

Background: Lignin is a plant cell wall polymer that inhibits enzymatic saccharification of polysaccharides for the production of biofuel.
Results: The adsorption of enzymes to lignin surfaces correlates to solvent-exposed hydrophobic clusters.
Conclusion: Hydrophobicity, not surface charge, identifies proteins that preferentially adsorb to lignin.
Significance: The method could be used to design improved cellulase cocktails to lower the cost of biofuel production.
Keywords: Cellulase, Enzyme Inhibitor, Glycoside Hydrolase, Protein Chemistry, Protein Engineering, Hydrophobic Interaction, Lignin
Abstract
The inhibitory action of lignin on cellulase cocktails is a major challenge to the biological saccharification of plant cell wall polysaccharides. Although the mechanism remains unclear, hydrophobic interactions between enzymes and lignin are hypothesized to drive adsorption. Here we evaluate the role of hydrophobic interactions in enzyme-lignin binding. The hydrophobicity of the enzyme surface was quantified using an estimation of the clustering of nonpolar atoms, identifying potential interaction sites. The adsorption of enzymes to lignin surfaces, measured using the quartz crystal microbalance, correlates to the hydrophobic cluster scores. Further, these results suggest a minimum hydrophobic cluster size for a protein to preferentially adsorb to lignin. The impact of electrostatic contribution was ruled out by comparing the isoelectric point (pI) values to the adsorption of proteins to lignin surfaces. These results demonstrate the ability to predict enzyme-lignin adsorption and could potentially be used to design improved cellulase cocktails, thus lowering the overall cost of biofuel production.
Introduction
Currently the cost of enzymes required for enzymatic digestion of biomass is a significant part of the total cost of producing lignocellulosic fuel; a recent techno-economic estimation attributes a cost of $1.47/gallon of ethanol produced from corn stover to enzyme production (1). The inhibitory effect of lignin is known to decrease the performance of cellulase cocktails and results in the need for high enzyme loadings. Nonproductive adsorption of cellulases to lignin is hypothesized to be a source of inhibition, yet the mechanisms driving enzyme adsorption have not been elucidated. This study is aimed at understanding the factors contributing to lignin inhibition and proposing solutions to advance the development of enzyme cocktails by reducing nonspecific binding to lignin.
Lignin is a polymer of cross-linked phenylpropane units, conferring hydrophobicity, structural rigidity, and microbial resistance to plant cell walls. The physical properties of lignin, which can vary among different plant sources and result from different pretreatment methods, can influence enzyme adsorption capacity. The hydrophilic carboxylic acid functionality in lignin samples correlates with enzymatic hydrolysis of lignocellulosic substrates, suggesting that some chemical properties of lignin may reduce enzyme adsorption to lignin (2). Attempts to genetically engineer plants for altered lignin biosynthesis can result in the incorporation of unusual phenylpropane units, such as coniferaldehyde, leading to an increase in hydrophobicity (3). This increased hydrophobicity in genetically engineered plants has led to a decrease in digestibility of the cell wall. Thus, although the mechanism of enzymatic inhibition by lignin is not fully understood, hydrophobicity appears to have a strong role.
Research of enzyme adsorption to lignin has focused on several mechanisms. The role of electrostatic interactions was investigated by comparing enzyme adsorption and protein pI values, but the results are inconclusive because both positively and negatively charged proteins were found to adsorb to lignin (4). The role of the carbohydrate-binding module (CBM),3 found in many cellulase enzymes and important for targeting enzyme to substrate, was shown to enhance enzyme adsorption to lignin (5). However, another study utilizing a mixture of cellulase enzymes found β-glucosidase, which does not have a CBM, preferentially adsorbs to lignin (6).
Enzymatic hydrolysis of lignocellulosic biomass can be enhanced by the inclusion of various protein and chemical additives. BSA has been used as a nonenzymatic protein additive to enhance glucose yields in enzymatic hydrolysis (7, 8), based on its propensity to nonspecifically bind hydrophobic steroid hormones, hemin, and fatty acids (9). A study assessing exposed hydrophobic regions of 112 soluble, monomeric proteins identifies BSA near the top for the amount of exposed hydrophobic surface regions (10). Yang and co-workers (7) showed that BSA adsorbs more strongly to pretreated corn stover, which contains both cellulose and lignin, than to model cellulose. Additionally, an enhancement of enzymatic hydrolysis upon the addition of BSA is seen for pretreated corn stover but not for model cellulose. The results imply that adsorption to lignin is the mechanism for inhibition and that added BSA binds to lignin, preventing cellulase binding.
Similar enhancements to enzymatic hydrolysis of lignocellulosic biomass have been seen upon the addition of nonionic surfactants such as Tween detergents, polyethylene glycol 4000, or dodecylbenzene sulfonic acid (11–15). Numerous mechanisms could lead to the enhancement effect including 1) increasing protein stability, 2) altering lignin structure, and 3) altering enzyme-lignin interaction. Addressing these proposed mechanisms, Eriksson et al. (12) found surfactants have very little effect on the thermal stability of Trichoderma reesei Cel7A but did enhance the hydrolysis of lignocellulose while not improving model cellulose hydrolysis. These results coincided with a decrease in enzyme adsorption to lignin. These observations are in agreement with BSA results, and support the role of surfactants in attenuating nonproductive enzyme adsorption to lignin. Furthermore, the results support the hypothesis that enzymes interact with lignin through a hydrophobic mechanism.
Although lignin adsorption is likely to have other dependences, if hydrophobicity is a significant contributor then hydrophobic surface properties could determine how strongly various enzymes adsorb to lignin. Additionally, protein engineers might have a clear and rational approach to mitigate these undesired interactions. Lijnzaad et al. developed a method to delineate contiguous hydrophobic patches on a protein surface (16). Briefly, all nonpolar atoms with nonzero solvent-accessible surface area (SASA) are assigned to be nodes on a graph, and edges are placed between nodes if there is exposed overlap between atoms. Jacak et al. (17) incorporated a similar method into the protein design software, Rosetta, adding a scoring function specifically designed to identify larger hydrophobic patches. The Rosetta hydrophobic patch score works by assigning a score to each identified patch, with scores increasing exponentially with increasing patch size.
Here we take a systematic approach to evaluate the surface properties of a select set of proteins for comparison to measured adsorption to lignin surfaces. The method developed by Jacak et al. is used to rank-order each protein by degree of surface hydrophobicity. Then the strength of the interactions between enzymes and lignin films is evaluated using quartz crystal microbalance with dissipation monitoring (QCM-D). QCM-D enables real-time measurements of enzyme adsorption to substrate films. Comparing surface properties for the studied set of enzymes to adsorption information provides information on how well the hydrophobic patch score predicts degree of binding. We describe enzyme interactions with lignin isolated from switchgrass via an organosolv process as an example of a biomass pretreatment process potentially useful in biofuel production.
EXPERIMENTAL PROCEDURES
Hydrophobic Surface Analysis
Surface properties of individual enzymes were evaluated using the protein design software, Rosetta (18, 19). Rosetta was used to identify and score clusters of hydrophobic atoms, referred to as hydrophobic patches (17). Additionally, hydrophobic and hydrophilic solvent-accessible surface area was computed for each structure using VADAR (Volume, Area, Dihedral Angle Reporter) (20). The molecular mass for each protein was computed based on the amino acid sequence using the ExPASy ProtParam tool (21).
