

Abstract
A green and mild protocol for the dehydrogenative olefination of cyclic enaminones was devised via palladium catalysis at room temperature using oxygen as the terminal oxidant. The synthetic utility of the olefinated cyclic enaminones afforded a series of unique 1,3,5-trisubstituted benzenes via an unanticipated Diels-Alder tandem reaction. The broad substrate scope and good yields achieved with this new protocol provide an alternative pathway for arene functionalization.
Keywords: arenes, C–H activation, cycloaddition, olefination, palladium
Introduction
1,3-Dienes and polyenes are important structural motifs found in many pharmaceutically active compounds and natural products, such as carotenes, vitamin A, bombykol, etc.[1] Synthesis of these compounds can be categorized into two classes: (1) Carbonyl olefination reactions,[2] represented by the Wittig reaction;[3] and (2) Cross olefination coupling, represented by the Heck reaction.[4] In the interests of atom economy, new methods that in addition generate minimal waste are highly desirable.[5]
Dehydrogenative cross-coupling of alkenes, arguably the ideal pathway for diene synthesis, surprisingly did not attract much attention until very recently. Examples of this kind are limited.[6] We recently developed a Pd-catalyzed cross dehydrogenative olefination of cyclic enaminones (Scheme 1a).[7] Despite the high yields and broad scope, our protocol requires stoichiometric amounts of a CuII oxidant and an additive, as well as elevated temperature (80 °C). This demand for sacrificial heavy metal oxidants is very common in the current field and is ironically contradictory to the goals of C–H functionalization (e.g. reducing heavy metal waste).[8] In search of alternative green oxidants, molecular oxygen is the ideal candidate because of its inexpensive and eco-friendly nature.[5]
Scheme 1.
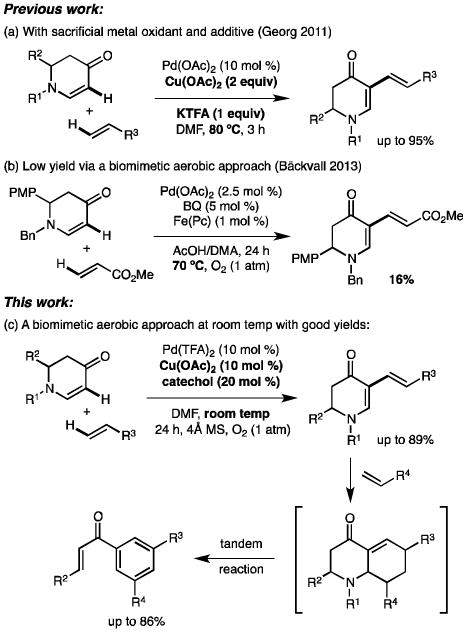
C–H alkenylation of cyclic enaminones.
The obstacle of applying O2 as the sole oxidant is the high activation energy and low concentration of O2 in solutions, which significantly impedes the direct oxidation of Pd0. To circumvent the high pressure of O2 used in early development,[5b, 8c] catalytic amounts of O2 activators, such as molybdovanadophosphoric acid (HPMoV) and benzoquinone, were introduced to promote oxidation in a biomimetic approach.[5a, 8c, 9] Mechanistically, this biomimetic strategy divides the oxidation process into several interconnected redox cycles and thereby effectively reduces the initial high energy barrier for electron transfer.[9] For example, Hosokawa discovered that inexpensive catechol and CuII could remarkably enhance PdII catalysis in the presence of O2 (Scheme 2).[10] Catechol is thought to act as o-quinone and that incorporates CuII as a ligand. With regard to dehydrogenative cross-couplings of alkenes, the use of O2 as a terminal oxidant is very rare and there has been no report of this type of reaction to proceed at room temperature.[11] Bäckvall reported a versatile biomimetic aerobic coupling between two alkenes under low catalyst loading.[11e] However, their method, involving acidic media, failed to deliver satisfactory outcome for cyclic enaminones (Scheme 1b).
Scheme 2.
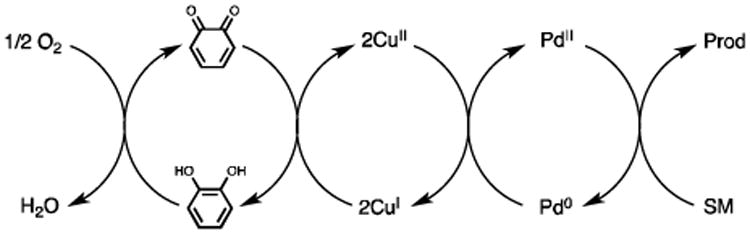
Biomimetic aerobic Pd catalysis with catechol.
In our continuous efforts in generating libraries for biological screening, we are particularly interested in functionalizing the non-aromatic cyclic enaminones as unique piperidine surrogates for alkaloid synthesis.[12] Our aforementioned protocol has furnished various alkenylated enaminones, but their synthetic utility had remained unexplored. In light of the recent advance in aerobic C–H functionalization, we first developed a greener and milder method for dehydrogenative olefination of cyclic enaminones at room temperature (Scheme 1c). Next, we envisioned the resulting dienes participating in a Diels-Alder reaction to gain quick access to hydroquinolines, a key structural motif in several major classes of alkaloids.[13] A serendipitous discovery of a Diels-Alder tandem reaction however led to the formation of a series of distinctive 1,3,5-trisubstituted benzenes, which are important structural motifs in material science[14] and medicinal chemistry[15] because of their unique symmetry. Herein, we disclose our recent efforts in improving the C–H olefination of cyclic enaminones and its unexpected application in the synthesis of 1,3,5-trisubstituted arenes.
Results and Discussion
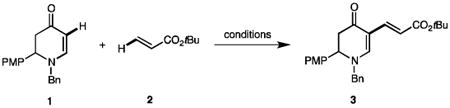
We started by probing aerobic conditions for the dehydrogenative olefination of enaminones under atmospheric pressure (Table 1). To better compare the optimization with our first protocol,[7] the same enaminone 1 and alkene 2 were chosen for the current optimization study. Compared to our reported result (87%, entry 1),[7] the yield dropped significantly to 27% under air without a CuII oxidant (entry 2). Applying pure O2 instead of air increased the yield to 44% (entry 3). Attempts to use 20 mol % of Cu(OAc)2 showed no improvement of the yield (entry 4). In order to activate O2, catalytic amounts of catechol were tested (entries 5–7). Initially, the temperature with Pd(OAc)2/Cu(OAc)2/catechol was set at 80 °C, but no beneficial effect was observed (entry 5). After examining various catalyst loadings and temperatures we were pleased to see a significant increase of yield to 78% in the presence of Pd(OAc)2 (10 mol %) with Cu(OAc)2 and catechol in a 1:1:2 ratio (entry 6). Remarkably, the coupling proceeded smoothly at room temperature. A similar increase on yield was also observed under air, albeit less optimal than that under O2 (entry 7). A series of common solvents were also assessed in addition to DMF (entries 8–10). Although no other solvents improved the olefination outcome, the relatively higher yields from polar solvents (e.g. DMSO, MeCN)indicate that their coordinating ability might help to extend the catalyst lifespan, reflecting on the higher yields. As Hosokawa reported the importance of interchangeable anionic ligands for the catalyst efficacy,[10a] we next evaluated a few anions (i.e. TFA−, OAc−, and Cl−, entries 11–13). The blend of Pd(TFA)2 and Cu(OAc)2 furnished a higher yield (81%, entry 11) compared to those from the OAc−/Cl− or TFA−/Cl− systems (entries 12–13). Presumably, the combination of TFA− and OAc− may sufficiently increase the electrophilicity of the PdII center for palladation, meanwhile balancing the level of acid byproducts (i.e. TFA and AcOH) not to decompose the acid-sensitive enaminones. We also investigated the role of bases and reaction concentrations (Table S6 in the Supporting Information). It soon became clear that bases inhibited the coupling by reducing the yield to 16% and more concentrated solutions (0.2–0.4 M) were favorable in general. Moreover, Stahl reported that molecular sieves were beneficial for Pd-catalyzed aerobic alcohol oxidation, because they could provide a heterogeneous surface that hindered bulk aggregation of Pd0 metal to increase the catalyst stability.[16] Indeed, the addition of 4Å MS promoted a full consumption of enaminone 1 and furnished an optimal isolated yield at 89% (entry 14).
Table 1. Optimization of aerobic dehydrogenative olefination of enaminones.
