

Abstract
Tumor necrosis factor α (TNFα) is a well-known mediator of inflammation in the context of obesity in adipose tissue. Its action appears to be directly linked to perturbations of the insulin pathway, leading to the development of insulin resistance. Visfatin has been suspected to be linked to insulin sensitivity, but the mechanism involved is still partly unknown. The aim of this study was to evaluate the role of visfatin in the impairment of the insulin pathway by TNFα activity in 3T3-L1 adipocytes and to unveil the mechanisms involved in such impairment.
We demonstrated in 3T3-L1 adipocytes that visfatin was involved in TNFα-mediated insulin resistance in adipocytes. Indeed, after TNFα treatment in 3T3-L1 cells, visfatin was downregulated, leading to decreased nicotinamide adenine dinucleotide (NAD+) concentrations in cells. This decrease was followed by a decrease in Sirt1 activity, which was linked to an increase in PTP1B expression. The modulation of PTP1B by visfatin was likely responsible for the observed decreases in glucose uptake and Akt phosphorylation in 3T3-L1 adipocytes.
Here, we demonstrated a complete pathway involving visfatin, NAD+, Sirt1, and PTP1B that led to the perturbation of insulin signaling by TNFα in 3T3-L1 adipocytes.
Keywords: adipocytes, insulin resistance, TNF, visfatin, sirtuin, NAD
Introduction
Tumor necrosis factor α (TNFα) has been proposed as the link between obesity and insulin resistance.1,2 Indeed, obesity is characterized by a low-grade inflammatory state, leading to the modulation of adipokine, chemokines, and cytokine expression including an increase in TNFα secretion by adipose tissue.3 The role of TNFα in insulin resistance is supported by the fact that obese mice lacking TNFα or its receptors are protected from the induction of insulin resistance.4 Molecular mechanisms involved in TNFα-dependent insulin resistance have begun to be unveiled. These mechanisms involve long-term effects mediated via transcriptional regulation of master regulators of adipocyte differentiation such as peroxisome proliferator-activated receptor γ (PPARγ) and CAAT/enhancer binding protein α (C/EBPα) as well as regulation of the expression of adipokines such as adiponectin, leptin, and interleukin 6 (IL-6), which deeply impact insulin sensitivity.5
Short-term effects of TNFα on insulin resistance have also been described. These effects occur through the blockage of insulin signaling.1,2 Indeed, TNFα notably inhibits insulin-stimulated insulin receptor (IR) and insulin receptor substrate 1 (IRS-1) phosphorylation of tyrosine residues by blocking phosphorylation of IRS-1 serine 307, inducing SOCS proteins6 and activating protein-tyrosine phosphatase 1B (PTP1B).7
PTP1B is a negative regulator of insulin signaling.8 Its expression, which is strongly correlated with its activity, is directly linked to the inflammatory state.9 In muscle and hepatic cells,10 in vitro PTP1B overexpression decreased IR and IRS-1 tyrosine phosphorylation, and consequently decreased glucose uptake. In 3T3-L1 adipocytes,11 the effect of PTP1B on IR and IRS-1 tyrosine phosphorylation was reproduced, but the impact on glucose uptake was more debatable, as Venable et al. reported no effect on this parameter,11 whereas Shimizu et al. observed a small but significant effect on glucose uptake.12 PTP1B−/− mice presented enhanced insulin sensitivity, resistance to high-fat feeding-induced obesity and increased phosphorylation of IR and IRS-1 in the liver and muscle after insulin injection.13,14 Recently, it has been reported that insulin-stimulated phosphorylation of IR and AKT under a high fat diet condition, is impaired in mice with an adipocyte-specific PTP1B deletion.15 In addition, PTP1B has been demonstrated to be involved in TNFα-mediated insulin resistance.7 Moreover, it has been described that Sirt1 could improve insulin sensitivity by repressing PTP1B transcription in skeletal muscles.16
Sirt1 is the mammalian ortholog of the yeast protein Sir2, which is associated with longevity control.17-19 This protein has deacetylase activity on lysine residues of histones.17 The deacetylase activity of Sirt1 also impacts non-histone protein substrates such as transcription factors or nuclear receptors, including PPARγ coactivator 1 α (PGC1α), nuclear receptor corepressor (NCoR), liver X receptor α (LXRα), forkhead box members of the class O (FOXO), nuclear factor-κB (NFκB), and p53,17 which are transcriptional regulators linked to metabolism, inflammation and cell survival. Several lines of evidence support the beneficial role of Sirt1 activation in the treatment of type 2 diabetes,20-22 as various effects of Sirt1 and/or its agonists on glucose homeostasis and insulin sensitivity have been reported in different tissues such as pancreas, liver, skeletal muscle, and adipose tissue.20,23,24 The activity of Sirt1 is NAD+-dependent;25 thus, NAD biosynthesis can be regarded as a key regulator of Sirt1 activity.19
In mammals, nicotinamide phosphoribosyltransferase (NAMPT) is a key enzyme of NAD+ biosynthesis that is found in the intra- or extracellular compartment.26-28 The extracellular form is also known as visfatin or pre-B-cell colony-enhancing factor (PBEF). This protein has been reported as an insulin-mimetic hormone,29,30 but these data remain controversial.27,31
Here, we show that visfatin is involved in TNFα-mediated insulin resistance in 3T3-L1 adipocytes. Indeed, after TNFα treatment in 3T3-L1 cells, visfatin was downregulated, leading to decreased NAD+ concentrations within cells. This decrease was followed by decreased Sirt1 activity, which was linked to an increase in PTP1B expression. This modulation of PTP1B by visfatin was likely responsible for the observed decreases in glucose uptake and Akt phosphorylation in 3T3-L1 adipocytes.
Results
TNFα downregulated visfatin mRNA levels
First, we evaluated the impact of TNFα treatment on visfatin expression in 3T3-L1 cells. TNFα treatment resulted in downregulation of visfatin mRNA expression in a dose- and time-dependent manner (Fig. 1). No modification of the quantity of visfatin secreted in the culture medium was observed (data not shown).