Enzyme Structures
Structural analysis using Rosetta requires modeled or experimentally determined structures. Experimentally determined protein structures were obtained from the Protein Data Bank (22), including the following: bovine serum albumin (PDB code 4f5s) (23), the catalytic domains of Acidothermus cellulolyticus endocellulase E1 (Cel5A, PDB code 1c0d) (24), T. reesei cellobiohydrolase I (Cel7A, PDB code 1cel) (25), the family 1 carbohydrate-binding module of T. reesei Cel7A (CBM1, PDB code 1cbh) (26), Thermomyces lanuginosus endo-1,4-β-xylanase (XynA, PDB code 1yna) (27), T. reesei eno-1,4-β-xylanase (XynII, PDB code 1enx) (28), and T. reesei acetyl xylan esterase (AxeI, PDB code 1qoz) (29).
Homology models were used for proteins or individual domains lacking an experimentally determined structure. α-l-Arabinofuranosidase B (Abfb) from Aspergillus niger shares 98% sequence identity with Aspergillus kawachii IFO4308 AbfB, which has an experimentally determined structure (PDB code 1wd3) (30). A homology model was obtained from the SWISS-MODEL Repository based on 1wd3 (31). The T. reesei AxeI CBM1 shares 69.5% sequence identity with the Cel7A CBM1. The Axe1 CBM1 sequence was modeled onto the T. reesei Cel7A CBM1 structure (PDB code 1cbh) using Rosetta. The A. niger β-glucosidase (BglI) shares 84% sequence identity with the Aspergillus aculeatus β-glucosidase, which has an experimentally determined structure (PDB code 4iib) (32). A sequence alignment was generated using MacVector (33), and the sequence for A. niger was threaded onto 4iib using Rosetta. Missing coordinates were built using SWISS-MODEL homology modeling tools (31).
Enzymes
BSA from Pierce was purchased from Thermo Scientific (Rockford, IL). A. niger β-glucosidase was purchased from Megazymes (Wicklow, Ireland). T. reesei XynII was purchased from Hampton Research (Aliso Viejo, CA). All purchased enzymes were desalted on HiPrep desalting columns (GE Healthcare) into 20 mm sodium acetate, pH 5.0, 100 mm sodium chloride buffer.
T. lanuginosus XynA was purchased from Sigma-Aldrich packaged as Pentopan® from Novozymes (34). Purification of XynA consisted of solubilizing the protein in 20 mm Tris buffer, pH 8.0, followed by centrifugation to remove the protein from the flour (Pentopan® is a baking additive) and subjecting the clarified supernatant to anion exchange chromatography on a Source15Q 10/100 Tricorn chromatography column (GE Healthcare) with a 0.0 to 1.0 m sodium chloride gradient in 20 mm Bis-Tris buffer, pH 8.0. Active fractions were pooled, concentrated by 5-kDa Vivaflow spin concentrators (Millipore), and subjected to size exclusion chromatography on a 26/60 Superdex 75 column in 20 mm sodium acetate, pH 5.0, 100 mm sodium chloride.
A. cellulolyticus endoglucanase E1 (Cel5A) was expressed in Escherichia coli BL21 as a truncated gene (CBM and linker delete) with a Tyr to Gly mutation at sequence position 245 (E1cdY245G). The protein was purified as described previously (24). T. reesei AxeI was expressed in Aspergillus awamori and purified using combinations of hydrophobic interaction, anion exchange, and size exclusion FPLC as described previously (34).
α-l-Arabinofuranosidase B (AbfB) from A. niger was expressed in and purified from A. awamori grown in CM-maltose medium at 30 °C, with shaking, for 6 days. Culture broth was filtered stepwise through Miracloth and 2.7-, 1.5-, 0.7-, and 0.45-μm filters, concentrated, and the buffer-exchanged into 20 mm Bis-Tris, pH 6.8, through a PES filter with 5-kDa cutoff (Pall Life Sciences). Buffer-exchanged broth proteins were separated by anion exchange using HiTrap Q Sepharose HP column (GE Healthcare) with a 0.0 to 1.0 m sodium chloride gradient in 20 mm Bis-Tris, pH 6.8. AbfB was followed by activity on o-nitrophenyl-α-l-arabinofuranoside (Sigma), and molecular mass as determined by SDS-Page on a 4 to 12% polyacrylamide gradient gel (Invitrogen). AbfB fractions were pooled and diluted in high salt buffer (4.0 m (NH4)2SO4 20 mm Bis-Tris, pH 6.8) to a final salt concentration of 2.0 m (NH4)2SO4 for purification by HiTrap phenyl Sepharose HP hydrophobic interaction column (GE Healthcare) with a gradient of 2.0 to 0 m (NH4)2SO4. AbfB fractions were pooled and subjected to size exclusion chromatography on a 26/50 Superdex 75 column in 20 mm sodium acetate, pH 5.0, 100 mm sodium chloride, 10% glycerol.
T. reesei Cel7A was expressed and purified as previously described (37). All enzymes were buffer-exchanged into 25 mΜ sodium citrate, pH 4.8, 50 mm sodium chloride using HiPrep desalting columns (GE Healthcare). All enzymes were brought to a final concentration of 5 μm.
Lignin Extraction
Switchgrass (Panicum virgatum) was collected from an established stand of Alamo variety grown in East Tennessee, air-dried, and comminuted in a 1-inch knife mill to give material ∼1–2” in length. Switchgrass fractionation was carried out by loading ∼430 g of switchgrass into a perforated Teflon basket and placing the basket in a Hastelloy C276 flowthrough pressure reactor. The reactor was sealed and placed under vacuum for 30 min. A single-phase mixture of methyl isobutyl ketone, ethanol, and water (16/34/50 w/w/w) in the presence of 0.1 m sulfuric acid as a catalyst was pulled into the reactor under vacuum and heated to 160 °C. Additional solvent was pumped through the system into a collection tank for 120 min at a rate sufficient to generate ∼7–8 liters of black liquor. Upon completion of the run, the solvent remaining in the reactor was carefully released into the collection tank and mixed with the black liquor collected during the run.
The black liquor was mixed with solid NaCl (10 g/100 ml of water contained in solvent mixture) in a separatory funnel, shaken, and allowed to stand for 30 min to generate aqueous and organic phases. The layers were separated, and the organic layer was washed once with ∼50% (v/v) water. The layers were separated and the organic layer was washed a second time with ∼75% (v/v) water. Lignin was isolated from the organic fraction by solvent removal on the rotary evaporator. The resulting lignin residue was triturated with diethyl ether. After decanting the ether, the lignin was placed under vacuum. The trituration step was repeated as necessary to give a free flowing brown powder. Ethanol contained in the combined aqueous fractions from the washing was removed on the rotary evaporator to precipitate a second lignin fraction that was isolated by filtration through a double layer of filter paper in a Büchner funnel and dried under vacuum to give a free flowing brown powder.
Lignin Thin Films
Thin films of lignin were used as substrates for the QCM-D studies. The QCM resonators consist of 5 MHz-AT cut quartz crystals sensors between two conductive gold layers with an upper coating of SiO2 (Q-Sense Style, Fil-Tech). The sensors were first cleaned with water and ethanol rinses followed by argon ion plasma treatments. Cleaned QCM-D sensors were then spin coated at 2,000 rpm for 60 s with 100 μl of 1 mg/ml of lignin dissolved in 9:1 dioxane:water.
Enzyme-Lignin Interactions Studied by QCM-D
A Q-Sense E4 (Biolin Scientific AB, Stockholm, Sweden) was used to study enzyme adsorption to lignin films deposited on the sensors. QCM-D measures both the change in frequency, Δf, and the change in dissipation, ΔD, of the quartz crystal. The temperature in our experiments was controlled to within ±0.02 °C by a Peltier element within the QCM instrument.
