

Abstract
Live-attenuated influenza vaccines (LAIV) have the potential to generate CD8 T cell immunity that may limit the virulence of an antigenically shifted influenza strain in a population lacking protective antibodies. However, current LAIVs exert limited T cell immunity restricted to the vaccine strains. One approach to improve LAIV-induced T cell responses could be to use specific adjuvants to enhance T cell priming by respiratory dendritic cells (rDCs), but this hypothesis has not been addressed. Here we studied the effect of the toll-like receptor (TLR)-3 ligand poly IC on CD8 T cell immunity and protection elicited by LAIVs. Mucosal treatment with poly IC shortly after vaccination enhanced rDC function, CD8 T cell formation, and production of neutralizing antibodies. This adjuvant effect of poly IC was dependent on amplification of TLR3 signaling by non-hematopoietic radio-resistant cells, and enhanced mouse protection to homosubtypic as well as heterosubtypic virus challenge. Our findings indicate that mucosal TLR3 ligation may be utilized to improve CD8 T cell responses to replicating vaccines, which has implications for protection in the absence of pre-existing antibody immunity.
Introduction
One of the major challenges faced by influenza vaccinology is to develop effective vaccines against a highly variable pathogen which causes seasonal epidemics that do not necessarily result in immunity to subsequent viral challenges (1). In addition, there is an urgent need to develop therapeutic and prophylactic strategies against putative pandemic influenza strains for which most of the human population lacks pre-existing antibody immunity. The development of live-attenuated influenza vaccines (LAIV) has only partially addressed these issues. LAIVs have limited viral replication which allows processing of viral core proteins encoding broadly conserved T cell epitopes (2), thus having the potential to generate broad CD8 T cell-based protection. While this has been consistently demonstrated in mouse models of infection (3, 4), LAIVs still induce sub-optimal cross-reactivity against subtypes of influenza viruses different from the vaccine strains in humans (5). However, the question of whether novel strategies can be developed to increase CD8 T cell immunity induced by LAIVs, and whether these strategies could improve vaccine protection and cross-reactivity has not been addressed.
The quantity and quality of vaccine-induced T cells is established during the innate phase of the immune response when migratory tissue-resident dendritic cells (DCs) encounter pathogen-derived antigens. Tissue DCs are myeloid cells that scan the skin and mucosal surfaces for antigens and that have the ability to process these antigens, transport them to tissue-draining lymph nodes, and prime antigen-specific naïve T cells (6). This process depends on DC maturation/activation which requires signaling through various innate immune receptors including TLRs. A substantial body of work indicates that TLR3+ respiratory DCs (rDCs) expressing CD103+ dominate the transport of influenza antigens to the lung-draining mediastinal lymph nodes (mLN) where they show an exceptional capacity for cross-priming of naïve T cells (7, 8).
Upon encountering with their cognate antigen, naïve T cells rapidly proliferate, and become effector cells with cytotoxic and helper capacity. These clonally expanded T cells, and are eventually massively eliminated during the contraction phase (9). Roughly, 2-5 % of effector T cells survive the contraction phase, giving rise to a small population of antigen-specific, tissue-resident as well as and circulating memory T cells (10). These memory T cell populations are maintained in the host for many months after infection, and in some instances, for the host's lifetime (10).
Polyinosinic-polycytidylic acid (poly IC) is a synthetic mimic of double-stranded RNA, a common subproduct of viral replication. Poly IC is recognized by both surface and cellular pattern-recognition receptors (PRRs) which include at least TLR-3 and melanoma differentiation-associated protein 5 (MDA-5) (11). Due to its ability to promote DC activation, poly IC has been extensively used as adjuvant of inactivated, DC-targeted, DNA, and subunit vaccines (12). However, the putative use of poly IC to boost immune protection generated by LAIVs has not been investigated because, due to their capacity to replicate in the host, LAIVs are believed to be intrinsically adjuvanted. In this study we sought to determine whether poly IC, used as adjuvant after mucosal administration of LAIV, could further potentiate rDC function and generation of vaccine-specific CD8 T cells. We observed that poly IC enhanced the activation and migration of antigen-bearing TLR3+ CD103+ rDCs to the mLNs resulting in significant generation of influenza-specific CD8 T cells and neutralizing antibodies. This, in turn, enhanced mice survival to lethal viral challenge. Loss of TLR3 function in knockout mice, abolished the adjuvant effect of poly IC which was dependent on CD8 T cell immunity but not on neutralizing antibodies. Finally, we demonstrate that poly IC-induced enhancement of CD8 T cell immunity requires amplification of TLR3 signaling by radio-resistant non-hematopoietic cells. Our findings underscore the importance of CD8 T cell responses for LAIV-induced immune protection, and provide the rationale for the use of TLR3 agonists to enhance influenza vaccine protection in a population lacking pre-existing antibody immunity.
Materials and Methods
Mice, reagents, and viruses
C57BL/6J and CD45.1+ coisogenic B6 mice were purchased from Jackson Laboratories and bred at the Heinrich Pette Institute animal facility. TLR3-/- mice (B6N.129S1-Tlr3tm1Flv/J), OT-I mice (C57BL/6-Tg(TcraTcrb)1100Mjb/J), and Langerin-DTR mice (B6.129S2-Cd207tm3Mal/J) were purchased from Jackson Laboratories. All the experiments described were performed with males between 8-10 weeks of age unless otherwise stated. Animal experiments were conducted according to the guidelines of the German animal protection law. All staff carrying out animal experiments passed training programs according to category B or C of the Federation of European Laboratory Animal Science Associations. Experimental vaccination with cold-adapted LAIV as well as viral challenge was achieved by inoculation of the virus solution in PBS directly to the nostrils of mice anesthetized with Isoflurane. Poly IC (Invivogen) was administered intranasally at the indicated time points and concentrations in a PBS solution. Influenza A/PR8/34 (H1N1), X-31 (H3N2), and PR8-GFP viruses were propagated in 10-day-old embryonated chicken eggs at 37 °C.
Generation of recombinant cold-adapted influenza vaccine
The plasmid-based system for rescue of recombinant influenza virus via reverse genetics has been previously described (13). Briefly, influenza segments cloned in ambisense pDZ plasmids were transfected into 293T cells using Lipofectamine 2000 (Invitrogen). 24 h after transfection, supernatants from 293T cells were used to coat influenza-permissive Madin-Darby canine kidney (MDCK) cells in the presence of 1% of TPCK trypsin (Sigma) at 33°C. Viral supernatants from MDCK cells were then inoculated into 9-day-old embryonated chicken eggs for three days at 33°C. Viral rescue was confirmed by PCR-based viral genome amplification and sequencing.