Figure 1. Time- and dose-dependent effects of TNFα on visfatin mRNA levels in 3T3-L1 adipocytes. Cells were harvested after treatment with TNFα at 15 ng/mL for 3, 6, 10, and 24 h or at 5, 10, 15, and 20 ng/mL for 24 h. Quantification of visfatin mRNA levels by real-time RT-PCR. Visfatin data were normalized to 18S rRNA.
TNFα-mediated downregulation of visfatin was linked to C/EBPα in 3T3-L1 adipocytes
We next attempted to identify the molecular mechanism involved in the regulation of visfatin expression by TNFα. Interestingly, as previously reported,32,33 we observed that visfatin expression was increased during the differentiation of preadipocytes to adipocytes (data not shown). This finding suggested that visfatin expression could be regulated by master regulators of adipocytes differentiation, i.e., PPARγ or C/EBPα. It is already known that PPARγ does not regulate visfatin expression in adipocytes (refs. 34 and 35 and personal unpublished data), but the impact of C/EBPα has never been reported. Interestingly, the expression of this transcription factor was strongly inhibited by TNFα treatment in 3T3-L1 cells at mRNA and protein levels (Fig. 2A), suggesting that decreased expression of C/EBPα could lead to decreased visfatin expression. To confirm the contribution of the decrease in C/EBPα expression to the downregulation of visfatin expression, siRNA designed against C/EBPα was transfected into 3T3-L1 adipocytes. This resulted in decreased C/EBPα mRNA levels (Fig. 2B) as well as decreased visfatin mRNA levels (Fig. 2C), confirming that C/EBPα expression has an impact on visfatin expression.

Figure 2. Transcriptional regulation of visfatin in 3T3-L1 adipocytes. (A) 3T3-L1 cells were incubated with or without TNFα (15 ng/mL) for 24 h. TNFα-mediated effects on C/EBPα were assessed at the mRNA level by quantitative RT-PCR and at the protein level by western blotting. mRNA quantification of C/EBPα was normalized to 18S rRNA. Protein quantification of C/EBPα is represented with regard to the quantity of β-actin. (B and C) 3T3-L1 adipocyte lysates were prepared from cells transfected with a control (non-targeted) siRNA or siRNA against C/EBPα. Quantification of C/EBPα (B) and visfatin (C) mRNA levels by quantitative RT-PCR. mRNA data were normalized to 18S rRNA. Data are presented as means ± SEM. *P < 0.05 (t test).
Visfatin downregulation by TNFα reduced NAD+ concentrations and Sirt1 activity in 3T3-L1 adipocytes
Physiological consequences of visfatin downregulation were next evaluated. While TNFα treatment had no effect on the secreted quantity of visfatin (data not shown), it significantly reduced the intracellular quantity of visfatin in 3T3-L1 adipocytes (Fig. 3A). Because this protein is the key enzyme of the NAD+ salvage pathway, we measured the concentration of NAD+. As anticipated, the concentration of NAD+ was decreased in TNFα-treated adipocytes (Fig. 3B). We also measured Sirt1 activity because its activity is strongly dependent on NAD+. Using a fluorescence-based assay, we observed a decrease in Sirt1 activity in cells incubated with TNFα (Fig. 3C). This reduction in Sirt1 activity was independent of Sirt1 mRNA levels, which were not modified by TNFα incubation (Fig. 3D). Altogether, these data strongly suggested that the decreased visfatin expression in TNFα-treated 3T3-L1 adipocytes resulted in decreased Sirt1 activity due to the reduced NAD+ concentrations in cells.

Figure 3. Downregulation of visfatin by TNFα leads to decreases in NAD+ concentrations and Sirt1 deacetylating activity in 3T3-L1 adipocytes. Cells were incubated with or without TNFα (15 ng/mL) for 24 h. (A and B) Intracellular concentrations of visfatin and NAD+. After incubation, cells were collected and processed for visfatin and NAD+ quantification as described in Materials and Methods. Values were determined in ng visfatin/mg of cellular protein and in ng NAD+/mg of cellular protein, respectively. Values are presented as means ± SEM. *P < 0.05 (t test). (C) Sirt1 activity in 3T3-L1 cells. Total cell lysates (20 μg) were submitted to a Sirt1 activity assay as described in Materials and Methods. Values are presented as means ± SEM. *P < 0.05 (t test). (D) Quantification of Sirt1 mRNA levels by quantitative RT-PCR. Sirt1 data were normalized to 18S rRNA. Data are presented as means ± SEM. *P < 0.05 (t test).
TNFα and Sirt1 modulation regulated PTP1B expression in 3T3-L1 adipocytes
In parallel to the regulation of visfatin, we also studied the impact of TNFα treatment on PTP1B expression in 3T3-L1 cells. Under our conditions, mRNA levels of PTP1B were significantly upregulated (P > 0.05; Figure 4A). This effect of TNFα treatment on PTP1B mRNA expression was accompanied by an upregulation of PTP1B protein expression, according to a time-dependent fashion (Fig. 4B). The effect of Sirt1 activity on the modulation of PTP1B expression in 3T3-L1 adipocytes was also studied. To this aim, cells were treated with SRT 1720 (10 μM) for 24 h. The mRNA levels of PTP1B were quantified in these different conditions. SRT 1720, a Sirt1 activator, repressed the expression of PTP1B (Fig. 4C), suggesting a direct role of Sirt1 activity in regulating PTP1B expression.