For all binding experiments, bare quartz sensors were characterized in both air and buffer solution to measure their fundamental frequencies. Following this, the sensors were coated with lignin and characterized to measure the new fundamental frequencies, allowing the mass of the lignin films to be calculated. Odd harmonic overtones were collected, and the third harmonic overtone was used to estimate the rates of adsorption.
Enzyme adsorption was monitored while flowing enzyme solution over the sensors for 25 min at a rate of 0.1 ml/min. Changes in dissipation were used to evaluate rigidity of the protein layer deposited on lignin surfaces. Changes in areal mass, Δm, were modeled using the Voigt viscoelastic model (38). The areal mass values obtained using the Voigt model generally agreed with the areal mass values obtained using the Sauerbrey equation, although the Sauerbrey equation did systematically under predict mass for about half of the proteins, which is a known limitation of the Sauerbrey model (39). The use of the Voigt viscoelastic model allowed for the estimation of the thickness for the adsorbed protein layer for A. niger BglI. The mass in all binding curves reached a plateau by 25 min, indicating saturation of binding. The binding capacity was therefore taken to be the areal mass value (ng/cm2) at 25 min.
Changes in frequency were fitted to an exponential decay function to model the initial enzyme adsorption rate as described by Turon et al. (40). The adsorption kinetics for nearly all proteins evaluated in this work did not fit to a single exponential. The use of a double-exponential equation improved the fit to the adsorption curve for many but not all of the proteins. Therefore, to provide a consistent method that could be applied to all proteins, the initial adsorption rate for each enzyme to the lignin films was determined using the limiting slope method (see Table 2). A range of greater than 20-fold is seen in the enzyme adsorption rates (Hz/min), with BSA as the fastest adsorbing protein and XynA as the slowest.
TABLE 2.
QCM-D measured adsorption parameters for proteins on lignin surfaces
Duplicate runs were performed for each enzyme, and the additional rates of adsorption and adsorbed mass after 25 min of protein injection are shown in parentheses.
| Adsorbed mass | Standard deviation of change in mass | Rate of adsorption | Standard deviation rate | |
|---|---|---|---|---|
| ng/cm2 | Hz/min | |||
| Bgl1 | 1155.6 (1170.6) | 10.6 | 8.2 (6.8) | 1.0 |
| BSA | 643.3 (631.7) | 8.2 | 21.3 (21.2) | 0.1 |
| Cel7A | 294.6 (285.8) | 6.2 | 8.3 (6.3) | 1.4 |
| Axe1 | 263.4 (245.3) | 12.8 | 10.1 (11.7) | 1.1 |
| E1 | 193.2 (192.5) | 0.5 | 3.9 (3.8) | 0.1 |
| AbfB | 170.5 (191.5) | 14.8 | 2.6 (1.5) | 0.8 |
| XynII | 172.2 (162.1) | 7.1 | 1.5 (2.2) | 0.5 |
| XynA | 69.7 (69.4) | 0.2 | 1.0 (0.6) | 0.3 |
Protein Isoelectric Point Determination
The pI for each protein was determined using a pH 3–10 isoelectric focusing (IEF) gel (Invitrogen) with a Novex IEF Marker 3–10 standard. The loading buffer was 0.01% bromphenol blue, 0.01% methyl red, and 10% glycerol. Electrophoresis conditions were 1 h at 100 V, 1 h at 200 V, and 0.5 h at 500 V. Proteins with more than one pI value, based on two or more bands in the IEF gel, were assigned a single pI value equal to the average of all determined pI values.
Analytical Ultracentrifugation
The hydrodynamic properties of BglI were analyzed by analytical ultracentrifugation using sedimentation velocity. BglI was diluted to an A280 of 0.5 in 100 mm sodium chloride, 30 mm sodium acetate, pH 5.0. The experiments were performed in a Beckman Coulter XL-A analytical ultracentrifuge at 45,000 r.p.m. and 21 °C. The biophysical properties were determined by using Ultrascan III software with two-dimensional spectrum analysis and a genetic algorithm (41).
Mass Spectrometry Analysis
BglI was supplied to Colorado State University Proteomics Facility, and 1 μl of the purified protein was mixed with 1 μl of 2,5-dihydroxy benzoic acid (10 mg/ml in 50% acetonitrile, 0.1% TFA). The mixture was spotted on the MALDI target and allowed to air dry. The sample was analyzed by an Ultraflex-TOF/TOF mass spectrometer (Bruker Daltonics, Billerica, MA) in positive ion, reflector mode using a 25 kV accelerating voltage. External calibration was done using a peptide calibration mixture (four to six peptides) on a spot adjacent to the sample. The raw data were processed in the FlexAnalysis software (version 3.3; Bruker Daltonics).
RESULTS
In this study we show that the software design program, Rosetta, can be used to predict protein adsorption to lignin by calculating the surface hydrophobicity. We also evaluated other physicochemical properties, such as protein size, experimentally measured pI, and total hydrophobic SASA, to determine the factors that influence enzyme binding to lignin. We select a set of proteins from families of industrially relevant cellulases or accessory biomass-degrading enzymes (Table 1). The enzymes have a diverse set of properties that allow a deeper investigation into the characteristics that drive adsorption to lignin. BSA is included because it has demonstrated enhancements in enzymatic degradation of lignocellulosic biomass when added to the enzymatic mixture.
TABLE 1.
Physical characteristics for the set of enzymes selected for in silico structure-based surface analysis and biophysical characterization using QCM-D
Major bands are shown in bold and underlined.
| Protein | Organism | Domain architecture | pI | Molecular mass | SASAHphob | Hydrophobic patch score |
|---|---|---|---|---|---|---|
| kDa | % | |||||
| β-Glucosidase (Bgl1) | A. niger | GH3 | 4.2 | 93.3 (117.5)a | 53.0b | 45.9b |
| Serum albumin | Bovine | BSA | 4.6 | 66.5 | 60.5 | 34.9 |
| Cellobiohydrolase (Cel7A) | T. reesei | GH7-CBM1 | 4.3 | 54.1 | 59.1 | 13.3 |
| Acetyl xylan esterase (Axe1) | T. reesei | Axe1-CBM1 | 6.8, 6.9, 7.1, 7.4 | 30.8 | 70.7 | 9.1 |
| Endocellulase (E1) | A. cellulolyticus | GH5 | 5.2 | 40.1 | 60.1 | 7.7 |
| Endoxylanase (XynA) | T. lanuginosus | GH11 | 3.8, 4.0 | 24.4 | 54.1 | 2.7 |
| Arabinofuranosidase (AbfB) | A. niger | GH54 | 3.6, 3.8 | 52.5 | 57.0 | 1.9 |
| Endoxylanase (XynII) | T. reesei | GH11 | 9 | 24.2 | 54.6 | 0.8 |
a Molecular mass for Bgl1 shown in parentheses was determined by mass spectrometry. Molecular masses for Bgl1 are for the monomeric species.
b SASA calculations and hydrophobic patch score for Bgl1 are for the dimeric species.
Analysis of Solvent-exposed Hydrophobic Patches
The simplest estimation of surface hydrophobicity of a protein structure is the hydrophobic SASA. The hydrophobic SASA of a protein can be a misleading metric because there is generally a positive correlation with the increasing size and thus increasing total surface. Evaluating the hydrophobic SASA as a percentage by normalizing by total SASA can give a more comparable metric of the relative amount of surface hydrophobicity, allowing for comparison of proteins of very different molecular masses.