Cell preparation and flow cytometry
Single cell suspensions were obtained from lungs cut into small fragments and digested for 45 min at 37°C with Collagenase D (2mg/ml) (Roche) in RPMI-1640 medium. Digested lungs were further disrupted by passage through a 70-mm nylon cell strainer (BD Biosciences). Red blood cells were lysed with BD Pharm Lysing Buffer (BD Biosciences).
Fc receptors were blocked with CD16/CD32 Fc Block antibody (BD Biosciences) followed by staining with fluorochrome-conjugated antibodies. A FACScanto II instrument (BD Biosciences) was used for flow cytometry. Staining of influenza-specific CD8 T cells was achieved by staining of H-2b-restricted NP366-374 specific T cells using commercial dextramers (Immudex). Analysis of data was performed with FlowJo software (Treestar).
Quantitative RT-PCR
For quantitative RT-PCR (qRT-PCR) analysis, RNA was isolated from the BAL using QIAamp Viral RNA Mini Kit (Qiagen) following the manufacturer's instructions. qRT-PCR was performed using 100 ng sample RNA and SYBR Green (Roche) in an Applied Biosystems Prism 7900HT instrument following the manufacturer's instructions. The specific sequences of the primers utilized are as follows: AArbor NP Fw: 5′ caagagtcagctggtgtgga 3′ and AArbor NP Rv: 5′ gcccagtacctgcttctcag 3′. To calculate the relative index of gene expression, we employed the 2-ΔCt method as described elsewhere.
Bone marrow chimeras
Bone marrow chimeras were generated as described previously (14). In brief, recipient mice were lethally irradiated (550 rad, 4 hr apart by a Caesium source) and reconstituted with 2× 106 bone marrow cells from coisogenic donor mice. Engraftment of donor cells was evaluated 4 weeks after reconstitution in peripheral blood by flow cytometry, and the experiments were performed 6 weeks after transplantation.
OT-I adoptive T cell transfer
Total T cells were purified from spleens of donor OT-I mice using magnetic bead separation procedures (Miltenyi Biotec) and following the instructions provided by the manufacturer. T cells were then labeled with Carboxyfluorescein succinimidyl ester (CFSE) (eBiosciences) as described elsewhere. 2× 106 CFSE-labeled T cells were adoptively transferred into recipient mice via retro-orbital injection.
Focus reduction neutralization assay
Neutralizing antibody titers in mouse serum were determined using serum reduction of viral foci formation in MDCK cells. Briefly, confluent monolayers of MDCKs were treated with two-fold serial dilutions of heat-inactivated sera and mixed at 1:1 ratio with 1000 pfu of PR8 or PR8-GFP. Twenty four hours post-incubation, samples were cold-fixated. To identify virus foci, cells were subjected to immunostaining using rabbit anti-influenza NP antibody or observed under the fluorescence microscope to assess GFP expression.
Results
Poly IC enhances protection exterted by LAIV
Attenuation of influenza viruses by cold adaptation, that is, restriction of viral replication to the upper respiratory tract without causing pulmonary pneumonia, is the basis of currently licensed LAIVs (15). To mimic vaccine formulations currently administered in humans we utilized reverse genetics to engineer a reassortant cold-adapted influenza vaccine harboring the hemagglutinin (HA) and neuraminidase (NA) of the influenza strain A/Puerto Rico/8/34 (H1N1) (hereafter referred to as PR8) on the background of the cold-adapted strain A/Ann Arbor/6/1960 (H2N2) (16). The recombinant H1N1 cold-adapted virus (hereafter referred to as CAV) grew in influenza-permissive cells at 33° C but not at 37° C confirming the temperature-restricted phenotype of the virus (Fig. 1A). Intranasal infection of C57BL/6J mice with up to 104 plaque-forming units (pfu) of CAV did not cause disease in agreement with the restriction of viral replication to the upper respiratory tract (Fig. 1B and (17)).
Figure 1. Effect of mucosal administration of poly IC on cold-adapted vaccine protection.
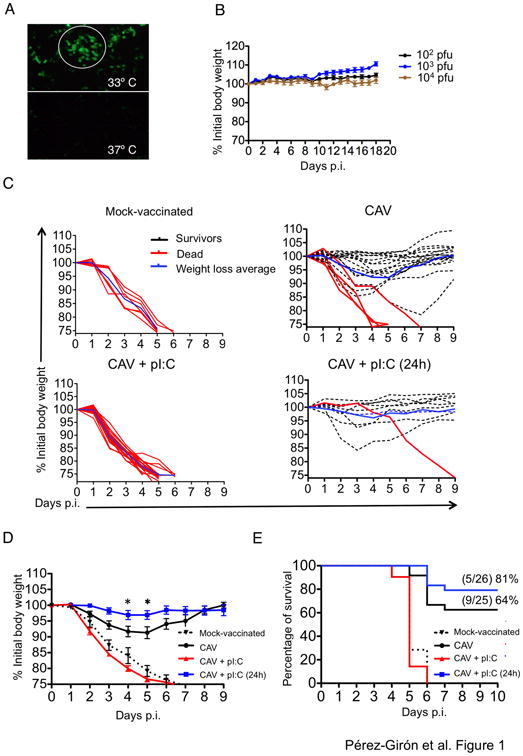
A. Cold-adapted influenza viruses (CAV) harboring HA and NA from the PR8 strain were rescued using reverse genetics protocols. Influenza-permissive Madin-Darby canine kidney (MDCK) cells were infected with CAV at a multiplicity of infection (MOI) of 0.1 for 48h. Cells were fixed and permeabilized and influenza virus was detected by immunofluorescence using anti-NP-FITC antibodies. The white circle indicates an area of viral replication. B. Vaccine safety was assessed in C57BL6/J mice by intranasal administration of the indicated CAV doses and monitored daily for weight loss and clinical signs for up to 20 days. Results represent an average from at least three independent experiments. C. Mice were vaccinated with 103 pfu of CAV in the presence of 20 μg of poly IC or poly IC administered 24h after vaccination. Control mice received PBS instead of vaccine (MOCK-vaccinated). At day 20 post-vaccination mice were challenged with 105 pfu of PR8 (100 × LD50). Mouse weight was monitored daily and mice were sacrificed when they reached the ethical endpoint of 25% loss of their initial weight as per animal approval guidelines. D. Average body weight of mice in all vaccine groups after lethal PR8 challenge. Asterisks denote statistical significance p< 0.05 as assessed by Two-Way ANOVA followed by a Bonferroni post-test. Results represent animals pooled from three independent experiments. E. Kaplan-Meier survival curve showing percentage of mice that survived to lethal influenza challenge in all the indicated conditions. Results represent animals pooled from three independent experiments.
