

Abstract
Immunohistochemistry (IHC) is a tool for visualizing protein expression employed as part of the diagnostic work-up for the majority of solid tissue malignancies. Existing IHC methods use antibodies tagged with fluorophores or enzyme reporters that generate colored pigments. Because these reporters exhibit spectral and spatial overlap when used simultaneously, multiplexed IHC is not routinely used in clinical settings. We have developed a method that uses secondary ion mass spectrometry to image antibodies tagged with isotopically pure elemental metal reporters. Multiplexed ion beam imaging (MIBI) is capable of analyzing up to 100 targets simultaneously over a five-log dynamic range. Here, we used MIBI to analyze formalin-fixed, paraffin-embedded (FFPE) human breast tumor tissue sections stained with ten labels simultaneously. The resulting data suggest that MIBI will provide new insights by integrating tissue microarchitecture with highly multiplexed protein expression patterns, and will be valuable for basic research, drug discovery and clinical diagnostics.
Introduction
Antibodies were first employed in tissue section analysis in 1942 to visualize pneumococcal antigens in organ biopsies from mice infused with live bacteria1. Since that time, immunohistochemistry (IHC) has become a mainstay of clinical diagnostics and basic research and is primarily used to assess the spatial distribution of one or two (rarely more) antigens in tissue sections. Despite the high specificity of many antibodies, the concentration of most antigens is insufficient to permit detection by conventional assays without signal amplification2-4. Signal amplification is typically achieved using multivalent, enzyme-linked secondary antibodies that bind the Fc-portion of the primary antibody. In bright-field microscopy, the most commonly used enzymatic reporter is horseradish peroxidase, typically used to oxidize 3,3′-diaminobenzidine (DAB), resulting in accumulation of a brown precipitate. Such non-linear enzymatic amplification can result in poor correlation with the target antigen concentration2,5.
Simultaneous detection of multiple antigens is subject to additional constraints that limit the utility of existing IHC-based analysis for predictive biomarker development in human clinical trials and clinical diagnostics. Colorimetric detection of four antigens has been reported using multiple enzyme-linked secondary antibodies, but in practice this approach is usually limited to two because of difficulties encountered in sample preparation and imaging2,6. Fluorescent labels used in the related immunofluorescence (IF) technique provide a higher signal-to-noise ratio and are more frequently used for simultaneous detection of multiple molecular targets. Practical limitations include the need for primary antibodies generated in dissimilar host species and for non-overlapping reporter emission spectra5. Thus, conventional IHC or IF methodologies do not support the robust generation of multiplexed, quantitative data needed to understand the relationship between tissue microarchitecture and expression at a molecular level.
Previous work by our lab, and others, have demonstrated the utility of elemental mass spectrometry in circumventing similar limitations encountered in fluorescence-based flow cytometry7-11. In this approach, termed, “mass cytometry”, cells stained with antibodies carrying isotopically pure, non-biological, elemental metal reporters are nebulized into single-cell droplets prior to sequential analysis via inductively-coupled plasma time-of-flight mass spectrometry. In principle, single-cell analysis of up to 100 parameters can be achieved without spectral overlap between channels11.
Here, we present a modality that uses secondary ion mass spectrometry to image metal isotope carrying antibodies. Multiplexed ion beam imaging (MIBI) is capable of analyzing samples stained simultaneously with up to 100 metal-isotope labeled antibodies and is compatible with standard formalin-fixed, paraffin-embedded (FFPE) tissue sections, the most common type of specimen in clinical repositories worldwide12. Depending on the element of interest, MIBI can achieve as low as parts-per-billion sensitivity with a dynamic range of 105 and resolution comparable to high-magnification light microscopy13-16. We used MIBI to image breast tumor tissue sections stained with clinically relevant metal-conjugated antibodies. The data generated from these experiments could be viewed both in a conventional imaging context as well by using high-dimensional quantitative immunophenotypic feature analysis compatible with higher levels of multiplexing and that can allow classification and unsupervised analysis of each biopsy.
Results
Performance assessment of MIBI
The workflow for MIBI is comparable to IF and IHC assays (Fig. 1). Instead of fluorophores or enzyme-conjugated reagents, biological specimens are incubated with primary antibodies coupled to stable lanthanides highly enriched for a single isotope (Fig. 1). Primary antibodies are combined in solution for simultaneous incubation with the specimen. The specimens prepared for MIBI are mounted in a sample holder and subjected to a rasterized oxygen duoplasmatron primary ion beam. As this ion beam strikes the sample lanthanide adducts of the bound antibodies are liberated as secondary ions. In this study, the secondary ions are subsequently analyzed via a magnetic sector mass spectrometer equipped with multiple detectors, permitting parallel detection of multiple lanthanide isotopes (mass-based reporters). The resultant data produces a two-dimensional map of the elemental distribution of each lanthanide, and thus each antibody and its corresponding epitope.
Figure 1. Workflow summary of multiplexed ion beam imaging (MIBI).