Alternatively, the location of hydrophobic surface areas can be more informative than measuring surface hydrophobicity because location can be used to identify hydrophobic patches that can act as interaction sites. An example of a large hydrophobic patch identified on the surface of the T. reesei Cel7A CBM family 1 domain is shown (Fig. 1, A and B). Here an algorithm that identifies and scores a protein based on the number and size of hydrophobic clusters, or patches, on the surface was used (17). The hydrophobic patch score increases exponentially with increasing patch size. This ensures that a larger protein, such as AbfB with multiple small hydrophobic patches, will receive a more equivalent hydrophobic patch score to much smaller proteins such as XynII or XynA, which also have very small, negligible hydrophobic patch sizes but fewer of them because of protein size. A protein with large hydrophobic patches, such as BSA, will receive a high score that distinguishes it from the other proteins, highlighting the presence of possible interaction sites. The hydrophobic patches identified in BSA are known to bind hydrophobic ligands (23).
FIGURE 1.
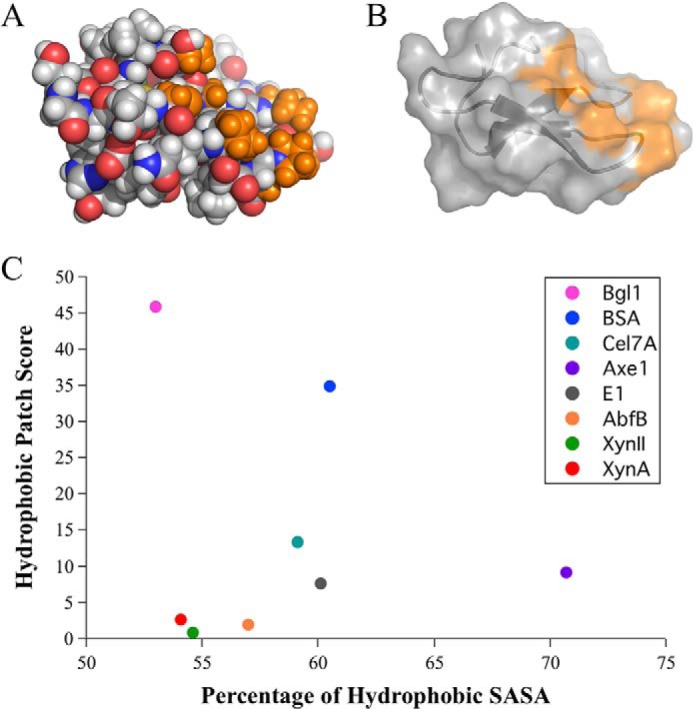
The total hydrophobic solvent-accessible surface area does not determine the presence of hydrophobic clusters. A, the family 1 carbohydrate-binding module (CBM1, PDB code 1cbh) from T. reesei Cel7A (GH7) is shown in sphere representation, with polar atoms colored red (oxygen) and blue (nitrogen), and nonpolar atoms shown in gray (carbon). Hydrogen atoms are shown in white. Nonpolar atoms from the largest hydrophobic cluster found on CBM1 are shown in orange. B, cartoon and surface representation of the CBM1 shown with the identified hydrophobic cluster highlighted in orange. C, hydrophobic patch scores are plotted against the percentage of hydrophobic SASA to show the lack of correlation between the percentage of hydrophobic SASA and the presence of large solvent-exposed hydrophobic patches as identified by the hydrophobic patch score.
Comparing the hydrophobic patch score to the percentage of hydrophobic SASA shows that the two do not have an apparent correlation (Fig. 1C). BglI, for example, has the lowest percentage of hydrophobic SASA at 53%, just below two xylanases, XynA and XynII. Given that approximately half the SASA for BglI is hydrophobic, if the hydrophobic SASA were evenly distributed there would likely be no clustering of hydrophobic surface area that could act as interaction sites. However, BglI also has the highest hydrophobic patch score for all investigated proteins, resulting from the presence of four large hydrophobic clusters. The two metrics therefore allow for very different analyses of surface hydrophobicity.
Hydrophobic Patch Scores Broken Down by Size of Patches
The hydrophobic patch score by Jacak et al. (17) was developed for the protein design software, Rosetta. The hydrophobic patch score was designed for the purpose of preventing the unintended formation of hydrophobic patches on the surface of computationally designed proteins, thus explicitly designing for enhanced solubility. As such, the objective function is mathematically designed to give large hydrophobic patches significantly higher scores.
The hydrophobic patches are placed into 50 Å2 bins according to size. The smallest patch size seen in the examined set of proteins is 50 Å2 or less, and the largest is close to 450 Å2. The count of each hydrophobic patch size is given for the individual proteins in Fig. 2A. The score increases exponentially per patch size, with a score of 0 for patches of 50 Å2, up to a score of 10.2 for patches of 450 Å2 (also shown in Fig. 2A). The total hydrophobic patch score, as shown below x axis in Fig. 2C, is the sum of the scores for every identified patch on the protein.
FIGURE 2.

The hydrophobic patch score broken down by the number and size of identified patches for each enzyme suggests a minimum patch size for preferential lignin adsorption. A, the individual hydrophobic patch score for each bin size is shown with red dots (right y axis). The count for each hydrophobic patch size found in the investigated set of proteins is shown with bars (left y axis). B, a surface representation of BSA with two of the four largest identified hydrophobic patches highlighted in orange, and side chains are shown in stick representation. C, the hydrophobic patch scores for each protein are listed in brackets under the protein name under the x axis. The count for each hydrophobic patch size is broken down for each protein. The patch sizes are binned by Å2 of solvent-accessible surface area. Larger patches (>350 Å2 of SASA) are only observed on A. niger BglI and BSA. Because the score increases exponentially for each bin size, the larger hydrophobic patch scores seen in BglI and BSA are explained by the presence of larger hydrophobic patches.
AbfB and XynII have only small patches of 0–50 Å2, receiving scores of 0, and 51–100 Å2, receiving scores of 0.16 per patch. The resulting scores for AbfB and XynII are very similar despite the fact that AbfB is more than twice as large as XynII (Fig. 2C and Table 1). On the other hand, the single patch found in BSA, sized 350 to 400 Å2, accounts for a large portion of the total hydrophobic patch score (Fig. 2B). The large patches, sized 400–450 Å2 on the BglI dimer also contribute significantly to the total hydrophobic patch score.
Evaluating Protein Adsorption to Lignin Films
The interaction of the selected set of proteins with lignin was investigated using QCM-D. QCM-D allows for real time monitoring of binding kinetics by measuring changes in the resonance frequency (Δf) that are proportional to changes in deposited mass on the sensor surface. After the protein injections, a change in resonance frequency was observed for all proteins, indicating enzyme adsorption to lignin films.
The Voigt viscoelastic model was used to estimate the change in deposited mass (ng/cm2) from the measured Δf (Fig. 3A). The total adsorbed mass for each protein was taken to be the adsorbed mass after 25 min, by which time all proteins had reached saturation of the lignin films. The total adsorbed mass at a point of saturation represents the binding capacity of the lignin for each protein. The proteins have greater than 15-fold difference in total adsorbed mass, with BglI displaying the greatest adsorption capacity to lignin and XynA displaying the least.
FIGURE 3.

A correlation exists between enzyme-lignin adsorption parameters and the number and size of hydrophobic patches on the surface of enzymes. A, QCM-D adsorption curves for a set of eight enzymes are shown. B, a linear correlation exists between the hydrophobic patch score for each protein and the total change in adsorbed mass on the lignin surfaces. The error bars are so small relative to the differences in areal mass that they are not visible in the graph. The error bars for areal mass for each protein are given in Table 2. C, a correlation is also seen for the initial adsorption rate for the seven monomeric proteins, determined by the slope of the initial linear portion of the adsorption curves and the hydrophobic patch score. The initial rate of adsorption for Bgl1 dimer unexpectedly lags the high adsorption capacity and the large hydrophobic patch score. This data point is shown but not included in the trend line. D, the percentage of hydrophobic SASA does not correlate with the binding capacity as determined by the total adsorbed mass. The error bars for total adsorbed mass are given in Table 2 because they are so small relative to the differences in adsorbed mass that they are not visible in the graph.
