

Abstract
We prepared 13 derivatives of N-(biphenyl-4′-yl)methyl (R)-2-acetamido-3-methoxypropionamide that differed in type and placement of a R-substituent in the terminal aryl unit. We demonstrated that the R-substituent impacted the compound’s whole animal and cellular pharmacological activities. In rodents, select compounds exhibited excellent anticonvulsant activities and protective indices (PI = TD50/ED50) that compared favorably with clinical antiseizure drugs. Compounds with a polar, aprotic R-substituent potently promoted Na+ channel slow inactivation and displayed frequency (use) inhibition of Na+ currents at low micromolar concentrations. The possible advantage of affecting these two pathways to decrease neurological hyperexcitability is discussed.
Introduction
(R)-N-Benzyl 2-acetamido-3-methoxypropionamide1 (lacosamide, (R)-1) is a first-in-class antiseizure drug (ASD) that is marketed worldwide for adjunctive treatment of partial-onset seizures.2 Whole animal pharmacological studies showed that (R)-1 displayed potent anticonvulsant activity1,3 in the maximal electroshock4 (MES), 6 Hz psychomotor,5 and hippocampal kindled6 seizure models. Whole-cell, patch-clamp electrophysiology studies indicated that (R)-1 reduced neuronal hyperexcitability by a mechanism that was consistent with its enhancing the transition of voltage-gated Na+ channels (VGSCs) to the slow-inactivated state.7−9 Recent structure–activity relationship studies demonstrated that modifying either the N-benzyl10,11 or the O-methoxy12,13 sites in (R)-1 provided analogues that retained excellent anticonvulsant activities. Importantly, we found that the two N-(biphenyl-4′-yl)methyl analogues, (R)-210 and (R)-3,11 when given orally to rats in the MES model, exhibited activities that exceeded those of (R)-1 (Figure 1). We further showed that the IC50 value for Na+ channel slow inactivation (the concentration at which half of the Na+ channels transitioned to the slow-inactivated state) in catecholamine A differentiated (CAD) cells14 was 29-fold lower (more potent) for (R)-3 than for (R)-1 (Figure 1).15 While these findings showed that introducing 4′-aryl substituents to (R)-1 provided pharmacologically potent compounds, they also showed that the 4′-aryl substituent affected animal behavioral neurotoxicity. Compound (R)-2 was significantly more neurotoxic than (R)-1,10 but (R)-3 was not (Figure 1).11
Figure 1.

Key pharmacological properties of (R)-1, (R)-2, and (R)-3.
In this study, we report the synthesis
and pharmacological activity
of 13 substituted N-(biphenyl-4′-yl)methyl
(R)-2-acetamido-3-methoxypropionamides (compound
class (R)-A) where the R-substituent
in the terminal aryl unit was systematically varied. The (R)-A’s R-substituent significantly impacted
the compound’s whole animal and cellular pharmacological activity.
Numerous compounds with a polar, aprotic R-substituent exhibited excellent
anticonvulsant and potent Na+ channel activities while
producing minimal behavioral neurotoxicities.
Results
Compound Selection
Thirteen (R)-A derivatives, (R)-4–(R)-16, containing different R-substituents
were synthesized and evaluated in animal and cellular pharmacological
models. The compounds were divided into two groups. One set, (R)-4–(R)-12, contained polar, aprotic R-substituents (i.e., F, Cl, Br, I, CN,
CF3, OCF3), and the second set, (R)-13–(R)-16, contained
polar, protic R-substituents (i.e., N(H)C(O)CH3, CH2OH, CO2H, NH3+Cl–). We expected that the CO2H substituent in (R)-15 and the NH3+Cl– substituent in (R)-16 were likely
ionized at the physiological-like pH values used in the evaluations.
When the R-substituent was Br, I, CN, CF3, N(H)C(O)CH3, CH2OH, CO2H, or NH3+Cl–, we installed the group at the 3″-position
of the terminal aryl ring ((R)-7, (R)-8, (R)-9,
(R)-10, (R)-13, (R)-14, (R)-15, (R)-16); when the R-substituent
was F, Cl, or OCF3, we prepared both the 3″ ((R)-3, (R)-5,
(R)-11) and the 4″ ((R)-4, (R)-6,
(R)-12) regioisomers. We expected that
the R-substituents would affect the compound’s biodistribution,
anticonvulsant activities, and ability to modulate Na+ channel
function.
Chemistry
Compounds (R)-4–(R)-16 were prepared by similar routes (Schemes 1–6) using the mixed anhydride coupling (MAC) method16 and employing (R)-N-tert-butoxycarbonylserine ((R)-45), (R)-2-acetamido-3-hydroxypropionic acid ((R)-69), or (R)-2-acetamido-3-methoxypropionic acid10,12,17 ((R)-70) as the carboxylic acid component. The choice of the carboxylic acid was dictated by the ease of purification of the intermediate products and the need to avoid competing side reactions in the latter stages of the synthesis. For compounds (R)-14–(R)-16, we removed the protecting group in the final step to facilitate the isolation of the desired polar products.
Scheme 1. Preparation of (Biphenyl-4-yl)methylamines 29–39.

Scheme 6. Synthesis of Compound (R)-16.

To prepare compounds (R)-4–(R)-13, (R)-N-tert-butoxycarbonylserine ((R)-45) was coupled with the requisite (biphenyl-4-yl)methylamine (29–39, 43, 44) using the MAC reagents, isobutyl chloroformate (IBCF) and N-methylmorpholine (NMM), to give the amides (R)-46–(R)-55 (Scheme 3). The crude (biphenyl-4-yl)methylamines were prepared by Suzuki coupling using tetrakis(triphenylphoshine)palladium(0).18 For 29–39, 4-bromobenzylamine (17) was reacted with the commercially available arylboronic acids (18–28) (Scheme 1), and for 43 and 44, p-aminomethylboronic acid (40) was coupled with the appropriate 1,3-dihalobenzene (41, 42). The serine hydroxyl group in (R)-46–(R)-55 was methylated (CH3I, Ag2O) to provide ethers (R)-56–(R)-65, respectively. Deprotection of the tert-butoxycarbonyl group in (R)-56–(R)-65 with acid (HCl/dioxane) followed by acetylation (AcCl, Et3N) gave the desired products (R)-4–(R)-13, respectively. For (R)-14 and (R)-15, we coupled (R)-69 with (biphenyl-4-yl)methylamines 37 and 38 (Scheme 5), and for (R)-16, we combined (R)-7010,12,17 with (3″-N-(tert-butoxycarbonyl)aminobiphenyl-4′-yl)methylamine (39) (Scheme 6). To facilitate the preparation of (R)-14–(R)-16, we first developed convenient synthetic procedures for (R)-69 and (R)-70 (Scheme 4). Commercially available d-serine benzyl ester hydrochloride ((R)-66) was selectively converted to (R)-2-N-acetamido-3-hydroxypropionate benzyl ester ((R)-67) using 0.9 equiv of AcCl at −20 °C. Compound (R)-67 was methylated (CH3I, Ag2O) at room temperature to give (R)-68, without racemization. Catalytic hydrogenation of (R)-67 and (R)-68 using 10% Pd/C in methanol provided the desired acids (R)-69 and (R)-70, respectively, which were used directly in the MAC reaction without purification.
Scheme 3. Synthesis of Compounds (R)-4–(R)-13.

Scheme 5. Synthesis of Compounds (R)-14 and (R)-15.

Scheme 4. Synthesis of (R)-69 and (R)-70.

Scheme 2. Preparation of (Biphenyl-4-yl)methylamines 43 and 44.

The enantiomeric purity of (R)-4–(R)-13 was assessed by detecting a single acetyl methyl peak and a single O-methyl peak in the 1H NMR spectrum (CDCl3) for each compound when a saturated solution of (R)-(−)-mandelic acid was added.19 In the cases of (R)-14–(R)-16, their poor solubility in CDCl3 prevented assessment of their optical purity by 1H NMR. Thus, we demonstrated with 1H NMR the enantiomeric purity of their immediate precursors (R)-73–(R)-75, respectively.
In the Experimental Section, we report the details (synthetic procedure, characterization) of the final step for all compounds evaluated in the anticonvulsant and cellular electrophysiology studies. In Supporting Information, we provide the experimental procedures and physical and spectroscopic properties for all synthetic compounds prepared in this study.