The efficacy of LAIV administered via mucosal route rely on the establishment of limited viral replication, which is necessary to trigger host immunity to the vaccine (15). We hypothesized that a timely intranasal administration of poly IC after the generation of sufficient antigen via viral replication could enhance vaccine-induced protection. To test this hypothesis we vaccinated groups of mice with CAV alone, CAV plus poly IC administered simultaneously, and CAV plus poly IC administered 24 h after vaccination. A group of MOCK-vaccinated mice that received vehicle (PBS) was used as control. We challenged mice at 20 days post-vaccination with a lethal dose of wt PR8 virus (100 × LD50). Analysis of virus-induced morbidity and mortality indicated that poly IC administration 24h after vaccination significantly improved vaccine protection. This group of mice, showed reduced weight loss after lethal influenza challenge compared to mice vaccinated with CAV alone (Fig. 1C and D), as well as enhanced survival (Fig. 1E). Conversely, simultaneous administration of poly IC with CAV completely abrogated the protective effect of the vaccine and all the mice challenged with wt virus died from the infection (Fig. 1C, D and E), in agreement with the ability of poly IC to induce a type I interferon (IFN-I)- dependent antiviral state and prevent vaccine replication (18).
Poly IC improves rDC function and antigen transport to lymphoid tissues
We next hypothesized that the improved antiviral protection observed in the animals treated with poly IC post-vaccination could be due to greater ferrying of vaccine antigen from the lung to the mLN, thereby enhancing the strenght of vaccine-induced adaptive immunity. DCs are regarded as the main cell type capable of transporting peripheral antigens to lymphoid tissues for priming of naïve T cells (6), therefore, we next sought to evaluate rDC responses to intranasal vaccination. rDCs were detected in the lungs of vaccinated mice as CD11c+ MHC class IIhi Siglec-F- cells in the low side-scatter (Supplementary Fig. 1).
We and others have previously demonstrated that intranasal vaccination results in significant increased numbers of CD11b+ rDCs both in the lungs and the mLN, likely due to infiltration and diferentiation of activated blood-borne monocytes (14, 19). However, only CD103+ rDCs transported influenza antigens from the lung to the mLNs, as shown by intracellular staining of viral nucleoprotein (NP) (data not shown), which was in agreement with previous reports (7, 8). To assess the effect of poly IC on antigenic transport by CD103+ rDCs, we engineered a recombinant CAV expressing the ovalbumin (OVA)-derived immunodominant peptide SIINFEKL inserted in the stalk of the viral neuraminidase, hereafter referred to as CAV-SIINFEKL. This vaccine efficiently induced the proliferation of CFSE-labeled naïve TCR-transgenic OVA-specific CD8 T cells (OT-I) that had been adoptively transferred to mice shortly after mucosal vaccination, indicating effective immunity against the heterologous epitope (Fig. 2A). Thus, we quantified the arrival of SIINFEKL-bearing CD103+ rDCs to the mLN of mice that received CAV-SIINFEKL alone, in combination with poly IC, or that were treated with poly IC 24h after vaccination. To this end, we used specific antibodies to detect H-2b-SIINFEKL complexes on the surface of DCs by flow cytometry. Our results indicated that mucosal treatment with poly IC 24h after vaccination with CAV-SIINFEKL significantly increased the arrival of peptide-bearing DCs to the mLNs compared with single vaccine administration so that at day 2 post-vaccination, 30% of total CD103+ migratory DCs expressed SIINFEKL-H-2b complexes on their surface (Fig. 2B). Conversely, simultaneous administration of CAV + poly IC prevented almost entirely the arrival of vaccine antigen to the mLN despite overt migration of CD103+ tDCs (Fig. 2B and data not shown), suggesting that poly IC-induced antiviral state abolished vaccine replication. To explore this possibility, we evaluated the RNA levels of vaccine-derived NP in the bronchoalveolar lavage (BAL) of mice subjected to all the vaccine regimens 48 hours post-vaccination. As shown in Figure 2C, simultaneous CAV+poly IC significantly abrogated vaccine replication compared to mice that received CAV alone as well as those that received CAV+poly IC 24 hours later. Of note, we observed significantly higher levels of vaccine RNA in the BAL of mice that received poly IC treatment 24 hours post-vaccination (Fig. 2C). Taken together, our findings indicate that in order to elicit antigen-specific responses, CD103+ rDCs need to be activated and loaded with sufficient antigenic material in the periphery, which is dependent on vaccine replication.
Figure 2. poly IC enhances migration of antigen-bearing DCs.
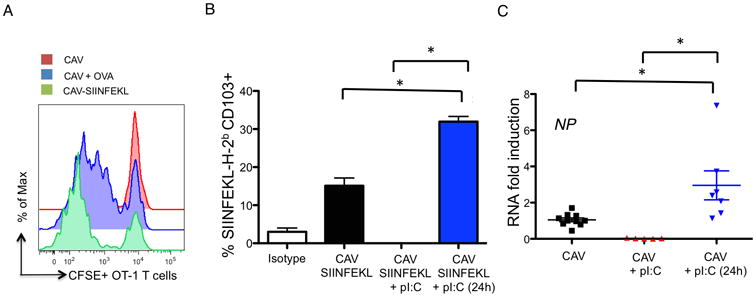
A. Mice vaccinated with the indicated regimens were infused with CFSE-labeled 2× 106 OT-I T cells 3 hours after vaccination. 4 days after vaccination OT-I T cell proliferation was determined in the mLNs by analysis of CFSE dilution in the CD3+ CD4- CD8+ T cell gate. Vaccination with CAV was used as a negative control while CAV + 2 μg of endotoxin-free OVA was used as positive control. B. Mice vaccinated with CAV-SIINFEKL alone, with simultaneous poly IC treatment, or treated with poly IC 24h after vaccination were sacrificed at day 3 post-vaccination. Antigen-bearing migratory CD103+ tDCs were identified as CD103+ H-2b-SIINFEKL+ cells. The graph depicted represents the average of three biological replicates. Asterisks denote statistical significance p<0.05 as assessed by Student's t test. C. Levels of vaccine-derived NP RNA were assessed in the BAL of vaccinated mice 48 h after vaccine administration using primers specific for the A/Ann Arbor/6/60 (H2N2) virus. PCR conditions and analysis are indicated in the Materials and Methods section.
Timely poly IC administration enhances vaccine-specific host immunity
We next assessed the effect of poly IC treatment on host immunity to LAIV. Due to the chief role of CD103+ rDCs on CD8 T cell priming, we first evaluated whether poly IC treatment enhanced vaccine-generated CD8 T cell immunity. We vaccinated mice with CAV, CAV plus simultaneous poly IC, and CAV plus intranasal poly IC 24 h after vaccination. Twenty days later, mice were challenged with wt PR8 and we quantified influenza-specific T cells in the lungs before and after the T cell contraction phase (Fig. 1A). We observed that administration of poly IC 24 h post-vaccination significantly increased the presence of CD8 T cells harboring TCRs specific for the H-2b-restricted immunodominant peptide NP366-374, which were readily detectable already at day 3 post-challenge (Fig. 3B). Also, vaccination with CAV +pIC (24h) resulted in improved maintenance of antigen-specific memory CD8 T cells after the contraction phase compared to CAV alone. The kinetics of NP-specific CD8 T cells in mice vaccinated with CAV plus simultaneous poly IC, mimicked that of MOCK-vaccinated mice, which suggests a primary CD8 T cell response to viral challenge, and therefore lack of pre-existing T cell memory (Fig. 3B and C). These data indicated that post-vaccine administration of poly IC, significantly increased the generation of antigen-specific CD8 T cells in the respiratory tract, presumably enhancing secondary responses to influenza virus at the natural portal of pathogen entry.