Hydrophobic Patch Scores Correlate with Protein-Lignin Adsorption Parameters
We compare the adsorption capacity and the initial adsorption rates to the various physiochemical properties of each protein. The hydrophobic patch score shows a strong correlation with binding capacity (Fig. 3B), with an R2 of 0.94 for all investigated proteins. The percentage of hydrophobic SASA does not trend with binding capacity, which is not surprising because the percentage of hydrophobic SASA does not trend with the hydrophobic patch score (Fig. 3D). A strong correlation is also seen between the hydrophobic patch score and the initial rate of adsorption for all monomeric proteins, with an R2 of 0.94 (Fig. 3C). The seven monomeric proteins, ranging in size from 24 to 66 kDa, display adsorption rates that trend with measured binding capacity and show similar linear correlations with the hydrophobic patch scores. The large dimeric protein BglI, with a molecular mass of 235 kDa, shows a slower initial adsorption rate that does not trend with the high binding capacity or the hydrophobic patch score. Because the hydrophobic patch score is designed to rank proteins based on the size and number of hydrophobic zones, these results suggest that hydrophobic interaction describes a dominant component of interaction energy between the proteins and lignin films. Interestingly, BSA and BglI display the largest patch sizes and also exhibit significantly higher binding capacities to lignin surfaces (Fig. 2A and Table 2).
Molecular mass does not trend with binding capacity, verifying that the observed correlations are not simply the result of the probability of finding larger patches or more hydrophobicity on the surface of larger proteins. Specifically, AbfB has a comparable hydrophobic patch score and binding capacity to both XynA and XynII, although the molecular mass of Abfb is approximately double the molecular mass of either endoxylanase (Tables 1 and 2 and Fig. 3B). AbfB, Cel7A,and BSA are comparable in molecular mass but display significantly different binding capacities and hydrophobic patch scores.
A. niger BglI Binding Agrees with Location of Hydrophobic Patches
The crystal structure determined for A. aculeatus β-glucosidase 1 (BglI) reveals a dimeric complex in the asymmetric unit cell (PDB code 4iib) (32). The dimeric interface buries 1450 Å2 of surface area and contains 25 hydrogen bonds. Further analysis using gel filtration chromatography shows that A. aculeatus BglI forms a dimer in solution as well. We investigate the oligomeric state of A. niger BglI using a native gel and sedimentation velocity analytical ultracentrifugation. The native gel shows no trace of the monomeric species, with a strong band for the dimer and a faint band for a higher order oligomeric species (Fig. 4). Sedimentation velocity results reveal a dominant presence of the dimeric species, with 95% of BglI appearing as a dimer (Table 3). The observed molecular mass of the native gel and analytical ultracentrifugation are higher than predicted based on sequence alone, thus the protein was further evaluated using mass spectrometry. The molecular mass was determined to be 117.5 kDa (Table 1) because of post-translational glycosylation. The dimeric structure for A. aculeatus BglI was therefore used to model A. niger BglI, including hydrophobic patch analysis.
FIGURE 4.
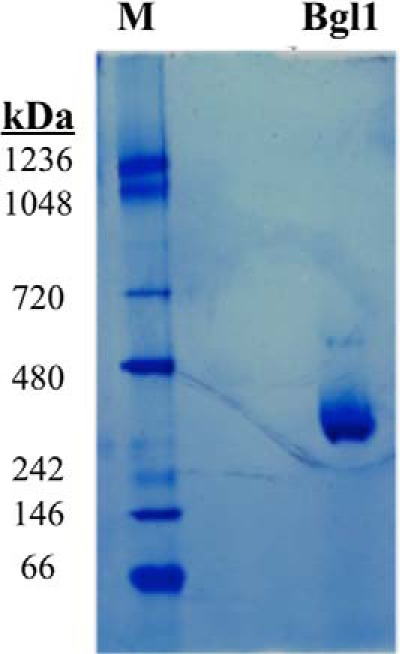
A. niger Bgl1 is a homodimer. Native PAGE of A. niger BglI is shown. 30 μg of BglI was run on a 2–12% Native PAGE (Invitrogen) and stained with SimplyBlue protein stain. The protein molecular mass aligns as a dimer, with no detectable band for monomeric species. There is, however, a band running at a higher molecular mass that may be a tetramer or higher order oligomeric species.
TABLE 3.
Sedimentation velocity analysis for A. niger BglI
Sedimentation coefficient distribution determined by van Holde Weishiet analysis using Ultrascan III. BglI was diluted to an A280 of 0.5 in 100 mm NaCl, 30 mm sodium acetate at pH 5.0 and centrifuged at 50,000 rpm. The sedimentation velocity data shown were determined using Ultrascan III software using two-dimensional spectrum analysis and genetic algorithm. The analytical ultracentrifugation data suggest that BglI is primarily a dimeric species with a molecular mass of 235 kDa and also contains 5% oligomers or other contaminants.
| Molecular Mass | Apparent S | S 20, W | Apparent D | D 20, W | f/f0 | Concentration |
|---|---|---|---|---|---|---|
| 2.35E+05 | 1.11E-12 | 1.08E-12 | 4.03E-07 | 4.04E-07 | 1.30E + 00 | 0.55829 (94.68%) |
| 1.58E+06 | 1.29E-12 | 1.25E-12 | 6.92E-08 | 6.93E-08 | 4.03E + 00 | 0.0069564 (1.18%) |
| 2.32E+05 | 1.58E-12 | 1.53E-12 | 5.76E-07 | 5.77E-07 | 9.17E-01 | 0.02439 (4.14%) |
The BglI was found to have the largest hydrophobic patch score for all proteins considered here and also displayed the highest binding capacity for lignin surfaces. The Voigt viscoelastic model employed here to estimate deposited mass on lignin surfaces is also used to estimate the thickness of deposited protein layers. The estimated thickness of the BglI layer on lignin was determined to be ∼114 Å after 25 min of protein injection (Fig. 5B). The BglI dimer forms an oblong structure with the identified hydrophobic patches located opposite each other on the long axis of the dimer. Interestingly, the distances between the hydrophobic patches are ∼125 and 118 Å, in close agreement with the estimated thickness of the protein layer (Fig. 5A). The shorter axis for the BglI dimer is ∼68 Å, a distance that does not fit the measured thickness of the BglI protein layer, thus precluding binding with the long, dimer axis parallel to lignin.
FIGURE 5.
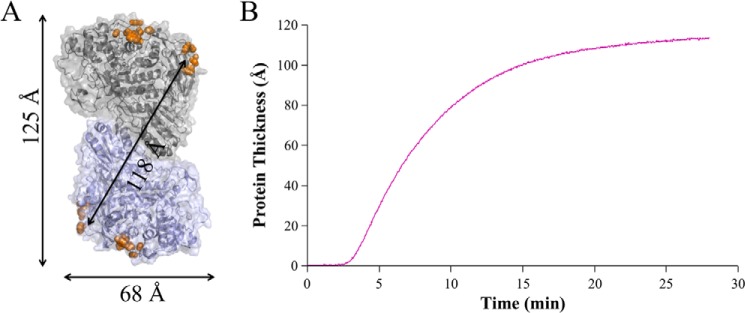
The distance of the dimeric structure between hydrophobic patches on A. niger BglI agrees with the thickness of the deposited protein layer as evaluated with QCM-D. A, the modeled BglI dimer is shown in cartoon and surface representation with the four largest hydrophobic patches highlighted with orange spheres. Distances between the hydrophobic patches (125 and 118 Å) are shown with arrows, and the monomer thickness across the short axis is shown below (68 Å). B, the estimated thickness of the BglI protein layer on the lignin surface is shown. The first 3 min is the flow of buffer only, followed by 25 min of protein injection.