Whole Animal Pharmacological Activity
Compounds (R)-4–(R)-16 were tested for anticonvulsant activity at the Anticonvulsant Screening Program (ASP) of the National Institute of Neurological Disorders and Stroke (NINDS), U.S. National Institutes of Health. Screening was performed using the established protocols and procedures at the ASP described by Stables and Kupferberg.20 We relied principally on two rodent models to assess anticonvulsant activity, the MES4 and the 6 Hz psychomotor5 seizure tests. The MES test is a model for generalized tonic–clonic seizure and is thought to provide an indication of a compound’s ability to prevent seizure spread when all neuronal circuits in the brain are maximally active. Correspondingly, the 6 Hz psychomotor test (32 mA) has been used as a model for therapy-resistant limbic seizures. Previous studies have shown that some compounds with minimal activity in the MES test are effective in the 6 Hz psychomotor model.5 The anticonvulsant data from the MES model4 (mice, ip; rat, po) and psychomotor 6 Hz (32 mA) seizure test for therapy-resistant limbic seizures5 (mice, ip) are summarized in Table 1 along with similar results obtained for (R)-1,1 (R)-2,10 (R)-3,11 and the ASDs phenytoin,21 phenobarbital,21 and valproate.21 For compounds that showed significant activity, we report the 50% effective dose (ED50) values from quantitative screening evaluations. We also include the median doses for 50% neurological impairment (TD50) in mice, using the rotorod test,22 and the behavioral toxicity effects observed in rats.23 The protective index (PI = TD50/ED50) for the test compounds, where possible, is also listed. Compounds (R)-2–(R)-6 and (R)-11 and (R)-12 were evaluated in the subcutaneous Metrazol (scMet) seizure model.24 No activity was observed below 100 mg/kg (data not shown). Similarly, we observed no activity in the scMet model for (R)-1 and structurally related compounds.1,10−13
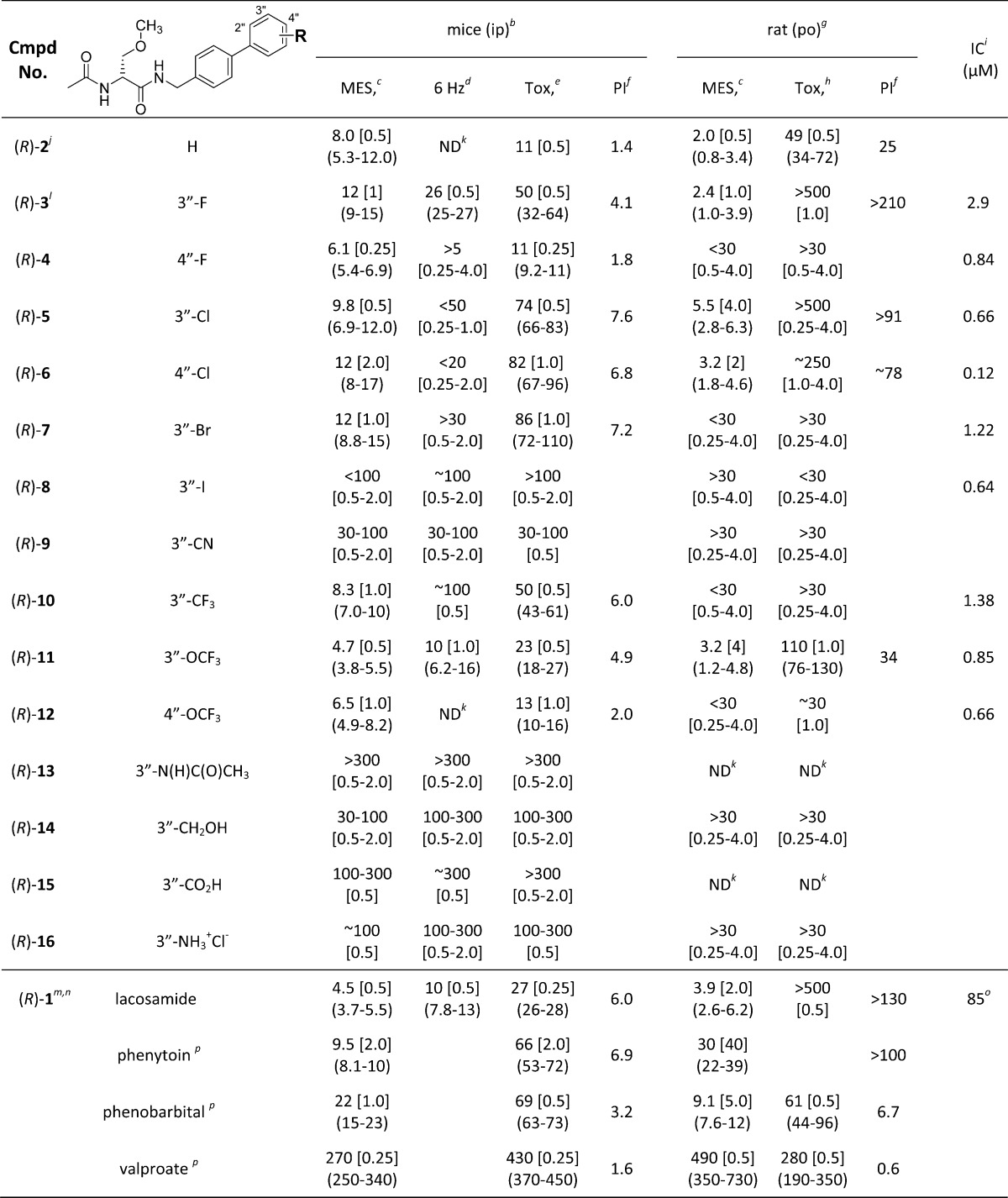
Table 1. Structure–Activity Relationship for (R)-4–(R)-16a.
The compounds were tested through the NINDS ASP.
The compounds were administered intraperitoneally. ED50 and TD50 values are in milligrams per kilogram. Numbers in parentheses are 95% confidence intervals. A dose–response curve was generated for all compounds that displayed sufficient activity. The dose–effect for these compounds was obtained at the “time of peak effect” (indicated in hours in the brackets).
MES = maximal electroshock seizure test.
6 Hz = 6 Hz psychomotor seizure test.
TD50 value determined from the rotorod test.
PI = protective index (TD50/ED50) in the MES test.
The compounds were administered orally. ED50 and TD50 values are in milligrams per kilogram. Numbers in parentheses are 95% confidence intervals. A dose–response curve was generated for all compounds that displayed sufficient activity. The dose–effect for these compounds was obtained at the “time of peak effect” (indicated in hours in the brackets).
Tox = behavioral toxicity.
IC50, concentration at which half of the Na+ channels have transitioned to a slow inactivated state.
Reference (10).
ND = not determined.
Reference (11).
Reference (1).
Reference (3).
Reference (9).
Reference (21).
The R-substituent in (R)-4–(R)-16 markedly affected the observed anticonvulsant activity in the MES and 6 Hz seizure tests (Table 1). We identified compounds with similar potency compared with the parent compound in this series, the unsubstituted N-(biphenyl-4′-yl)methyl (R)-2-acetamido-3-methoxypropionamide ((R)-2),10 and the ASD (R)-1,1 while other compounds showed little or no activity. The most potent anticonvulsant compounds in the MES test contained a polar, aprotic R-substituent (i.e., F ((R)-3, (R)-4); Cl ((R)-5, (R)-6); Br ((R)-7); CF3 ((R)-10); OCF3 ((R)-11, (R)-12). The MES ED50 values in mice (ip) ranged from 4.7 to 12 mg/kg and in the rat (po) from 2.4 to <30 mg/kg. The corresponding ED50 values for (R)-2 in mice (ip) and rats (po) were 8.0 and 2.0 mg/kg,10 and for (R)-1 they were 4.5 and 3.9 mg/kg.1 We observed little differences in the anticonvulsant activities in the MES test in mice (ip) for the 3″-substituted F, Cl, and OCF3 compounds ((R)-3, (R)-5, (R)-11) compared with their 4″-substituted regioisomers ((R)-4, (R)-6, (R)-12) (MES ED50 (mice, ip, mg/kg): (R)-3, 12; (R)-4, 6.1; (R)-5, 9.8; (R)-6, 12; (R)-11, 4.7; (R)-12, 6.5). The 3″-CN substituted compound ((R)-9) exhibited only moderate activity in the MES seizure model (mice, ip, mg/kg): (R)-9, 30–100). A pronounced decrease in activity was seen for compounds (R)-13–(R)-16 containing a polar, protic R-substituent (i.e., N(H)C(O)CH3 ((R)-13); CH2OH ((R)-14); CO2H ((R)-15); NH3+Cl– ((R)-16). Most of these compounds showed little activity in the MES seizure model in mice (ip), with only (R)-14 exhibiting modest activity (ED50, mice, ip, mg/kg: (R)-13, >300; (R)-14, 30–100; (R)-15, 100–300; (R)-16, ∼100).
We found that the neurotoxicity of (R)-A in the rotorod (mice, ip) and behavioral (rat, po) tests varied with the nature of the R-substituent and its position in the terminal aryl ring. Many of the compounds ((R)-3, (R)-5–(R)-11, (R)-13–(R)-16) were significantly less neurotoxic in mice than the parent, unsubstituted compound (R)-2 (TD50 = 11 mg/kg)10 and exhibited comparable or less neurotoxicity than the ASD (R)-1 (TD50 = 27 mg/kg).1 Thus, the excellent activities in the MES test (mice, ip) of (R)-3, (R)-5, (R)-6, and (R)-10 (ED50 (mg/kg), (R)-3, 12; (R)-5, 9.8; (R)-6, 12; (R)-7, 12; (R)-10, 8.3) coupled with their low neurotoxicities (TD50 (mg/kg), (R)-3, 50; (R)-5, 74; (R)-6, 82; (R)-7, 86; (R)-10, 50) provided compounds with PIs (4.1–7.6) that were notably higher than that of the parent, unsubstituted compound (R)-2 (PI = 1.4) and that were similar to that of (R)-1 (PI = 6.0). When R was either F or OCF3, we found that the 3″-derivative was less neurotoxic than the 4″-substituted isomer (TD50 (mg/kg), (R)-3 (3″-F), 50; (R)-4 (4″-F), 11; (R)-11 (3″-OCF3), 23; (R)-12 (4″-OCF3), 13) and provided higher PI values (PI, (R)-3 (3″-F), 4.1; (R)-4 (4″-F), 1.8; (R)-11 (3″-OCF3), 4.9; (R)-12 (4″-OCF3), 2.0); however, when R was Cl, there was little difference in the neurotoxicity (TD50 (mg/kg), (R)-5 (3″-Cl), 74; (R)-6 (4″-Cl), 82) and in the PI values (PI, (R)-5 (3″-Cl), 7.6; (R)-6 (4″-Cl), 6.8) for the regioisomers. A drop in neurotoxicity was observed for (R)-13–(R)-16, the four compounds with polar, protic R-substituents (TD50 of 100 to >300 mg/kg). These compounds showed moderate or little protection in the seizure models. Taken together, these findings demonstrated the importance of the R-substituent in (R)-A in eliciting the optimal responses in the animal models. We found that specific substituents and their placement in the terminal aryl ring afforded compounds ((R)-5 (3″-Cl), (R)-6 (4″-Cl), (R)-7 (3″-Br), (R)-10 (3″-CF3), (R)-11 (3″-OCF3)) with excellent anticonvulsant activities and improved PI values compared with the parent, unsubstituted compound, (R)-2,10 in the mouse (ip). Similar results were observed in the rat (po) (PI: (R)-2, 25;10 (R)-3, >210;11 (R)-5, >91; (R)-6, ∼78; (R)-11, 34).
