA synthetase-interacting multifunctional protein-3. Proc Natl Acad Sci U S A. 2011;108:19635–40. doi: 10.1073/pnas.1103922108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.LaRiviere FJ, Wolfson AD, Uhlenbeck OC. Uniform binding of aminoacyl-tRNAs to elongation factor Tu by thermodynamic compensation. Science. 2001;294:165–8. doi: 10.1126/science.1064242. [DOI] [PubMed] [Google Scholar]
- 39.Lee JW, Beebe K, Nangle LA, Jang J, Longo-Guess CM, et al. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50–5. doi: 10.1038/nature05096. [DOI] [PubMed] [Google Scholar]
- 40.Leonard SE, Reddie KG, Carroll KS. Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem Biol. 2009;4:783–99. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]
- 41.Levine RL, Mosoni L, Berlett BS, Stadtman ER. Methionine residues as endogenous antioxidants in proteins. Proc Natl Acad Sci U S A. 1996;93:15036–40. doi: 10.1073/pnas.93.26.15036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L, Boniecki MT, Jaffe JD, Imai BS, Yau PM, et al. Naturally occurring aminoacyl-tRNA synthetases editing-domain mutations that cause mistranslation in Mycoplasma parasites. Proc Natl Acad Sci U S A. 2011;108:9378–83. doi: 10.1073/pnas.1016460108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ling J, Reynolds N, Ibba M. Aminoacyl-tRNA synthesis and translational quality control. Annu Rev Microbiol. 2009;63:61–78. doi: 10.1146/annurev.micro.091208.073210. [DOI] [PubMed] [Google Scholar]
- 44.Ling J, Soll D. Severe oxidative stress induces protein mistranslation through impairment of an aminoacyl-tRNA synthetase editing site. Proc Natl Acad Sci U S A. 2010;107:4028–33. doi: 10.1073/pnas.1000315107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo S, Levine RL. Methionine in proteins defends against oxidative stress. FASEB J. 2009;23:464–72. doi: 10.1096/fj.08-118414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meyerovich M, Mamou G, Ben-Yehuda S. Visualizing high error levels during gene expression in living bacterial cells. Proc Natl Acad Sci U S A. 2010;107:11543–8. doi: 10.1073/pnas.0912989107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miranda I, Rocha R, Santos MC, Mateus DD, Moura GR, et al. A genetic code alteration is a phenotype diversity generator in the human pathogen Candida albicans. PLoS One. 2007;2:e996. doi: 10.1371/journal.pone.0000996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moskovitz J. Methionine sulfoxide reductases: ubiquitous enzymes involved in antioxidant defense, protein regulation, and prevention of aging-associated diseases. Biochim Biophys Acta. 2005;1703:213–9. doi: 10.1016/j.bbapap.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 49.Moura G, Pinheiro M, Silva R, Miranda I, Afreixo V, et al. Comparative context analysis of codon pairs on an ORFeome scale. Genome Biol. 2005;6:R28. doi: 10.1186/gb-2005-6-3-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moura GR, Carreto LC, Santos MA. Genetic code ambiguity: an unexpected source of proteome innovation and phenotypic diversity. Curr Opin Microbiol. 2009;12:631–7. doi: 10.1016/j.mib.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 51.Namy O, Galopier A, Martini C, Matsufuji S, Fabret C, Rousset JP. Epigenetic control of polyamines by the prion [PSI+] Nat Cell Biol. 2008;10:1069–75. doi: 10.1038/ncb1766. [DOI] [PubMed] [Google Scholar]
- 52.Netzer N, Goodenbour JM, David A, Dittmar KA, Jones RB, et al. Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nature. 2009;462:522–6. doi: 10.1038/nature08576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nureki O, Vassylyev DG, Tateno M, Shimada A, Nakama T, et al. Enzyme structure with two catalytic sites for double-sieve selection of substrate. Science. 1998;280:578–82. doi: 10.1126/science.280.5363.578. [DOI] [PubMed] [Google Scholar]
- 54.O'Farrell PH. The suppression of defective translation by ppGpp and its role in the stringent response. Cell. 1978;14:545–57. doi: 10.1016/0092-8674(78)90241-6. [DOI] [PubMed] [Google Scholar]
- 55.Parisien M, Yi C, Pan T. Rationalization and prediction of selective decoding of pseudouridine-modified nonsense and sense codons. RNA. 2012;18:355–67. doi: 10.1261/rna.031351.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parker J, Johnston TC, Borgia PT. Mistranslation in cells infected with the bacteriophage MS2: direct evidence of Lys for Asn substitution. Mol Gen Genet. 1980;180:275–81. doi: 10.1007/BF00425839. [DOI] [PubMed] [Google Scholar]
- 57.Patil A, Chan CT, Dyavaiah M, Rooney JP, Dedon PC, Begley TJ. Translational infidelity-induced protein stress results from a deficiency in Trm9-catalyzed tRNA modifications. RNA Biol. 2012;9:990–1001. doi: 10.4161/rna.20531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pendergast AM, Venema RC, Traugh JA. Regulation of phosphorylation of aminoacyltRNA synthetases in the high molecular weight core complex in reticulocytes. J Biol Chem. 1987;262:5939–42. [PubMed] [Google Scholar]