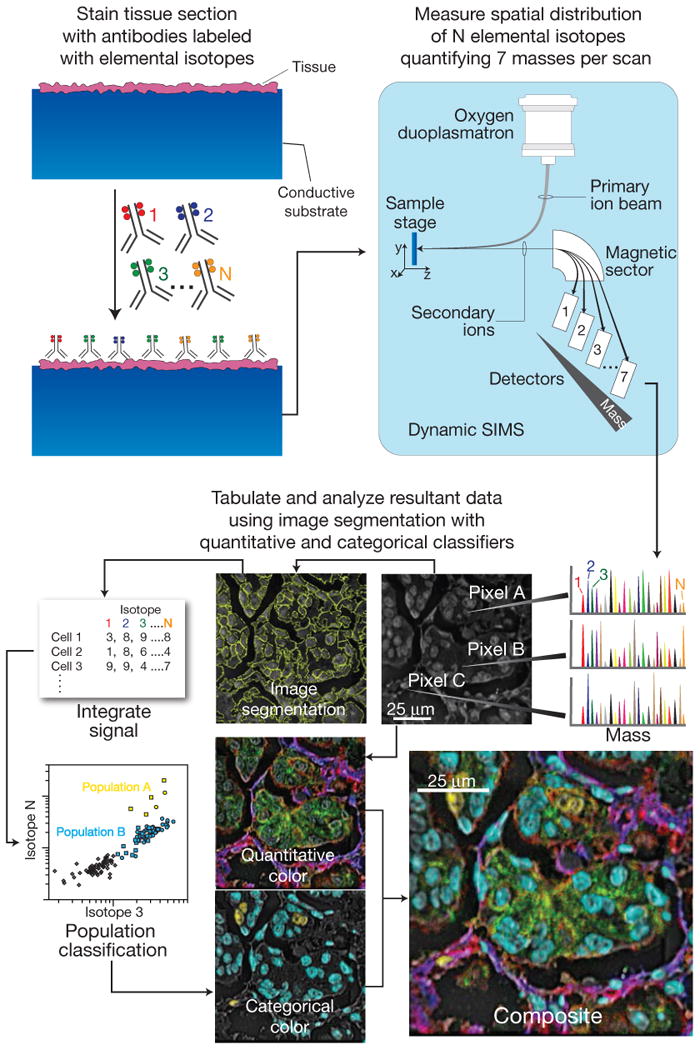
Biological specimens, such as FFPE tissue or cell suspensions, are immobilized on a conductive substrate, such as indium tin oxide coated glass or silicon wafer. Samples are subsequently stained with antibodies conjugated to unique transition element isotope reporters, dried, and loaded under vacuum for MIBI analysis. The sample surface is rasterized with a primary ion beam (O-) that sputters the antibody-specific isotope reporters native to the sample surface as secondary ions. Metal conjugated antibodies are quantified via replicate scans of the same field of view, where up to seven metals reporters are measured with each scan, and the detectors are mass calibrated in between each scan cycle. Regions of interest demarcating nuclear and cytosolic compartments of each cell are integrated, tabulated, and categorized. Composite images comprised of pseudo-colored categorical features and quantitative three-color overlays are constructed to summarize multidimensional expression data.
As part of preliminary validation studies, peripheral blood mononuclear cells (PBMC) stained with seven metal isotope-conjugated primary antibodies (CD3, CD4, CD8, CD14, CD19, CD45, HLA-DR) were assessed in parallel using mass cytometry and MIBI (Fig. 2). Mass cytometry was performed on the PBMC suspension as described previously7. For MIBI, cells were immobilized on a poly-l-lysine-coated silicon wafer, dried under vacuum, and subsequently analyzed using a NanoSIMS 50L™ mass spectrometer. Sequential 50-μm fields were each scanned for 5 min (Fig. 2a). 10-μm overlapping regions of each field were aligned to construct a composite mosaic using an automated script in Matlab. The resultant mosaic was segmented into single-cell regions of interest (ROIs) using the CD45 channel17. To extract single cell expression data for each antibody, the ion count for each channel was integrated for each cell ROI. To achieve this mosaic, 1190 fields were imaged for 5 min per field (99 h total scan time). However, as discussed below, orders-of-magnitude improvements in throughput can be achieved with appropriate sample preparation and instrumentation modifications.
Figure 2. Analysis of PBMCs stained with metal-conjugated antibodies using mass cytometry and MIBI.
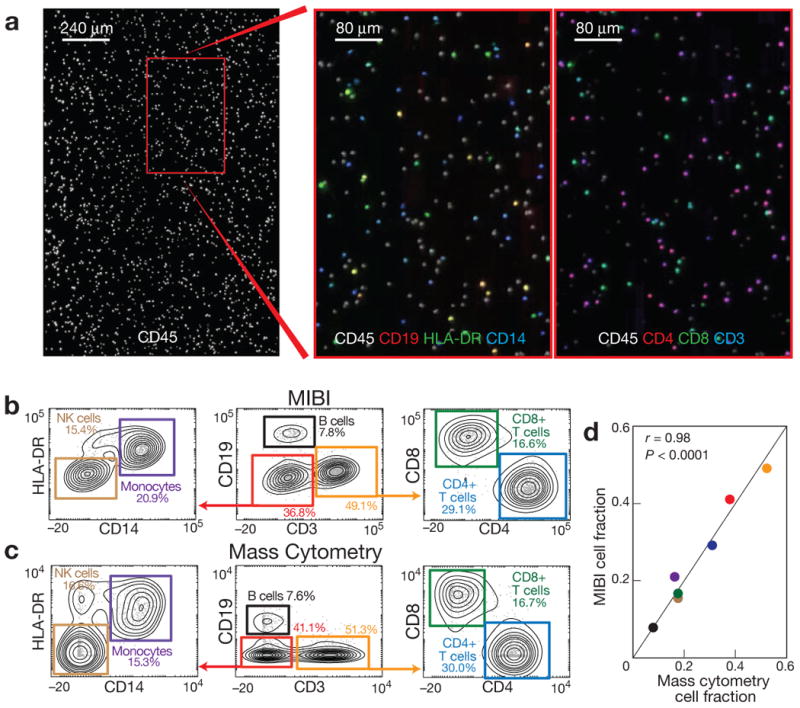
(a) PBMCs stained with seven antibodies were immobilized on a silicon wafer and imaged using MIBI. Single cell regions of interest were segmented using CD45 surface expression and integrated for each antibody. (b, c) Hierarchical gating of the resultant data yielded comparable values for seven cell populations relative to those found by mass cytometry. Biaxial plots are arcsinh(x/5) scaled. (d) Pearson correlation of the relative abundance of each cell population demonstrated strong agreement between the two methods (r = 0.98, P < 0.0001, two-tailed t-test).
Mass cytometry and MIBI produced comparable results and qualitative patterns of expression when analyzed via traditional biaxial plots (Fig. 2b) with marker intensities determined by MIBI demonstrating a dynamic range of 105. Additionally, both platforms yielded quantitatively similar frequencies for seven manually gated cell populations (Fig. 2c), with three of these populations differing by less than 1% between platforms (B-cells, CD8+ T-cells, CD4+ T-cells). Altogether, using PBMCs as a test case, MIBI yielded both qualitatively and quantitatively equivalent results as a conventional analytical platform while also revealing spatial features of protein expression at the subcellular level.
Ten color imaging of human breast tumor tissue sections
In order to utilize MIBI for analysis of tissue sections acquired in a diagnostic setting, we sought to verify the performance of metal-conjugated versions of the antibodies used in conventional IHC staining by comparing staining behavior of metal-conjugated vs. unmodified primary antibodies. Secondary staining of serial sections from a single FFPE human breast tumor tissue block treated with metal-conjugated or unmodified primary antibodies for Ki67 or estrogen receptor alpha (ER) demonstrate positive nuclear staining of comparable intensity and similar levels of background staining (Fig. 3a), indicating that the metal conjugation did not materially affect specific and non-specific staining behavior.