Three of the more active (R)-A compounds were tested in the rat hippocampal seizure model (ip).6 This is a test of partial complex or temporal lobe seizures, the most common and most drug-resistant type of adult focal epilepsy.25−27 We obtained ED50 values (mg/kg) for (R)-3, (R)-5, and (R)-11 of 13, 36, and 1.7, respectively. The ED50 value for (R)-11 was 8-fold lower than that for (R)-1 (14 mg/kg).3
Whole-Cell, Patch-Clamp Electrophysiology
Compounds of interest were characterized for their abilities to interact with VGSCs by whole-cell patch-clamp electrophysiological studies. In neurons, VGSCs are responsible for action potential generation and propagation.28−30 These channels undergo inactivation in response to activity over two distinct time courses and mechanisms: fast inactivation occurs over a few milliseconds, whereas slow inactivation occurs over several hundred milliseconds. Inactivation is reversible, but for a time, these channels are unable to produce current in response to stimulus.31 The function of several ASDs, such as carbamazepine and (R)-1, has been attributed to their ability to inactivate VGSCs and reduce synaptic activity.7−9,32 We assessed the ability of the (R)-A compounds to alter Na+ channel kinetics of fast inactivation and slow inactivation and also for frequency (use) dependence. These studies were carried out in several cell models: CAD cells allow for rapid characterization of compounds; rat cortical neurons allow for characterization in cells related to those that generate epileptic phenotypes in humans; and HEK293 cells allow for characterization of activity on single VGSC isoforms.
A. 3″- and 4″-Polar, Aprotic Substituted N-(Biphenyl-4′-yl)methyl (R)-Acetamido-3-methoxypropionamide Derivatives in CAD Cells
We previously showed that (R)-3 inhibited several parameters of VGSC activities in neuronal model CAD cells.15 Here, we tested whether (R)-4–(R)-16 would result in similar action. Initially, we focused on (R)-A compounds with polar, aprotic R-substituents (i.e., R = F, Cl, Br, I, CF3, OCF3) at either the 3″ or the 4″ position of the terminal aryl ring, since these compounds exhibited the greatest anticonvulsant activities (Table 1).
CAD cells are abundant and easy to use.14 These cells express endogenous tetrodotoxin-sensitive Na+ currents that display rapid activation and inactivation kinetics upon membrane depolarization14 and likely comprise a majority of NaV1.7 channels, with minor contributions by NaV1.1, NaV1.3, and NaV1.9 channels.9,33,34 Importantly, we earlier demonstrated that the Na+ channel properties of (R)-1 in CAD cells9 were similar to those reported in cultured neurons and in mouse N1E-115 neuroblastoma cells.7
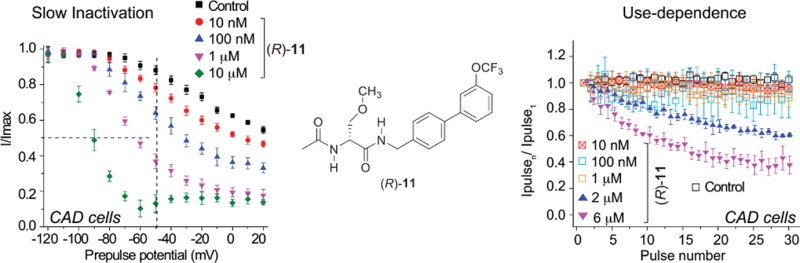
First, we examined (R)-3,15 (R)-5, (R)-7, (R)-8, (R)-10, and (R)-11, compounds with a 3″-polar, aprotic R-substituent (R = F, Cl, Br, I, CF3, OCF3), for their ability to transition Na+ channels to the slow-inactivated state. CAD cells were held at −80 mV and conditioned to potentials ranging from −120 to +20 mV (in +10 mV increments) for 5 s. Then fast-inactivated channels were allowed to recover for 150 ms at a hyperpolarized pulse to −120 mV, and then the fraction of channels available was tested by a single depolarizing pulse, to 0 mV, for 15 ms (Figure 2A, left). Brief hyperpolarization to −120 mV allows channel recovery from fast inactivation while limiting recovery from slow inactivation such that the property of slow inactivation can be isolated. Representative slow inactivation traces, at −120 and −50 mV, from CAD cells treated with 0.1% DMSO (control) or 10 μM (R)-11 are shown in Figure 2A (right). At −50 mV, a majority of the channels undergo steady-state inactivation that involves contributions from slow- and fast-inactivating pathways.28,35 This voltage is also near the resting membrane potential (RMP) and approaches the action potential firing threshold for CNS neurons,36 where slow inactivation appears to be physiologically relevant during sustained subthreshold depolarizations.37 Finally, changes in the Na+ channel availability near −50 mV can affect the overlap of Na+ current activating and inactivating under steady-state conditions.28,38 For these reasons, we utilized −50 mV conditioning pulses to compare pharmacological effects on slow inactivation and to develop concentration–response curves and IC50 values for each tested compound (Figure 2B–G). The slow inactivation IC50 values are shown in Figure 2H and summarized in Table 1. Compared with the reported IC50 calculated value of 85 μM for slow inactivation induced by (R)-1,9 the IC50 values for these 3″-substituted (R)-A compounds were considerably lower. For example, the IC50 values for (R)-3 and (R)-8 were 29- and 133-fold lower than (R)-1. Collectively, these data indicate that these (R)-A derivatives more effectively induced Na+ channel transition to a slow-inactivated state than did (R)-1.
Figure 2.

Effects of (R)-A compounds with a polar, aprotic substituent at the 3″-position on the steady-state, slow inactivation state of Na+ currents in CAD cells. (A) Voltage protocol for slow inactivation. Currents were evoked by 5 s prepulses between −120 and +20 mV, and then fast-inactivated channels were allowed to recover for 150 ms at a hyperpolarized pulse to −120 mV. The fraction of channels available at 0 mV was analyzed. Representative current traces from CAD cells were taken in the absence (control, 0.1% DMSO) or presence of 10 μM (R)-11. The black and red traces represent the currents evoked at −120 and −50 mV, respectively (also highlighted in the voltage protocol). (B–G) Summary of steady-state, slow activation curves for CAD cells treated with 0.1% DMSO (control) or with various concentrations of the indicated compounds. (H) The concentrations at which half of the channels have transitioned to the slow-inactivated state (see text for details), the SI IC50, are indicated. Data are from four to seven cells per condition.
A similar series of experiments were undertaken to investigate the effects of (R)-A compounds with polar, aprotic substituents (R = F, Cl, OCF3) at the 4″-aryl site on Na+ current slow inactivation. We observed pronounced, concentration-dependent slow inactivation for (R)-4, (R)-6, and (R)-12 (Supporting Information, Figure S1B–D). As for the 3″-substituted compounds, the IC50 values of slow inactivation were calculated by fitting the concentration responses of slow inactivation against the V1/2 of slow inactivation (Table 1; Supporting Information, Figure S1E). The IC50 values for (R)-4, (R)-6, and (R)-12 were 101-, 708-, and 129-fold lower than for (R)-1.
We next asked if the polar, aprotic (R)-A derivatives could enhance steady-state fast inactivation in CAD cells. For these studies we used a protocol (Figure 3A) designed to induce a fast-inactivated state, similar to that previously described.33 Cells were held at −80 mV, stepped to inactivating prepulse potentials ranging from −120 to −10 mV (in 10 mV increments) for 500 ms. Then the cells were stepped to 0 mV for 20 ms to measure the available current. A 500 ms conditioning pulse was used because it allowed all of the endogenous channels to transition to a fast-inactivated state at all potentials assayed. In Figure 3, we provide the steady-state, fast inactivation curves of Na+ currents (representative family of current traces shown in Figure 3A from control (0.1% DMSO) and Figure 3B–G for (R)-A derivatives with a polar, aprotic 3″-substituent). (R)-3-, (R)-5-, (R)-7-, (R)-8-, (R)-10-, and (R)-11-treated CAD neurons were well fitted with a single Boltzmann function (R2 > 0.969 for all conditions). The V1/2 inactivation value for 0.1% DMSO-treated cells was −68.3 ± 3.1 mV (n = 5), which was significantly different from that of (R)-8 (10 μM) treated cells (−76.5 ± 1.3 mV; n = 5; p < 0.05; Student’s t-test; see Figure 3E). Compared with the ∼8.2 mV shift in V1/2 of fast inactivation in the hyperpolarizing direction observed in the presence of (R)-8, the shifts caused by the other five compounds were not significantly different from control (Figure 3B–D,F,G) (p < 0.05 vs 0.1% DMSO (control); Student’s t-test). We also found that (R)-1 did not affect Na+ channel fast inactivation.9
Figure 3.

Effects of (R)-A compounds with a polar, aprotic substituent at the 3″-position on fast inactivation and steady-state activation state of Na+ currents in CAD cells. (A) Voltage protocol for fast inactivation (left) and activation (right). Representative families of currents in response to these protocols are illustrated. (B–G) Representative Boltzmann fits for steady-state fast inactivation and activation for CAD cells treated with 0.1% DMSO (control) and various concentrations of the indicated compounds are shown. Values for V1/2, the voltage of half-maximal inactivation and activation and the slope factors (k) were derived from Boltzmann distribution fits to the individual recordings and were averaged to determine the mean (±SEM) voltage dependence of steady-state inactivation and activation, respectively. The V1/2 and k of steady-state fast inactivation or activation other than (R)-8 were not different among any of the conditions tested (p > 0.05, one-way ANOVA). Data are from four to six cells per condition.
The effects of (R)-A derivatives with 4″-polar, aprotic substituents (F, Cl, OCF3) on Na+ current fast inactivation was also investigated (Supporting Information, Figure S2). Steady-state, fast inactivation curves of Na+ currents from 0.1% DMSO- (control) and (R)-4-, (R)-6-, and (R)-12-treated CAD cells were well fitted with a single Boltzmann function (R2 > 0.989 for all conditions). The V1/2 inactivation value for 0.1% DMSO-treated cells was not significantly different from any of the conditions (p > 0.05; Student’s t-test; Supporting Information Figure S2B–D).