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Entry[a] | [Pd] (mol %) | [Cu] (mol %) | Atm. | Additive (mol %) | Solvent | Temp (°C) | Yield (%) [b] |
| 1 | Pd(OAc)2 (10) | Cu(OAc)2 (200) | N2 | KTFA (100) | DMF | 80 | 87 (81[c]) |
| 2 | Pd(OAc)2 (10) | – | air | KTFA (100) | DMF | 80 | 27 |
| 3 | Pd(OAc)2 (10) | – | O2 | KTFA (100) | DMF | 80 | 44 |
| 4 | Pd(OAc)2 (10) | Cu(OAc)2 (20) | O2 | KTFA (100) | DMF | 80 | 41 |
| 5 | Pd(OAc)2 (5) | Cu(OAc)2 (5) | O2 | catechol (10) | DMF | 80 | 40 |
| 6 | Pd(OAc)2 (10) | Cu(OAc)2 (10) | O2 | catechol (20) | DMF | rt | 78 |
| 7 | Pd(OAc)2 (10) | Cu(OAc)2 (10) | air | catechol (20) | DMF | rt | 66 |
| 8 | Pd(OAc)2 (10) | Cu(OAc)2 (10) | O2 | catechol (20) | DMSO | rt | 62 |
| 9 | Pd(OAc)2 (10) | Cu(OAc)2 (10) | O2 | catechol (20) | MeCN | rt | 65 |
| 10 | Pd(OAc)2 (10) | Cu(OAc)2 (10) | O2 | catechol (20) | THF | rt | 39 |
| 11 | Pd(TFA)2 (10) | Cu(OAc)2 (10) | O2 | catechol (20) | DMF | rt | 81 |
| 12 | PdCl2 (10) | Cu(OAc)2 (10) | O2 | catechol (20) | DMF | rt | 57 |
| 13 | Pd(TFA)2 (10) | CuCl2 (10) | O2 | catechol (20) | DMF | rt | 27 |
| 14 | Pd(TFA)2 (10) | Cu(OAc)2 (10) | O2 | catechol (20) + 4Å MS | DMF | rt | 91 (89[c]) |
Other conditions: 1 (0.10 mmol), 2 (0.40 mmol), balloon (1 atm), solvent (0.5 mL), 24 h (PMP = para-methoxyphenyl).
1H NMR yields with Ph3SiMe (1.0 equiv) as the internal standard.
Isolated yield. (Detailed optimization is in the Supporting Information.)
Next, we embarked on examining the scope of the reaction (Table 2). Acrylates were excellent alkene sources providing yields of up to 89% (3–6). Vinyl phosphonate, vinyl ketone, acrylamide, and styrene were all well tolerated (7–10). In contrast, disubstituted alkenes gave significantly lower yields (11–13), possibly due to steric hindrance from the substituents that might hinder the migratory insertion. Interestingly, double bond isomerization, common in prior reports,[17] was not observed when more than one β-hydrogen was present in the alkenes. Only conjugated (to enaminone) dienes (12 and 13) were isolated. This is contrary to our earlier olefination protocol,[7] where unconjugated (to enaminone) dienes were favored at an elevated temperature (80 °C). We speculate that the conformer after migratory insertion might favor the β-hydride elimination to furnish a conjugated diene, whereas at a higher temperature a subsequent re-insertion of the Pd–H species could be promoted to reconstruct the Pd intermediate, which eventually furnishes an unconjugated diene as a major product presumably due to its thermal stability. Moreover, the viability of allyl acetate (representing the class of unactivated alkenes) was examined. Unfortunately, it failed to afford any desired product.
Table 2.
Scope of aerobic dehydrogenative olefination of enaminones. [a]
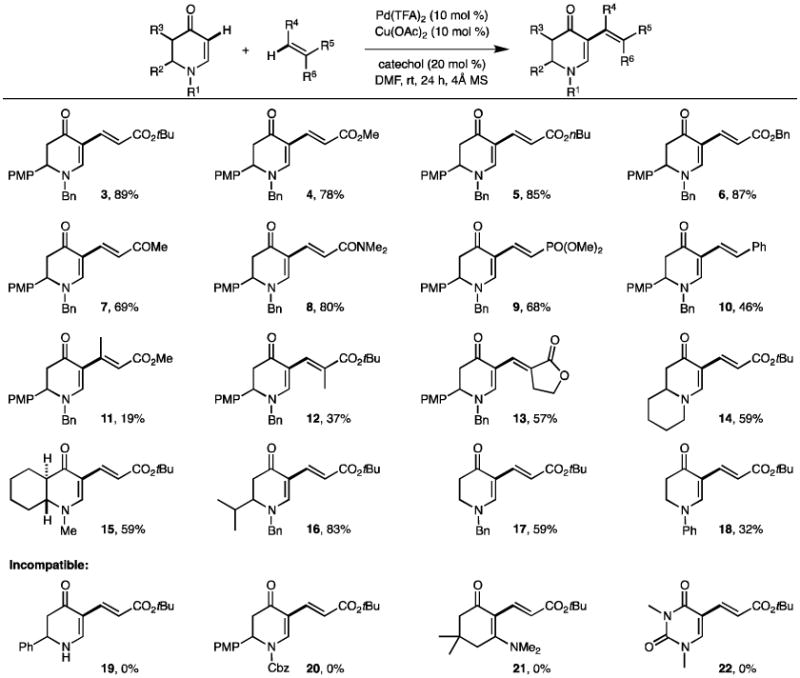
|
Conditions: enaminone (0.10 mmol), alkene (0.40 mmol), Pd(TFA)2 (10 mol %), Cu(OAc)2 (10 mol %), catechol (20 mol %) and 4Å molecular sieves (ca. 30 mg) under O2 (balloon) in DMF (0.5 mL) at rt for 24 h. Isolated yield.
A collection of enaminones was assessed under the optimized conditions as well (Table 2). Bicyclic, electron-rich enaminones offered moderate yields (14–15). Replacing the 2-aryl group (i.e. PMP) with an alkyl group (i.e. iPr) retained a good yield (83%, 16). However, removing the 2-PMP group caused a significant yield decline (to 57%, 17). The reason behind this observation is yet to be elucidated. As we reported before,[7, 18] N-phenylenaminone is a less effective substrate possibly due to its attenuated nucleophilicity. Indeed, it afforded a lower yield (32%, 18) compared to the N-benzyl analogue (57%, 17). It is worth mentioning that the N-H enaminone, N-Cbz enaminone, and E-enaminone were again all incompatible under the current aerobic conditions (19–21), albeit consistent with our previous studies.[18-19] Our attempts on the uracil scaffold (22) were unfortunately not successful either. Nonetheless, we have devised a greener and milder method to alkenylate cyclic enaminones with comparable yields to our first protocol. This is also the first example of cross-dehydrogenative couplings of alkenes taking place at room temperature.
To explore the synthetic utility of our new protocol, we proposed a Diels-Alder reaction to transform the alkenylated enaminones to hydroquinoline analogues, a common structural feature in many alkaloids.[13] In fact, similar amino-substituted dienes have been employed to regio- and stereoselectively construct the octahydroquinoline scaffold in the Comins group.[20] Surprisingly, our attempts on the Diels-Alder reaction between diene 23 and dienophile 24 resulted in the formation of compound 27 instead of the anticipated 25 (Scheme 3). Presumably, the multiple electron-withdrawing substituents and the amino leaving group in cycloadduct 25 promoted aromatization with O2 present in the reaction.[20a, 20c, 21] Afterwards a retro-Michael fragmentation released methylamine[22] to afford 1,3,5-trisubstituted benzene 27.
Scheme 3.
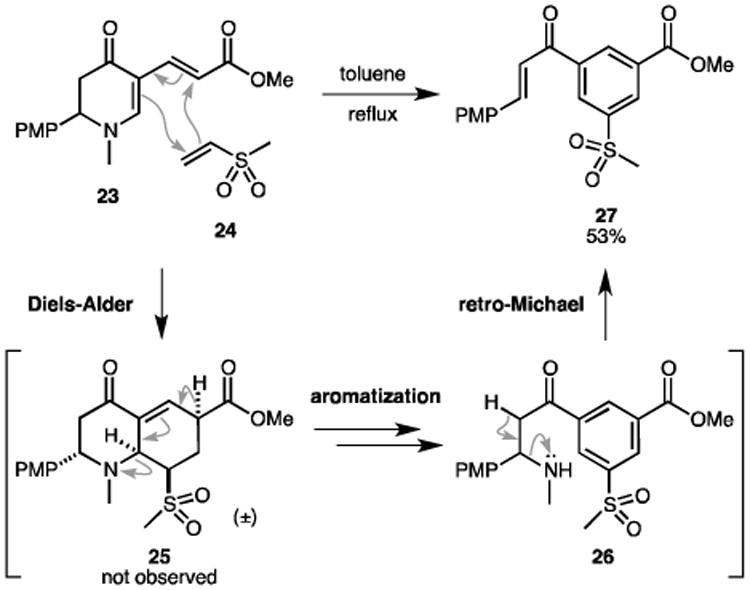
Formation of 1,3,5-trisubstituted benzenes.