Protein Surface Charge Shows No Correlation with Protein-Lignin Adsorption
Elevating pH has been shown to increase enzymatic saccharification of lignocellulose and decrease cellulase binding to lignin (42), although it is not clear how much of this effect is from changes in the lignin or the enzymes. We investigate the role of electrostatic interactions at constant pH by comparing protein surface charge, as measured by pI, to binding capacity on the lignin surfaces (Table 1). Proteins that display multiple bands on the IEF gel were given a single, average pI value for comparison to QCM-D-determined binding capacity. Our set of proteins includes enzymes with pI values ranging from 3.6 for AbfB to ∼9 for XynII. Two endoxylanase enzymes from glycoside hydrolase family 11, XynA and XynII, have significantly different pI values (3.8 and 4.0 for XynA and 9 for XynII) but are nearly identical in molecular masses and hydrophobic patch scores.
Comparison between the pI for each protein and the binding capacity to lignin reveals no apparent correlation, with an R2 of 0.06 (Fig. 6). XynII and XynA have comparable binding capacity to lignin films. Conversely, BSA and Cel7A have similar pI values, with 4.6 for BSA and a range of 4.3 to 4.7 for Cel7A, yet the binding capacity of BSA for lignin films is more than twice that of Cel7A.
FIGURE 6.
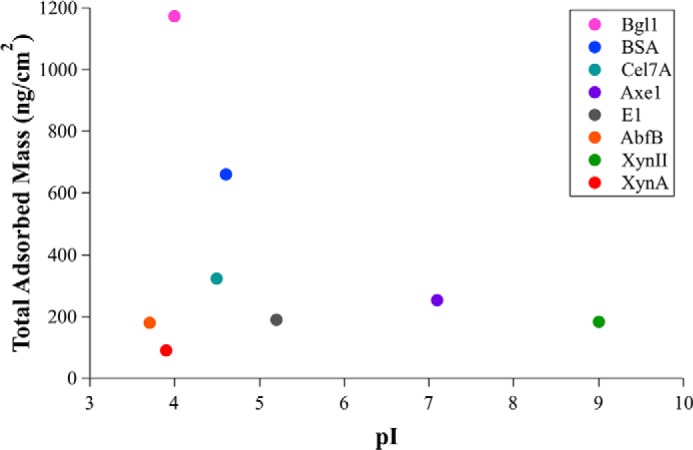
There is no correlation between the pI and measured binding capacity to lignin surfaces for the investigated proteins. pI values were averaged for enzymes with multiple bands in the IEF gels to obtain single pI values to compare with adsorbed mass values. The error bars for total adsorbed mass for each protein are given in Table 2 because they are so small relative to the differences in total adsorbed mass that they are not visible in the graph.
DISCUSSION
Enzymatic degradation of lignocellulosic biomass is a promising renewable source of liquid fuel provided cost reductions can be achieved. Cellulase preparation required to efficiently digest sugars from lignocellulosic biomass remains a sizable portion of the total cost, prompting research investments in identifying avenues to improve cellulase efficiency and decrease costs. The loss of enzymatic activity caused by the presence of lignin has therefore spurred considerable interest.
Multiple hypotheses regarding enzyme adsorption to lignin have been proposed, although the mechanism has remained elusive. Here, a systematic approach was used to investigate the role of hydrophobic interaction in enzyme-lignin adsorption. The hydrophobic patch score correlates surprisingly well with the measured enzyme adsorption to lignin. One protein, BglI, shows a lagging initial binding rate yet reaches the highest overall level of binding. BglI differs from the other proteins in two major facets; it forms a dimer in solution and has a pair of large distal hydrophobic patch regions. The other proteins are known to be monomeric (investigated by x-ray crystallography (28, 30), gel filtration chromatography (43–45), native polyacrylamide gel (46), and both analytical ultracentrifugation and gel filtration chromatography (47)) and, excepting BSA, presented similar patterns of small, random hydrophobic patching on their surface (Fig. 2C). Interaction energies are generally multifaceted, and the apparent dominance of the hydrophobic component could obscure other energetic contributions. Still, these results suggest that hydrophobic interaction accounts for much of the interaction energy between proteins and lignin.
A. niger BglI provides a unique opportunity to investigate a highly important cellulase enzyme that has been shown to preferentially adsorb to lignin when included in enzyme cocktails (6). BglI adsorption to lignin surfaces investigated here using QCM-D also shows higher adsorption capacity compared with other proteins, including BSA and T. reesei Cel7A. Interestingly, the distance from the lignin surface of the BglI layer is in agreement with the length of the BglI dimer if BglI adsorbs at either of the identified hydrophobic patches. Further, BglI has been shown to irreversibly adsorb to lignin yet still maintain activity (6). The hydrophobic patches identified here are far from the active site, allowing BglI to bind lignin while leaving the active sites available. β-Glucosidase enzymes act on soluble substrate, unlike other cellulase enzymes. Thus, perhaps β-glucosidases, lacking a CBM domain, in fact benefit from adsorption to lignin, anchoring them near substrate yet not obscuring the active site.
Many cellulase enzymes are multidomain proteins, with a catalytic domain defining the function and a CBM targeting the enzyme to substrate. Because CBMs concentrate enzymes onto substrates, they might also drive enzyme adsorption to lignin. Rahikainen et al. (5) compared adsorption to lignin of the multidomain T. reesei Cel7A to the isolated catalytic domain of Cel7A with CBM and linker removed. Using QCM-D, they found that the full-length Cel7A adsorbed to lignin films faster and to a greater extent than catalytic domain only.
Here, two proteins containing a family 1 CBM are investigated, including T. reesei Cel7A. Hydrophobic patch analysis identifies the largest patch on the Cel7A CBM, not the catalytic domain, despite the fact that the CBM is much smaller. Hydrophobic patch analysis offers a possible explanation as to why the CBM increases adsorption to lignin for T. reesei Cel7A.
CBMs, divided into numerous families based on protein fold, display a variety of substrate specificities (47, 48). In some cases a multivalent effect has been observed, where single cellulase enzymes contain multiple CBMs to increase association with target substrates (49, 50). The presence of multiple CBMs may also be deleterious for enzymatic function depending on the substrate and the enzymatic mechanism, as seen with cellulosomes on pretreated biomass (51). Thus, evaluating the extent of enzyme adsorption to lignin for various CBM families and for enzymes containing multiple CBMs may be important for future cellulase engineering efforts.
Elevating pH is known to enhance enzymatic saccharification of lignocellulosic biomass and decrease enzyme adsorption to lignin (42, 52). Here, the role of protein surface charge at a constant pH of 4.8 was investigated. Measured pI values did not show a correlation with enzyme adsorption to lignin. Although it remains unclear whether altering pH has a larger effect on the enzyme or the lignin, protein surface charge does not identify which enzymes will preferentially adsorb. It is worth noting, however, that although neither endoglucanase strongly adsorbed to lignin compared with the other proteins studied, XynII has a pI of ∼9 and adsorbs more strongly to lignin compared with XynA with a pI of 3.9. Thus, engineering a protein to have a more positive surface charge may serve to attenuate lignin adsorption.