Because changes in current amplitudes can result from changes in channel gating, we tested whether the nine 3″- and 4″-polar, aprotic (R)-A derivatives ((R)-4–(R)-12) could alter voltage-dependent activation properties of Na+ currents in CAD cells. Activation changes for the (R)-A derivative treated CAD cells were measured by whole-cell ionic conductances by comparing their midpoints (V1/2) and slope factors (k) in response to changes in command voltage (Figure 3A, top right). Representative traces in response to the voltage protocol are shown in Figure 3A (bottom right). Boltzmann fits for 0.1% DMSO (control) and various concentrations of (R)-3, (R)-5, (R)-7, (R)-8, (R)-10, and (R)-11 are shown in Figure 3B–G and those for (R)-4, (R)-6, and (R)-12 in Supporting Information, Figure S2B–D. An analysis of the six 3″-substituted compounds ((R)-3, (R)-5, (R)-7, (R)-8, (R)-10, (R)-11) V1/2 and k values showed that, with the exception of two (R)-8 concentrations, there were no changes in the steady-state activation properties of Na+ currents between CAD cells treated with 0.1% DMSO (control) or with any of the tested compounds (Figure 3C–G). The V1/2 steady-state activation value for 0.1% DMSO-treated (control) cells was −12.8 ± 0.5 mV (n = 4), which was significantly different from that of (R)-8-treated cells at 100 nM (−24.6 ± 3.9 mV, n = 4) and at 300 nM (−20.7 ± 2.4 mV, n = 5) (p < 0.05; Student’s t-test; see Figure 3E). Similarly, we found that the V1/2 or k values for steady state activation for 0.1% DMSO-treated (control) cells were not significantly different from those of the 4″-substituted (R)-A compound (10 μM) treated CAD cells ((R)-4, (R)-6, and (R)-12; p > 0.05; Student’s t-test; Supporting Information Figure S2B–D). These data indicate that, except for (R)-8, polar, aprotic (R)-A derivatives did not affect a closed channel’s transition to an open conformation.
Finally, we tested whether (R)-4–(R)-12 could elicit a frequency (use) dependent block of Na+ currents in CAD cells. Use dependency represents an ability to bind more efficiently to a channel undergoing repeated transitions between closed, open, and inactivated conformations. The ability to block Na+ currents in an activity- or use-dependent manner is a useful property for ASDs, since it allows for preferential decreases in Na+ channel availability during high-frequency (seizure-like) but not low-frequency firing.39 Several theories have been proposed to account for the complexities of use-dependent inhibition, including the modulated receptor hypothesis,40 which postulates that the receptor on the Na+ channels can exist in multiple configurations and the affinity and binding rates of drugs depend on channel’s state, which in turn can depend on voltage. The response to voltage changes can be viewed as reflecting voltage-sensitive shifts in equilibrium between conducting, unblocked channels and nonconducting, blocked channels. The modulated-receptor hypothesis postulates shifts in equilibrium as the result of a variable-affinity receptor and modified inactivation gate kinetics in drug-complexed channels. We tested compounds (R)-5, (R)-7, (R)-8, (R)-10, and (R)-11 at a range of concentrations from below to above the determined slow inactivation IC50 values. A train of 30 test pulses (20 ms to −10 mV) was delivered from a holding potential of −80 mV at 10 Hz (Figure 4A). The available current in control cells (0.1% DMSO) and cells in the presence of the (R)-A compounds was calculated by dividing the peak current at any given pulse (pulseN) by the peak current in response to the initial pulse (pulse1). Representative currents elicited by the voltage protocol are illustrated for control and 5 μM (R)-5-treated cells (Figure 4A). Compounds (R)-5, (R)-10, and (R)-11 exhibited a statistically significant concentration-dependent frequency (use) inhibition of Na+ currents (Figure 4B,E,F) by the last pulse, compared with control (0.1% DMSO). The peak current was ∼50% lower in the presence of 5 μM (R)-5, ∼18% lower in the presence of 10 μM (R)-10, and ∼65% lower in the presence of 6 μM (R)-11. These test concentrations were 7.1–7.6 times higher than their slow inactivation IC50 values. By comparison, as we previously reported, there was a 45% diminution of Na+ currents in the presence of 15 μM (R)-3 (Figure 4B, purple circles), a concentration that is 5.2 times higher than the slow inactivation IC50 value. Similarly, the traces for (R)-7 and (R)-8 (Figure 4C,D) suggested frequency (use) inhibition of the Na+ currents at a 10 μM concentration, but the values did not reach statistical significance. We did not test these (R)-A compounds at higher concentrations.
Figure 4.

Effect of (R)-A compounds with a polar, aprotic substituent at the 3″-position on frequency (use) dependent block of Na+ currents in CAD cells. (A) The frequency dependence of the block was examined by holding cells at the hyperpolarized potential of −80 mV and evoking currents at 10 Hz by 20 ms test pulses to −10 mV (inset middle). Representative overlaid traces are illustrated by pulses 1 and 30 for control (predrug) and in the presence of (R)-5 (5 μM). (B–F) Summary of average frequency (use) dependent decrease in current amplitude (±SEM) produced by control (0.1% DMSO) or by the presence of various concentrations of the indicated compounds (p > 0.05, one-way ANOVA with Dunnett’s post hoc test). Data are from three to six cells per condition. As a comparison, a use-dependent block imposed by (R)-3 is also shown.15 Compounds (R)-3, (R)-5, (R)-10, and (R)-11 caused a significant decrease (indicated by ∗) in current amplitude compared with 0.1% DMSO-treated cells (control) (p < 0.05, one-way ANOVA with Dunnett’s post hoc test). Note the rapid frequency-dependent facilitation of the block by (R)-3 and (R)-5, which was observed beginning as early as pulse 4. Data are from three to six cells per condition.
We also examined whether the three polar, aprotic 4″-substituted derivatives (R)-4, (R)-6, and (R)-12 affected frequency (use) dependent block of Na+ currents using a single concentration of 1 μM, a concentration at which near maximal effects on slow inactivation were observed (IC50 (μM): (R)-4, 0.82; (R)-6, 0.12; (R)-12, 0.66; Supporting Information Figure S1). Only (R)-4 and (R)-6 exhibited small but statistically significant reduction in frequency (use) dependent bock of Na+ currents; by the last pulse, compared with control, the peak current was ∼15% lower in the presence of 1 μM (R)-4 and ∼16% lower in the presence of 1 μM (R)-6 (Supporting Information, Figure S3B).
B. Polar, Aprotic and Polar, Protic N-(Biphenyl-4′-yl)methyl (R)-2-Acetamido-3-methoxypropionamide Derivatives in Rat Embryonic Cortical Neurons
Next, we tested many of the compounds with 3″-polar, aprotic R-substituents (R = F, Cl, Br, I, CF3, OCF3) in rat embryonic cortical neurons. These cells express channel subtypes (NaV1.1, NaV1.2, NaV1.3, NaV1.6) that participate in seizure propagation,41 and we assumed contributions from all four CNS NaV isoforms to the observed effects. Slow inactivation, fast inactivation, steady-state activation, and frequency (use) dependence of Na+ currents in rat cortical neurons grown for 7–10 days in vitro were examined using protocols described earlier.42 Because of the reduced availability of cortical neurons and their viability under the patch-clamp electrophysiology conditions, we used only 10 μM (R)-A compounds. Furthermore, because the CAD cell patch-clamp electrophysiology data for the 3″- and 4″-substituted isomers were similar, we examined only the 3″-substituted derivatives. Using cortical neurons, we also evaluated several (R)-A compounds with 3″-polar, protic R-substituents (R = N(H)C(O)CH3, CO2H, NH3+Cl–) to try to correlate their activities on VGSCs with their diminished anticonvulsant activities.
First, we determined the ability of the (R)-A derivatives with a 3″-polar, aprotic R-substituent ((R)-3, (R)-5, (R)-7, (R)-8, (R)-10, (R)-11) to modulate the VGSC transition to a slow-inactivated state. Cortical cells were held at −70 mV and conditioned to potentials ranging from −100 to +20 mV (in +10 mV increments) for 5 s. Then fast-inactivated channels were allowed to recover for 1000 ms at a hyperpolarized pulse to −70 mV, and the fraction of channels available was tested by a single depolarizing pulse, to −10 mV, for 20 ms (Figure 5A, left). The hyperpolarization pulse allows channel recovery from fast inactivation while limiting recovery from slow inactivation. Representative traces that illustrate the extent of slow inactivation observed at −50 mV are compared with the prepulse at −100 mV in the absence or presence of (R)-5 in Figure 5A, with complete slow inactivation curves (normalized peak vs prepulse potential) shown in Figure 5B. For the 0.1% DMSO (control) treated cells (−50 mV), 0.82 ± 0.03 fractional unit (n = 5) of the Na+ current was available, suggesting a small fraction (0.18 ± 0.03, calculated as 1 minus the normalized INa) of the channels transitioned to a nonconducting (slow-inactivated) state (Figure 5B,C). Compared with control (0.1% DMSO), all of the 3″-polar, aprotic substituted (R)-A compounds at 10 μM caused significant increase in the maximal fraction of current unavailable by depolarization, with maximal induction observed in the presence of (R)-11: 0.82 ± 0.02 (n = 5; p < 0.01, Mann–Whitney U test). The effectiveness of these (R)-A compounds to promote slow inactivation in cortical neurons at 10 μM approximated the IC50 values observed for slow inactivation in CAD cells where the order of potency was (R)-5, (R)-8 > (R)-11 > (R)-7, (R)-10 > (R)-3 (Figure 2, Table 1). When we tested the (R)-A derivatives with a 3″-polar, protic R-substituent ((R)-13, (R)-15, (R)-16), only (R)-15 caused an increase in the extent of slow inactivation (Supporting Information, Figure S4A,C,E) while neither (R)-13 nor (R)-16 (Supporting Information, Figure S4B,E) was different from 0.1% DMSO (control) treated neurons.
Figure 5.

Effects of (R)-A compounds with a polar, aprotic substituent at the 3″-position on steady-state, slow inactivation state of Na+ currents in embryonic cortical neurons. (A) Voltage protocol for slow inactivation. Currents were evoked by 5 s prepulses between −100 and +20 mV (in +10 mV increments), and then fast-inactivated channels were allowed to recover for 1000 ms at a hyperpolarized pulse to −70 mV before testing for the fraction of available channels for 20 ms at −10 mV. The fraction of channels available at −10 mV was analyzed. Shown are representative current traces from cortical neurons in the absence (control, 0.1% DMSO) or presence of 10 μM (R)-5. The black and gray traces represent the currents evoked at −100 and −50 mV, respectively (highlighted in the voltage protocol as thick, dashed line). (B) Summary of steady-state, slow-activation curves for neurons treated with 0.1% DMSO (control) or 10 μM (R)-5. Significant drug-induced slow inactivation was evident at voltages more depolarizing than −80 mV in neurons treated with (R)-5. (C) Summary of the available current fraction at −50 mV for neurons treated with DMSO (control) or 10 μM (R)-3, (R)-5, (R)-7, (R)-8, (R)-10, and (R)-11. Asterisks (∗) indicate statistically significant differences in available current fraction between control (0.1% DMSO) and the indicated compounds (p < 0.05, Student’s t-test). Data are from five to six cells per condition.