Figure 4. Regulation of PTP1B expression by TNFα and a Sirt1 activator in 3T3-L1 adipocytes. Cells were harvested after treatment with TNFα at 15 ng/mL for 3, 6, 10, and 24 h or at 5, 10, 15, and 20 ng/mL for 24 h. (A) Quantification of PTP1B mRNA levels by real-time RT-PCR. PTP1B data were normalized to 18S rRNA. Data are presented as means ± SEM. Data were compared among groups (Student t test), and those with no common superscript letter are significantly different; P < 0.05. (B) Cells were incubated with TNFα at 15 ng/mL for 3, 6, 10, and 24 h. Total cell lysates (40 μg) were subjected to SDS-PAGE and immunoblotted with PTP1B or β-actin antibodies. The western blot is representative of three independent experiments. (C) Cells were treated with or without SRT 1720 (10 μM) for 24 h. PTP1B mRNA was quantified using real-time RT-PCR, and data were normalized to 18S rRNA. Data are presented as means ± SEM. *P < 0.05 (t test).
Visfatin inhibition led to a decrease in NAD+ concentrations and an increase in PTP1B expression
To establish the causative link between the regulation of visfatin and the expression of PTP1B, two strategies were used: one based on RNAi to decrease visfatin expression and the second based on the use of a chemical inhibitor called FK866.36 3T3-L1 cells were incubated with TNFα alone or together with FK866 at 1 or 10 nM. As reported in Figure 5A, TNFα incubation reduced NAD+ concentrations in cells. Cotreatment with TNFα and FK866 dose-dependently decreased the intracellular concentrations of NAD+ relative to TNFα treatment alone. This decrease in NAD+ levels was paralleled by an induction of PTP1B mRNA and protein levels (Fig. 5B and C). Similarly, siRNA designed against visfatin together with TNFα treatment significantly decreased NAD+ concentrations relative to TNFα treatment combined with non-targeted siRNA (Fig. 5D). This effect was associated with increased PTP1B mRNA and protein expression in the case of TNFα, which was exacerbated in presence of siRNA against visfatin (Fig. 5E and F). Together, these data suggested that visfatin inhibition via RNAi or chemical inhibition induced the expression of PTP1B.

Figure 5. Inhibition of visfatin decreases NAD+ concentrations and induces PTP1B expression in 3T3-L1 adipocytes. (A–C) Cells were incubated with or without TNFα (15 ng/mL) and in the presence of the visfatin inhibitor FK866 at 1 and 10 nM for 24 h. (A) After incubation, cells were collected and processed for NAD+ quantification as described in Materials and Methods. Values were determined in ng NAD+/mg of cellular proteins. (B) PTP1B mRNA levels were quantified using real-time RT-PCR, and data were normalized to 18S rRNA. Data are presented as means ± SEM. Data were compared among groups (Student t test), and those with no common superscript letter are significantly different; P < 0.05. (C) Total cell lysates (40 μg) were subjected to SDS-PAGE and immunoblotted with PTP1B or β-actin antibodies. The western blot is representative of three independent experiments. (D–F) Cells transfected with control (non-targeted) siRNA or siRNA against visfatin were incubated with or without TNFα (15 ng/mL) for 24 h. (D) 3T3-L1 cells were collected and processed for NAD+ quantification as described in Materials and Methods. Values were determined in ng NAD+/mg of cellular proteins. (E) PTP1B mRNA levels were quantified using real-time RT-PCR, and data were normalized to 18S rRNA. Data are presented as means ± SEM. Data were compared among groups (Student t test), and those with no common superscript letter are significantly different; P < 0.05. (F) Total cell lysates (40 μg) were subjected to SDS-PAGE and immunoblotted with PTP1B or β-actin antibodies. The western blot is representative of three independent experiments.
Visfatin inhibition led to decreased glucose uptake and Akt phosphorylation
To study the involvement of visfatin in TNFα-mediated effects on glucose metabolism, we measured 2-deoxyglucose uptake in 3T3-L1 adipocytes treated with TNFα alone or pretreated with FK866. TNFα treatment led to a 28% decrease in insulin-stimulated glucose transport compared with transport in control cells (Fig. 6A). Incubation with FK866 followed by TNFα treatment led to a 29% decrease in insulin-stimulated glucose uptake compared with transport after TNFα treatment alone. Together, these data suggested that visfatin inhibition reinforced the decrease in glucose uptake mediated by TNFα.
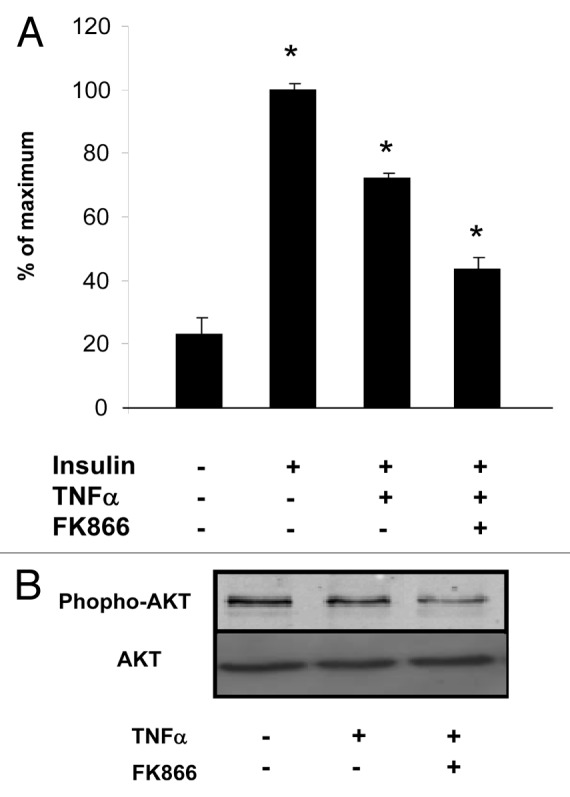
Figure 6. Glucose uptake is reduced by visfatin inhibition in 3T3-L1 adipocytes. (A) Adipocytes were incubated with or without TNFα (15 ng/mL) and in the presence of FK866 at 1 nM for 24 h. Cells were serum-starved for 1 h before a 30 min stimulation with insulin (0 and 170 nM). 2-deoxy-D-[3H]glucose uptake was measured as described in Materials and Methods. The uptake measurements were performed in triplicates and normalized to protein concentrations. Results (means ± SEM) are expressed as percentage of maximum uptake. (B) Akt phosphorylation is reduced by visfatin inhibition in differentiated 3T3-L1 cells. Adipocytes were incubated with or without TNFα (15 ng/mL) and in the presence of FK866 at 1 nM for 24 h. Total cell lysates (40 μg) were subjected to SDS-PAGE and immunoblotted with phospho-AKT or AKT antibodies. The western blot is representative of three independent experiments.