Figure 3. 10-color imaging of human breast tumors using MIBI.
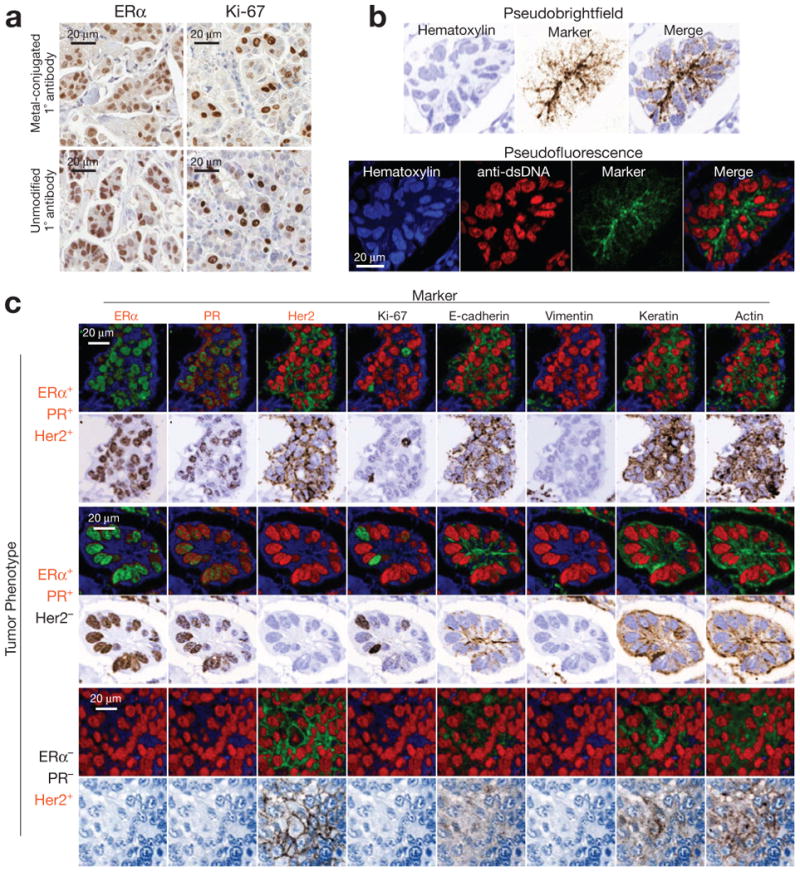
(a) Avidity of primary antibodies is unaffected by metal-conjugation. To access the effect of metal conjugation on antibody avidity, immunoperoxidase staining of serial sections from a single human breast tumor were stained with metal-conjugated or unmodified primary antibodies for Ki67 or ER-alpha. Positive-staining nuclei of comparable intensity were present in similar numbers when using metal-conjugated or unmodified primary antibodies. (b) Visual representation of MIBI data. Single channel ion data can be color mapped and merged to construct pseudo-brightfield or pseudo-darkfield images resembling conventional immunoperoxidase or immunofluorescence staining, respectively. (c) 10-color imaging of human breast tumors. FFPE tissue sections from three different patients were analyzed using MIBI. HER2, ER, and PR are expressed appropriately with respect to the known immunophenotype of each specimen. ER, PR, and Ki67 demonstrate well-demarcated nuclear positivity, while e-cadherin, actin, HER2, and keratin expression is appropriately membranous. Field of view = 80 μm.
Finally, to assess the overall performance of MIBI in a diagnostic imaging application FFPE breast tumor tissue sections from three different patients with different immunophenotypic profiles were analyzed. ER, progesterone receptor (PR), and HER2 positivity were verified in a clinical IHC lab using validated reagents. For MIBI, tumor sections were mounted on poly-l-lysine-coated silicon wafers, deparaffinized, and subjected to heat-induced epitope retrieval prior to overnight staining with metal-conjugated antibodies for dsDNA, ER, progesterone receptor (PR), e-cadherin, Ki67, vimentin, actin, keratin, and HER2. Conveniently, a hematoxylin counterstain can be readily detected by measuring its elemental aluminum content. The following day the sections were washed, counterstained with hematoxylin, and dehydrated via graded ethanol series.
Using the MIBI analysis, conventional high-resolution images can be generated using FFPE tissues. Ten marker images were acquired in two consecutive scans of the same 80-μm field of view, each scan lasting 25 min. Hematoxylin, ER, PR, Ki67, e-cadherin, her2, and dsDNA were acquired during the first scan, and keratin, vimentin, and actin during the second. Pseudo-brightfield images mimicking traditional DAB staining were constructed by encoding hematoxylin on a white-to-blue scale while putting the desired marker on a white to brown scale (Fig. 3b, top). Pseudo-fluorescence images mimicking three-color immunofluorescence were constructed using a red-encoded dsDNA channel, a blue-encoded hematoxylin channel, and a green encoded marker channel (Fig. 3b, bottom). Pseudo-brightfield and pseudo-fluorescence composites for each antibody within a single field of view are shown for each of the three tissue sections (Fig. 3c). Comparison of HER2-, ER-, and PR-positivity across the three specimens demonstrates appropriate expression with respect to immunophenotypes established by conventional IHC staining. Sections expressing ER and PR demonstrate well-demarcated nuclear staining, scattered Ki67-positive nuclei, and intense positive staining for vimentin in mesenchymal cells. HER2+ sections demonstrate strong membrane staining. E-cadherin, actin, and keratin also demonstrate appropriate subcellular staining patterns.
Image segmentation and feature extraction
In order to further explore the utility of the information inherent in the quantitatively multiplexed images in this study, image segmentation was performed so that cellular features could be analyzed and compared. Hematoxylin and dsDNA channels for each tumor were segmented using CellProfiler in order to extract summary statistics describing subcellular expression18,19 (Fig. 4a). The pixel intensity for each marker and subcellular ROI demonstrate distinctly different distributions with respect to the known immunophenotype of each tumor (Supplementary Fig. S1). Mean pixel intensities were quantified for each marker within nuclear, cytoplasmic, and cellular ROIs for each cell. Biaxial scatter plots demonstrate marker coexpression matching the known immunophenotype for each tumor (Fig. 4b). Triple-positive and ER+PR+ tumors demonstrate appropriate nuclear co-expression of ER and PR that is absent in the HER2+ER−PR− tumor. Subpopulations of keratin, e-cadherin-positive ductal cells are distinctly segregated from vimentin-positive mesenchymal cells.