We next asked if the (R)-A derivatives that contained a 3″-polar R-substituent that was either aprotic ((R)-3, (R)-5, (R)-6, (R)-7, (R)-8, (R)-10, (R)-11) or protic ((R)-13, (R)-15, (R)-16) could enhance steady-state fast inactivation. To investigate, we used a protocol (Figure 6A, left) designed to induce a fast-inactivated state, similar to that previously described.42 Neurons were held at −70 mV, stepped to inactivating prepulse potentials ranging from −120 to −10 mV (in 10 mV increments) for 500 ms. Then the cells were stepped to 0 mV for 20 ms to measure the available current. A 500 ms conditioning pulse was used because it allowed all of the endogenous channels to transition to a fast-inactivated state at all potentials assayed. Steady-state, fast inactivation curves of Na+ currents (representative family of control current traces shown in Figure 6B, left) from control (0.1% DMSO) treated and (R)-3-, (R)-5-, (R)-6-, (R)-7-, (R)-8-, (R)-10-, and (R)-11-treated cortical neurons were well fitted with a single Boltzmann function (R2 > 0.983 for all conditions). The V1/2 inactivation value for 0.1% DMSO-treated cells was −53.2 ± 1.2 mV (n = 4), which was significantly different from that of (R)-5 (−59.9 ± 3.1 mV; n = 5), (R)-7 (−58.3 ± 1.4 mV; n = 4), and (R)-11 (−60.1 ± 1.2 mV; n = 5) treated cells (p < 0.05 vs control (0.1% DMSO); Student’s t-test; Figure 6C,D); none of the other compounds ((R)-3, (R)-6, (R)-8, (R)-10) were different from controls (0.1% DMSO). The slope values were unchanged between control and any of the conditions tested. By comparison, we did not see evidence to indicate that the (R)-A compounds with 3″-polar, protic R-substituents, (R)-13, (R)-15, and (R)-16, affected V1/2 or k values of steady-state fast inactivation (Figure 6D).
Figure 6.
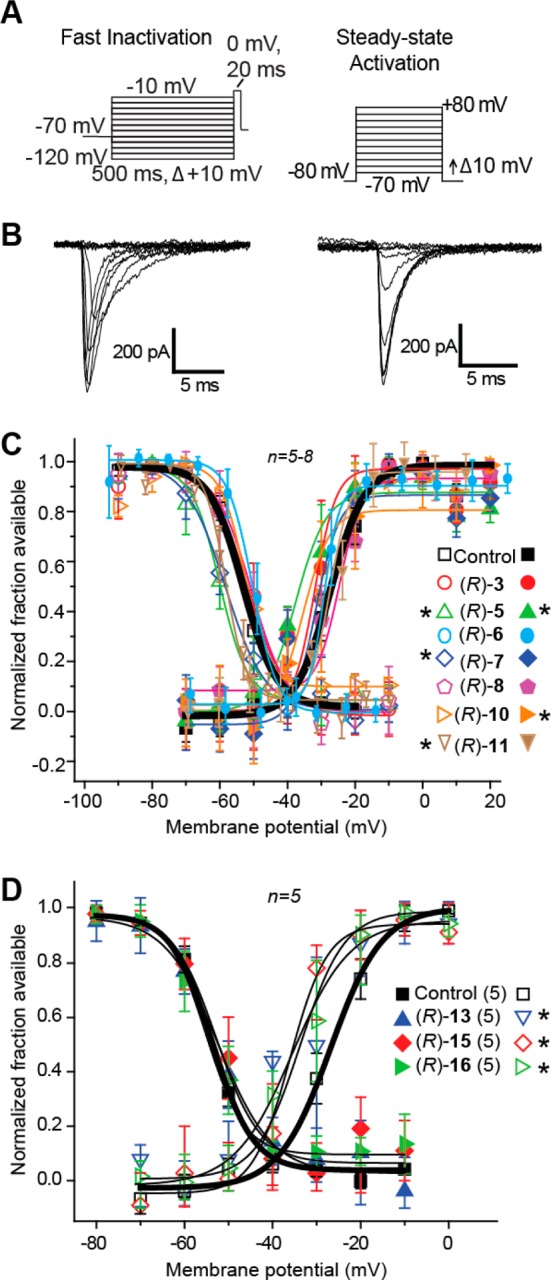
Effects of (R)-A compounds with a polar, aprotic or a polar, protic substituent at the 3″-position on fast inactivation and steady-state activation states of Na+ currents in cortical embryonic neurons. (A) Voltage protocol for fast inactivation (left) and activation (right). (B) Representative family of currents in response to these protocols is illustrated. (C, D) Representative Boltzmann fits for steady-state fast inactivation and activation for cortical neurons treated with 0.1% DMSO (control) and various concentrations of the indicated compounds are shown. Values for V1/2, the voltage of half-maximal inactivation and activation, and the slope factors (k) were derived from Boltzmann distribution fits to the individual recordings and were averaged to determine the mean (±SEM) voltage dependence of steady-state inactivation and activation, respectively. Statistically significant differences between control and fast inactivation or activation are indicated by asterisks (p < 0.05, one-way ANOVA). Data are from five to eight cells per condition.
Next, we tested whether (R)-A compounds that contained polar R-substituents, aprotic or protic, at the 3″-position could alter voltage-dependent activation properties of Na+ currents in cortical neurons. Compound-treated cortical neuron activation changes were measured by whole-cell ionic conductances by comparing their midpoints (V1/2) and slope factors (k) in response to command voltage changes (Figure 6A, right). Representative traces for the 0.1% DMSO-treated neurons (control) in response to the voltage protocol are shown in Figure 6B (right). Boltzmann fits for DMSO (control) and the (R)-A compounds with polar, aprotic and polar, protic R-substituents are shown in Figure 6C and Figure 6D, respectively. The V1/2 value for steady-state activation for 0.1% DMSO-treated (control) neurons was −27.3 ± 3.3 mV (n = 5), which was significantly different from those of (R)-5 (−37.5 ± 4.9 mV; n = 5), (R)-10 (−35.4 ± 5.4 mV; n = 5), (R)-11 (−33.2 ± 1.9 mV; n = 4), (R)-13 (−39.0 ± 2.6 mV; n = 4), (R)-15 (−36.8 ± 3.0 mV; n = 5), and (R)-16 (−33.4 ± 3.2 mV; n = 5) (p < 0.05 vs control; Student’s t-test; Figure 6C,D). The hyperpolarizing shifts induced by these compounds suggest that in the presence of the compounds, NaV currents are likely to activate earlier (i.e., at less-depolarized potentials).
Finally, we tested whether the (R)-A derivatives containing polar R-substituents, aprotic or protic, at the 3″-position could affect frequency (use) dependent block of Na+ currents. Unlike the data obtained with CAD cells, none of the compounds tested at a 10 μM concentration in cortical neurons affected frequency (use) dependent block of Na+ currents, compared with controls (0.1% DMSO) (Supporting Information, Figure S5).
C. (R)-N-(3″-Chlorobiphenyl-4′-yl)methyl (R)-2-Acetamido-3-methoxypropionamide ((R)-5) in HEK293 Cells
In order to assess the relative effect of an (R)-A compound on Na+ channel isoforms, we examined the Na+ currents in HEK293 cells that stably express CNS (hNaV1.1 and rNaV1.3), peripheral nervous system (hNaV1.7), or cardiac (hNaV1.5) isoforms. Compound (R)-5 (10 μM) was chosen for study because it displayed excellent anticonvulsant activity in mice (ip) and rats (po) (Table 1), and it modulated Na+ currents in CAD cells and rat embryonic cortical neurons.
Properties of slow inactivation, fast inactivation, steady-state activation, and frequency (use) dependent inhibition were examined in the four cell lines using voltage protocols illustrated in Figure 7A,K. The results from these experiments are summarized in Table 2. Notably, (R)-5 exhibited similar, but not identical, effects on these biophysical properties irrespective of Na+ channels, indicating that this compound exhibited little isoform specificity. We observed that (R)-5 transitioned the four Na+ channel subtypes to the slow-inactivated state and that differences were observed in the degree to which (R)-5 affected Na+ channel fast inactivation, fast activation, and the frequency (use) inhibition of Na+ currents.
Figure 7.

Analysis of (R)-5 on electrophysiological properties of NaV1.1, NaV1.3, NaV1.7, and NaV1.5 currents in HEK293 cells. (A, K) Voltage protocols for examining slow inactivation, fast inactivation, activation, and frequency (use) dependent block. NaV1.5 uses hyperpolarized protocols because of differences in the hyperpolarized activation of this isoform. (B, E, H, L) Summary of steady-state slow-activation curves for HEK293 cells treated with 0.1% DMSO (control) or 10 μM (R)-5. Insets illustrate representative current traces from HEK293 cells in the absence (control, 0.1% DMSO) or presence of 10 μM (R)-5. The black and colored traces represent the currents evoked at −120 and −50 mV, respectively (highlighted in the voltage protocol). (C, F, I, M) Representative Boltzmann fits for steady-state fast inactivation and activation for HEK293 cells treated with 0.1% DMSO (control) or 10 μM (R)-5. Values for V1/2, the voltage of half-maximal inactivation and activation, and the slope factors (k) were derived from Boltzmann distribution fits to the individual recordings and were averaged to determine the mean (±SEM) voltage dependence of steady-state inactivation and activation, respectively. (D, G, J, N) Summary of average frequency (use) dependent decrease in current amplitude over time (±SEM) produced by control (0.1% DMSO) or 10 μM (R)-5. Data are from five to six cells per condition.