The impact on insulin signaling was assessed at the downstream level by evaluating the phosphorylation of Akt. Compared with that in control cells, TNFα treatment decreased Akt phosphorylation. Pretreatment with FK866 followed by TNFα treatment markedly impaired Akt phosphorylation (Fig. 6B).
Discussion
The perturbation of insulin signaling that notably occurs during obesity is a complex phenomenon implying several mechanisms and proteins. Among these factors, TNFα appears to be a master disruptor of insulin signaling. More recently, visfatin and sirtuin family members and phosphatases such as PTP1B have also been shown to play crucial roles, but the link between all these partners was still partly unknown.
In the present study, we showed that TNFα treatment resulted in downregulation of visfatin gene expression as well as its intracellular protein levels in 3T3-L1 adipocytes. This regulation of visfatin by TNFα has already been reported in mice.32,37 Surprisingly, some studies in humans reported an inverse correlation between visfatin and TNFα levels in plasma,38 although these data are still controversial.39 The origin of this species-specific regulation deserves further attention. In mice, the expression of visfatin after TNFα treatment has been quantified in adipose tissue, whereas in human studies, plasma correlations between visfatin and TNFα were reported. This could explain the discrepancy, as other tissues and/or cell types such as skeletal muscle, liver, bone marrow, and lymphocytes secrete visfatin.39-42 Our data suggest the involvement of C/EBPα in the regulation of visfatin by TNFα. This assumption was confirmed by RNAi experiments (Fig. 2B). However, in silico analysis of the mouse visfatin promoter did not suggest the localization of a C/EBPα responsive element (data not shown), suggesting that this regulation could be indirect. This assertion remains to be elucidated.
Going further, we showed that TNFα-mediated downregulation of visfatin in 3T3-L1 cells led to decreased intracellular NAD+ concentrations, as previously reported in other models,26,43,44 resulting in decreased Sirt1 activity because this enzyme is highly NAD+-dependent.25 It is noteworthy that inhibition of Sirt1 in adipocytes led to a decrease in insulin sensitivity.23 Indeed, knockdown of Sirt1 inhibited insulin-stimulated glucose transport in adipocytes in particular by inhibiting insulin signaling. Thus, due to decreased NAD+ concentrations and subsequently decreased Sirt1 activity, visfatin could be linked to insulin sensitivity.
In parallel, we also observed an induction of PTP1B (mRNA and protein), which is involved in TNFα-mediated insulin resistance in myocytes.7 This regulation has already been reported9 at the mRNA level after a short (4 h) incubation of 3T3-L1 adipocytes with TNFα and confirmed for a longer (17 to 36 h) incubation at the protein level. These authors reported a role of NFκB in this regulation. Interestingly, in our experiments, we noted a lag between TNFα-mediated visfatin and PTP1B expression. Three hours after incubation with TNFα, PTP1B, but not visfatin, was upregulated in 3T3-L1 cells. One hypothesis is that this lag may be explained by a sequential response to TNFα. Indeed, we can speculate that the regulation of PTP1B by TNFα occurs in two steps. In the first step, NFκB regulates the expression of PTP1B as reported by Zabolotny et al.,9 and in a second step, the regulation of PTP1B is achieved by the visfatin/NAD+/Sirt1 pathway, as suggested by our data. These assumptions will require additional experiments.
To establish a link between the decrease in Sirt1 activity and the increase in PTP1B expression, we used SRT 1720, a Sirt1 agonist, to demonstrate that Sirt1 activation led to downregulation of PTP1B expression. It is noteworthy that this result is fully in agreement with the study of Sun et al.,16 who demonstrated the regulation of PTP1B by Sirt1 and its consequences in term of insulin sensitivity in C2C12 cells. In contrast, Yoshizaki et al. did not reproduce this inverse correlation between Sirt1 and PTP1B in adipocytes.23 This discrepancy could be due to differences in term of incubation time (48 h incubation in the experiments by Yoshizaki et al.23 vs. 24 h in our conditions and in the experiments by Sun et al.16).
We next wanted to demonstrate a link between visfatin and PTP1B. Through two approaches (RNAi and chemical inhibition), we showed that decrease expression or activation of visfatin resulted in a decrease in intracellular NAD+ concentrations and an increase in PTP1B expression, strongly suggesting a role of visfatin in PTP1B expression via Sirt1 activity. To our knowledge, this is the first report that highlights the role of visfatin in the regulation of PTP1B. Finally, the impact of chemical inhibition of visfatin reinforced the mechanism of TNFα-mediated insulin resistance as measured by glucose uptake and Akt phosphorylation, suggesting that the decrease in visfatin activity, in addition to its downregulation (via TNFα treatment), is directly involved in TNFα-mediated insulin resistance.
Although the insulin-mimetic activity of visfatin is still highly controversial,27,31,45 the impact of visfatin on glucose uptake and metabolism appears more evident,29,30,43,46,47 notably via NAD+ production and the regulation of pancreatic β-cell function.27 Here, we confirmed that visfatin is involved in the control of glucose metabolism via NAD+, and for the first time, we identified a Sirt1/PTP1B pathway that mediated visfatin effects in mice adipocytes. In addition, our model is fully compatible with experiments that demonstrated an effect of visfatin on the phosphorylation of IR and IRS-1.30,47,48 Indeed, this effect could be due to PTP1B, which is known to modulate the phosphorylation level of these proteins.8,10,12 To the best of our knowledge, this assumption has never been asserted, but it reconciles the findings of most of the studies. In fact, when visfatin expression decreased in response to TNFα, PTP1B expression increased, and IR and IRS-1 were dephosphorylated, leading to decreased glucose uptake and Akt phosphorylation.