Available methods to synthesize 1,3,5-trisubstituted benzenes mostly depend on commercially available 1,3,5-trihalobenzenes (or alike) as precursors with an established substitution pattern.[14, 23] There is a lack of general, regioselective methods to generate unsymmetrical 1,3,5-trisubstituted benzenes, which are more valuable in medicinal chemistry than the symmetrical ones.[15] Encouraged by our results, we considered the potential of the Diels-Alder tandem reaction to devise an alternative useful method to synthesize 1,3,5-trisubstituted benzenes. We hence tested a series of conditions for the Diels-Alder tandem reaction (Tables S7–S9 in the Supporting Information). After screening solvents, microwave techniques, temperatures, reaction time, and stoichiometry, we found that 8.0 equiv of dienophiles in toluene at 160 °C for 24 h delivered the optimal outcome.
The scope of this tandem reaction was then investigated (Table 3). A wide range of electron-deficient dienophiles (e.g. vinyl sulfone, vinyl ketone, acrylates, acrylonitrile, vinyl phosphonate) was viable under the conditions with yields up to 86% (27–33). However, maleimide-generating arene 34 was only obtained in a low yield (16%). Although styrene was not reactive as a dienophile, the flexibility of our protocol allowed us to install styrene beforehand (as R3 in diene 10) via the dehydrogenative olefination, and the tandem reaction thereafter could furnish biphenyls 35 and 36. When acrylic acid was used, expected compound 40 was not detected (Scheme 4a). Instead, a decarboxylation occurred to form 37. Enaminone diene 14 was also subjected to the tandem reaction. Surprisingly, compound 38 was obtained as the final product (Scheme 4b). Due to the unique structural feature in adduct 42, we postulate that a retro-Mannich reaction took place instead of a retro-Michael fragmentation. It is worth noting that the Diels-Alder reaction exhibited excellent regioselectivity, furnishing a distinctive 1,3,5-trisubstitution pattern. In addition, no octahydroquinoline products (such as 25) were left after any of these transformations.
Table 3. Synthesis of 1,3,5-trisubstituted benzenes via the Diels-Alder tandem reaction[a].
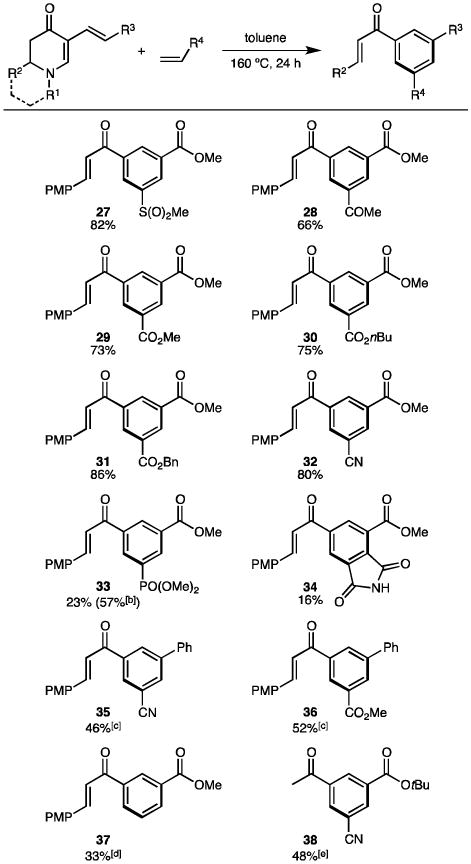
|
Conditions: 23 (0.07 mmol, R1 = Me, R2 = PMP, R3 = CO2Me), dienophile (0.56 mmol) in toluene (1 mL) at 160 °C, 24 h. Isolated yields.
Based on recovered 23.
10 was used as the diene (R1 = Bn, R2 = PMP, R3 = Ph).
Acrylic acid was used as the dienophile.
14 was used as the diene.
Scheme 4.
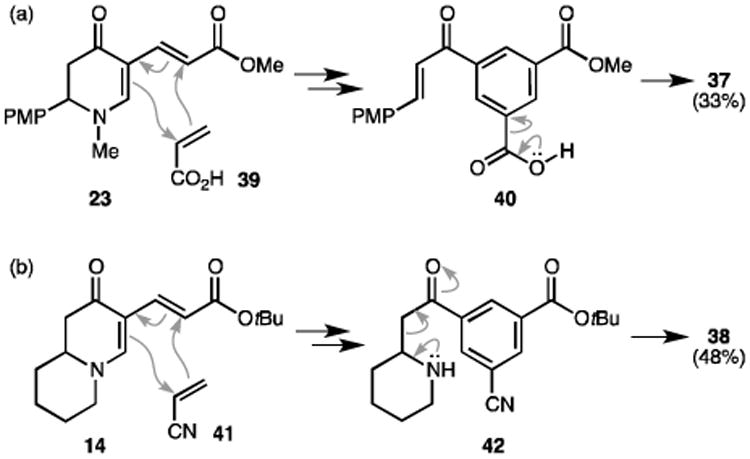
Tandem reactions featuring (a) decarboxylation and (b) retro-Mannich fragmentation.
The advantages of the new tandem reaction in regioselectively functionalizing arenes are three-fold:
A series of electron-withdrawing groups (EWGs) are compatible. Due to the nature of the dehydrogenative alkenylation and the normal-electron-demand Diels-Alder reaction, electron-deficient alkenes are preferred in both reactions (Tables 2 and 3). In particular, functionalities such as sulfone (27) and phosphonate (33) can now be installed onto the phenyl ring from commercially available vinyl reagents in one step, which eliminates the prefunctionalization needed in other methods.[24]
The new protocol provides a direct pathway to synthesize unsymmetrical 1,3,5-EWG-benzenes. The synthesis of this class of arenes is not easy via conventional approaches. For instance, the classics electrophilic substitution would suffer severe deactivation by additional EWGs. Metal-catalyzed/mediated cross-couplings would require meticulously 1,3,5-prefunctionalized benzenes with different (pseudo)halogens.[15] As to C–H functionalization, meta-selectivity has just started to gain more attention with limited conditions and substrate scope.[25]
The distribution of functional groups can also be easily controlled (e.g. symmetrical 28 vs. unsymmetrical 32). Remarkably, several products, such as 38, have three orthogonal functionalities that might be modified in a chemoselective manner. In addition, compounds 35 and 36 have also demonstrated the flexibility of changing the sequence of alkenes used in each step to afford satisfactory yield.
Lastly, we also probed the feasibility of a “one-pot, two-step” synthesis of 1,3,5-trisubstituted benzene 43 directly from enaminone 1 (Scheme 5). Cyclic enaminone 1 was sequentially subjected to the aerobic olefination conditions, followed by the Diels-Alder conditions with no purification in between, which eventually furnished 51% of the desired product 43. Albeit unoptimized, our preliminary results indeed reveal the potential of simply changing alkenes and raising temperatures to generate diverse trisubstituted benzenes in a very convenient manner.
Scheme 5.
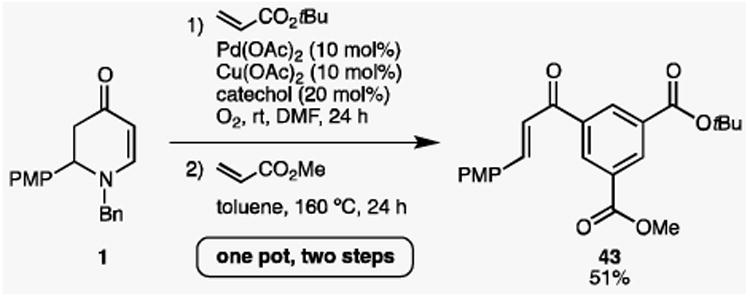
One-pot synthesis of trisubstituted benzene 43.
It is worth noting that most of the products (27–37) are also chalcones, a major class of flavonoids that exhibit a broad spectrum of pharmacological activities (e.g. anti-inflammatory, antiproliferative, antioxidant, and anticancer effects).[26] Previous chalcone syntheses, such as Claisen-Schmidt condensation[27] or Pd-catalyzed cross-coupling methods,[28] paid most attention to constructing the vinylketone linkage of chalcones. Their utilization has been mostly limited by the availability of aryl coupling precursors. In contrast, our approach focuses on the regioselective diversification of arenes, and therefore does not depend as much on the availability of aryl precursors. We expect that the capability to synthesize unique 1,3,5-trisubstituted benzenes shall facilitate the generation of distinctive chalcone libraries for medicinal study.
Conclusion
We improved the dehydrogenative olefination method for cyclic enaminones via a biomimetic approach. The new protocol uses O2 as the terminal oxidant and significantly reduces the heavy metal oxidant Cu(OAc)2 to a catalytic level. This aerobic C–H olefination reaction shows comparable scope and proceeds smoothly at room temperature, which is the first time reported for the dehydrogenative cross-couplings of alkenes.
Our synthetic development of olefinated cyclic enaminones unveiled a Diels-Alder tandem reaction, which led to a series of distinctive 1,3,5-trisubstituted benzenes, including chalcones, with good yields. This unexpected transformation shall offer an alternative approach to synthesize both symmetrical and unsymmetrical 1,3,5-trisubstituted benzenes and chalcones for material science and medicinal study respectively.