Table 2. Comparative Current Densities, Cell Capacitances, and Boltzmann Parameters of Voltage Dependence of Channel Activation and Steady-State Fast Inactivation Curves for Control-Treated and (R)-N-(3″-Chlorobiphenyl-4′-yl)methyl 2-Acetamido-3-methoxypropionamide ((R)-5) Treated HEK293 Cells Expressing NaV1.1, NaV1.3, NaV1.7, or NaV1.5 Channelsa.
| voltage dependence of activationc |
voltage dependence of
fast inactivation |
|||||
|---|---|---|---|---|---|---|
| condition | extent of slow inactivation (at −50 mV)b | V1/2 (mV) | slope (mV/e-fold) | V1/2 (mV) | slope (mV/e-fold) | use-dependent inhibitiond |
| NaV1.1 | ||||||
| control | 0.17 ± 0.04 (6) | –24.29 ± 1.52 (5) | 4.92 ± 0.60 (5) | –64.29 ± 3.63 (6) | 4.93 ± 1.48 (6) | 1.02 ± 0.08 (5) |
| 10 μM (R)-5 | 0.77 ± 0.03 (6)* | –22.43 ± 2.94 (6) | 4.34 ± 1.78 (5) | –70.00 ± 4.67 (6) | 6.21 ± 2.11 (6) | 0.81 ± 0.07 (6)* |
| NaV1.3 | ||||||
| control | 0.04 ± 0.02 (5) | –29.56 ± 4.94 (5) | 4.07 ± 1.96 (5) | –65.39 ± 7.77 (5) | 5.55 ± 1.28 (5) | 1.12 ± 0.19 (5) |
| 10 μM (R)-5 | 0.71 ± 0.09 (5)* | –25.44 ± 2.01 (4) | 4.47 ± 1.56 (4) | –63.39 ± 7.01 (5) | 6.81 ± 2.67 (5) | 1.03 ± 0.18 (5) |
| NaV1.7 | ||||||
| control | 0.07 ± 0.04 (5) | –27.15 ± 4.01 (5) | 3.61 ± 1.24 (5) | –61.09 ± 2.99 (5) | 4.83 ± 1.12 (5) | 1.03 ± 0.09 (5) |
| 10 μM (R)-5 | 0.80 ± 0.03 (5)* | –27.27 ± 1.98 (5) | 4.21 ± 1.43 (5) | –78.01 ± 3.69 (5)* | 5.52 ± 1.98 (5) | 0.92 ± 0.06 (5) |
| NaV1.5 | ||||||
| control | 0.18 ± 0.04 (5) | –55.67 ± 3.19 (5) | 3.16 ± 1.75 (5) | –96.52 ± 1.60 (5) | 5.34 ± 0.87 (4) | 1.05 ± 0.16 (5) |
| 10 μM (R)-5 | 0.80 ± 0.03 (5)* | –57.12 ± 4.71 (5) | 3.69 ± 1.69 (7) | –109.11 ± 1.76 (5) | 8.92 ± 0.82 (7)* | 0.62 ± 0.31 (5) |
N values are indicated in parentheses. The protocols used for these parameters are illustrated in Figure 7. Asterisks represent statistically significant differences compared to control within each tested group (p < 0.05, Student’s t test).
The extent of slow inactivation was calculated as 1 minus the normalized peak INa and denotes the fraction of channels that have transitioned to a nonconducting (slow-inactivated) state at −50 mV.
Values for V1/2, the voltage of half-maximal activation, and slope were derived from Boltzmann distribution fits to the individual recordings and averaged to determine the mean and standard error of the mean (±SEM).
Fraction of current remaining at the end of the 30-pulse train, normalized to the first pulse.
Discussion
Recently, we merged the structures of functionalized amino acids (FAAs)1,7−9 and α-aminoamides (AAAs),43 two classes of compounds that have shown excellent anticonvulsant activities in seizure models, to give compounds (R)-B and reported on their anticonvulsant activities (Figure 8).11 The substituted (R)-A compounds described herein are examples of (R)-B where R′ is a methoxymethyl moiety (R′ = CH2OCH3) and the X-Y-Z unit is a single bond; they are also 4′-aryl derivatives of (R)-1. In 2010, we showed that the unsubstituted N-(biphenyl-4′-yl)methyl derivative (R)-2 exhibited excellent anticonvulsant activity but that seizure protection in mice and rats was accompanied by appreciable neurotoxicity (Figure 1, Table 1; MES ED50 (mg/kg), 8.0 (mice, ip), 2.0 (rat, po); TD50 (mg/kg), 11 (mice, ip), 49 (rat, po)).10 Subsequently, we showed that the corresponding 3″-F-substituted analogue (R)-3 retained high anticonvulsant activity but that neurotoxicity was reduced (Figure 1, Table 1; MES ED50 (mg/kg), 12 (mice, ip), 2.4 (rat, po); TD50 (mg/kg), 50 (mice, ip), rat >500 (rat, po)).11 While the PI value in mice for (R)-3 was higher than for (R)-2, it was still below that of the ASD (R)-1 (PI (mice, ip): (R)-2, 1.4; (R)-3, 4.1; (R)-1, 6.0).1 In this study, we explored the effect of the terminal aryl R-substituent in (R)-A on anticonvulsant activity, neurotoxicity, and Na+ channel function.
Figure 8.
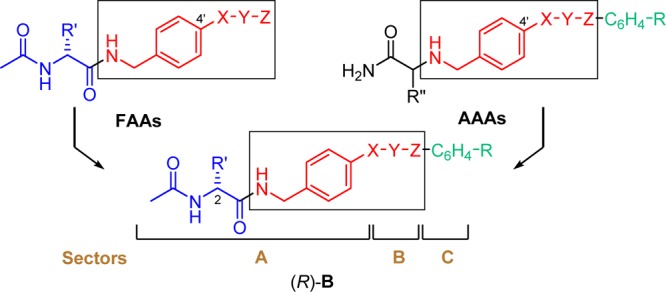
Overlay of pharmacophores in FAAs and AAAs to give (R)-B.
We found that the placement of polar, aprotic R-substituents on the terminal aryl ring of (R)-A provided compounds with excellent anticonvulsant activities and that these activities were only modestly affected by the site of aryl substitution (3″ vs 4″). In addition, we found that several (R)-A derivatives with polar, aprotic R-substituents had improved PI values over (R)-2 (PI = 1.4) but that the PI values varied with the R-substituent and the site of substitution. For the 3″-Cl and 4″-Cl derivatives ((R)-5, (R)-6), high PI values were obtained regardless of the site of substitution (PI (mice, ip): (R)-5 (3″), 7.6; (R)-6 (4″), 6.8) and that the values exceeded that of (R)-2 (PI = 1.4) and even (R)-1 (PI = 6.0). Correspondingly, for the 3″- and 4″-F derivatives ((R)-3, (R)-4) and the 3″- and 4″-OCF3 compounds ((R)-11, (R)-12), high PI values were observed for only the 3″-substituted isomers (PI (mice, ip): (R)-3 (3″-F), 4.1; (R)-4 (4″-F), 1.8; (R)-11 (3″-OCF3), 4.9; (R)-12 (4″-OCF3), 2.0). Correspondingly, when a polar, protic R-substituent was placed in the terminal aryl ring in (R)-A to give (R)-13–(R)-16, we observed minimal or no anticonvulsant activity. Accompanying this loss of activity was a reduction in neurotoxicity, suggesting that the activity loss resulted from either reduced levels of these compounds in the CNS or a reduced interaction with the receptor(s) responsible for the seizure protection and toxicity or both. When we tested whether (R)-13, (R)-15, and (R)-16 modulated Na+ currents in rat embryonic cortical neurons, only (R)-15 (R = 3″-CO2H) displayed notable activity (Supporting Information, Figure S4). This latter finding indicated that the diminished anticonvulsant activities for (R)-13 and (R)-16 may have resulted, in part, from their inability to promote Na+ channel slow inactivation during the seizure test.
Both FAAs7−9 and AAAs44 likely exert their anticonvulsant activities by interacting with Na+ channels. The FAA, (R)-1, has been shown to preferentially promote the transition of Na+ channels to the slow inactivation state without affecting fast inactivation.7−9 We demonstrated that (R)-3 displayed potent Na+ channel slow inactivation in CAD cells that exceeded (R)-1 (IC50 (μM): (R)-3, 2.9; (R)-1, 85).15 Moreover, (R)-3 displayed notable frequency (use) dependent inhibition of Na+ currents at concentrations (15 μM) above its IC50 value (Figure 4B).15 In this study, we determined the Na+ channel properties of the (R)-A compounds in three different cell types (CAD cells, rat embryonic cortical neurons, HEK293-transfected cells). Overall, we found it difficult to compare the effects of individual (R)-A compounds across the different cell systems. For several compounds, we observed differences in the three cell types in their ability to affect Na+ channel fast inactivation and fast activation and frequency (use) inhibition of Na+ currents. We have tentatively attributed these differences to the different channel compositions, expression levels, auxiliary proteins, and origins (mouse, rat, human) in the CAD, cortical, and HEK293 cell systems and the test (R)-A concentrations used in the studies. Accordingly, the experimental results in any given cell system were used to identify interesting compounds, and direct comparisons were only made within data sets from the same cell type.
We found that (R)-4–(R)-8 and (R)-10–(R)-12, compounds that contained polar, aprotic R-substituents, potently transitioned CAD cell Na+ channels to the slow-inactivated state (IC50 = 0.12–2.9 μM) and that Na+ channel fast inactivation was not observed (Figures 2, 3; Supporting Information Figures S1, S2). Furthermore, we observed that many compounds ((R)-4–(R)-6, (R)-10, (R)-11) showed frequency (use) dependent inhibition of Na+ currents, provided that concentrations higher than the slow inactivation IC50 value were employed (Figure 4; Supporting Information, Figure S3). We9 and others7 have not observed notable frequency (use) inhibition of Na+ currents for (R)-1 at 100 μM. Our finding that frequency (use) inhibition for selected (R)-A compounds was observed only at concentrations ≥5-fold higher than their slow inactivation IC50 values in CAD cells led us to retest (R)-1 at concentrations above its IC50 value (85 μM). At 500 μM (R)-1, we observed noticeable frequency (use) inhibition of Na+ currents (Figure 9) similar to that seen for the (R)-A compounds (Figure 4). Significantly, this concentration is higher than the reported human therapeutic (R)-1 plasma concentrations (20–50 μM),45 suggesting that frequency (use) inhibition of Na+ currents may not be a clinically important pathway for its drug function. In contrast, we found that several substituted (R)-A derivatives, at low micromolar concentrations, promoted Na+ channel slow inactivation and frequency (use) dependent inhibition of Na+ currents. This finding provides a potential comparative pharmacological advantage for these compounds, since both mechanisms are proven pathways to control neuronal hyperexcitability in the epileptic neuron.7−9,39
Figure 9.