In summary, the current study establishes a link between TNFα, visfatin, NAD+, Sirt1, and PTP1B in adipocytes. We demonstrated that the decrease in C/EBPα induced by TNFα leads to visfatin inhibition, which participates in the TNFα-mediated perturbation of the insulin pathway and glucose uptake via an NAD+/Sirt1/PTP1B pathway. The implication for visfatin in this pathway brings new perspective concerning its role in adipocytes and more generally in cell metabolism.
Materials and Methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Invitrogen, and fetal bovine serum (FBS) was obtained from PAA Laboratories. Isobutylmethylxanthine, dexamethasone and insulin were purchased from Sigma-Aldrich. TRIzol reagent, random primers and Moloney murine leukemia virus reverse transcriptase were obtained from Invitrogen. SYBR Green reaction buffer was purchased from Eurogentec. Anti-C/EBPα antibody was from Santa-Cruz Biotechnology, Inc. Anti-β-actin antibody was from Sigma-Aldrich. Anti-PTP1B antibody, anti-AKT and anti-phospho-AKT(Ser473) antibodies were from Millipore SAS. Horseradish peroxidase-linked anti-rabbit or anti-mouse were from Thermo Fisher Scientific. Unless otherwise specified, all other reagents were purchased from Sigma-Aldrich.
Cell culture
3T3-L1 preadipocytes (ATCC) were seeded in 3.5-cm diameter dishes at a density of 15 × 104 cells/well. Cells were grown in DMEM supplemented with 10% FBS at 37 °C in a 5% CO2 humidified atmosphere as previously reported.49 To induce differentiation, two-day postconfluent 3T3-L1 preadipocytes (day 0) were stimulated for 48 h with 0.5 mmol/L isobutylmethylxanthine, 0.25 µmol/L dexamethasone, and 1 µg/mL insulin in DMEM supplemented with 10% FBS. The cultures were then continued with DMEM supplemented with 10% FBS and 1 µg/mL of insulin. All treatments were performed on day 8. The data are the mean of three independent experiments, each performed in triplicate.
RNA isolation and qPCR
Total cellular RNA was extracted from 3T3-L1 cells and mice epididymal fat pads using TRIzol reagent as previously reported.50,51 The cDNA was synthesized from 1 µg of total RNA in 20 µL using random primers and Moloney murine leukemia virus reverse transcriptase. Real-time quantitative RT-PCR analyses were performed using the Mx3005P Real-Time PCR System (Stratagene) as previously reported.52,53 The primers used were as follows: for visfatin, 5′-ACAACCCGGC CACATGAA-3′ and 5′-CAGAAAAAAT GCACAGCTGA ACA-3′; for PTP1B, 5′-ATGGAAGAAG CCCAGAGGAG-3′ and 5′-GTGCCCACAT GTGTTTGGTA-3′; for Sirt1, 5′-GCTTCATGAT GGCAAGTGG-3′ and 5′-TCGTGGAGAC ATTTTTAATC AGG-3′; for C/EBPα, 5′-AGCAACGAGT ACCGGGTACG-3′ and 5′-TGTTTGGCTT TATCTCGGCT C-3′; and for 18S, 5′-CGCCGCTAGA GGTGAAATTC T-3′ and 5′-CATTCTTGGC AAATGCTTTC G-3′. For each condition, expression was quantified in duplicate, and 18S mRNA was used as the endogenous control in the comparative cycle threshold (CT) method.
NAD+ quantification
Control cells and cells were pre-treated with FK 866 (dissolved in ethanol; Cayman Chemical) for 24 h at the final concentration of 1 and 10 nM, followed by an incubation with or without TNFα (15 ng/mL) for an additional 24 h. NAD+ quantification was performed using a colorimetric method according to the manufacturer’s instructions (NAD+/NADH Quantification Kit, BioVision).
Western blot
Differentiated adipocytes were treated with FK 866 (1 and 10 nM) or with siRNA against visfatin for 24 h and then incubated with TNFα (15 ng/mL) for an additional 24 h. Whole cell lysates were obtained using a lysis buffer (20 mM Tris, pH 7.4, 150 mM NaCl, 10 mM EDTA, 10 mM Na4P2O7, 100 mM NaF, 1% Triton X100).54 Proteins (40 µg) were boiled for 5 min in Laemmli buffer and loaded onto a 10% SDS-PAGE gel for migration (200 V for 1 h). After blocking with 5% (w/v) bovine serum albumin (BSA) in Tris-buffered saline (137 mmol/L NaCl, 20 mmol/L Tris, pH 7.6) plus 0.05% Tween 20 (v/v) (TBST) solution, the membrane was incubated overnight at 4 °C with the primary antibody. Then, proteins were transferred onto a polyvinyldilene difluoride membrane (100 V for 1 h). The membrane was blocked for 1 h at room temperature in TBST with 5% BSA. Primary antibody was incubated with the membrane in TBST buffer overnight at 4 °C. The membrane was washed three times with the TBST solution and incubated with the secondary antibody. After three washings with the TBST solution, the bound HRP-conjugated antibody was detected by chemiluminescence using Immobilon western chemiluminescent HRP substrate (Millipore). The resulting luminescence was detected on an autoradiography film. Quantifications were performed with ImageJ software.
Sirt1 activity
Cells treated with or without TNFα (15 ng/mL) for 24 h were recovered. Sirt1 activity was determined using a deacetylation assay kit (Fluor de Lys-SIRT1, BIOMOL, Plymouth Meeting, PA) according to the manufacturer instructions. Briefly, 20 μg of protein was incubated with 25 μl of Fluor de Lys-SIRT1 substrate (100 μmol/L) and NAD+ (500 μmol/L) for 30 min at 37 °C. The reaction was stopped by the addition of 50 μL of developer reagent and nicotinamide (2 mmol/L), and fluorescence was then monitored for 30 min at 360 nm (excitation) and 460 nm (emission).