Figure 4. Quantitative analysis of tumor immunophenotype.
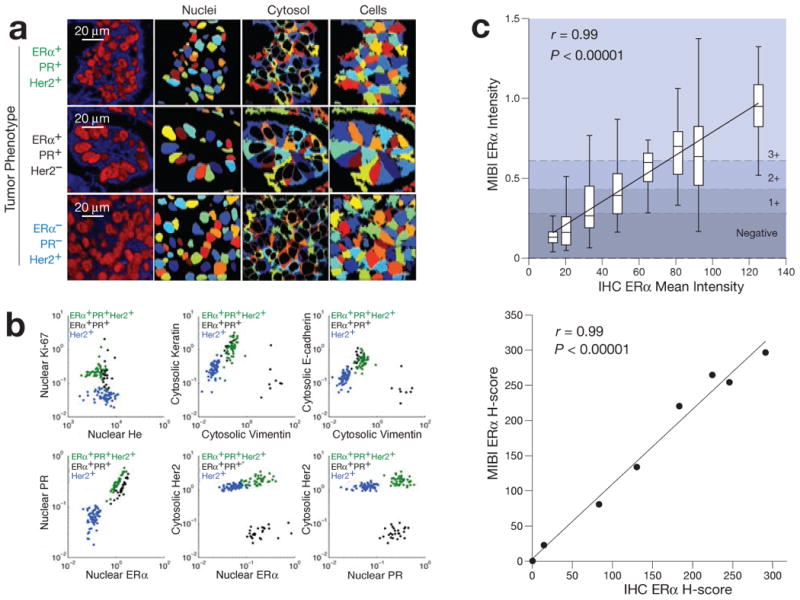
(a) For quantitative single cell analysis, ion images are segmented into ROIs demarcating nuclear and cytoplasmic compartments. (b) Examination of the resultant data using conventional biaxial scatter plots demonstrates quantitative expression patterns matching the known immunophenotype of each respective tumor. Each point represents the mean pixel intensity for each respective cell ROI. Biaxial plots are log scaled. (c) Comparison of ER staining by IHC and MIBI. ER staining intensity of a cohort of breast tumors analyzed using IHC were compared with corresponding values attained using MIBI. Linear regression analysis comparing mean intensities (Top) and H-scores (Bottom) using the two methods demonstrates strong, robust agreement between the two methods (r = 0.99, P < 0.0001).
Quantitative accuracy was further assessed by comparing MIBI to an FDA-approved quantitative image analysis (QIA) workflow for determining the staining intensity of scanned IHC tissue sections. QIA was used to quantify ER nuclear expression in a cohort of breast tumors. Tumor-containing regions were manually annotated and subsequently analyzed using an automated algorithm optimized for determining immunoperoxidase nuclear staining intensity. The resultant data included mean intensity, as well as an overall H-score. Linear regression analysis comparing mean ER nuclear staining intensity by MIBI or IHC demonstrated robust agreement between the two methods (Fig. 4c top, r = 0.99, P < 0.00001). Cutoffs for MIBI staining intensity were calculated with the resultant linear equation (MIBI = 0.064 + 0.0073*IHC) using the respective values for negative, 1+, 2+, and 3+ employed by QIA and subsequently used to calculate an overall H-score. Linear regression analysis comparing IHC and MIBI H-scores also demonstrate strong, robust agreement (Fig. 4c bottom, r = 0.99, P <0.00001) with a slope near unity (m = 1.06 ± 0.06SD). The strong correlation between H-scores derived using the two methods suggests that MIBI not only captured the mean overall staining intensity, but also was able to accurately capture the biological variability of ER expression. This implies that the true distribution of staining intensity was valid and accurately recapitulated. These results also suggest that, at least within the context of its application here, MIBI analysis is not materially affected by sample-to-sample matrix effects that can arise when using a bioanalysis platform. Furthermore, comparison of ER staining intensity in serial sections treated with either all nine antibodies or ER only show that the quantitative accuracy of this method is unaffected by multiplexing (Supplementary Fig. S2).
Integrated histological and immunophenotypic features of multidimensional MIBI data can be visualized by generating composite images that combine quantitative (continuous) cytoplasmic and categorical (positive or negative) nuclear expression patterns (Fig. 5). Hormone-receptor-positive regions within the epithelial compartment, showing variable non-nuclear expression of actin (red) and e-cadherin (green), can be distinguished from interspersed mesenchymal cells co-expressing actin (red) and vimentin (blue). Approximately 8% of cells are seen to be Ki67+. Unlike conventional chromogenic IHC, which is not well-suited to detecting co-localization of multiple markers, MIBI analysis readily demonstrates ER+PR+ (aqua) or ER+PR+Ki67+ (yellow) subpopulations.
Figure 5. Composite representation of multidimensional MIBI data using categorical and quantitative colorization.
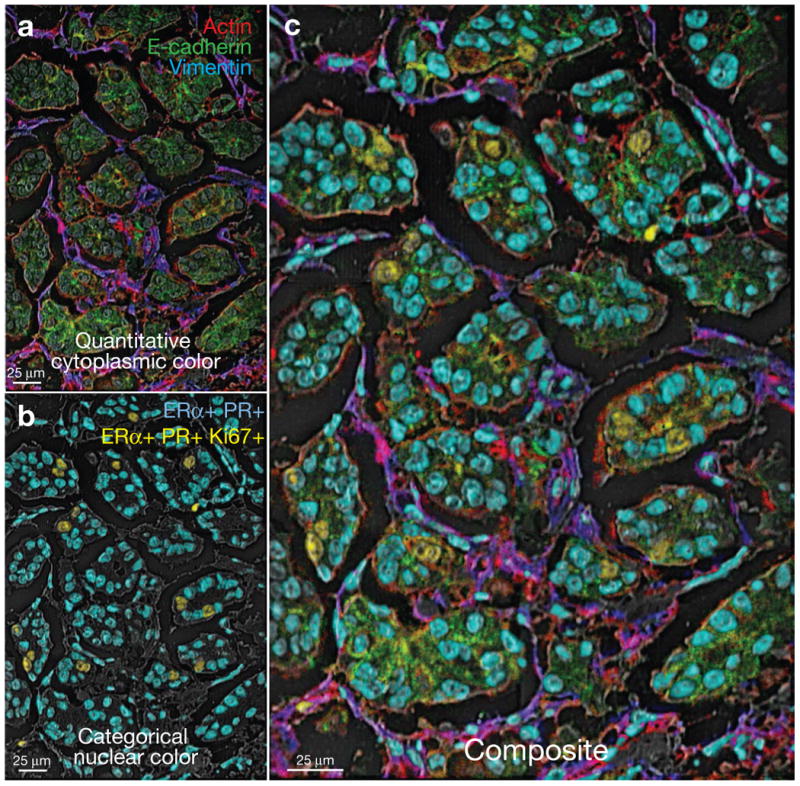
(a) Quantitative colorization of cytoplasmic features. Green-encoded e-cadherin, red-encoded actin, and blue-encoded vimentin channels were merged to generate a quantitative representation of protein expression and colocalization. (b) Categorical colorization of nuclei. Subpopulations of ER+PR+Ki67+ positive or ER+PR+ positive nuclei are pseudo-colored yellow or aqua, respectively. (c) Multidimensional data are summarized in a composite image illustrating quantitative and categorical expression patterns.