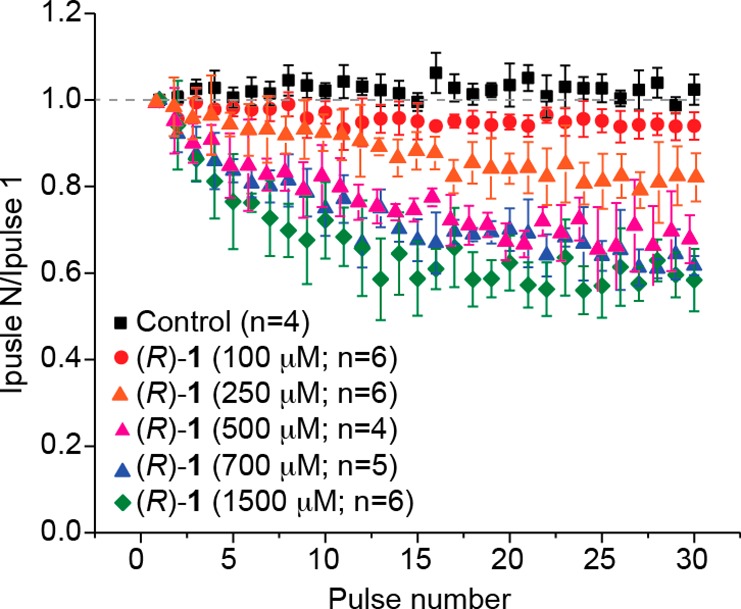
Effect of (R)-1 in CAD cells on frequency (use) dependent block. Summary of average frequency (use) dependent current amplitude decrease (±SEM) produced by control (0.1% DMSO) or by the presence of various concentrations of (R)-1 is shown (p > 0.05, one-way ANOVA with Dunnett’s post hoc test). Concentrations greater than 500 μM (R)-1 caused a significant inhibition of use-dependence starting at about pulse 8 (p < 0.05, one-way ANOVA with Dunnett’s post hoc test). Data are from four to six cells per condition.
To better approximate CNS VGSC activities, we tested many of the (R)-A compounds with a 3″-polar, aprotic R-substituent in rat embryonic cortical neurons that expressed the NaV1.1, NaV1.2, NaV1.3, and NaV1.6 channels. We observed that (R)-3, (R)-5, (R)-7, (R)-8, (R)-10, and (R)-11 all promoted slow inactivation at 10 μM (Figure 5) and their relative effectiveness mirrored the slow inactivation IC50 values measured in CAD cells (Figure 2). Differences were found between the cortical neurons and the CAD cells on the ability of these compounds to affect fast inactivation and their ability to inhibit frequency (use) dependent inhibition of Na+ currents (Figures 3, 4, 6; Supporting Information, Figure S5). We have not explored the factors that contributed to these differences.
There are nine Na+ channel isoforms. Despite structural similarity, the isoforms’ distribution and biophysical properties (e.g., kinetics of activation and inactivation, voltage, and frequency (use) dependency) are distinct, each having specific functions.39,41,46 Thus, we evaluated (R)-5 in HEK293 cells expressing hNaV1.1, rNaV1.3, hNaV1.5, or hNaV1.7 channels. The patch-clamp electrophysiology protocols for the cells containing hNaV1.5 were different from those used for hNaV1.1, rNaV1.3, and hNaV1.7 (Figure 7A,K). We found that 10 μM (R)-5 transitioned the Na+ channels to the slow-inactivated state regardless of channel type (Figure 7B,E,H,L). Correspondingly, differences in the fast inactivation profiles were observed for the four isoforms. Compound (R)-5 did not affect rNaV1.3 fast inactivation compared with 0.1% DMSO (control), while a depolarizing shift was found for the hNaV1.1-, hNaV1.5-, and hNaV1.7-transfected cells, with the most pronounced shift observed in hNaV1.7-expressing cells (Figure 7C,F,I,M). Finally, we observed notable frequency (use) inhibition of Na+ currents for the transfected hNaV1.5 cells (Figure 7N), lesser amounts for the hNaV1.1- and hNaV1.7-expressing cells (Figure 7D,J), and no inhibition for the rNaV1.3-expressing cells (Figure 7G). Together these findings showed that (R)-5, and perhaps the other polar, aprotic R-substituted (R)-A derivatives, displayed little Na+ channel isoform specificity. Rather, (R)-5 appears to act as a functionally selective Na+ channel inhibitor47 that controls hyperexcitability upon excitation by transitioning a greater fraction of Na+ channels to their slow-inactivated state and by inhibiting Na+ currents in a frequency (use) dependent manner.
Conclusions
The successful merger of FAAs and AAAs (Figure 8) provided substituted N-(biphenyl-4′-yl)methyl (R)-2-acetamido-3-methoxypropionamide derivatives ((R)-A). Select (R)-A compounds displayed low ED50 values and high PI values that, in rodents, compared favorably with most clinical ASDs. We found compounds that potently transitioned Na+ channels into the slow-inactivated state and displayed frequency (use) inhibition of Na+ currents at low micromolar concentrations. The activities of (R)-As along with their pharmacokinetic properties are being investigated further.
Experimental Section
General Methods
Melting points were determined in open capillary tubes using a Thomas-Hoover melting point apparatus and are uncorrected. Optical rotations were obtained on a Jasco P-1030 polarimeter at the sodium D line (589 nm) using a 1 dm path length cell. NMR spectra were obtained at 400 MHz (1H) and 100 MHz (13C) using TMS as an internal standard. Chemical shifts (δ) are reported in parts per million (ppm) from tetramethylsilane. Low-resolution mass spectra were obtained with a BioToF-II-Bruker Daltonics spectrometer by Dr. S. Habibi at the University of North Carolina, Department of Chemistry. The high-resolution mass spectrum was performed on a Bruker Apex-Q 12 Telsa FTICR spectrometer by Dr. S. Habibi. Microanalyses were performed by Atlantic Microlab, Inc. (Norcross, GA). Reactions were monitored by analytical thin-layer chromatography (TLC) plates (Aldrich, catalog no. Z12272-6) and analyzed with 254 nm UV light. The mixtures were purified by flash column chromatography using silica gel (Dynamic Adsorbents Inc., catalog no. 02826-25). All chemicals and solvents were reagent grade and used as obtained from commercial sources without further purification. Yields reported are for purified products and were not optimized. Compounds were checked by TLC, 1H and 13C NMR, MS, and elemental analyses. The analytical results are within ±0.40% of the theoretical value. The NMR and the analytical data confirmed that the purity of the products was ≥95%.
General Procedure for the Deprotection and Acetylation of (R)-N-(Biphenyl-4-yl)methyl 2-N-(tert-Butoxycarbonyl)amino-3-methoxypropionamide Derivatives (Method 1)
A CH2Cl2 solution (0.1–0.3 M) of the (R)-N-(biphenyl-4-yl)methyl 2-N-(tert-butoxycarbonyl)amino-3-methoxypropionamide derivative was treated with 4 M HCl in dioxane (3–4 equiv) at room temperature (2–6 h). The reaction mixture was evaporated in vacuo. The resulting residue was dissolved in CH2Cl2 (0.1–0.3 M), and then triethylamine (2–3 equiv) and acetyl chloride (1.0–1.2 equiv) were carefully added at 0 °C. The resulting solution was stirred at room temperature (2–4 h). The resulting solution was washed with an aqueous 10% citric acid solution followed by a saturated aqueous NaHCO3 solution. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by column chromatography on SiO2 and/or recrystallized with EtOAc/hexanes.
(R)-N-(4″-Fluorobiphenyl-4′-yl)methyl 2-Acetamido-3-methoxypropionamide ((R)-4)
Using method 1, (R)-56 (3.17 g, 7.9 mmol) and 4 M HCl (6.90 mL, 27.6 mmol) followed by Et3N (2.42 mL, 17.3 mmol) and AcCl (0.61 mL, 8.7 mmol) gave (R)-4 as a white solid (2.50 g, 92%): Rf = 0.40 (MeOH/CH2Cl2 1/20); mp 189–190 °C; [α]26D −17.8° (c 1.0, CHCl3); 1H NMR (CDCl3) δ 2.04 (s, 3H), 3.40 (s, 3H), 3.44–3.48 (m, 1H), 3.83 (dd, J = 3.9, 9.0 Hz, 1H), 4.45–4.59 (m, 3H), 6.43–5.46 (br d, 1H), 6.81–6.85 (m, 1H), 7.12 (t, J = 8.6 Hz, 2H), 7.32 (d, J = 7.6 Hz, 2H), 7.49–7.54 (m, 4H); addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-4 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (DMSO-d6) δ 22.6, 41.8, 52.7, 58.2, 72.1, 115.7 (d, J = 21.3 Hz, 2C), 126.5, 127.6, 128.5 (d, J = 8.0 Hz, 2C), 136.4 (d, J = 2.9 Hz), 137.6, 138.6, 161.8 (d, J = 242.4 Hz), 169.5, 169.8; LRMS (ESI+) 367.1 [M + Na]+ (calcd for C19H21FN2O3Na+ 367.1). Anal. (C19H21FN2O3) C, H, F, N.
(R)-N-(3″-Chlorobiphenyl-4′-yl)methyl 2-Acetamido-3-methoxypropionamide ((R)-5)
Using method 1, (R)-57 (4.00 g, 9.6 mmol) and 4 M HCl (8.36 mL, 33.4 mmol) followed by Et3N (2.93 mL, 21.0 mmol) and AcCl (0.74 mL, 10.5 mmol) gave (R)-5 as a white solid (2.85 g, 83%): Rf = 0.40 (MeOH/CH2Cl2 1/20); mp 172–173 °C; [α]26D −15.8° (c 1.1, CHCl3); 1H NMR (CDCl3) δ 2.00 (s, 3H), 3.37 (s, 3H), 3.49 (dd, J = 7.0, 9.0 Hz, 1H), 3.78 (dd, J = 4.0, 9.0 Hz, 1H), 4.43 (1/2HH′q, J = 5.8, 15.0 Hz, 1H), 4.49 (1/2HH′q, J = 5.8, 15.0 Hz, 1H), 4.62–4.67 (m, 1H), 6.71 (d, J = 6.8 Hz, 1H), 7.15–7.18 (m, 1H), 7.30–7.52 (m, 8H); addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-5 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.3, 43.3, 52.7, 59.2, 72.1, 125.3, 127.3, 127.5, 128.1, 130.2, 134.8, 137.9, 139.1, 142.6, 170.3, 170.6; the remaining aromatic peak was not detected and is believed to overlap with the observed signals; LRMS (ESI+) 383.1 [M + Na]+ (calcd for C19H21ClN2O3Na+ 383.1). Anal. (C19H21ClN2O3) C, H, Cl, N.