Glucose uptake
3T3-L1 preadipocytes were differentiated into adipocytes in 12-well plates for eight days as already described.55 After induction of differentiation, cells were maintained in medium supplemented with insulin. At day 8, cells were pretreated with FK866 (1 nM) for 24 h and then treated with TNFα (15 ng/mL) for 24 h. One hour before performing the experiment, cell medium was replaced by serum-free medium. Cells were then incubated in 1 mL/well of phosphate-buffered saline (PBS) containing 170 nM insulin for 30 min at 37 °C. After washing in PBS buffer, cells were incubated in 1 mL of PBS containing 0.1 mM 2-deoxyglucose and 1 µCi/mL 2-deoxy-D-[3H]glucose for 5 min. Cells were then washed 3 times in ice-cold PBS solubilized in 200 µL of 0.1 N NaOH. Radioactivity was quantified by liquid scintillation counting. Protein quantification was also performed using the BCA method. The uptake measurement was performed in triplicates. Glucose uptake values were normalized to protein concentrations.
Visfatin ELISA
Control cells and cells treated for 24 h with TNFα (15 ng/mL) were recovered. Visfatin quantification was performed using a sandwich ELISA method according to the manufacturer’s instructions (Mouse Visfatin/PBEF ELISA Kit, CircuLEX, MBL International).
RNA interference
3T3-L1 differentiated cells were seeded in 6-well plates and transfected with either visfatin siRNA or a non-targeting siRNA, following the manufacturer’s instructions (Dharmacon, Inc.). Briefly, cells were transfected overnight using a mixture of 100 nM siRNA and 2 µL of DharmaFECT reagent/well. Next, the media were replaced with complete media.
Statistical analysis
The data are expressed as means ± SEM. Significant differences between control and treated groups were determined by the unpaired Student t test or ANOVA using Statview software (SAS Institute). P values less than 0.05 were considered significant.
Acknowledgments
This work was supported by grants from INRA and INSERM.
Disclosure of Potential Conflict of Interest
The authors declare that they have no conflict of interest
References
- 1.Cawthorn WP, Sethi JK. TNF-alpha and adipocyte biology. FEBS Lett. 2008;582:117–31. doi: 10.1016/j.febslet.2007.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nieto-Vazquez I, Fernández-Veledo S, Krämer DK, Vila-Bedmar R, Garcia-Guerra L, Lorenzo M. Insulin resistance associated to obesity: the link TNF-alpha. Arch Physiol Biochem. 2008;114:183–94. doi: 10.1080/13813450802181047. [DOI] [PubMed] [Google Scholar]
- 3.Tourniaire F, Romier-Crouzet B, Lee JH, Marcotorchino J, Gouranton E, Salles J, Malezet C, Astier J, Darmon P, Blouin E, et al. Chemokine Expression in Inflamed Adipose Tissue Is Mainly Mediated by NF-κB. PLoS One. 2013;8:e66515. doi: 10.1371/journal.pone.0066515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389:610–4. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 5.Ruan H, Miles PD, Ladd CM, Ross K, Golub TR, Olefsky JM, Lodish HF. Profiling gene transcription in vivo reveals adipose tissue as an immediate target of tumor necrosis factor-alpha: implications for insulin resistance. Diabetes. 2002;51:3176–88. doi: 10.2337/diabetes.51.11.3176. [DOI] [PubMed] [Google Scholar]
- 6.Pirola L, Johnston AM, Van Obberghen E. Modulation of insulin action. Diabetologia. 2004;47:170–84. doi: 10.1007/s00125-003-1313-3. [DOI] [PubMed] [Google Scholar]
- 7.Nieto-Vazquez I, Fernández-Veledo S, de Alvaro C, Rondinone CM, Valverde AM, Lorenzo M. Protein-tyrosine phosphatase 1B-deficient myocytes show increased insulin sensitivity and protection against tumor necrosis factor-alpha-induced insulin resistance. Diabetes. 2007;56:404–13. doi: 10.2337/db06-0989. [DOI] [PubMed] [Google Scholar]
- 8.Koren S, Fantus IG. Inhibition of the protein tyrosine phosphatase PTP1B: potential therapy for obesity, insulin resistance and type-2 diabetes mellitus. Best Pract Res Clin Endocrinol Metab. 2007;21:621–40. doi: 10.1016/j.beem.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 9.Zabolotny JM, Kim YB, Welsh LA, Kershaw EE, Neel BG, Kahn BB. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J Biol Chem. 2008;283:14230–41. doi: 10.1074/jbc.M800061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Egawa K, Maegawa H, Shimizu S, Morino K, Nishio Y, Bryer-Ash M, Cheung AT, Kolls JK, Kikkawa R, Kashiwagi A. Protein-tyrosine phosphatase-1B negatively regulates insulin signaling in l6 myocytes and Fao hepatoma cells. J Biol Chem. 2001;276:10207–11. doi: 10.1074/jbc.M009489200. [DOI] [PubMed] [Google Scholar]
- 11.Venable CL, Frevert EU, Kim YB, Fischer BM, Kamatkar S, Neel BG, Kahn BB. Overexpression of protein-tyrosine phosphatase-1B in adipocytes inhibits insulin-stimulated phosphoinositide 3-kinase activity without altering glucose transport or Akt/Protein kinase B activation. J Biol Chem. 2000;275:18318–26. doi: 10.1074/jbc.M908392199. [DOI] [PubMed] [Google Scholar]
- 12.Shimizu S, Maegawa H, Egawa K, Shi K, Bryer-Ash M, Kashiwagi A. Mechanism for differential effect of protein-tyrosine phosphatase 1B on Akt versus mitogen-activated protein kinase in 3T3-L1 adipocytes. Endocrinology. 2002;143:4563–9. doi: 10.1210/en.2002-220517. [DOI] [PubMed] [Google Scholar]