Experimental Section
General Information
All reactions were carried out in clear 2-dram vials used without drying. All reagents and anhydrous solvents were purchased and directly used without further purification or drying. Flash column chromatography was carried out on silica gel (230–400 mesh). TLC was conducted on 250 micron, F254 silica gel plates. 1H NMR spectra were recorded at 400 MHz and 13C NMR spectra at 100 MHz with complete proton decoupling. Chemical shifts are reported as ppm relative to chloroform (CHCl3: 7.26 ppm for 1H, 77.16 ppm for 13C). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), coupling constants (Hz) and integration. IR spectra of solids were obtained by dissolving the sample in CH2Cl2 on a NaCl plate. High-resolution mass spectrometry was performed on an ESI-TOF instrument. Melting points were uncorrected.
Preparation of Starting Materials
For details on the synthesis of starting materials, see the Supporting Information.
General Procedures for the Dehydrogenative Aerobic Olefination of Cyclic Enaminones
In a 2-dram vial, cyclic enaminone (0.10 mmol) was mixed with Pd(TFA)2 (3.3 mg, 0.01 mmol), Cu(OAc)2 (1.8 mg, 0.01 mmol), catechol (2.2 mg, 0.02 mmol) and 4Å molecular sieves (ca. 30 mg). To the mixture was added alkene (0.40 mmol), followed by DMF (0.5 mL). The vial was purged with O2 and then sealed with an O2 balloon attached. After stirred at room temperature for 24 h, the reaction was diluted with acetone (2 mL). The mixture was filtered through Celite, and the filter cake was washed with acetone (20 mL). The filtrate was then concentrated under reduced pressure and purified by flash column chromatography on silica gel.
(E)-1-Benzyl-5-(2-(t-butoxycarbonyl)vinyl)-2-(4-methoxyphenyl)-2,3-dihydropyridin-4(1H)-one (3)
Prepared by the general procedure described above and 35.1 mg (89%) was isolated as a light yellow solid (mp 64–68 °C). Analytical data are consistent with those from our previous report.[7]
(E)-1-Benzyl-5-(2-(methoxycarbonyl)vinyl)-2-(4-methoxyphenyl)-2,3-dihydropyridin-4(1H)-one (4)
Prepared by the general procedure described above and 28.0 mg (78%) was isolated as a yellowish solid (mp 52–55 °C). Analytical data are consistent with those from our previous report.[7]
(E)-1-Benzyl-5-(2-(n-butoxycarbonyl)vinyl)-2-(4-methoxyphenyl)-2,3-dihydropyridin-4(1H)-one (5)
Prepared by the general procedure described above and 34.2 mg (85%) was isolated as a yellowish solid (mp 102–104 °C). Analytical data are consistent with those from our previous report.[7]
(E)-5-(2-(Benzoxycarbonyl)vinyl)-1-benzyl-2-(4-methoxyphenyl)-2,3-dihydropyridin-4(1H)-one (6)
Prepared by the general procedure described above and 37.4 mg (87%) was isolated as a yellowish solid (mp 49–52 °C). Analytical data are consistent with those from our previous report.[7]
(E)-1-Benzyl-2-(4-methoxyphenyl)-5-(3-oxobut-1-enyl)-2,3-dihydropyridin-4(1H)-one (7)
Prepared by the general procedure described above and 23.7 mg (69%) was isolated as a yellowish solid (mp 49–52 °C). Analytical data are consistent with those from our previous report.[7]
(E)-1-Benzyl-5-(2-(dimethylcarbamoyl)vinyl)-2-(4-methoxyphenyl)-2,3-dihydropyridin-4(1H)-one (8)
Prepared by the general procedure described above and 29.4 mg (80%) was isolated as a yellow solid (mp 51–53 °C). Analytical data are consistent with those from our previous report.[7]
(E)-1-Benzyl-5-(2-(diethoxyphosphinyl)vinyl)-2-(4-methoxyphenyl)-2,3-dihydropyridin-4(1H)-one (9)
Prepared by the general procedure described above and 27.4 mg (68%) was isolated as a yellow wax. 1H NMR (400 MHz, CDCl3) δ 7.50 (s, 1H), 7.42–7.34 (m, 3H), 7.18–7.07 (m, 4H), 6.95 (dd, J = 24.7, 17.1 Hz, 1H), 6.87 (d, J = 8.5 Hz, 2H), 6.52 (dd, J = 22.2, 17.0 Hz, 1H), 4.51 (t, J = 6.8 Hz, 1H), 4.42 (d, J = 14.9 Hz, 1H), 4.25 (d, J = 14.9 Hz, 1H), 3.81 (s, 3H), 3.72 (d, J = 3.3 Hz, 3H), 3.69 (d, J = 3.2 Hz, 3H), 2.92 (dd, J = 16.4, 7.3 Hz, 1H), 2.70 (dd, J = 16.3, 6.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 188.2, 160.0, 157.3, 145.4 (d, J = 8.0 Hz), 134.7, 129.6, 129.4, 128.9, 128.3, 128.0, 114.8, 106.7 (d, J = 22.0 Hz), 104.5 (d, J = 189.5 Hz), 60.1, 58.2, 55.5, 52.3 (d, J = 5.3 Hz), 52.3 (d, J = 5.3 Hz), 44.3; FTIR (NaCl, cm-1) 3156, 2953, 2930, 2851, 1654, 1600, 1513, 1457, 1442, 1392, 1358, 1298, 1194, 1179, 1059, 1035, 991, 868, 832; HRMS (ESI+) m/e calculated for [M+Na]+ C23H26NO5PNa: 450.1441, found 450.1447.
(E)-1-Benzyl-2-(4-methoxyphenyl)-5-styryl-2,3-dihydropyridin-4(1H)-one (10)
Prepared by the general procedure described above and 17.0 mg (46%) was isolated as a yellow solid (mp 54–56 °C). Analytical data are consistent with those from our previous report.[7]
(E)-1-Benzyl-5-(2-(methoxycarbonyl)-1-methylvinyl)-2-(4-methoxyphenyl)-2,3-dihydropyridin-4(1H)-one (11)
Prepared by the general procedure described above and 7.0 mg (19%) was isolated as a waxy solid. Analytical data are consistent with those from our previous report.[7]
(E)-1-Benzyl-5-(2-(tert-butoxycarbonyl)propenyl)-2-(4-methoxyphenyl)-2,3-dihydropyridin-4(1H)-one (12)
Prepared by the general procedure described above and 12.2 mg (37%) was isolated as a waxy solid. 1H NMR (400 MHz, CDCl3) δ 7.58 (s, 1H), 7.50 (d, J = 0.9 Hz, 1H), 7.39–7.33 (m, 3H), 7.22–7.12 (m, 4H), 6.88 (d, J = 8.6 Hz, 2H), 4.54 (t, J = 7.0 Hz, 1H), 4.41 (d, J = 14.9 Hz, 1H), 4.26 (d, J = 14.9 Hz, 1H), 3.81 (s, 3H), 2.92 (dd, J = 16.4, 7.1 Hz, 1H), 2.73 (dd, J = 16.5, 6.9 Hz, 1H), 1.90 (d, J = 1.2 Hz, 3H), 1.50 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 188.5, 168.4, 159.9, 154.5, 135.4, 131.9, 130.0, 129.3, 128.7, 128.4, 128.0, 123.0, 114.7, 107.3, 80.0, 60.1, 58.1, 55.5, 43.5, 28.4, 15.2; FTIR (NaCl, cm-1) 3035, 2981, 1640, 1594, 1513, 1466, 1442, 1391, 1368, 1299, 1176, 1121, 1035, 833; HRMS (ESI+) m/e calculated for [M+Na]+ C27H31NO4Na: 456.2145, found 456.2144.