Discussion
In this study, we have presented and validated elemental-mass-based multiplexed IHC analysis that circumvents the limitations associated with conventional staining methods relying on optical absorbance or fluorescence signals. This method can be used on virtually any vacuum-compatible specimen, including FFPE tissue. FFPE tissue is the most common type of specimen, with an estimated 1 billion blocks stored in clinical repositories globally11. In validating this method we were able to demonstrate an almost quantitatively identical immunophenotypic analysis of PBMCs compared to a more conventional approach (Fig. 2) as well as equivalent (staining pattern and intensity) imaging of FFPE breast tumors with different immunophenotypes with the additional benefit of simultaneous staining of ten or more markers (Fig. 3). Additionally, marker multiplexing and image segmentation permitted quantitative feature extraction describing cellular and subcellular expression, that in aggregate, revealed immunophenotypes of cell subpopulations that could be related back to the original clinical pathology of the tissue (Fig. 4). The quantitative accuracy of MIBI was demonstrated via side-by-side comparison with an FDA-approved QIA IHC platform. Finally, novel approaches in combinatorial false (or pseudo-) coloring of images could distill the high-dimensional analysis down to a rapidly interpretable single image in which multiple phenotypes could be represented by single colors in an automated fashion (Fig. 5).
MIBI has a number of advantages over conventional IHC techniques. Background signal due to autofluorescence is absent and the dynamic range presented here is already 105, exceeding immunofluorescence and chromogenic IHC by 100-fold and 1000-fold, respectively2,20,21. Because the mass accuracy can resolve less than a fraction of a Dalton at even the lowest resolution (Supplementary Fig. S3) no spectral overlap is observed between mass adjacent elemental reporters. Moreover, the reporter panel can be designed such that neither the residual isotopic contaminants (Supplementary Fig. S3 and Supplementary Table S1) nor the metal oxide abducts (Supplementary Figs. S4 and S5) interfere materially with the reporter masses associated with each antibody probe, obviating the need for channel compensation in the experiments herein. Assay linearity is improved relative to both chromogenic IHC and IF because neither secondary labeling nor amplified detection are required2. Meanwhile, relatively conventional methods are used for immunoreactions, and because mass tags do not degrade, samples are stable indefinitely, permitting remote preparation together with a centralized reading facility.
Immunofluorescence-based high-level multiplexing assays have been reported previously. Multispectral imaging with careful selection of antibodies, secondary antibodies, fluorescent dyes and filter cubes can be used to achieve multiplexing up to seven simultaneous labels, although such performance is rarely achieved, and requires much optimization22. Sequential methods for multiplexing, sometimes referred to as “dye cycling”, have been described that use the general approach of repeated cycles of primary staining (with or without secondary staining), imaging, and then quenching or bleaching or otherwise removing each cycle's fluorescent reporters. Methods for erasing the signals have included low-pH antibody elutions, high-temperature fluorophore denaturation, antibody stripping, or photobleaching23-28. These approaches have several shortcomings not found with MIBI. Some methods of dye cycling lead to accumulative structural changes that alter epitope antigenicity23. The techniques typically use just a few primary antibodies per round of staining, making the iterative methodology labor-intensive and time-consuming. Repeated processing of tissue sections can lead to altered histology that reduces the accuracy of image co-registration across staining cycles. In contrast, samples prepared for MIBI are stained with all antibodies in a single step. Markers are acquired in parallel in a single imaging session without any additional sample processing. Furthermore, histological stains, such as hematoxylin, are also detectable with this technique and can be overlaid with antibody expression data making the representations created here indistinguishable from conventional IHC-based pathological analyses.
Other mass-based reporter systems have been used to image FFPE tissues before29. These have taken two forms: 1) matrix-assisted laser desorption ionization (MALDI) MS, and 2) laser-ablation (LA) inductively coupled plasma (ICP) MS. MALDI-MS imaging, while adept at analyzing macromolecules, has inherent limitations due to its requirement for a crystalline chemical matrix30-32. The matrix combined with the instrument sensitivity reduces achievable resolution (>5 μm) and obscures the signal from elemental reporters. LA-ICP-MS imaging, on the other hand, offers many of the same benefits as MIBI (dynamic range, multiplexing of isotopic reporters). However, laser spot size and instrument sensitivity in previous studies have been limited in elemental reporter-based imaging assays. As such, imaging studies of FFPE breast carcinoma using commercially available LA-ICP-MS systems have yielded images with around 100-μm resolution33– a factor of 103 less than the MIBI analysis presented here. Altogether, such practical constraints would limit the wide application of previously reported mass-based techniques in a clinical setting as compared to MIBI.
One obstacle to broader application of this method is sample throughput. For antigens expressed at levels similar to those analyzed here, a 100-μm field of view for seven antibodies at a resolution comparable to light microscopy (200–300 nm) can be acquired in as little as 5 min. At this rate, 500-μm field of view would require approximately 2 h to image. Larger numbers of antibodies could be acquired with repeat scans, though this would increase the scan time proportionally, however. In future work, we expect to ameliorate the majority of these issues by employing newly developed primary ion beam sources as well as new instrument configurations. Next-generation oxygen ion sources with higher current densities and 50-nm beam spot sizes have recently become commercially available theoretically permitting ∼20-fold faster image acquisition than the current implementation described here34. This new ion beam source will provide lateral resolution comparable to confocal microscopy and axial resolution that exceeds it34. Furthermore, new instrument configurations utilizing a time-of-flight (TOF) mass analyzer would permit parallel quantification of all mass reporters in a given experiment without a cyclic analysis. Taken together, gains from these achievable next-generation instrument configurations should reduce the duty cycle for 50–100 targets (mass reporters) in a 500 μm field of view to 5–10 min when imaged at resolutions equal to light microscopy.