- 13.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–8. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 14.Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim YB, Sharpe AH, et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 2000;20:5479–89. doi: 10.1128/MCB.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Owen C, Czopek A, Agouni A, Grant L, Judson R, Lees EK, Mcilroy GD, Göransson O, Welch A, Bence KK, et al. Adipocyte-specific protein tyrosine phosphatase 1B deletion increases lipogenesis, adipocyte cell size and is a minor regulator of glucose homeostasis. PLoS One. 2012;7:e32700. doi: 10.1371/journal.pone.0032700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun C, Zhang F, Ge X, Yan T, Chen X, Shi X, Zhai Q. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metab. 2007;6:307–19. doi: 10.1016/j.cmet.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 17.Feige JN, Auwerx J. Transcriptional targets of sirtuins in the coordination of mammalian physiology. Curr Opin Cell Biol. 2008;20:303–9. doi: 10.1016/j.ceb.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blander G, Guarente L. The Sir2 family of protein deacetylases. Annu Rev Biochem. 2004;73:417–35. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 19.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 20.Imai S, Kiess W. Therapeutic potential of SIRT1 and NAMPT-mediated NAD biosynthesis in type 2 diabetes. Front Biosci (Landmark Ed) 2009;14:2983–95. doi: 10.2741/3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–42. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–22. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 23.Yoshizaki T, Milne JC, Imamura T, Schenk S, Sonoda N, Babendure JL, Lu JC, Smith JJ, Jirousek MR, Olefsky JM. SIRT1 exerts anti-inflammatory effects and improves insulin sensitivity in adipocytes. Mol Cell Biol. 2009;29:1363–74. doi: 10.1128/MCB.00705-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nan YM, Wu WJ, Fu N, Liang BL, Wang RQ, Li LX, Zhao SX, Zhao JM, Yu J. Antioxidants vitamin E and 1-aminobenzotriazole prevent experimental non-alcoholic steatohepatitis in mice. Scand J Gastroenterol. 2009;44:1121–31. doi: 10.1080/00365520903114912. [DOI] [PubMed] [Google Scholar]
- 25.van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1) J Biol Chem. 2004;279:28873–9. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- 26.Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem. 2004;279:50754–63. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- 27.Revollo JR, Grimm AA, Imai S. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr Opin Gastroenterol. 2007;23:164–70. doi: 10.1097/MOG.0b013e32801b3c8f. [DOI] [PubMed] [Google Scholar]
- 28.Dahl TB, Holm S, Aukrust P, Halvorsen B. Visfatin/NAMPT: a multifaceted molecule with diverse roles in physiology and pathophysiology. Annu Rev Nutr. 2012;32:229–43. doi: 10.1146/annurev-nutr-071811-150746. [DOI] [PubMed] [Google Scholar]
- 29.Moschen AR, Kaser A, Enrich B, Mosheimer B, Theurl M, Niederegger H, Tilg H. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol. 2007;178:1748–58. doi: 10.4049/jimmunol.178.3.1748. [DOI] [PubMed] [Google Scholar]
- 30.Xie H, Tang SY, Luo XH, Huang J, Cui RR, Yuan LQ, Zhou HD, Wu XP, Liao EY. Insulin-like effects of visfatin on human osteoblasts. Calcif Tissue Int. 2007;80:201–10. doi: 10.1007/s00223-006-0155-7. [DOI] [PubMed] [Google Scholar]
- 31.Arner P. Visfatin--a true or false trail to type 2 diabetes mellitus. J Clin Endocrinol Metab. 2006;91:28–30. doi: 10.1210/jc.2005-2391. [DOI] [PubMed] [Google Scholar]
- 32.Kralisch S, Klein J, Lossner U, Bluher M, Paschke R, Stumvoll M, Fasshauer M. Hormonal regulation of the novel adipocytokine visfatin in 3T3-L1 adipocytes. J Endocrinol. 2005;185:R1–8. doi: 10.1677/joe.1.06211. [DOI] [PubMed] [Google Scholar]
- 33.MacLaren R, Cui W, Cianflone K. Visfatin expression is hormonally regulated by metabolic and sex hormones in 3T3-L1 pre-adipocytes and adipocytes. Diabetes Obes Metab. 2007;9:490–7. doi: 10.1111/j.1463-1326.2006.00625.x. [DOI] [PubMed] [Google Scholar]
- 34.Lv Q, Wang Y, Wang W, Wang L, Zhou X. Effect of pioglitazone on visfatin expression in 3T3-L1 adipocytes and SD rats. Endocr Res. 2009;34:130–41. doi: 10.3109/07435800903287061. [DOI] [PubMed] [Google Scholar]
- 35.Mayi TH, Duhem C, Copin C, Bouhlel MA, Rigamonti E, Pattou F, Staels B, Chinetti-Gbaguidi G. Visfatin is induced by peroxisome proliferator-activated receptor gamma in human macrophages. FEBS J. 2010;277:3308–20. doi: 10.1111/j.1742-4658.2010.07729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63:7436–42. [PubMed] [Google Scholar]