(E)-1-Benzyl-2-(4-methoxyphenyl)-5-((2-oxodihydro furan-3(2H)-ylidene)methyl)-2,3-dihydropyridin-4(1H)-one (13)
Prepared by the general procedure described above and 21.2 mg (57%) was isolated as a yellowish solid (mp 59–62 °C). Analytical data are consistent with those from our previous report.[7]
(E)-3-(2-(tert-Butoxycarbonyl)vinyl)-7,8,9,9a-tetra-hydro-1H-quinolizin-2(6H)-one (14)
Prepared by the general procedure described above and 15.5 mg (59%) was isolated as a yellowish solid (mp 122–124 °C). Analytical data are consistent with those from our previous report.[7]
(E)-(trans)-3-(2-(tert-Butoxycarbonyl)vinyl)-1-methyl-4a,5,6,7,8,8a-hexahydroquino-lin-4(1H)-one (15)
Prepared by the general procedure described above and 16.4 mg (59%) was isolated as a yellowish solid (mp 146–148 °C). Analytical data are consistent with those from our previous report.[7]
(E)-1-Benzyl-5-(2-(tert-butoxycarbonyl)vinyl)-2-isopropyl-2,3-dihydropyridin-4(1H)-one (16)
Prepared by the general procedure described above and 27.0 mg (83%) was isolated as a yellow wax. 1H NMR (400 MHz, CDCl3) δ 7.43–7.35 (m, 4H), 7.25 (d, J = 7.0 Hz, 2H), 7.08 (d, J = 15.6 Hz, 1H), 6.50 (d, J = 15.6 Hz, 1H), 4.58 (d, J = 15.0 Hz, 1H), 4.48 (d, J = 15.1 Hz, 1H), 3.32–3.22 (m, 1H), 2.63 (dd, J = 16.6, 7.6 Hz, 1H), 2.47 (dd, J = 16.6, 2.3 Hz, 1H), 2.22 (dq, J = 13.4, 6.7 Hz, 1H), 1.46 (s, 9H), 0.98 (d, J = 6.8 Hz, 3H), 0.92 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 188.7, 168.6, 155.9, 138.9, 135.6, 129.4, 128.9, 127.6, 113.1, 105.9, 79.2, 61.8, 59.8, 37.5, 29.7, 28.4, 19.8, 18.1; FTIR (NaCl, cm-1) 3052, 2975, 2932, 1685, 1654, 1594, 1496, 1455, 1439, 1391, 1367, 1319, 1271, 1241, 1151, 1093, 990, 859; HRMS (ESI+) m/e calculated for [M+Na]+ C22H29NO3Na: 378.2040, found 378.2038.
(E)-1-Benzyl-5-(2-(tert-butoxycarbonyl)vinyl)-2,3-dihydropyridin-4(1H)-one (17)
Prepared by the general procedure described above and 17.7 mg (59%) was isolated as a yellowish oil. Analytical data are consistent with those from our previous report.[7]
(E)-5-(2-(tert-Butoxycarbonyl)vinyl)-1-phenyl-2,3-dihydropyridin-4(1H)-one (18)
Prepared by the general procedure described above and 9.0 mg (32%) was isolated as a yellowish solid (mp 150–152 °C). Analytical data are consistent with those from our previous report.[7]
General Procedures for the Synthesis of 1,3,5-Trisubstituted Benzenes via the Tandem Reaction
In a 2-dram vial, alkenylated cyclic enaminone (0.07 mmol) was mixed with alkene (0.56 mmol) and toluene (1 mL). The vial was then sealed and stirred at 160 °C. After 24 h, the reaction mixture was cooled, concentrated under reduced pressure, and then purified by flash chromatography on silica gel.
(E)-Methyl 3-(3-(4-Methoxyphenyl)acryloyl)-5-(methylsulfonyl)benzoate (27)
Prepared by the general procedure described above and 15.0 mg (82%) was isolated as a yellow solid (mp 185–187 °C). 1H NMR (400 MHz, CDCl3) δ 8.89 (s, 1H), 8.77 (s, 1H), 8.72 (s, 1H), 7.89 (d, J = 15.4 Hz, 1H), 7.65 (d, J = 7.8 Hz, 2H), 7.42 (d, J = 15.5 Hz, 1H), 6.96 (d, J = 7.8 Hz, 2H), 4.02 (s, 3H), 3.88 (s, 3H), 3.15 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.6, 164.9, 162.5, 147.3, 142.2, 140.4, 134.0, 132.5, 131.8, 131.1, 131.0, 127.1, 118.1, 114.7, 55.6, 53.1, 44.5; FTIR (NaCl, cm-1) 3055, 2987, 1732, 1665, 1592, 1571, 1513, 1422, 1325, 1308, 1212, 1174, 1149, 1060, 1030, 984, 961, 896, 828; HRMS (ESI+) m/e calculated for [M+Na]+ C19H18O6SNa: 397.0716, found 397.0716.
(E)-Methyl 3-Acetyl-5-(3-(4-methoxyphenyl)acryloyl)benzoate (28)
Prepared by the general procedure described above and 17.5 mg (66%) was isolated as a yellow solid (mp 72–74 °C). 1H NMR (400 MHz, CDCl3) δ 8.83 (s, 1H), 8.78 (s, 1H), 8.75 (s, 1H), 7.86 (d, J = 15.5 Hz, 1H), 7.65 (d, J = 7.7 Hz, 2H), 7.46 (d, J = 15.4 Hz, 1H), 6.96 (d, J = 7.8 Hz, 2H), 4.00 (s, 3H), 3.87 (s, 3H), 2.72 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.8, 188.7, 165.9, 162.3, 146.4, 139.5, 137.9, 133.4, 132.9, 132.0, 131.5, 130.8, 127.3, 118.7, 114.7, 55.6, 52.8, 27.1; FTIR (NaCl, cm-1) 3055, 2987, 1728, 1693, 1664, 1599, 1572, 1513, 1422, 1361, 1195, 1173, 1031, 985, 829; HRMS (ESI+) m/e calculated for [M+Na]+ C20H18O5Na: 361.1046, found 361.1044.
(E)-Dimethyl 5-(3-(4-Methoxyphenyl)acryloyl)isophthalate (29)
Prepared by the general procedure described above and 20.3 mg (73%) was isolated as a yellow solid (mp 111–113 °C). 1H NMR (400 MHz, CDCl3) δ 8.88 (s, 1H), 8.84 (s, 2H), 7.88 (d, J = 15.4 Hz, 1H), 7.66 (d, J = 7.6 Hz, 2H), 7.46 (d, J = 15.3 Hz, 1H), 6.97 (d, J = 8.7 Hz, 2H), 4.01 (s, 6H), 3.89 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 188.6, 165.8, 162.3, 146.3, 139.4, 134.2, 133.4, 131.4, 130.8, 127.4, 118.8, 114.7, 55.6, 52.8; FTIR (NaCl, cm-1) 3055, 2987, 2956, 1730, 1664, 1601, 1572, 1513, 1422, 1208, 1173, 1032, 1002, 896, 830; HRMS (ESI+) m/e calculated for [M+Na]+ C20H18O6Na: 377.0996, found 377.1005.
(E)-1-Butyl 3-Methyl 5-(3-(4-Methoxyphenyl) acryloyl)isophthalate (30)
Prepared by the general procedure described above and 23.2 mg (75%) was isolated as a yellow wax. 1H NMR (400 MHz, CDCl3) δ 8.85 (s, 1H), 8.82 (s, 2H), 7.86 (d, J = 15.3 Hz, 1H), 7.64 (d, J = 8.1 Hz, 2H), 7.45 (d, J = 15.3 Hz, 1H), 6.96 (d, J = 8.2 Hz, 2H), 4.40 (t, J = 7.2 Hz, 2H), 4.00 (s, 3H), 3.87 (s, 3H), 1.81 (m, 2H), 1.50 (h, J = 7.4, 6.9 Hz, 2H), 1.00 (t, J = 8.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 188.7, 165.8, 165.4, 162.2, 146.3, 139.3, 134.1, 133.4, 133.3, 131.8, 131.3, 130.7, 127.4, 118.8, 114.7, 65.8, 55.6, 52.8, 30.9, 19.4, 13.9; FTIR (NaCl, cm-1) 3053, 2962, 2936, 1725, 1664, 1601, 1572, 1513, 1464, 1443, 1424, 1386, 1288, 1204, 1173, 1109, 1065, 1032, 986, 828; HRMS (ESI+) m/e calculated for [2M+Na]+ C46H48O12Na: 815.3038, found 815.3044.
(E)-1-Benzyl 3-Methyl 5-(3-(4-Methoxyphenyl)acryloyl)isophthalate (31)
Prepared by the general procedure described above and 24.0 mg (86%) was isolated as a yellow wax. 1H NMR (400 MHz, CDCl3) δ 8.88 (s, 1H), 8.85 (s, 1H), 8.82 (s, 1H), 7.85 (d, J = 15.5 Hz, 1H), 7.64 (d, J = 7.8 Hz, 2H), 7.50–7.37 (m, 6H), 6.96 (d, J = 7.8 Hz, 2H), 5.44 (s, 2H), 3.99 (s, 3H), 3.87 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 188.6, 165.8, 165.2, 162.2, 146.3, 139.4, 135.7, 134.2, 133.6, 133.5, 131.5, 131.4, 130.8, 128.8, 128.7, 128.6, 127.4, 118.8, 114.7, 67.6, 55.6, 52.8; FTIR (NaCl, cm-1) 3055, 2987, 1728, 1664, 1601, 1588, 1572, 1513, 1422, 1201, 1173, 1030, 986, 896, 829; HRMS (ESI+) m/e calculated for [M+Na]+ C26H22O6Na: 453.1309, found 453.1314.