In addition to instrument improvements, lanthanide metal antibody conjugation protocols achieve labeling efficiencies of 100–200 metal atoms per antibody8. The development of new nanoparticle-based reagents are projected to enable the attachment of up to 10,000 metal atoms per antibody. Such gains in labeling efficiency could further reduce scan times by as much as two orders of magnitude, such that a tumor microarray containing 1,000 600 μm core specimens could be imaged in as little as 1.5 h.
As a consequence of using antibodies for protein detection, MIBI inherits many of the issues that can limit the utility of conventional IHC. Poorly characterized reagents can exhibit non-specific binding, and some epitopes are difficult to target with antibody-based approaches. However, we expect that reagents can be developed which extends the capability of MIBI to other arenas and away from antibody-based analysis, such as in situ hybridization and subcellular metabolic analysis. The extended applications of MIBI taken with the gains permitted by relatively minor modifications of existing analytical systems introduce the prospect of a practical, multiplexed imaging platform that integrates tissue histology, protein expression, gene expression, and metabolism on a subcellular level. The basic science utility of such a system is evident, and clinical deployment of this technology would extend multiplexed expression analysis typically restricted to flow cytometry of cell suspensions (such as blood) to any solid tissue. Given the transformative effect that flow cytometry has shown in the diagnosis, staging, and treatment of hematopoietic malignancies, the present approach, applied to solid tissue samples could provide new insight into disease pathogenesis, address pathway activation status, explore tumor heterogeneity, document effects of therapeutic interventions, and, it is to be hoped, improve patient outcomes.
Online Methods
Substrate preparation
Silicon wafers (Silicon Valley Microelectronics) were diced into 18mm2 pieces, rinsed two times with methanol, and polished with a cotton-tipped applicator. Cleaned substrates were subsequently immersed in 2% poly-l-lysine solution (Sigma-Aldrich) for 10 min and baked at 60 °C for 1 hr.
Antibodies
A summary of antibodies, reporter isotopes, and concentrations can be found in Table S1 and S2. Metal conjugated primary antibodies were prepared 100 μg at a time using the MaxPAR antibody conjugation kit (DVS Sciences, Toronto, Canada) according to the manufacturer's recommended protocol. Following labeling, antibodies were diluted in Candor PBS Antibody Stabilization solution (Candor Bioscience GmbH, Wangen, Germany) to 0.4 mg mL-1 and stored long-term at 4 °C.
Cells
Unmatched human peripheral blood was purchased from the Stanford Blood Bank according to an IRB-approved protocol. All blood samples were collected in heparin sulfate anticoagulant, stored at room temperature for 4–6 hrs, and then separated over Ficoll-Paque Plus (Amersham Biosciences) using Accuspin tubes (Sigma-Aldrich, St. Louis, MO) to remove erythrocytes, platelets, and granulocytes. Cells were frozen in FCS with 10% DMSO. Cells were rested at 37 °C, 5% CO2 for 1 h in RPMI with 10% FCS (supplemented with 2mM EDTA in the case of frozen samples), 1X L-glutamine and 1X penicillin with streptomycin (Invitrogen).
Staining of peripheral blood mononuclear cells
Cellular staining protocols were based on procedures previously described. Briefly, after resting cells for 1 hr, surface marker antibodies were added yielding 100 μL final reaction volumes and incubated at room temperature for 30 min. Following incubation, cells were washed two times with cell staining media and split into two aliquots. For mass cytometry analysis, cells were permeabilized with 4 °C methanol for 10 min at 4 °C, washed twice with cell staining media to remove residual methanol, and then stained with 1 mL of 1:4000 191,193Ir DNA intercalator diluted in PBS with 1.6% PFA for 20 min at room temperature. Cells were then washed once with cell staining media, once with PBS, and then diluted in dH2O to approximately 106 cells mL-1 prior to analysis. For MIBI analysis, 50 μL of cells diluted in PBS to approximately 107 cells mL-1 were placed on silicon substrate and allowed to adhere for 20 min. The substrate was then gently rinsed with PBS, fixed for 5 min in PBS with 2% glutaraldehyde, and rinsed twice with dH2O. Lastly, samples were dehydrated via a graded ethanol series, air dried at room temperature, and stored in a vacuum desiccator for at least 24 hrs prior to analysis.
Breast tumor tissue sections
Tissue sections (4 μm thickness) were cut from FFPE tissue blocks of human breast tumor using a microtome, mounted on poly-l-lysine-coated silicon substrate for MIBI analysis or a glass slide for immunoperoxidase (IPOX) staining. Silicon-mounted sections were baked at 65 °C for 15 min, deparaffinized in xylene, and rehydrated via a graded ethanol series. The sections were then immersed in epitope retrieval buffer (10 mM sodium citrate, pH 6) and placed in a pressure cooker for 30 min (Electron Microscopy Sciences, Hatfield, PA). The sections were subsequently rinsed twice with dH2O and once with wash buffer (TBS, 0.1% Tween, pH 7.2). Residual buffer was removed by gently touching the surface with a lint-free tissue prior to incubating with blocking buffer for 30 min (TBS, 0.1% Tween, 3% BSA, 10% donkey serum, pH 7.2). Blocking buffer was subsequently removed and the sections were stained overnight at 4 °C in a humidified chamber. The following morning, the sections were rinsed twice in wash buffer, postfixed for 5 min (PBS, 2% glutaraldehyde), rinsed in dH2O, and stained with Harris hematoxylin for 10s. Finally, the sections were dehydrated via graded ethanol series, air dried at room temperature, and then stored in a vacuum desiccator for at least 24 hrs prior to imaging. Antigen retrieval was performed using a Decloaking Chamber (Biocare Medical, Concord, CA) with citrate buffer at pH 6.0, 125 °C and pressure to 15 psi. The total time slides were in the chamber was 45 min. Incubations with primary antibodies were performed at room temperature overnight in a humidified chamber. Normal goat serum was used for blocking. Biotinylated goat anti-rabbit (1:1000) was the secondary antibody used with a Vectastain ABC Kit Elite and a Peroxidase Substrate Kit DAB (Vector Labs, Burlingame, CA) used for amplification and visualization of signal, respectively. Tissues known to contain each assessed antigen were used as positive controls.