Figure 3. Effect of poly IC on vaccine-induced adaptive immunity.
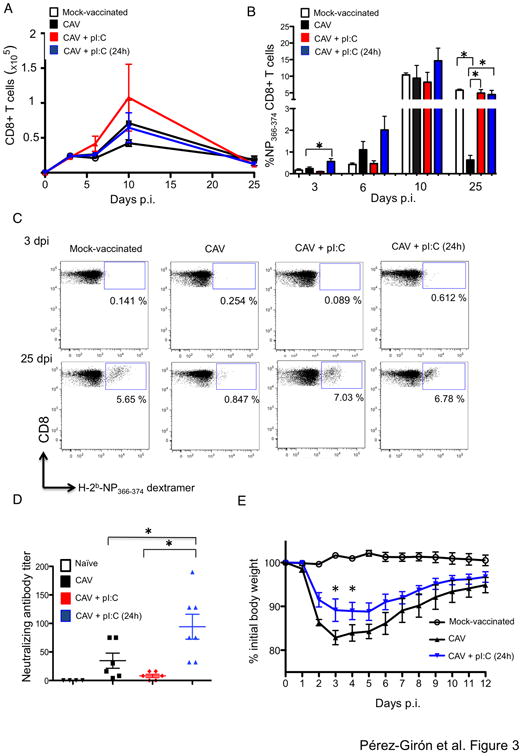
A. Kinetics of total CD8 T cells in the lungs of mice subjected to the indicated vaccine regimens after viral challenge with 103 pfu of PR8. Graphs represent total number of CD8 T cells at the indicated time points in the T cell gate. At least 5 mice per time-point are shown. B. Mice vaccinated intranasally with the indicated regimens were challenged with 103 pfu of PR8. Mice were sacrificed at days 3, 6, 10, and 25 post-infection and NP366-374 antigen specific CD8+ T cells were determined in the lung by multicolor flow cytometry. FITC-conjugated dextramers harboring the immunodominant peptide NP366-374 were utilized to detect vaccine-specific CD8 T cells. Graphs indicate the percentage of dextramer+ cells in the CD8 T cell gate (CD3+ CD4- CD8+). C. Representative plots illustrating the percentage of antigen-specific CD8 T cells in the lungs at days 3 and 25 post-challenge. D. Mice vaccinated with the indicated regimens were bled at day 20 post-vaccination. Pre-immune mice (naïve) are shown for baseline levels. Serum samples were utilized in a focus reduction neutralization assay as described in Materials and Methods. Neutralization titers are represented as the reciprocal of the last dilution at which infection was completely blocked. E. Mice were vaccinated with 103 pfu of CAV alone or with CAV+ poly IC 24 h later. Control mice received PBS instead of vaccine (MOCK-vaccinated). At day 20 post-vaccination mice were challenged with 106 pfu of X-31 virus. Mouse weight was monitored daily and mice were sacrificed when they reached the ethical endpoint of 25% loss of their initial weight. Asterisks denote statistical significance p< 0.05 as assessed by Two-Way ANOVA followed by a Bonferroni post-test.
Based on the recently discovered link between the strenght of the innate immune response and the magnitude of antibody production (20), we reasoned that poly IC treatment could also result in enhanced production of influenza neutralizing antibodies. To test this hypothesis we collected serum samples from mice vaccinated with CAV alone, as well as with simultaneous or post-vaccine administered poly IC at day 20 after immunization. The presence of influenza neutralizing antibodies was determined in serum by focus reduction neutralization assay. Our results indicated that a single administration of CAV resulted in a modest neutralization titer (34.67 ± 13.26) which as expected was not increased by simultaneous administration of CAV + poly IC. However post-vaccine poly IC treatment resulted in three-fold enhancement of influenza neutralizing antibodies in vaccinated mice (Fig. 3D). These results suggested that post-vaccine poly IC treatment engaged both arms of the host immune response likely due to improved induction of innate immunity and enhanced arrival of vaccine antigen to the lymphoid tissues.
A presumable advantage of stronger CD8 T cell immunity is improved vaccine cross-reactivity to heterologous virus. The rationale for this is that T cell epitopes are mostly derived from internal viral proteins that undergo little host antibody pressure, and thus, are highly conserved among distantly related influenza strains (21). To test whether poly IC treatment improved vaccine-induced heterosubtypic immunity, we vaccinated mice with CAV, and 24 h post-vaccination we administered intranasal poly IC or vehicle control (PBS). Twenty days after vaccination we challenged mice with recombinant X-31 virus, a recombinant influenza vaccine candidate harboring six internal segments from PR8 and the HA and NA of the pandemic A/Hong Kong/2/68 (H3N2) (22). Analysis of weight loss in vaccinated mice indicated that poly IC treatment significantly reduced morbidity associated with heterosubtypic X-31 infection (Fig. 3E), which indicated that poly IC not only improved early protection to homosubtypic viral challenge but also improved vaccine-induced heterosubtypic immunity.
The adjuvant effect of poly IC is dependent on TLR3
We next sought to determine the molecular mechanisms responsible for the adjuvant effect of poly IC. To do so, we first determined the effect of TLR3 function on vaccine protection. Wt mice as well as TLR3-/- mice were vaccinated with CAV and treated wih poly IC or PBS 24h post-vaccination. Twenty days later, mice were challenged with PR8 and morbidity/mortality was assessed. As expected, poly IC increased protection of wt mice and significantly reduced influenza associated weight loss. However, loss of TLR3 function completely abolished the adjuvant effect of poly IC (Fig. 4A). Interestingly, in the absence of poly IC treatment, TLR3-/- mice showed significant reduced morbidity when compared with vaccinated wt mice, suggesting that TLR3 signaling may participate in the induction of an antiviral state in vaccinated mice, and thus, may prevent vaccine replication to some extent. Strikingly, loss of TLR3 function did not prevent the ability of poly IC to enhance neutralizing antibody production (Fig. 4B), suggesting that the adjuvant effect of poly IC relies on its capacity to enhance the formation of vaccine-specific CD8 T cells, which was in agreement with previous findings (12).
Figure 4. Role of TLR3 on the adjuvant effect of poly IC.