(E)-Methyl 3-Cyano-5-(3-(4-methoxyphenyl)acryloyl)benzoate (32)
Prepared by the general procedure described above and 16.6 mg (80%) was isolated as a light yellow solid (mp 257–259 °C). 1H NMR (400 MHz, CDCl3) δ 8.82 (s, 1H), 8.49 (s, 1H), 8.45 (s, 1H), 7.87 (d, J = 15.4 Hz, 1H), 7.65 (d, J = 7.2 Hz, 2H), 7.38 (d, J = 15.4 Hz, 1H), 6.97 (d, J = 6.8 Hz, 2H), 4.01 (s, 3H), 3.88 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.3, 164.7, 162.5, 147.3, 140.0, 136.3, 135.8, 133.0, 132.1, 130.9, 127.1, 117.9, 117.4, 114.8, 113.8, 55.6, 53.2; FTIR (NaCl, cm-1) 3156, 2957, 2932, 2254, 1794, 1731, 1662, 1600, 1571, 1513, 1465, 1444, 1382, 1307, 1287, 1225, 1173, 1096, 1034, 987, 827; HRMS (ESI+) m/e calculated for [M+Na]+ C19H15NO4Na: 344.0893, found 344.0891.
(E)-Methyl 3-(Dimethoxyphosphoryl)-5-(3-(4-methoxyphenyl)acryloyl)benzoate (33)
Prepared by the general procedure described above and 6.0 mg (23%) was isolated as a yellow wax. 1H NMR (400 MHz, CDCl3) δ 8.83 (s, 1H), 8.64 (d, J = 12.1 Hz, 1H), 8.60 (d, J = 12.1 Hz, 1H), 7.87 (d, J = 15.6 Hz, 1H), 7.65 (d, J = 8.6 Hz, 2H), 7.44 (d, J = 15.5 Hz, 1H), 6.96 (d, J = 8.6 Hz, 2H), 3.99 (s, 3H), 3.88 (s, 3H), 3.83 (d, J = 11.1 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 188.5, 165.7, 162.3, 146.6, 139.4 (d, J = 13.9 Hz), 136.4 (d, J = 10.9 Hz), 135.7 (d, J = 11.0 Hz), 133.2 (d, J = 3.0 Hz), 131.4 (d, J = 15.2 Hz), 130.9, 129.1 (d, J = 191.5 Hz), 127.3, 118.6, 114.7, 55.6, 53.2 (d, J = 5.8 Hz), 52.9; FTIR (NaCl, cm-1) 3055, 2987, 1738, 1638, 1512, 1422, 1174, 1033, 896; HRMS (ESI+) m/e calculated for [M+Na]+ C20H21O7PNa: 427.0917, found 427.0929.
(E)-4-Methoxycarbonyl-6-(3-(4-methoxyphenyl)acryloyl)isoindoline-1,3-dione (34)
Prepared by the general procedure described above and 4.4 mg (16%) was isolated as a bright yellow solid (mp 165–167 °C). 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 8.56 (s, 1H), 7.89 (d, J = 15.5 Hz, 2H), 7.65 (d, J = 8.7 Hz, 2H), 7.40 (d, J = 15.5 Hz, 1H), 6.97 (d, J = 8.7 Hz, 2H), 4.05 (s, 3H), 3.88 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 187.5, 165.9, 165.0, 164.5, 162.7, 147.7, 144.0, 134.7, 134.4, 132.7, 131.0, 130.8, 127.0, 125.4, 118.1, 114.8, 55.7, 53.4; FTIR (NaCl, cm-1) 3413, 3055, 2987, 1783, 1744, 1638, 1513, 1421, 1173, 896; HRMS (ESI+) m/e calculated for [M+Na]+C20H15NO6Na: 388.0792, found 388.0791.
(E)-3-Cyano-5-(3-(4-methoxyphenyl)acryloyl)-1,1′-biphenyl (35)
Prepared by the general procedure described above and 10.5 mg (46%) was isolated as a yellow solid (mp 61–63°C). 1H NMR (400 MHz, CDCl3) δ 8.42 (s, 1H), 8.24 (s, 1H), 8.06 (s, 1H), 7.88 (d, J = 15.5 Hz, 1H), 7.70–7.62 (m, 4H), 7.51 (m, 3H), 7.40 (d, J = 15.7 Hz, 1H), 6.98 (d, J = 7.8 Hz, 2H), 3.89 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 188.4, 162.4, 146.7, 143.3, 140.1, 138.3, 134.0, 131.2, 130.8, 130.5, 129.4, 129.0, 127.3, 127.2, 118.6, 118.3, 114.7, 113.6, 55.6; FTIR (NaCl, cm-1) 3155, 2929, 2254, 1794, 1710, 1663, 1597, 1571, 1513, 1465, 1382, 1342, 1308, 1288, 1206, 1173, 1095, 1049, 1032, 985, 827; HRMS (ESI+) m/e calculated for [M+Na]+ C23H17NO2Na: 362.1151, found 362.1153.
(E)-3-Methoxycarbonyl-5-(3-(4-methoxyphenyl)acryloyl)-1,1′-biphenyl (36)
Prepared by the general procedure described above and 13.0 mg (52%) was isolated as a yellow solid (mp 124–126 °C). 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 8.48 (s, 1H), 8.42 (s, 1H), 7.86 (d, J = 15.5 Hz, 1H), 7.73–7.62 (m, 4H), 7.52–7.40 (m, 4H), 6.96 (d, J = 8.0 Hz, 2H), 4.00 (s, 3H), 3.87 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 189.7, 166.6, 162.1, 145.8, 142.3, 139.5, 139.4, 132.1, 131.3, 131.3, 130.6, 129.2, 128.4, 128.2, 127.6, 127.4, 119.4, 114.6, 55.6, 52.6; FTIR (NaCl, cm-1) 3055, 2987, 1723, 1662, 1599, 1572, 1513, 1438, 1422, 1349, 1197, 1173, 1048, 1031, 983, 896, 828; HRMS (ESI+) m/e calculated for [M+Na]+ C24H20O4Na: 395.1254, found 395.1254.
(E)-Methyl 3-(3-(4-Methoxyphenyl)acryloyl)benzoate (37)
Prepared by the general procedure described above and 7.6 mg (33%) was isolated as an off-white solid (mp 93–95 °C). 1H NMR (400 MHz, CDCl3) δ 8.65 (s, 1H), 8.29–8.14 (m, 2H), 7.83 (d, J = 15.5 Hz, 1H), 7.61 (dd, J = 20.1, 8.0 Hz, 3H), 7.44 (d, J = 15.5 Hz, 1H), 6.95 (d, J = 8.0 Hz, 2H), 3.97 (s, 3H), 3.87 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 189.5, 166.4, 161.9, 145.5, 138.8, 133.4, 132.7, 130.6, 130.4, 129.4, 128.9, 127.4, 119.2, 114.5, 55.4, 52.4; FTIR (NaCl, cm-1) 3155, 2984, 2955, 2929, 1793, 1722, 1660, 1595, 1572, 1512, 1466, 1382, 1305, 1291, 1208, 1173, 1096, 1034, 830; HRMS (ESI+) m/e calculated for [M+Na]+ C18H16O4Na: 319.0941, found 319.0946.
tert-Butyl 3-Acetyl-5-cyanobenzoate (38)
Prepared by the general procedure described above and 8.1 mg (48%) was isolated as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 8.71 (s, 1H), 8.42 (s, 1H), 8.37 (s, 1H), 2.67 (s, 3H), 1.63 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 195.3, 163.0, 138.2, 136.8, 135.1, 134.1, 133.0, 117.4, 113.7, 83.4, 28.2, 26.8; FTIR (NaCl, cm-1) 3055, 2986, 2931, 2238, 1721, 1698, 1639, 1552, 1422, 1371, 1330, 1158, 1129, 978, 896, 844; HRMS (ESI+) m/e calculated for [M+Na]+ C14H15NO3Na: 268.0944, found 268.0943.
“One-Pot, Two-Step” Procedures for the Synthesis of Benzene 43
In a 2-dram vial, cyclic enaminone 1 (0.10 mmol) was mixed with Pd(TFA)2 (3.3 mg, 0.01 mmol), Cu(OAc)2 (1.8 mg, 0.01 mmol), catechol (2.2 mg, 0.02 mmol) and 4Å molecular sieves (ca. 30 mg). To the mixture was added t-butyl acrylate (0.40 mmol), followed by DMF (0.5 mL). The vial was purged with O2 and then sealed with an O2 balloon attached. After stirred at room temperature for 24 h, the reaction mixture was filtered through Celite, and the filter cake was washed with acetone (20 mL). The filtrate was then concentrated and mixed with methyl acrylate (0.80 mmol) and toluene (1 mL). The vial was then sealed and stirred at 160 °C. After 24 h, the reaction mixture was cooled, concentrated, and then purified by flash chromatography on silica gel to yield 20.1 mg (51%) of product 43 as a yellow wax. 1H NMR (400 MHz, CDCl3) δ 8.81–8.76 (m, 3H), 7.85 (d, J = 15.6 Hz, 1H), 7.64 (d, J = 8.6 Hz, 2H), 7.45 (d, J = 15.5 Hz, 1H), 6.96 (d, J = 8.7 Hz, 2H), 3.99 (s, 3H), 3.87 (s, 3H), 1.64 (s, 9H); 13C NMR (100 MHz, CDCl3) 188.9, 166.0, 164.5, 162.2, 146.2, 139.2, 134.0, 133.4, 133.3, 133.0, 131.2, 130.7, 127.4, 118.9, 114.7, 82.5, 55.6, 52.8, 28.3; HRMS (ESI+) m/e calculated for [M+H]+ C23H25O6: 397.1646, found 397.1650.