MIBI analysis
MIBI analysis was performed with a NanoSIMS 50L mass spectrometer (Cameca) using an O- primary ion beam supplied by an oxygen duoplasmatron source. The primary optics, secondary optics, and mass spectrometer were tuned prior to each experiment. The seven detector trolleys were tuned to the elemental peak corresponding to each metal isotope conjugated antibody using antibody master mixes that had been air dried on silicon. Images containing more than seven channels were acquired by recalibrating the detector trolleys between repeat scans of the same field. The detectors were tuned to the following masses for the first imaging cycle: detector 1 – 27Al, detector 2 – 139La, detector 3 – 143Nd, detector 4 – 147Sm, detector 5 – 158Gd, detector 6 – 166Er, detector 7 – 176Yb. The following settings were used for the second imaging cycle: detector 4 – 154Sm, detector 5 – 162Dy, detector 6 – 168Er. All data were taken in positive ion mode using D1 aperture 2, D0 aperture 0 or 3, and L1 voltage of approximately 1500 V. Because resolving the metal isotopes of interest requires only unit resolution, entrance slit 0 and aperture slit 0 were used in order to maximize ion transmission to the detectors (Fig. S1 and S2). ROIs identified on serial sections using brightfield microscopy were located using the CCD camera in the NanoSIMS analysis chamber. Ion images were acquired over a 50–100 μm fields of view with pixel dwell times between 2–10 ms and up to 10 repeat scans over a single area. Total scan time for a single field of view ranged between 5–25 min. Larger areas were constructed by stitching together multiple contiguous fields of view into a single mosaic.
Mass cytometry measurement
Cell events were collected on a CyTOF mass cytometer as previously described6. With detection in dual counting mode using the ‘data’ calibration, cell length was set to range from 10–75 with a convolution threshold of 100. A detector stability delay of 20 s was used and all samples were diluted such that the acquisition rate was less than 500 cells per second.
PBMC mosaic stitching
The MIBI PBMC data was collected in a series of 1200 individual square 50 μm (128 pixel) tiles, arranged in a 40x30 rectangle. The relative positions of the tiles were determined using the log-transformed CD45 images. The reported offset between adjacent tiles was 40 μm in both the x- and y-directions, but the actual offset was observed to vary due to imprecision in the stage's location. To account for this, each tile was initially placed according to its reported offset, and then moved around 1–20 pixels in both the x- and y-directions to multiple different positions. At each location, the correlation in the overlap area between the new tile and previous tile was computed. The tile was then assigned to the position that maximized the correlation of the overlapped areas.
PBMC image segmentation
The log-transformed mosaic of CD45 tiles was convolved with a 2-dimensional Gaussian kernel with standard deviation of 3 pixels, and then thresholded at a density of 1. Each continuous region with density greater than this threshold was preliminarily labeled as an individual cell. The next step was to separate into their constituent singlets any sets of multiple cells that were close enough to be initially labeled as single cells. To do this, for each preliminary cell, the two points on the boundary were identified between which there was the maximum ratio of distance along the boundary to Euclidean distance (the “pinch points”). When this ratio exceeded 0.42 (a heuristic cutoff), the preliminary cell was separated into two cells with a new border segment between the pinch points. This process was iterated over all cells, and repeated with each new preliminary cell created, until no cells had pinch points that exceeded this separation criteria17.
Once the cell boundaries were determined, the raw values of each channel measured were summed within each boundary to create a table of total ion intensity on a per-cell basis. The number of pixels within each cell was also calculated as a measure of cell size. This table was equivalent to an .fcs file such as from a standard mass cytometry experiment.
Data analysis
To filter out doublets and debris, singlets were gated from the mass cytometry PBMCs by applying standard cell-length by DNA and then cell-length by CD45 gates; a singlet gate using cell area by CD45 was applied to the MIBI PBMCs. The subsequent gating scheme for both the MIBI and CyTOF processed PBMCs is shown in Figs. 2a and c, respectively.
Supplementary Material
Acknowledgments
We thank Neil Hubbard, Carmina Espiritu, Sandra Rost, Linda Rangell, and the Genentech Human Tissue labs for assistance in preparing and processing tissue sections. We also thank Astraea Jager for technical support with the CyTOF and antibody labeling. M.A. is supported by the Stanford Molecular Imaging Scholars program through the US National Institutes of Health (NIH 5R25CA11868107). S.C.B. is supported by the Damon Runyon Cancer Research Foundation Fellowship (DRG-2017-09) and US National Institutes of Health (1K99 GM104148-01). This work was supported by grants from the US National Science Foundation (0922648), the US National Institutes of Health (0158 G KB065, 1R01CA130826, 5U54CA143907, HHSN272200700038C, N01-HV-00242, 41000411217, 5-24927, P01 CA034233-22A1, P01 CA034233-22A1, PN2EY018228, RFA CA 09-009, RFA CA 09-011, U19 AI057229, U54CA149145), the California Institute for Regenerative Medicine (DR1-01477, RB2-01592), the European Commission (HEALTH.2010.1.2-1), the US Food and Drug Administration (HHSF223201210194C: BAA-12-00118), and the US Department of Defense (W81XWH-12-1-0591 OCRP-TIA NWC).
Footnotes
Author Contributions: M.A., S.B., and R.F. conducted experiments and wrote the manuscript. M.H. designed and fabricated reagents. C.H. assisted in data acquisition and experimental design. A.B. and R.L. prepared tissue sections, performed IHC, and assisted in writing the manuscript. J.L., S.L., S.Z., and Y.N. prepared tissue sections, performed IHC, and assisted in optimizing protocols used in MIBI analysis. G.N. assisted in experimental design and wrote the manuscript.