- 59.Reynolds NM, Lazazzera BA, Ibba M. Cellular mechanisms that control mistranslation. Nat Rev Microbiol. 2010;8:849–56. doi: 10.1038/nrmicro2472. [DOI] [PubMed] [Google Scholar]
- 60.Reynolds NM, Ling J, Roy H, Banerjee R, Repasky SE, et al. Cell-specific differences in the requirements for translation quality control. Proc Natl Acad Sci U S A. 2010;107:4063–8. doi: 10.1073/pnas.0909640107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rodnina MV. Quality control of mRNA decoding on the bacterial ribosome. Adv Protein Chem Struct Biol. 2012;86:95–128. doi: 10.1016/B978-0-12-386497-0.00003-7. [DOI] [PubMed] [Google Scholar]
- 62.Ruan B, Palioura S, Sabina J, Marvin-Guy L, Kochhar S, et al. Quality control despite mistranslation caused by an ambiguous genetic code. Proc Natl Acad Sci U S A. 2008;105:16502–7. doi: 10.1073/pnas.0809179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Santos MA, Cheesman C, Costa V, Moradas-Ferreira P, Tuite MF. Selective advantages created by codon ambiguity allowed for the evolution of an alternative genetic code in Candida spp. Mol Microbiol. 1999;31:937–47. doi: 10.1046/j.1365-2958.1999.01233.x. [DOI] [PubMed] [Google Scholar]
- 64.Santos MA, Tuite MF. The CUG codon is decoded in vivo as serine and not leucine in Candida albicans. Nucleic Acids Res. 1995;23:1481–6. doi: 10.1093/nar/23.9.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schimmel P. Development of tRNA synthetases and connection to genetic code and disease. Protein Sci. 2008;17:1643–52. doi: 10.1110/ps.037242.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sideri TC, Stojanovski K, Tuite MF, Grant CM. Ribosome-associated peroxiredoxins suppress oxidative stress-induced de novo formation of the [PSI+] prion in yeast. Proc Natl Acad Sci U S A. 2010;107:6394–9. doi: 10.1073/pnas.1000347107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smyth RP, Davenport MP, Mak J. The origin of genetic diversity in HIV-1. Virus Res. 2012;169:415–29. doi: 10.1016/j.virusres.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 68.Sorensen MA. Charging levels of four tRNA species in Escherichia coli Rel(+) and Rel(-) strains during amino acid starvation: a simple model for the effect of ppGpp on translational accuracy. J Mol Biol. 2001;307:785–98. doi: 10.1006/jmbi.2001.4525. [DOI] [PubMed] [Google Scholar]
- 69.Stapulionis R, Deutscher MP. A channeled tRNA cycle during mammalian protein synthesis. Proc Natl Acad Sci U S A. 1995;92:7158–61. doi: 10.1073/pnas.92.16.7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Suzuki G, Shimazu N, Tanaka M. A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science. 2012;336:355–9. doi: 10.1126/science.1219491. [DOI] [PubMed] [Google Scholar]
- 71.Swanson R, Hoben P, Sumner-Smith M, Uemura H, Watson L, Soll D. Accuracy of in vivo aminoacylation requires proper balance of tRNA and aminoacyl-tRNA synthetase. Science. 1988;242:1548–51. doi: 10.1126/science.3144042. [DOI] [PubMed] [Google Scholar]
- 72.True HL, Berlin I, Lindquist SL. Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature. 2004;431:184–7. doi: 10.1038/nature02885. [DOI] [PubMed] [Google Scholar]
- 73.True HL, Lindquist SL. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature. 2000;407:477–83. doi: 10.1038/35035005. [DOI] [PubMed] [Google Scholar]
- 74.Wen D, Vecchi MM, Gu S, Su L, Dolnikova J, et al. Discovery and investigation of misincorporation of serine at asparagine positions in recombinant proteins expressed in Chinese hamster ovary cells. J Biol Chem. 2009;284:32686–94. doi: 10.1074/jbc.M109.059360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wiltrout E, Goodenbour JM, Frechin M, Pan T. Misacylation of tRNA with methionine in Saccharomyces cerevisiae. Nucleic Acids Res. 2012;40:10494–506. doi: 10.1093/nar/gks805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yewdell JW, Nicchitta CV. The DRiP hypothesis decennial: support, controversy, refinement and extension. Trends Immunol. 2006;27:368–73. doi: 10.1016/j.it.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 77.Yewdell JW, Reits E, Neefjes J. Making sense of mass destruction: quantitating MHC class I antigen presentation. Nat Rev Immunol. 2003;3:952–61. doi: 10.1038/nri1250. [DOI] [PubMed] [Google Scholar]
- 78.Zaborske JM, Narasimhan J, Jiang L, Wek SA, Dittmar KA, et al. Genome-wide analysis of tRNA charging and activation of the eIF2 kinase Gcn2p. J Biol Chem. 2009;284:25254–67. doi: 10.1074/jbc.M109.000877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zaher HS, Green R. Fidelity at the molecular level: lessons from protein synthesis. Cell. 2009;136:746–62. doi: 10.1016/j.cell.2009.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]