A model system of organosolv-extracted lignin from switchgrass was used to probe enzyme-lignin interactions independent of enzyme-cellulose interactions. Both the method of extraction as well as the plant source for lignin can result in different chemical properties that may alter enzyme-lignin interactions (35, 36, 53). Considering alternate sources of lignin and different extraction methods could build upon this work.
Understanding the mechanisms that drives enzyme adsorption to lignin promises to help engineering efforts to mitigate these undesired interactions. Detailed structural analysis affords a deeper understanding of enzyme-lignin interactions, as well as the importance of various physicochemical properties and structural regions. The approach presented here offers the added benefit of identifying specific protein regions and sequence positions for future investigation.
This work was supported by the U.S. Department of Energy under Contract DE-AC36–08GO28308 with the National Renewable Energy Laboratory and by the Department of Energy Office of Energy Efficiency and Renewable Energy, Bioenergy Technologies Office. This work was also supported by the Center for Direct Catalytic Conversion of Biomass to Biofuels (C3Bio), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0000997.
- CBM
- carbohydrate-binding module
- PDB
- Protein Data Bank
- AbfB
- α-l-arabinofuranosidase B
- QCM-D
- quartz crystal microbalance with dissipation monitoring
- IEF
- isoelectric focusing
- SASA
- solvent-accessible surface area.
REFERENCES
- 1. Klein-Marcuschamer D., Oleskowicz-Popiel P., Simmons B. A., Blanch H. W. (2012) The challenge of enzyme cost in the production of lignocellulosic biofuels. Biotechnol. Bioeng. 109, 1083–1087 [DOI] [PubMed] [Google Scholar]
- 2. Nakagame S., Chandra R. P., Kadla J. F., Saddler J. N. (2011) Enhancing the enzymatic hydrolysis of lignocellulosic biomass by increasing the carboxylic acid content of the associated lignin. Biotechnol. Bioeng. 108, 538–548 [DOI] [PubMed] [Google Scholar]
- 3. Grabber J. H. (2005) How do lignin composition, structure, and cross-linking affect degradability? A review of cell wall model studies. Crop Sci. 45, 820–831 [Google Scholar]
- 4. Nakagame S., Chandra R. P., Kadla J. F., Saddler J. N. (2011) The isolation, characterization and effect of lignin isolated from steam pretreated Douglas-fir on the enzymatic hydrolysis of cellulose. Bioresour. Technol. 102, 4507–4517 [DOI] [PubMed] [Google Scholar]
- 5. Rahikainen J. L., Martin-Sampedro R., Heikkinen H., Rovio S., Marjamaa K., Tamminen T., Rojas O. J., Kruus K. (2013) Inhibitory effect of lignin during cellulose bioconversion: the effect of lignin chemistry on non-productive enzyme adsorption. Bioresour. Technol. 133, 270–278 [DOI] [PubMed] [Google Scholar]
- 6. Haven M. O., Jørgensen H. (2013) Adsorption of β-glucosidases in two commercial preparations onto pretreated biomass and lignin. Biotechnol. Biofuels 6, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brethauer S., Studer M. H., Yang B., Wyman C. E. (2011) The effect of bovine serum albumin on batch and continuous enzymatic cellulose hydrolysis mixed by stirring or shaking. Bioresour. Technol. 102, 6295–6298 [DOI] [PubMed] [Google Scholar]
- 8. Yang B., Wyman C. E. (2006) BSA treatment to enhance enzymatic hydrolysis of cellulose in lignin containing substrates. Biotechnol. Bioeng. 94, 611–617 [DOI] [PubMed] [Google Scholar]
- 9. Zunszain P. A., Ghuman J., Komatsu T., Tsuchida E., Curry S. (2003) Crystal structural analysis of human serum albumin complexed with hemin and fatty acid. BMC Struct. Biol. 3, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lijnzaad P., Berendsen H. J., Argos P. (1996) Hydrophobic patches on the surfaces of protein structures. Proteins 25, 389–397 [DOI] [PubMed] [Google Scholar]
- 11. Qing Q., Yang B., Wyman C. E. (2010) Impact of surfactants on pretreatment of corn stover. Bioresour. Technol. 101, 5941–5951 [DOI] [PubMed] [Google Scholar]
- 12. Eriksson T., Borjesson J., Tjerneld F. (2002) Mechanism of surfactant effect in enzymatic hydrolysis of lignocellulose. Enzyme Microb. Tech. 31, 353–364 [Google Scholar]
- 13. Ballesteros I., Oliva J. M., Carrasco J., Cabanas A., Navarro A. A., Ballesteros M. (1998) Effect of surfactants and zeolites on simultaneous saccharification and fermentation of steam-exploded poplar biomass to ethanol. Appl. Biochem. Biotechnol. 70–72, 369–381 [DOI] [PubMed] [Google Scholar]
- 14. Kaar W. E., Holtzapple M. T. (1998) Benefits from Tween during enzymic hydrolysis of corn stover. Biotechnol. Bioeng. 59, 419–427 [DOI] [PubMed] [Google Scholar]
- 15. Kurakake M., Ooshima H., Kato J., Harano Y. (1994) Pretreatment of bagasse by nonionic surfactant for the enzymatic hydrolysis. Bioresour. Technol. 49, 247–251 [Google Scholar]
- 16. Lijnzaad P., Berendsen H. J., Argos P. (1996) A method for detecting hydrophobic patches on protein surfaces. Proteins 26, 192–203 [DOI] [PubMed] [Google Scholar]
- 17. Jacak R., Leaver-Fay A., Kuhlman B. (2012) Computational protein design with explicit consideration of surface hydrophobic patches. Proteins 80, 825–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuhlman B., Baker D. (2000) Native protein sequences are close to optimal for their structures. Proc. Natl. Acad. Sci. U.S.A. 97, 10383–10388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rohl C. A., Strauss C. E., Misura K. M., Baker D. (2004) Protein structure prediction using Rosetta. Methods Enzymol. 383, 66–93 [DOI] [PubMed] [Google Scholar]
- 20. Willard L., Ranjan A., Zhang H., Monzavi H., Boyko R. F., Sykes B. D., Wishart D. S. (2003) VADAR: a web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. 31, 3316–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wilkins M. R., Gasteiger E., Bairoch A., Sanchez J. C., Williams K. L., Appel R. D., Hochstrasser D. F. (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552 [DOI] [PubMed] [Google Scholar]
- 22. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bujacz A. (2012) Structures of bovine, equine and leporine serum albumin. Acta Crystallogr. D Biol. Crystallogr 68, 1278–1289 [DOI] [PubMed] [Google Scholar]
- 24. Baker J. O., McCarley J. R., Lovett R., Yu C. H., Adney W. S., Rignall T. R., Vinzant T. B., Decker S. R., Sakon J., Himmel M. E. (2005) Catalytically enhanced endocellulase Cel5A from Acidothermus cellulolyticus. Appl. Biochem. Biotechnol. 121–124, 129–148 [PubMed] [Google Scholar]
- 25. Divne C., Ståhlberg J., Reinikainen T., Ruohonen L., Pettersson G., Knowles J. K., Teeri T. T., Jones T. A. (1994) The three-dimensional crystal structure of the catalytic core of cellobiohydrolase I from Trichoderma reesei. Science 265, 524–528 [DOI] [PubMed] [Google Scholar]