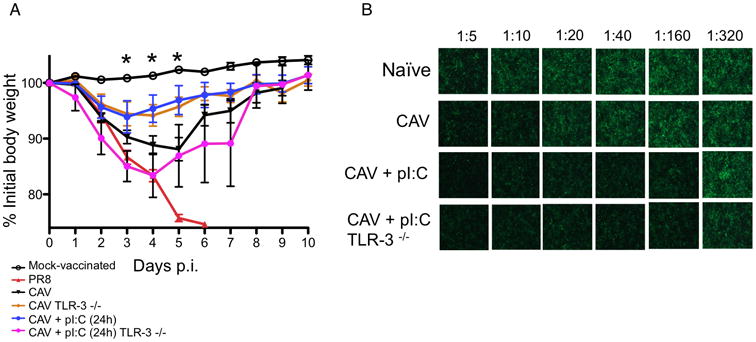
A. Wild-type or TLR-3-/- mice were vaccinated with 103 pfu alone or with poly IC administered 24 hours after vaccination. Control mice received PBS instead of vaccine (MOCK-vaccinated). At day 20 post-vaccination mice were challenged with 105 pfu of PR8. Infection control mice were infected with PR8 in the absence of vaccination. Mouse weight was monitored daily and mice were sacrificed when they reached the ethical endpoint of 25% loss of their initial weight. Results represent individual animals and average from at least three independent experiments. Asterisks denote statistical significance p< 0.05 as assessed by Two-Way ANOVA followed by a Bonferroni post-test. B. Mouse serum at the indicated dilutions was used to coat confluent MDCK cells that were later infected with PR8-GFP at an MOI of 1. Neutralization of virus infection was assessed by evaluation of GFP expression. Naïve (pre-immune) mice were also bled for evaluation of baseline antibody levels.
The adjuvant effect of poly IC requires amplification by radio-resistant cells
To determine the cellular compartments responsible for the amplification of TLR3 signaling in vivo, we next engineered bone marrow chimeric mice in which loss of TLR3 function was targeted to specific cell types. First, we reconstituted irradiated coisogenic CD45.1 mice with a 75:25 mix of Langerin-DTR: wt bone marrow (Lang-WT mice) or a 75:25 mix of Langerin-DTR: TLR3-/- bone marrow (Lang-TLR3-/-). In this model, intranasal administration of DT depleted lung-resident DTR-expressing langerin+ TLR3+ CD103+ DCs, allowing us to study vaccine immunity in the presence of either wt or TLR3-/- CD103+ rDCs (Supplementary Fig. 2). In order to compare vaccine-induced T cell immunity using commercially available dextramers which harbor immunodominant peptides derived from the PR8 strain, we utilized the X-31 candidate vaccine strain (22). Mice were vaccinated with X-31 and treated with poly IC or MOCK-treated (PBS) 24h later. Twenty days after infection we compared the generation of CD8 T cells specific for the NP366-374 epitope between DT-treated Lang-WT and Lang-TLR3-/- mice. Our results indicated that loss of TLR3 function in CD103+ rDCs did not prevent poly IC-induced increase of antigen-specific CD8 T cells, indicating that the effect of poly IC on T cell immunity did not depend on TLR3-mediated sensing of the adjuvant by CD103+ rDCs (Fig. 5A). Thus, we next engineered bone marrow chimeric mice in which TLR3 was specifically absent in hematopoietic cells. These wtTLR3-/- mice were then vaccinated intranasally with or without post-vaccine poly IC treatment. The generation of influenza-specific CD8 T cells was then compared between wtTLR3-/- mice and wt controls (wtwt) 20 days after vaccination. We observed that loss of TLR3 in the hematopoietic compartment did not prevent the enhancement of virus-specific CD8 T cell formation in both chimeras after poly IC treatment, strongly suggesting that the adjuvant effect of poly IC required signal amplification by radio-resistant stromal cells (Fig. 5B). To test this possibility we vaccinated wt as well as TLR3-/- mice with X-31, and again, evaluated the generation of NP-specific CD8 T cells in the lung 20 days later. Loss of TLR3 function in both the hematopoietic and stromal compartments completely abolished the effect of poly IC in the generation of CD8 T cells (Fig. 5C), suggesting that amplification of TLR3 signaling by radio-resistant stromal cells in the lung, is necessary for the adjuvant effect of poly IC. This hypothesis was further confirmed when we engineered reverse chimeras in which loss of TLR3 signaling was restricted to the radio-resistant compartment (TLR3-/-wt). In these mice, poly IC treatment after vaccination did not result in greater generation of memory CD8 T cells in the lung, confirming the requirement of radio-resistant cells for amplification of TLR3 signaling (Fig. 5D).
Figure 5. Cell types responsible for amplification of TLR3 signaling.
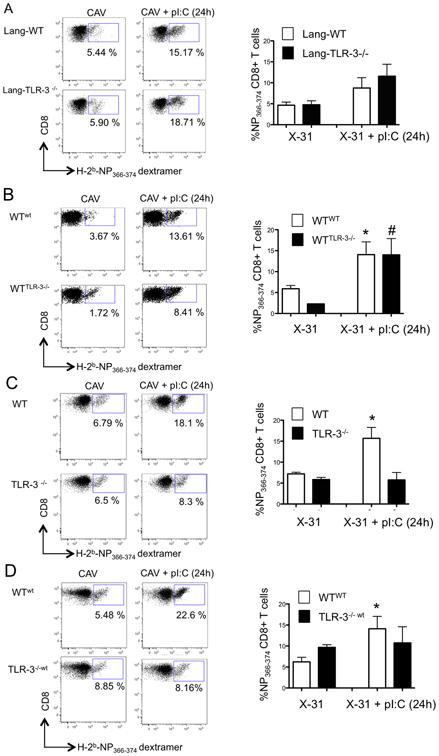
A. Bone marrow chimeric mice were engineered as described in Materials and Methods. Left panel: Lang-wt (Langerin-DTR 75%: wt 25%) and Lang-TLR3-/- (Langerin-DTR 75%: TLR3-/- 25%) were treated intranasally with 50 ng of diphteria toxin (DT). Daily doses were administered starting treatment at day -2 before vaccination and until day 2 post-vaccination. Mice were vaccinated with 500 pfu of X-31 or X-31+ poly IC 24 h later. At day 20 post-vaccination NP366-374-specific CD8 T cells were determined by dextramer staining in the lungs. Left panels show representative plots, and right panel shows the average percentage of dextramer+ CD8 T cells from triplicate samples. B. Bone marrow chimeric mice reconstituted with bone marrow from wild type (WTwt) or TLR-3-/- mice (WTTLR-3-/-) were vaccinated with 3 × 103 pfu of x-31 virus or x-31+ poly IC 24 h later. At day 20 post-vaccination NP366-374-specific CD8 T cells were determined by dextramer staining in the lungs. Left panels show representative plots, and right panel shows the average percentage of dextramer+ CD8 T cells from triplicate samples. C. wt and TLR3-/- mice were vaccinated with X-31 as described above and NP-specific CD8 T cells were asessed in the lungs at day 20 post-vaccination. Left panels show representative plots, and right panel shows the average percentage of dextramer+ CD8 T cells from triplicate samples. D. WT and TLR3-/- mice were lethally irradiated and reconstituted with bone marrow cells from coisogenic CD45.1+ donor mice. Upon reconstitution mice were vaccinated with 3 × 103 pfu of x-31 virus or x-31+ poly IC 24 h later. At day 20 post-vaccination NP366-374-specific CD8 T cells were determined by dextramer staining in the lungs. Left panels show representative plots, and right panel shows the average percentage of dextramer+ CD8 T cells from triplicate samples.