Supplementary Material
Acknowledgments
We greatly appreciate the financial support from the National Institutes of Health (GM081267) and the University of Minnesota through the Vince and McKnight Endowed Chairs (to G.I.G.). We also thank Prof. Jin-Quan Yu and Prof. Dale L. Boger for kindly providing equipment for GC-MS and LC-MS analyses respectively.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/adsc.201######.
References
- 1.Wang G, Mohan S, Negishi Ei. P Natl Acad Sci USA. 2011;108:11344–11349. doi: 10.1073/pnas.1105155108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Majewski M, Gleave DM. Tetrahedron Lett. 1989;30:5681–5684. [Google Scholar]
- 3.Maryanoff BE, Reitz AB. Chem Rev. 1989;89:863–927. [Google Scholar]
- 4.Cabri W, Candiani I. Acc Chem Res. 1995;28:2–7. [Google Scholar]
- 5.a) Shi Z, Zhang C, Tang C, Jiao N. Chem Soc Rev. 2012;41:3381–3430. doi: 10.1039/c2cs15224j. [DOI] [PubMed] [Google Scholar]; b) Punniyamurthy T, Velusamy S, Iqbal J. Chem Rev. 2005;105:2329–2363. doi: 10.1021/cr050523v. [DOI] [PubMed] [Google Scholar]
- 6.Shang X, Liu ZQ. Chem Soc Rev. 2013;42:3253–3260. doi: 10.1039/c2cs35445d. [DOI] [PubMed] [Google Scholar]
- 7.Yu YY, Niphakis MJ, Georg GI. Org Lett. 2011;13:5932–5935. doi: 10.1021/ol202677g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Yeung C, Dong V. Chem Rev. 2011;111:1215–1292. doi: 10.1021/cr100280d. [DOI] [PubMed] [Google Scholar]; b) Liu C, Zhang H, Shi W, Lei A. Chem Rev. 2011;111:1780–1824. doi: 10.1021/cr100379j. [DOI] [PubMed] [Google Scholar]; c) Le Bras J, Muzart J. Chem Rev. 2011;111:1170–1214. doi: 10.1021/cr100209d. [DOI] [PubMed] [Google Scholar]; d) Cho SH, Kim JY, Kwak J, Chang S. Chem Soc Rev. 2011;40:5068–5083. doi: 10.1039/c1cs15082k. [DOI] [PubMed] [Google Scholar]
- 9.Piera J, Bäckvall JE. Angew Chem. 2008;120:3558–3576. [Google Scholar]; Angew Chem Int Ed. 2008;47:3506–3523. doi: 10.1002/anie.200700604. [DOI] [PubMed] [Google Scholar]
- 10.a) Kawamura Y, Kawano Y, Matsuda T, Ishitobi Y, Hosokawa T. J Org Chem. 2009;74:3048–3053. doi: 10.1021/jo802807w. [DOI] [PubMed] [Google Scholar]; b) Minami K, Kawamura Y, Koga K, Hosokawa T. Org Lett. 2005;7:5689–5692. doi: 10.1021/ol052377l. [DOI] [PubMed] [Google Scholar]
- 11.a) Hatamoto Y, Sakaguchi S, Ishii Y. Org Lett. 2004;6:4623–4625. doi: 10.1021/ol047971u. [DOI] [PubMed] [Google Scholar]; b) Xu YH, Chok YK, Loh TP. Chem Sci. 2011;2:1822–1825. [Google Scholar]; c) Bai Y, Zeng J, Cai S, Liu XW. Org Lett. 2011;13:4394–4397. doi: 10.1021/ol201734w. [DOI] [PubMed] [Google Scholar]; d) Xu YH, Lu J, Loh TP. J Am Chem Soc. 2009;131:1372–1373. doi: 10.1021/ja8084548. [DOI] [PubMed] [Google Scholar]; e) Gigant N, Bäckvall JE. Chem Eur J. 2013;19:10799–10803. doi: 10.1002/chem.201301771. [DOI] [PubMed] [Google Scholar]
- 12.a) Tsukanov SV, Comins DL. Angew Chem. 2011;123:8785–8787. [Google Scholar]; Angew Chem Int Ed. 2011;50:8626–8628. doi: 10.1002/anie.201103596. [DOI] [PubMed] [Google Scholar]; b) Davis FA, Xu H, Zhang JY. J Org Chem. 2007;72:2046–2052. doi: 10.1021/jo062365t. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Michael JP, de Koning CB, van der Westhuyzen CW. Org Biomol Chem. 2005;3:836–847. doi: 10.1039/b418062c. [DOI] [PubMed] [Google Scholar]; d) Back TG, Hamilton MD, Lim VJJ, Parvez M. J Org Chem. 2005;70:967–972. doi: 10.1021/jo048284j. [DOI] [PubMed] [Google Scholar]; e) Niphakis MJ, Gay BC, Hong KH, Bleeker NP, Georg GI. Biorg Med Chem. 2012;20:5893–5900. doi: 10.1016/j.bmc.2012.07.044. [DOI] [PubMed] [Google Scholar]; f) Leighty MW, Georg GI. ACS Med Chem Lett. 2011;2:313–315. doi: 10.1021/ml1003074. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Niphakis MJ, Georg GI. J Org Chem. 2010;75:6019–6022. doi: 10.1021/jo101051w. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Comins DL, Sahn JJ. Org Lett. 2005;7:5227–5228. doi: 10.1021/ol052068v. [DOI] [PubMed] [Google Scholar]
- 13.a) Saha M, Carter RG. Org Lett. 2013;15:736–739. doi: 10.1021/ol303272w. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sasaki Y, Kato D, Boger DL. J Am Chem Soc. 2010;132:13533–13544. doi: 10.1021/ja106284s. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Santarem M, Vanucci-Bacque C, Lhommet G. J Org Chem. 2008;73:6466–6469. doi: 10.1021/jo801150e. [DOI] [PubMed] [Google Scholar]; d) Yuan ZQ, Ishikawa H, Boger DL. Org Lett. 2005;7:741–744. doi: 10.1021/ol050017s. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kozmin SA, Iwama T, Huang Y, Rawal VH. J Am Chem Soc. 2002;124:4628–4641. doi: 10.1021/ja017863s. [DOI] [PubMed] [Google Scholar]; f) Danieli B, Lesma G, Luzzani M, Passarella D, Silvani A. Tetrahedron. 1996;52:11291–11296. [Google Scholar]
- 14.a) Stackhouse PJ, Wilson A, Lacey D, Hird M. Liq Cryst. 2010;37:1191–1203. [Google Scholar]; b) Fitié CFC, Roelofs WSC, Kemerink M, Sijbesma RP. J Am Chem Soc. 2010;132:6892–6893. doi: 10.1021/ja101734g. [DOI] [PubMed] [Google Scholar]; c) Miller TM, Neenan TX. Chem Mater. 1990;2:346–349. [Google Scholar]; d) Pang J, Tao Y, Freiberg S, Yang XP, D'Iorio M, Wang S. J Mater Chem. 2002;12:206–212. [Google Scholar]; e) Yamaguchi Y, Ochi T, Miyamura S, Tanaka T, Kobayashi S, Wakamiya T, Matsubara Y, Yoshida ZI. J Am Chem Soc. 2006;128:4504–4505. doi: 10.1021/ja057751r. [DOI] [PubMed] [Google Scholar]
- 15.a) Isaacs RCA, Newton CL, Cutrona KJ, Mercer SP, Payne LS, Stauffer KJ, Williams PD, Cook JJ, Krueger JA, Lewis SD, Lucas BJ, Lyle EA, Lynch JJ, McMasters DR, Naylor-Olsen AM, Michener MT, Wallace AA. Bioorg Med Chem Lett. 2011;21:1536–1540. doi: 10.1016/j.bmcl.2010.12.105. [DOI] [PubMed] [Google Scholar]; b) Georgsson J, Sköld C, Plouffe B, Lindeberg G, Botros M, Larhed M, Nyberg F, Gallo-Payet N, Gogoll A, Karlén A, Hallberg A. J Med Chem. 2005;48:6620–6631. doi: 10.1021/jm050280z. [DOI] [PubMed] [Google Scholar]; c) Lindman S, Lindeberg G, Nyberg F, Karlén A, Hallberg A. Biorg Med Chem. 2000;8:2375–2383. doi: 10.1016/s0968-0896(00)00169-3. [DOI] [PubMed] [Google Scholar]; d) Liu B, Shetty RS, Moffett KK, Kelly MJ. Tetrahedron Lett. 2011;52:1680–1684. [Google Scholar]
- 16.Steinhoff BA, King AE, Stahl SS. J Org Chem. 2006;71:1861–1868. doi: 10.1021/jo052192s. [DOI] [PubMed] [Google Scholar]
- 17.a) Yu YY, Georg GI. Chem Commun. 2013;49:3694–3696. doi: 10.1039/c3cc41130c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miyasaka M, Hirano K, Satoh T, Miura M. J Org Chem. 2010;75:5421–5424. doi: 10.1021/jo101214y. [DOI] [PubMed] [Google Scholar]; c) Min M, Kim Y, Hong S. Chem Commun. 2013;49:196–198. doi: 10.1039/c2cc37676h. [DOI] [PubMed] [Google Scholar]; d) Gigant N, Gillaizeau I. Org Lett. 2012;14:3304–3307. doi: 10.1021/ol301249n. [DOI] [PubMed] [Google Scholar]
- 18.a) Kim YW, Niphakis MJ, Georg GI. J Org Chem. 2012;77:9496–9503. doi: 10.1021/jo301531k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yu YY, Bi L, Georg GI. J Org Chem. 2013;78:6163–6169. doi: 10.1021/jo400830t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Bi L, Georg GI. Org Lett. 2011;13:5413–5415. doi: 10.1021/ol202202a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ge H, Niphakis MJ, Georg GI. J Am Chem Soc. 2008;130:3708–3709. doi: 10.1021/ja710221c. [DOI] [PubMed] [Google Scholar]
- 20.a) Comins DL, Kuethe JT, Miller TM, Fevrier FC, Brooks CA. J Org Chem. 2005;70:5221–5234. doi: 10.1021/jo050559n. [DOI] [PubMed] [Google Scholar]; b) Kuethe JT, Brooks CA, Comins DL. Org Lett. 2003;5:321–323. doi: 10.1021/ol027308a. [DOI] [PubMed] [Google Scholar]; c) Kuethe JT, Comins DL. Tetrahedron Lett. 2003;44:4179–4182. [Google Scholar]
- 21.Bodwell GJ, Hawco KM, da Silva RP. Synlett. 2003:179–182. [Google Scholar]
- 22.In the presence of an excess amount of dienophile (e.g. methyl acrylate), methylamine could readily undergo an aza-Michael addition, the product of which was detected by LC-MS. See the Supporting Information for more details.