References
- 1.Coons AH, Creech HJ, Jones RN, Berliner E. The demonstration of pneumococcal antigen in tissues by the use of fluorescent antibody. J Immunol. 1942;45:159–170. [Google Scholar]
- 2.Rimm DL. What brown cannot do for you. Nat Biotechnol. 2006;24:914–916. doi: 10.1038/nbt0806-914. [DOI] [PubMed] [Google Scholar]
- 3.Anagnostou VK, et al. Analytic variability in immunohistochemistry biomarker studies. Cancer Epidemiol Biomarkers Prev. 2010;19:982–991. doi: 10.1158/1055-9965.EPI-10-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bordeaux J, et al. Antibody validation. BioTechniques. 2010;48:197–209. doi: 10.2144/000113382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCabe A, Dolled-Filhart M, Camp RL, Rimm DL. Automated quantitative analysis (AQUA) of in situ protein expression, antibody concentration, and prognosis. J Natl Cancer Inst. 2005;97:1808–1815. doi: 10.1093/jnci/dji427. [DOI] [PubMed] [Google Scholar]
- 6.Hasui K, et al. Double autoimmunostaining with glycine treatment. J Histochem Cytochem. 2003;51:1169–1176. doi: 10.1177/002215540305100907. [DOI] [PubMed] [Google Scholar]
- 7.Bendall SC, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lou X, et al. Polymer-based elemental tags for sensitive bioassays. Angew Chem Int Ed Engl. 2007;46:6111–6114. doi: 10.1002/anie.200700796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ornatsky OI, et al. Development of analytical methods for multiplex bio-assay with inductively coupled plasma mass spectrometry. J Anal Atom Spectrom. 2008;23:463–469. doi: 10.1039/b710510j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ornatsky O, et al. Highly multiparametric analysis by mass cytometry. J Immunol Methods. 2010;361:1–20. doi: 10.1016/j.jim.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 11.Bandura DR, et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal Chem. 2009;81:6813–6822. doi: 10.1021/ac901049w. [DOI] [PubMed] [Google Scholar]
- 12.Blow N. Tissue preparation: Tissue issues. Nature. 2007;448:959–963. doi: 10.1038/448959a. [DOI] [PubMed] [Google Scholar]
- 13.Lechene C, et al. High-resolution quantitative imaging of mammalian and bacterial cells using stable isotope mass spectrometry. J Biol. 2006;5:20. doi: 10.1186/jbiol42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Senyo SE, et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature. 2013;493:433–436. doi: 10.1038/nature11682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steinhauser ML, et al. Multi-isotope imaging mass spectrometry quantifies stem cell division and metabolism. Nature. 2012;481:516–519. doi: 10.1038/nature10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams P. Biological imaging using secondary ions. J Biol. 2006;5:18. doi: 10.1186/jbiol49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gordon A, et al. Single-cell quantification of molecules and rates using open-source microscope-based cytometry. Nat Meth. 2007;4:175–181. doi: 10.1038/nmeth1008. [DOI] [PubMed] [Google Scholar]
- 18.Carpenter AE, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamentsky L, et al. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics. 2011;27:1179–1180. doi: 10.1093/bioinformatics/btr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bodo J, Durkin L, Hsi ED. Quantitative In Situ Detection of Phosphoproteins in Fixed Tissues Using Quantum Dot Technology. J Histochem Cytochem. 2009;57:701–708. doi: 10.1369/jhc.2009.953547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Camp RL, Chung GG, Rimm DL. Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat Med. 2002;8:1323–1327. doi: 10.1038/nm791. [DOI] [PubMed] [Google Scholar]
- 22.Tsurui H, et al. Seven-color fluorescence imaging of tissue samples based on Fourier spectroscopy and singular value decomposition. J Histochem Cytochem. 2000;48:653–662. doi: 10.1177/002215540004800509. [DOI] [PubMed] [Google Scholar]
- 23.Glass G, Papin JA, Mandell JW. SIMPLE: a sequential immunoperoxidase labeling and erasing method. J Histochem Cytochem. 2009;57:899–905. doi: 10.1369/jhc.2009.953612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wählby C, Erlandsson F, Bengtsson E, Zetterberg A. Sequential immunofluorescence staining and image analysis for detection of large numbers of antigens in individual cell nuclei. Cytometry. 2002;47:32–41. [PubMed] [Google Scholar]
- 25.Pirici D, et al. Antibody elution method for multiple immunohistochemistry on primary antibodies raised in the same species and of the same subtype. J Histochem Cytochem. 2009;57:567–575. doi: 10.1369/jhc.2009.953240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedenberger M, Bode M, Krusche A, Schubert W. Fluorescence detection of protein clusters in individual cells and tissue sections by using toponome imaging system: sample preparation and measuring procedures. Nat Protoc. 2007;2:2285–2294. doi: 10.1038/nprot.2007.320. [DOI] [PubMed] [Google Scholar]
- 27.Schubert W, et al. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat Biotechnol. 2006;24:1270–1278. doi: 10.1038/nbt1250. [DOI] [PubMed] [Google Scholar]
- 28.Schubert W, Gieseler A, Krusche A, Hillert R. Toponome mapping in prostate cancer: detection of 2000 cell surface protein clusters in a single tissue section and cell type specific annotation by using a three symbol code. J Proteome Res. 2009;8:2696–2707. doi: 10.1021/pr800944f. [DOI] [PubMed] [Google Scholar]
- 29.Wu B, Becker JS. Imaging of elements and molecules in biological tissues and cells in the low-micrometer and nanometer range. Int J Mass Spectrom. 2011;307:112–122. [Google Scholar]
- 30.Seeley EH, Caprioli RM. Imaging mass spectrometry: Towards clinical diagnostics. Proteomics Clin Appl. 2008;2:1435–1443. doi: 10.1002/prca.200800013. [DOI] [PubMed] [Google Scholar]
- 31.Aerni HR, Cornett DS, Caprioli RM. High-throughput profiling of formalin-fixed paraffin-embedded tissue using parallel electrophoresis and matrix-assisted laser desorption ionization mass spectrometry. Anal Chem. 2009;81:7490–7495. doi: 10.1021/ac900974j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caprioli RM. Perspectives on imaging mass spectrometry in biology and medicine. Proteomics. 2008;8:3679–3680. doi: 10.1002/pmic.200890064. [DOI] [PubMed] [Google Scholar]
- 33.Giesen C, et al. Multiplexed Immunohistochemical Detection of Tumor Markers in Breast Cancer Tissue Using Laser Ablation Inductively Coupled Plasma Mass Spectrometry. Anal Chem. 2011;83:8177–8183. doi: 10.1021/ac2016823. [DOI] [PubMed] [Google Scholar]
- 34.Smith NS, Tesch PP, Martin NP, Kinion DE. A high brightness source for nano-probe secondary ion mass spectrometry. Appl Surf Sci. 2008;255:1606–1609. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.