Discusion
With the notable exception of the polio and yellow fever vaccines, most if not all existing prophylactic vaccines exert their protective effect by enhancing antibody responses, including inactivated influenza vaccines (23). However, in order to protect the human population against pandemic as well as antigenically shifted influenza strains, research efforts are needed to develop vaccines that elicit cross-reactive CD8 T cell memory (24) in addition to humoral responses. A number of previous studies have demonstrated that poly IC as adjuvant of inactivated and subunit-based influenza vaccines improves not only antibody responses, but also vaccine-specific T cell immunity (12, 25, 26). Unfortunately, even in the presence of adjuvants, non-replicating influenza vaccines generate very little T cell response (15, 27). Alternatively, simultaneous administration of experimental LAIVs with alpha-galactosil ceramide or chitosan, resulted in enhanced protection to influenza challenge (28, 29), but the involvement of memory T cell responses in vaccine protection was not assessed by these studies. Here, we sought to determine whether mucosal administration of poly IC, could be utilized to boost T cell immunity elicited by LAIVs, via local activation of respiratory DCs, and the generation of broadly conserved T cell epitopes by viral replication (2). We observed that poly IC administered shortly after vaccination enhanced the activation and migratory ability of CD103+ rDCs, the main cross-presenting DC population in the periphery (8). Enhanced rDC function resulted in greater transport of vaccine antigen to the lymphoid tissue and increased generation of vaccine-specific CD8 T cells. Our findings are consistent with previous studies demonstrating that optimization of the amount of antigen that tissue DCs carry to the lymph nodes is essential for proper CD8 T cell responses due to the fact that peptides bound to MHC class I dissociate quickly (30), a situation that is especially limiting in vaccination conditions due to the overall low production of vaccine-derived antigen (31). Of note, poly IC treatment not only enhanced CD8 T cell formation, but also increased production of influenza neutralizing antibodies. The combination of cross-reactive CD8 T cells and neutralizing antibodies has been previously proposed by others as a surrogate of adjuvant strength based upon the idea that it reflects enhanced clonal expansion of naïve T cell pools (32). Remarkably, despite the fact that co-administration of poly IC with LAIV triggered DC migration, the low presence of influenza antigen on migratory DCs prevented sufficient T cell priming. Simultaneous Poly IC+LAIV treatment prevented LAIV replication likely by the ability of poly IC to induce a IFN-I-dependent antiviral state (18). Surprisingly, poly IC administered 24h post-vaccination significantly enhanced vaccine replication. We hypothesize that this may be due to inflammation-dependent protection of viral replication (33, 34), a common phenomenon in the upper-respiratory tract that is especially relevant in the context of influenza and bacterial co-infections (35). Research efforts in our laboratory to elucidate the molecular mechanisms responsible for this effect of poly IC are under way. It is therefore possible, that the enhanced transport of vaccine antigen from the lungs to the mLNs observed in the LAIV+ poly IC (24h) group, may be due not only to enhanced DC function, but also to greater generation of vaccine antigen. In any case, our findings highlight the requirement of sufficient vaccine replication in the upper respiratory tract for the generation of influenza T cell epitopes derived from the viral core proteins (36, 37). In this regard, it is tempting to speculate that the strong induction of type I IFNs observed in humans vaccinated with LAIV in comparison, for example, with individuals vaccinated with YF17D, may prevent sufficient accumulation of influenza antigen by migratory DCs, and thus, may preclude optimal T cell immunity (27).
Previous studies have underscored the effect of poly IC on maintenance of memory T cells, and this effect has been atributted to type I IFN-dependent upregulation of IL-15, TLR3 ligation, and upregulation of pro-inflammatory cytokines such as RANTES and IP-10 at early points after poly IC administration (38, 39). Our results indicated that TLR3 function was required for the adjuvant effect of poly IC but was dispensable for neutralizing antibody production. These results strongly suggest that the increase of vaccine protection elicited by poly IC is dependent on its capacity to generate higher levels of CD8 T cells and not of neutralizing antibodies. These findings are in agreement with previous reports indicating that CD8 T cells can alone generate protective immunity to secondary influenza challenge in the absence of antibodies (40).
Finally, using bone marrow chimeras in which loss of TLR3 function was targeted to specific cell compartments, we identified radio-resistant non-hematopoietic cells as the primary cell type responsible for amplification of TLR3 signaling. These results presumably reflect both the requirement of TLR3 signal amplification for efficient T cell priming (26, 41) and for maintenance of antigen-specific memory T cells (42). These observations are also consistent with the requirement of type I IFN signaling for the generation of CD8 T cells (38, 43). It is also very likely that the observed effects of poly IC at inducing DC activation and CD8 T cell immunity may require signal amplification by additional cellular sensors such as MDA-5 and RIG-I (44, 45), which is the topic of current investigations in our laboratory.
Our study highlights that a timely mucosal poly IC treatment increases CAV-induced CD8 T cell immunity, which play a major protective role after influenza infection. In order to induce efficient CD8 T cell immunity, CAV must replicate in the upper respiratory tract, produce sufficient antigenic material, and allow rDCs to load these antigens and transport them to the lymphoid tissue for T cell priming. Future design of novel adjuvants targeted to coordinate vaccine replication with DC migration could improve T cell memory and protection elicited by mucosal influenza vaccines.
Supplementary Material
Acknowledgments
We thank Ursula Müller and Hasso Münd for excellent technical assistance. J.V. P-G. is a recipient of a postdoctoral fellowship by the Cajamadrid Foundation, J.L.G.C. is a recipient of a Humboldt-Bayer postdoctoral fellowship.
This work was partially supported by funds from the Leibniz Association and the Leibniz Center of Infection (to C. M-F.) and grant U01AI095611 from the NIAID Mucosal Immunity Study Team (MIST) Program (to A. G-S.) and by the Center for Research in Influenza Pathogenesis (CRIP), an NIAID funded Center of Excellence in Influenza Research and Surveillance (CEIRS, HHSN266200700010C) and NIAID contracts HHSN272200900032C and HHSN272201000054C (to A.G-S.).