- 23.a) Karpuk E, Schollmeyer D, Meier H. Eur J Org Chem. 2007:1983–1990. [Google Scholar]; b) Majchrzak MW, Zobel JN, Obradovich DJ, Peterson GA. Org Prep Proced Int. 1997;29:361–364. [Google Scholar]
- 24.a) Yuan G, Zheng J, Gao X, Li X, Huang L, Chen H, Jiang H. Chem Commun. 2012;48:7513–7515. doi: 10.1039/c2cc32964f. [DOI] [PubMed] [Google Scholar]; b) Alonso DA, Fuensanta M, Nájera C, Varea M. J Org Chem. 2005;70:6404–6416. doi: 10.1021/jo050852n. [DOI] [PubMed] [Google Scholar]; c) Bardelle C, Coleman T, Cross D, Davenport S, Kettle JG, Ko EJ, Leach AG, Mortlock A, Read J, Roberts NJ, Robins P, Williams EJ. Bioorg Med Chem Lett. 2008;18:5717–5721. doi: 10.1016/j.bmcl.2008.09.087. [DOI] [PubMed] [Google Scholar]; d) Pilgram K, Korte F. Tetrahedron. 1964;20:177–187. [Google Scholar]; e) Dvorak CA, Liu C, Shelton J, Kuei C, Sutton SW, Lovenberg TW, Carruthers NI. ACS Med Chem Lett. 2012;3:637–639. doi: 10.1021/ml3000676. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Liao TB, Ling Y, Chen ZX, Zhou YM, Weng LH. Chem Commun. 2010;46:1100–1102. doi: 10.1039/b917987a. [DOI] [PubMed] [Google Scholar]; g) Garczarek P, Janczak J, Zoń J. J Mol Struct. 2013;1036:505–509. [Google Scholar]
- 25.a) Dai HX, Li G, Zhang XG, Stepan AF, Yu JQ. J Am Chem Soc. 2013;135:7567–7571. doi: 10.1021/ja400659s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Robbins DW, Hartwig JF. Angew Chem. 2013;125:967–971. doi: 10.1002/anie.201208203. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2013;52:933–937. doi: 10.1002/anie.201208203. [DOI] [PubMed] [Google Scholar]; c) Hofmann N, Ackermann L. J Am Chem Soc. 2013;135:5877–5884. doi: 10.1021/ja401466y. [DOI] [PubMed] [Google Scholar]; d) Truong T, Daugulis O. Angew Chem. 2012;124:11845–11847. doi: 10.1002/anie.201206568. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2012;51:11677–11679. doi: 10.1002/anie.201206568. [DOI] [PubMed] [Google Scholar]; e) Leow D, Li G, Mei TS, Yu JQ. Nature. 2012;486:518–522. doi: 10.1038/nature11158. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Lee EY, Park J. Chemcatchem. 2011;3:1127–1129. [Google Scholar]; g) Cornella J, Righi M, Larrosa I. Angew Chem. 2011;123:9601–9604. doi: 10.1002/anie.201103720. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:9429–9432. doi: 10.1002/anie.201103720. [DOI] [PubMed] [Google Scholar]; h) Saidi O, Marafie J, Ledger AEW, Liu PM, Mahon MF, Kociok-Koehn G, Whittlesey MK, Frost CG. J Am Chem Soc. 2011;133:19298–19301. doi: 10.1021/ja208286b. [DOI] [PubMed] [Google Scholar]; i) Chen B, Hou XL, Li YX, Wu YD. J Am Chem Soc. 2011;133:7668–7671. doi: 10.1021/ja201425e. [DOI] [PubMed] [Google Scholar]; j) Duong HA, Gilligan RE, Cooke ML, Phipps RJ, Gaunt MJ. Angew Chem. 2011;123:483–486. doi: 10.1002/anie.201004704. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:463–466. [Google Scholar]; k) Zhou Y, Zhao J, Liu L. Angew Chem. 2009;121:7262–7264. [Google Scholar]; Angew Chem Int Ed. 2009;48:7126–7128. doi: 10.1002/anie.200902762. [DOI] [PubMed] [Google Scholar]; l) Phipps RJ, Gaunt MJ. Science. 2009;323:1593–1597. doi: 10.1126/science.1169975. [DOI] [PubMed] [Google Scholar]; m) Yue W, Li Y, Jiang W, Zhen Y, Wang Z. Org Lett. 2009;11:5430–5433. doi: 10.1021/ol9023198. [DOI] [PubMed] [Google Scholar]; n) Murphy JM, Liao X, Hartwig JF. J Am Chem Soc. 2007;129:15434–15435. doi: 10.1021/ja076498n. [DOI] [PubMed] [Google Scholar]
- 26.a) Nowakowska Z. Eur J Med Chem. 2007;42:125–137. doi: 10.1016/j.ejmech.2006.09.019. [DOI] [PubMed] [Google Scholar]; b) Patil CB, Mahajan SK, Katti SA. J Pharm Sci Res. 2009;1:11–22. [Google Scholar]; c) Dimmock JR, Elias DW, Beazely MA, Kandepu NM. Curr Med Chem. 1999;6:1125–1150. [PubMed] [Google Scholar]
- 27.a) Claisen L, Claparede A. Ber Dtsch Chem Ges. 1881;14:2460–2468. [Google Scholar]; b) Schmidt JG. Ber Dtsch Chem Ges. 1881;14:1459–1461. [Google Scholar]
- 28.a) Eddarir S, Cotelle N, Bakkour Y, Rolando C. Tetrahedron Lett. 2003;44:5359–5363. [Google Scholar]; b) Wu XF, Neumann H, Beller M. Angew Chem. 2010;122:5412–5416. [Google Scholar]; Angew Chem Int Ed. 2010;49:5284–5288. doi: 10.1002/anie.201002155. [DOI] [PubMed] [Google Scholar]; c) Wu XF, Neumann H, Spannenberg A, Schulz T, Jiao H, Beller M. J Am Chem Soc. 2010;132:14596–14602. doi: 10.1021/ja1059922. [DOI] [PubMed] [Google Scholar]; d) Wu XF, Neumann H, Beller M. Chem Asian J. 2012;7:282–285. doi: 10.1002/asia.201100630. [DOI] [PubMed] [Google Scholar]; e) Unoh Y, Hirano K, Satoh T, Miura M. J Org Chem. 2013;78:5096–5102. doi: 10.1021/jo400716e. [DOI] [PubMed] [Google Scholar]; f) Bianco A, Cavarischia C, Guiso M. Eur J Org Chem. 2004:2894–2898. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.