- 37.Li L, Yang G, Shi S, Yang M, Liu H, Boden G. The adipose triglyceride lipase, adiponectin and visfatin are downregulated by tumor necrosis factor-alpha (TNF-alpha) in vivo. Cytokine. 2009;45:12–9. doi: 10.1016/j.cyto.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 38.Chang YC, Chang TJ, Lee WJ, Chuang LM. The relationship of visfatin/pre-B-cell colony-enhancing factor/nicotinamide phosphoribosyltransferase in adipose tissue with inflammation, insulin resistance, and plasma lipids. Metabolism. 2010;59:93–9. doi: 10.1016/j.metabol.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 39.Varma V, Yao-Borengasser A, Rasouli N, Bodles AM, Phanavanh B, Lee MJ, Starks T, Kern LM, Spencer HJ, 3rd, McGehee RE, Jr., et al. Human visfatin expression: relationship to insulin sensitivity, intramyocellular lipids, and inflammation. J Clin Endocrinol Metab. 2007;92:666–72. doi: 10.1210/jc.2006-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Curat CA, Wegner V, Sengenès C, Miranville A, Tonus C, Busse R, Bouloumié A. Macrophages in human visceral adipose tissue: increased accumulation in obesity and a source of resistin and visfatin. Diabetologia. 2006;49:744–7. doi: 10.1007/s00125-006-0173-z. [DOI] [PubMed] [Google Scholar]
- 41.Garten A, Petzold S, Barnikol-Oettler A, Körner A, Thasler WE, Kratzsch J, Kiess W, Gebhardt R. Nicotinamide phosphoribosyltransferase (NAMPT/PBEF/visfatin) is constitutively released from human hepatocytes. Biochem Biophys Res Commun. 2010;391:376–81. doi: 10.1016/j.bbrc.2009.11.066. [DOI] [PubMed] [Google Scholar]
- 42.Samal B, Sun Y, Stearns G, Xie C, Suggs S, McNiece I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. 1994;14:1431–7. doi: 10.1128/mcb.14.2.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song HK, Lee MH, Kim BK, Park YG, Ko GJ, Kang YS, Han JY, Han SY, Han KH, Kim HK, et al. Visfatin: a new player in mesangial cell physiology and diabetic nephropathy. Am J Physiol Renal Physiol. 2008;295:F1485–94. doi: 10.1152/ajprenal.90231.2008. [DOI] [PubMed] [Google Scholar]
- 44.Busso N, Karababa M, Nobile M, Rolaz A, Van Gool F, Galli M, Leo O, So A, De Smedt T. Pharmacological inhibition of nicotinamide phosphoribosyltransferase/visfatin enzymatic activity identifies a new inflammatory pathway linked to NAD. PLoS One. 2008;3:e2267. doi: 10.1371/journal.pone.0002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wanecq E, Prévot D, Carpéné C. Lack of direct insulin-like action of visfatin/Nampt/PBEF1 in human adipocytes. J Physiol Biochem. 2009;65:351–9. doi: 10.1007/BF03185930. [DOI] [PubMed] [Google Scholar]
- 46.Skop V, Kontrova K, Zidek V, Sajdok J, Pravenec M, Kazdova L, Mikulik K, Zidkova J. Autocrine effects of visfatin on hepatocyte sensitivity to insulin action. Physiol Res. 2010;59:615–8. doi: 10.33549/physiolres.931845. [DOI] [PubMed] [Google Scholar]
- 47.Kang YS, Song HK, Lee MH, Ko GJ, Han JY, Han SY, Han KH, Kim HK, Cha DR. Visfatin is upregulated in type-2 diabetic rats and targets renal cells. Kidney Int. 2010;78:170–81. doi: 10.1038/ki.2010.98. [DOI] [PubMed] [Google Scholar]
- 48.Dahl TB, Yndestad A, Skjelland M, Øie E, Dahl A, Michelsen A, Damås JK, Tunheim SH, Ueland T, Smith C, et al. Increased expression of visfatin in macrophages of human unstable carotid and coronary atherosclerosis: possible role in inflammation and plaque destabilization. Circulation. 2007;115:972–80. doi: 10.1161/CIRCULATIONAHA.106.665893. [DOI] [PubMed] [Google Scholar]
- 49.Gouranton E, Yazidi CE, Cardinault N, Amiot MJ, Borel P, Landrier JF. Purified low-density lipoprotein and bovine serum albumin efficiency to internalise lycopene into adipocytes. Food Chem Toxicol. 2008;46:3832–6. doi: 10.1016/j.fct.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 50.Landrier JF, Gouranton E, El Yazidi C, Malezet C, Balaguer P, Borel P, Amiot MJ. Adiponectin expression is induced by vitamin E via a peroxisome proliferator-activated receptor gamma-dependent mechanism. Endocrinology. 2009;150:5318–25. doi: 10.1210/en.2009-0506. [DOI] [PubMed] [Google Scholar]
- 51.Landrier JF, Malezet-Desmoulins C, Reboul E, Marie Lorec A, Josephe Amiot M, Borel P. Comparison of different vehicles to study the effect of tocopherols on gene expression in intestinal cells. Free Radic Res. 2008;42:523–30. doi: 10.1080/10715760802098859. [DOI] [PubMed] [Google Scholar]
- 52.Hassan M, El Yazidi C, Landrier JF, Lairon D, Margotat A, Amiot MJ. Phloretin enhances adipocyte differentiation and adiponectin expression in 3T3-L1 cells. Biochem Biophys Res Commun. 2007;361:208–13. doi: 10.1016/j.bbrc.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 53.Landrier JF, Gouranton E, Reboul E, Cardinault N, El Yazidi C, Malezet-Desmoulins C, André M, Nowicki M, Souidi M, Borel P. Vitamin E decreases endogenous cholesterol synthesis and apo-AI-mediated cholesterol secretion in Caco-2 cells. J Nutr Biochem. 2010;21:1207–13. doi: 10.1016/j.jnutbio.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 54.Gouranton E, Thabuis C, Riollet C, Malezet-Desmoulins C, El Yazidi C, Amiot MJ, Borel P, Landrier JF. Lycopene inhibits proinflammatory cytokine and chemokine expression in adipose tissue. J Nutr Biochem. 2011;22:642–8. doi: 10.1016/j.jnutbio.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 55.Marcotorchino J, Gouranton E, Romier B, Tourniaire F, Astier J, Malezet C, Amiot MJ, Landrier JF. Vitamin D reduces the inflammatory response and restores glucose uptake in adipocytes. Mol Nutr Food Res. 2012;56:1771–82. doi: 10.1002/mnfr.201200383. [DOI] [PubMed] [Google Scholar]