Abreviations used in this article
- LAIV
live attenuated influenza vaccine
- CAV
cold-adapted vaccine
- rDCs
respiratory dendritic cells
- poly IC
Polyinosinic-polycytidylic acid
- PRR
pattern recognition receptor
- mLNs
mediastinal lymph nodes
- pfu
plaque forming units
- MOI
multiplicity of infection
- LD50
lethal dose for 50% of the population
- DT
diphtheria toxin
- DTR
diphtheria toxin receptor
Footnotes
Disclosures: The authors have no conflicting financial interests.
References
- 1.Medina RA, García-Sastre A. Influenza A viruses: new research developments. Nat Rev Microbiol. 2011;9:590–603. doi: 10.1038/nrmicro2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas PG, Brown SA, Keating R, Yue W, Morris MY, So J, Webby RJ, Doherty PC. Hidden epitopes emerge in secondary influenza virus-specific CD8+ T cell responses. J Immunol. 2007;178:3091–3098. doi: 10.4049/jimmunol.178.5.3091. [DOI] [PubMed] [Google Scholar]
- 3.Epstein SL, Kong WP, Misplon JA, Lo CY, Tumpey TM, Xu L, Nabel GJ. Protection against multiple influenza A subtypes by vaccination with highly conserved nucleoprotein. Vaccine. 2005;23:5404–5410. doi: 10.1016/j.vaccine.2005.04.047. [DOI] [PubMed] [Google Scholar]
- 4.Slütter B, Pewe LL, Kaech SM, Harty JT. Lung airway-surveilling CXCR3(hi) memory CD8(+) T cells are critical for protection against influenza A virus. Immunity. 2013;39:939–948. doi: 10.1016/j.immuni.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nabel GJ, Fauci AS. Induction of unnatural immunity: prospects for a broadly protective universal influenza vaccine. Nat Med. 2010;16:1389–1391. doi: 10.1038/nm1210-1389. [DOI] [PubMed] [Google Scholar]
- 6.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 7.GeurtsvanKessel CH, Willart MA, van Rijt LS, Muskens F, Kool M, Baas C, Thielemans K, Bennett C, Clausen BE, Hoogsteden HC, Osterhaus AD, Rimmelzwaan GF, Lambrecht BN. Clearance of influenza virus from the lung depends on migratory langerin+CD11b- but not plasmacytoid dendritic cells. J Exp Med. 2008;205:1621–1634. doi: 10.1084/jem.20071365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helft J, Manicassamy B, Guermonprez P, Hashimoto D, Silvin A, Agudo J, Brown BD, Schmolke M, Miller JC, Leboeuf M, Murphy KM, García-Sastre A, Merad M. Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J Clin Invest. 2012;122:4037–4047. doi: 10.1172/JCI60659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pepper M, Jenkins MK. Origins of CD4(+) effector and central memory T cells. Nat Immunol. 2011;12:467–471. doi: 10.1038/ni.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woodland DL, Kohlmeier JE. Migration, maintenance and recall of memory T cells in peripheral tissues. Nat Rev Immunol. 2009;9:153–161. doi: 10.1038/nri2496. [DOI] [PubMed] [Google Scholar]
- 11.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 12.Ichinohe T, Watanabe I, Ito S, Fujii H, Moriyama M, Tamura SI, Takahashi H, Sawa H, Chiba J, Kurata T, Sata T, Hasegawa H. Synthetic double-stranded RNA poly(I:C) combined with mucosal vaccine protects against influenza virus infection. J Virol. 2005;79:2910–2919. doi: 10.1128/JVI.79.5.2910-2919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pleschka S, Jaskunas R, Engelhardt OG, Zürcher T, Palese P, Garcia-Sastre A. A plasmid-based reverse genetics system for influenza A virus. J Virol. 1996;70:4188–4192. doi: 10.1128/jvi.70.6.4188-4192.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Munoz-Fontela C, Pazos M, Delgado I, Murk W, Mungamuri SK, Lee SW, García-Sastre A, Moran TM, Aaronson SA. p53 Serves as a Host Antiviral Factor That Enhances Innate and Adaptive Immune Responses to Influenza A Virus. J Immunol. 2011;187:6428–6436. doi: 10.4049/jimmunol.1101459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cox RJ, Brokstad KA, Ogra P. Influenza virus: immunity and vaccination strategies. Comparison of the immune response to inactivated and live, attenuated influenza vaccines. Scand J Immunol. 2004;59:1–15. doi: 10.1111/j.0300-9475.2004.01382.x. [DOI] [PubMed] [Google Scholar]
- 16.Chan W, Zhou H, Kemble G, Jin H. The cold adapted and temperature sensitive influenza A/Ann Arbor/6/60 virus, the master donor virus for live attenuated influenza vaccines, has multiple defects in replication at the restrictive temperature. Virology. 2008;380:304–311. doi: 10.1016/j.virol.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 17.Falcon AM, Fernandez-Sesma A, Nakaya Y, Moran TM, Ortin J, Garcia-Sastre A. Attenuation and immunogenicity in mice of temperature-sensitive influenza viruses expressing truncated NS1 proteins. J Gen Virol. 2005;86:2817–2821. doi: 10.1099/vir.0.80991-0. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in detente. Science. 2006;312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 19.Ballesteros-Tato A, Leon B, Lund FE, Randall TD. Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8(+) T cell responses to influenza. Nat Immunol. 2010;11:216–224. doi: 10.1038/ni.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, Ravindran R, Stewart S, Alam M, Kwissa M, Villinger F, Murthy N, Steel J, Jacob J, Hogan RJ, Garcia-Sastre A, Compans R, Pulendran B. Programming the magnitude and persistence of antibody responses with innate immunity. Nature. 2011;470:543–547. doi: 10.1038/nature09737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grebe KM, Yewdell JW, Bennink JR. Heterosubtypic immunity to influenza A virus: where do we stand? Microbes Infect. 2008;10:1024–1029. doi: 10.1016/j.micinf.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bodewes R, Kreijtz JH, Baas C, Geelhoed-Mieras MM, de Mutsert G, van Amerongen G, van den Brand JM, Fouchier RA, Osterhaus AD, Rimmelzwaan GF. Vaccination against human influenza A/H3N2 virus prevents the induction of heterosubtypic immunity against lethal infection with avian influenza A/H5N1 virus. PLoS ONE. 2009;4:e5538. doi: 10.1371/journal.pone.0005538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sallusto F, Lanzavecchia A, Araki K, Ahmed R. From vaccines to memory and back. Immunity. 2010;33:451–463. doi: 10.1016/j.immuni.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doherty PC, Turner SJ, Webby RG, Thomas PG. Influenza and the challenge for immunology. Nat Immunol. 2006;7:449–455. doi: 10.1038/ni1343. [DOI] [PubMed] [Google Scholar]