- 26. Kraulis J., Clore G. M., Nilges M., Jones T. A., Pettersson G., Knowles J., Gronenborn A. M. (1989) Determination of the three-dimensional solution structure of the C-terminal domain of cellobiohydrolase I from Trichoderma reesei: a study using nuclear magnetic resonance and hybrid distance geometry-dynamical simulated annealing. Biochemistry 28, 7241–7257 [DOI] [PubMed] [Google Scholar]
- 27. Gruber K., Klintschar G., Hayn M., Schlacher A., Steiner W., Kratky C. (1998) Thermophilic xylanase from Thermomyces lanuginosus: high-resolution x-ray structure and modeling studies. Biochemistry 37, 13475–13485 [DOI] [PubMed] [Google Scholar]
- 28. Törrönen A., Rouvinen J. (1995) Structural comparison of two major endo-1,4-xylanases from Trichoderma reesei. Biochemistry 34, 847–856 [DOI] [PubMed] [Google Scholar]
- 29. Hakulinen N., Tenkanen M., Rouvinen J. (2000) Three-dimensional structure of the catalytic core of acetylxylan esterase from Trichoderma reesei: insights into the deacetylation mechanism. J. Struct. Biol. 132, 180–190 [DOI] [PubMed] [Google Scholar]
- 30. Miyanaga A., Koseki T., Matsuzawa H., Wakagi T., Shoun H., Fushinobu S. (2004) Crystal structure of a family 54 α-l-arabinofuranosidase reveals a novel carbohydrate-binding module that can bind arabinose. J. Biol. Chem. 279, 44907–44914 [DOI] [PubMed] [Google Scholar]
- 31. Kiefer F., Arnold K., Künzli M., Bordoli L., Schwede T. (2009) The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 37, D387–D392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Suzuki K., Sumitani J., Nam Y. W., Nishimaki T., Tani S., Wakagi T., Kawaguchi T., Fushinobu S. (2013) Crystal structures of glycoside hydrolase family 3 β-glucosidase 1 from Aspergillus aculeatus. Biochem. J. 452, 211–221 [DOI] [PubMed] [Google Scholar]
- 33. Rastogi P. A. (2000) MacVector. Integrated sequence analysis for the Macintosh. Methods Mol. Biol. 132, 47–69 [DOI] [PubMed] [Google Scholar]
- 34. Selig M. J., Knoshaug E. P., Adney W. S., Himmel M. E., Decker S. R. (2008) Synergistic enhancement of cellobiohydrolase performance on pretreated corn stover by addition of xylanase and esterase activities. Bioresour. Technol. 99, 4997–5005 [DOI] [PubMed] [Google Scholar]
- 35. El Hage R., Brosse N., Sannigrahi P., Ragauskas A. (2010) Effects of process severity on the chemical structure of miscanthus ethanol organosolv lignin. Polym. Degrad. Stabil. 95, 997–1003 [Google Scholar]
- 36. Erdtman H. (1972) Lignins: occurrence, formation, structure and reactions J. Polym. Sci. Part B Polym. Lett. 10, 228–230 [Google Scholar]
- 37. Jeoh T., Michener W., Himmel M. E., Decker S. R., Adney W. S. (2008) Implications of cellobiohydrolase glycosylation for use in biomass conversion. Biotechnol. Biofuels 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Voinova M. V., Rodahl M., Jonson M., Kasemo B. (1999) Viscoelastic acoustic response of layered polymer films at fluid-solid interfaces: continuum mechanics approach. Phys. Scripta 59, 391–396 [Google Scholar]
- 39. Dutta A. K., Nayak A., Belfort G. (2008) Viscoelastic properties of adsorbed and cross-linked polypeptide and protein layers at a solid-liquid interface. J. Colloid Interface Sci. 324, 55–60 [DOI] [PubMed] [Google Scholar]
- 40. Turon X., Rojas O. J., Deinhammer R. S. (2008) Enzymatic kinetics of cellulose hydrolysis: a QCM-D study. Langmuir 24, 3880–3887 [DOI] [PubMed] [Google Scholar]
- 41. Demeler B. (2005) in UltraScan: A Comprehensive Data Analysis Software Package for Analytical Ultracentrifugation Experiments (Scott D. J., Harding S. E., Rowe A. J., eds), pp. 210–230, Royal Society of Chemistry, Cambridge, UK [Google Scholar]
- 42. Lou H., Zhu J. Y., Lan T. Q., Lai H., Qiu X. (2013) pH-induced lignin surface modification to reduce nonspecific cellulase binding and enhance enzymatic saccharification of lignocelluloses. ChemSusChem 6, 919–927 [DOI] [PubMed] [Google Scholar]
- 43. Pingali S. V., O'Neill H. M., McGaughey J., Urban V. S., Rempe C. S., Petridis L., Smith J. C., Evans B. R., Heller W. T. (2011) Small angle neutron scattering reveals pH-dependent conformational changes in Trichoderma reesei cellobiohydrolase I: implications for enzymatic activity. J. Biol. Chem. 286, 32801–32809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sundberg M., Poutanen K. (1991) Purification and properties of 2-acetylxylan esterases of Trichoderma reesei. Biotechnol. Appl. Bioc. 13, 1–11 [Google Scholar]
- 45. Sakon J., Adney W. S., Himmel M. E., Thomas S. R., Karplus P. A. (1996) Crystal structure of thermostable family 5 endocellulase E1 from Acidothermus cellulolyticus in complex with cellotetraose. Biochemistry 35, 10648–10660 [DOI] [PubMed] [Google Scholar]
- 46. Shrivastava S., Shukla P., Deepalakshmi P. D., Mukhopadhyay K. (2013) Characterization, cloning and functional expression of novel xylanase from Thermomyces lanuginosus SS-8 isolated from self-heating plant wreckage material. World J. Microbiol. Biotechnol. 29, 2407–2415 [DOI] [PubMed] [Google Scholar]
- 47. Lebowitz J., Lewis M. S., Schuck P. (2002) Modern analytical ultracentrifugation in protein science: a tutorial review. Protein Sci. 11, 2067–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cuskin F., Flint J. E., Gloster T. M., Morland C., Baslé A., Henrissat B., Coutinho P. M., Strazzulli A., Solovyova A. S., Davies G. J., Gilbert H. J. (2012) How nature can exploit nonspecific catalytic and carbohydrate binding modules to create enzymatic specificity. Proc. Natl. Acad. Sci. U.S.A. 109, 20889–20894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boraston A. B., Bolam D. N., Gilbert H. J., Davies G. J. (2004) Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. J. 382, 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brunecky R., Alahuhta M., Xu Q., Donohoe B. S., Crowley M. F., Kataeva I. A., Yang S. J., Resch M. G., Adams M. W., Lunin V. V., Himmel M. E., Bomble Y. J. (2013) Revealing nature's cellulase diversity: the digestion mechanism of Caldicellulosiruptor bescii CelA. Science 342, 1513–1516 [DOI] [PubMed] [Google Scholar]
- 51. Resch M. G., Donohoe B. S., Baker J. O., Decker S. R., Bayer E. A., Beckham G. T., Himmel M. E. (2013) Fungal cellulases and complexed cellulosomal enzymes exhibit synergistic mechanisms in cellulose deconstruction. Energ. Environ. Sci. 6, 1858–1867 [Google Scholar]
- 52. Rahikainen J. L., Evans J. D., Mikander S., Kalliola A., Puranen T., Tamminen T., Marjamaa K., Kruus K. (2013) Cellulase-lignin interactions: the role of carbohydrate-binding module and pH in non-productive binding. Enzyme Microb. Tech. 53, 315–321 [DOI] [PubMed] [Google Scholar]
- 53. El Hage R., Brosse N., Chrusciel L., Sanchez C., Sannigrahi P., Ragauskas A. (2009) Characterization of milled wood lignin and ethanol organosolv lignin from miscanthus. Polym. Degrad. Stabil. 94, 1632–1638 [Google Scholar]