- 25.Asahi-Ozaki Y, Itamura S, Ichinohe T, Strong P, Tamura SI, Takahashi H, Sawa H, Moriyama M, Tashiro M, Sata T, Kurata T, Hasegawa H. Intranasal administration of adjuvant-combined recombinant influenza virus HA vaccine protects mice from the lethal H5N1 virus infection. Microbes Infect. 2006;8:2706–2714. doi: 10.1016/j.micinf.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 26.Jelinek I, Leonard JN, Price GE, Brown KN, Meyer-Manlapat A, Goldsmith PK, Wang Y, Venzon D, Epstein SL, Segal DM. TLR3-specific double-stranded RNA oligonucleotide adjuvants induce dendritic cell cross-presentation, CTL responses, and antiviral protection. J Immunol. 2011;186:2422–2429. doi: 10.4049/jimmunol.1002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakaya HI, Wrammert J, Lee EK, Racioppi L, Marie-Kunze S, Haining WN, Means AR, Kasturi SP, Khan N, Li GM, McCausland M, Kanchan V, Kokko KE, Li S, Elbein R, Mehta AK, Aderem A, Subbarao K, Ahmed R, Pulendran B. Systems biology of vaccination for seasonal influenza in humans. Nat Immunol. 2011;12:786–795. doi: 10.1038/ni.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kopecky-Bromberg SA, Fraser KA, Pica N, Carnero E, Moran TM, Franck RW, Tsuji M, Palese P. Alpha-C-galactosylceramide as an adjuvant for a live attenuated influenza virus vaccine. Vaccine. 2009;27:3766–3774. doi: 10.1016/j.vaccine.2009.03.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Zhang W, Liu F, Zheng M, Zheng D, Zhang T, Yi Y, Ding Y, Luo J, Dai C, Wang H, Sun B, Chen Z. Intranasal immunization with live attenuated influenza vaccine plus chitosan as an adjuvant protects mice against homologous and heterologous virus challenge. Arch Virol. 2012;157:1451–1461. doi: 10.1007/s00705-012-1318-7. [DOI] [PubMed] [Google Scholar]
- 30.Zinkernagel RM, Doherty PC. The discovery of MHC restriction. Immunol Today. 1997;18:14–17. doi: 10.1016/s0167-5699(97)80008-4. [DOI] [PubMed] [Google Scholar]
- 31.Henrickson SE, Perro M, Loughhead SM, Senman B, Stutte S, Quigley M, Alexe G, Iannacone M, Flynn MP, Omid S, Jesneck JL, Imam S, Mempel TR, Mazo IB, Haining WN, Von Andrian UH. Antigen availability determines CD8(+) T cell-dendritic cell interaction kinetics and memory fate decisions. Immunity. 2013;39:496–507. doi: 10.1016/j.immuni.2013.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stephenson I, Bugarini R, Nicholson KG, Podda A, Wood JM, Zambon MC, Katz JM. Cross-reactivity to highly pathogenic avian influenza H5N1 viruses after vaccination with nonadjuvanted and MF59-adjuvanted influenza A/Duck/Singapore/97 (H5N3) vaccine: a potential priming strategy. J Infect Dis. 2005;191:1210–1215. doi: 10.1086/428948. [DOI] [PubMed] [Google Scholar]
- 33.Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KCF, Welch M, Schreiber RD, Carlos de la Torre J, Oldstone MBA. Persistent LCMV Infection Is Controlled by Blockade of Type I Interferon Signaling. Science. 2013;340:207–211. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013;340:202–207. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mina MJ, Klugman KP. Pathogen replication, host inflammation, and disease in the upper respiratory tract. Infect Immun. 2013;81:625–628. doi: 10.1128/IAI.01460-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mueller SN, Langley WA, Carnero E, García-Sastre A, Ahmed R. Immunization with live attenuated influenza viruses that express altered NS1 proteins results in potent and protective memory CD8+ T-cell responses. J Virol. 2010;84:1847–1855. doi: 10.1128/JVI.01317-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turner SJ, Diaz G, Cross R, Doherty PC. Analysis of clonotype distribution and persistence for an influenza virus-specific CD8+ T cell response. Immunity. 2003;18:549–559. doi: 10.1016/s1074-7613(03)00087-6. [DOI] [PubMed] [Google Scholar]
- 38.Huber JP, Farrar JD. Regulation of effector and memory T-cell functions by type I interferon. Immunology. 2011;132:466–474. doi: 10.1111/j.1365-2567.2011.03412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kohlmeier JE, Reiley WW, Perona-Wright G, Freeman ML, Yager EJ, Connor LM, Brincks EL, Cookenham T, Roberts AD, Burkum CE, Sell S, Winslow GM, Blackman MA, Mohrs M, Woodland DL. Inflammatory chemokine receptors regulate CD8(+) T cell contraction and memory generation following infection. J Exp Med. 2011;208:1621–1634. doi: 10.1084/jem.20102110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Epstein SL, Lo CY, Misplon JA, Bennink JR. Mechanism of protective immunity against influenza virus infection in mice without antibodies. J Immunol. 1998;160:322–327. [PubMed] [Google Scholar]
- 41.Schulz O, Diebold SS, Chen M, Näslund TI, Nolte MA, Alexopoulou L, Azuma YT, Flavell RA, Liljeström P, Reis e Sousa C. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature. 2005;433:887–892. doi: 10.1038/nature03326. [DOI] [PubMed] [Google Scholar]
- 42.Hervas-Stubbs S, Olivier A, Boisgerault F, Thieblemont N, Leclerc C. TLR3 ligand stimulates fully functional memory CD8+ T cells in the absence of CD4+ T-cell help. Blood. 2007;109:5318–5326. doi: 10.1182/blood-2006-10-053256. [DOI] [PubMed] [Google Scholar]
- 43.Ramos HJ, Davis AM, Cole AG, Schatzle JD, Forman J, Farrar JD. Reciprocal responsiveness to interleukin-12 and interferon-alpha specifies human CD8+ effector versus central memory T-cell fates. Blood. 2009;113:5516–5525. doi: 10.1182/blood-2008-11-188458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, Cella M, Gilfillan S, Colonna M. Cutting edge: polyinosinic: polycytidylic acid boosts the generation of memory CD8 T cells through melanoma differentiation-associated protein 5 expressed in stromal cells. J Immunol. 2010;184:2751–2755. doi: 10.4049/jimmunol.0903201. [DOI] [PubMed] [Google Scholar]
- 45.Schneider-Ohrum K, Giles B, Weirback H. Adjuvants that stimulate TLR3 or NLPR3 pathways enhance the efficiency of influenza virus-like particle vaccines in aged mice. Vaccine. 2011;29:9081–9092. doi: 10.1016/j.vaccine.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.