(R)-N-(4″-Chlorobiphenyl-4′-yl)methyl 2-Acetamido-3-methoxypropionamide ((R)-6)
Using method 1, (R)-58 (2.80 g, 6.7 mmol), and 4 M HCl (6.70 mL, 26.7 mmol) followed by Et3N (2.10 mL, 14.7 mmol) and AcCl (0.52 mL, 7.4 mmol) gave (R)-6 as a white solid (1.83 g, 76%): Rf = 0.40 (MeOH/CH2Cl2 1/20); mp 215–216 °C; [α]26D −16.9° (c 1.1, CHCl3); 1H NMR (CDCl3) δ 2.03 (s, 3H), 3.39 (s, 3H), 3.44–3.48 (m, 1H), 3.82 (dd, J = 4.0, 9.2 Hz, 1H), 4.45–4.61 (m, 3H), 6.45–6.49 (br d, 1H), 6.86–6.91 (m, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.39 (d, J = 8.4 Hz, 2H), 7.48–7.52 (m, 4H); addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-6 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.4, 43.4, 52.7, 59.3, 71.9, 127.5, 128.2, 128.5, 129.2, 133.7, 137.5, 139.3, 139.4, 170.3, 170.5; LRMS (ESI+) 361.2 [M + H]+ (calcd for C19H21ClN2O3H+ 361.2). Anal. (C19H21ClN2O3) C, H, Cl, N.
(R)-N-(3″-Bromobiphenyl-4′-yl)methyl 2-Acetamido-3-methoxypropionamide ((R)-7)
Using method 1, 4 M HCl in dioxane (3.5 mL), (R)-59 (1.30 g, 2.8 mmol), Et3N (862 mg, 8.5 mmol), and AcCl (268 mg, 3.4 mmol) gave compound (R)-7 as a white solid (620 mg, 54%): Rf = 0.44 (1:20 MeOH/CH2Cl2); mp 171–173 °C; [α]26D −17.3° (c 1.0, CHCl3); 1H NMR (CDCl3) δ 2.00 (s, 3H), 3.37 (s, 3H), 3.45–3.49 (m, 1H), 3.79 (dd, J = 4.0, 9.0 Hz, 1H), 4.40–4.55 (m, 2H), 4.56–4.66 (m, 1H), 6.60 (d, J = 6.4 Hz, 1H), 6.98–7.08 (br s, 1H), 7.24–7.35 (m, 3H), 7.39–7.56 (m, 4H), 7.68 (s, 1H); addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-7 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.1, 43.1, 52.5, 59.1, 71.8, 122.9, 125.6, 127.3, 127.9, 130.0, 130.2, 130.3, 137.6, 138.8, 142.7, 170.1, 170.3; LRMS (ESI+) 405.0 [M + H]+ (100%), 407.0 [M + 2 + H]+ (100%); HRMS (ESI+) 405.0814 [M + H]+ (calcd for C19H2179BrN2O3H+ 405.0814). Anal. (C19H21BrN2O3) C, H, Br, N.
(R)-N-(3″-Iodobiphenyl-4′-yl)methyl 2-Acetamido-3-methoxypropionamide ((R)-8)
Using method 1, 4 M HCl in dioxane (2.0 mL), (R)-60 (670 mg, 1.3 mmol), Et3N (515 mg, 3.9 mmol), and AcCl (123 mg, 1.6 mmol) gave compound (R)-8 as a white solid (515 mg, 87%): Rf = 0.41 (1:20 MeOH/CH2Cl2); mp 161–163 °C; [α]26D −11.0° (c 1.0, CHCl3); 1H NMR (CDCl3) δ 2.05 (s, 3H), 3.41 (s, 3H), 3.43–3.50 (m, 1H), 3.84 (dd, J = 4.3, 9.3 Hz, 1H), 4.46–4.55 (m, 2H), 4.57–4.60 (m, 1H), 6.45 (d, J = 6.8 Hz, 1H), 6.83 (t, J = 4.8 Hz, 1H), 7.17 (t, J = 7.8 Hz, 1H), 7.34 (d, J = 8.2 Hz, 2H), 7.48–7.55 (m, 3H), 7.68 (d, J = 7.8 Hz, 1H), 7.92 (s, 1H); addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-8 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.2, 43.2, 52.4, 59.1, 71.6, 94.8, 126.3, 127.4, 127.9, 130.4, 136.0, 136.2, 137.6, 138.9, 142.9, 170.0, 170.3; HRMS (ESI+) 453.0675 [M + H]+ (calcd for C19H21IN2O3H+ 453.0673). Anal. (C19H21IN2O3) C, H, I, N.
(R)-N-(3″-Cyanobiphenyl-4′-yl)methyl 2-Acetamido-3-methoxypropionamide ((R)-9)
Using method 1, (R)-61 (3.10 g, 7.6 mmol) and 4 M HCl (9.5 mL) followed by Et3N (2.30 g, 22.7 mmol) and AcCl (0.71 g, 9.1 mmol) gave the desired product (R)-9 (2.00 g, 75%) as a white solid: Rf = 0.42 (CH2Cl2/CH3OH 19/1); mp 122–123 °C; [α]24.5D −16.0° (c 1.0, CHCl3); 1H NMR (CDCl3) δ 2.03 (s, 3H), 3.40 (s, 3H), 3.47–3.52 (m, 1H), 3.79–3.84 (m, 1H), 4.48–4.54 (m, 2H), 4.59–4.63 (m, 1H), 6.56–6.59 (br d, 1H), 7.03–7.07 (br t, 1H), 7.36 (d, J = 8.4 Hz, 2H), 7.48–7.64 (m, 4H), 7.76–7.82 (m, 2H); addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-9 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.4, 43.3, 52.7, 59.3, 71.9, 113.2, 119.0, 127.5, 128.3, 129.8, 130.8, 130.9, 131.6, 138.2, 138.5, 142.9, 170.4, 170.6; LRMS (ESI+) 352.2 [M + H]+ (calcd for C20H22N3O3+ 352.2); HRMS (ESI+) 352.1660 [M + H]+ (calcd for C20H22N3O3+ 352.1661). Anal. (C20H21N3O3) C, H, N.
(R)-N-(3″-Trifluoromethylbiphenyl-4′-yl)methyl 2-Acetamido-3-methoxypropionamide ((R)-10)
Using method 1, 4 M HCl in dioxane (3.0 mL), (R)-62 (1.34 g, 3.0 mmol), Et3N (896 mg, 8.9 mmol), and AcCl (348 g, 4.4 mmol) gave compound (R)-10 as a white solid (1.02 g, 88%): Rf = 0.42 (1:20 MeOH/CH2Cl2); mp 161–162 °C; [α]24D −13.7° (c 1.0, CHCl3); 1H NMR (CDCl3) δ 2.03 (s, 3H), 3.40 (s, 3H), 3.49 (dd, J = 4.3, 9.3 Hz, 1H), 3.81 (dd, J = 4.3, 9.3 Hz, 1H), 4.45–4.58 (m, 2H) 4.61–4.64 (m, 1H), 6.58 (d, J = 7.0 Hz, 1H), 7.04 (t, J = 5.3 Hz, 1H), 7.36 (d, J = 8.2 Hz, 2H), 7.51–7.62 (m, 4H), 7.73 (d, J = 7.4 Hz, 1H), 7.80 (s, 1H); addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-10 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (CDCl3) δ 23.1, 43.1, 52.5, 59.1, 71.7, 123.7 (q, J = 4.0 Hz), 124.0 (q, J = 4.0 Hz), 124.1 (q, J = 272.0 Hz), 126.0, 127.4, 129.2, 130.2, 131.2 (q, J = 32.0 Hz), 137.9, 138.9, 141.4, 170.1, 170.4; MS (ESI+) 395.2 [M + H]+ (calcd for C20H22F3N2O3+ 395.2); HRMS (ESI+) 395.1583 [M + H]+ (calcd for C20H22F3N2O3+ 395.1582). Anal. (C20H21F3N2O3) C, H, F, N.
(R)-N-(3″-Trifluoromethoxybiphenyl-4′-yl)methyl 2-Acetamido-3-methoxypropionamide ((R)-11)
Using method 1, (R)-63 (2.14 g, 4.6 mmol) and 4 M HCl (4.00 mL, 16.0 mmol) followed by Et3N (1.40 mL, 10.1 mmol) and AcCl (0.36 mL, 5.0 mmol) gave (R)-11 as a white solid (1.69 g, 90%): Rf = 0.40 (MeOH/CH2Cl2 1/20); mp 139–140 °C; [α]26D −13.5° (c 1.3, CHCl3); 1H NMR (CDCl3) δ 2.03 (s, 3H), 3.40 (s, 3H), 3.47 (dd, J = 7.4, 9.2 Hz, 1H), 3.82 (dd, J = 4.0, 9.2 Hz, 1H), 4.46–4.61 (m, 3H), 6.49 (d, J = 6.4 Hz, 1H), 6.89–6.93 (m, 1H), 7.18–7.21 (m, 1H), 7.33–7.54 (m, 7H); addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-11 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (DMSO-d6) δ 22.5, 41.7, 52.7, 58.2, 72.1, 119.1, 119.6, 120.1 (q, J = 254.9 Hz), 125.7, 126.7, 127.7, 130.8, 136.8, 139.5, 142.3, 149.0, 169.4, 169.8; LRMS (ESI+) 433.1 [M + Na]+ (calcd for C20H21F3N2O4Na+ 433.1). Anal. (C20H21F3N2O4) C, H, F, N.
(R)-N-(4″-Trifluoromethoxybiphenyl-4′-yl)methyl 2-Acetamido-3-methoxypropionamide ((R)-12)
Using method 1, (R)-64 (0.81 g, 1.7 mmol) and 4 M HCl (1.49 mL, 6.0 mmol) followed by Et3N (0.53 mL, 3.8 mmol) and AcCl (0.13 mL, 1.9 mmol) gave (R)-12 as a white solid (0.59 g, 84%): Rf = 0.40 (MeOH/CH2Cl2 1/20); mp 194–195 °C; [α]26D −14.3° (c 0.9, CHCl3); 1H NMR (CDCl3) δ 2.02 (s, 3H), 3.39 (s, 3H), 3.48 (dd, J = 7.4, 9.2 Hz, 1H), 3.81 (dd, J = 4.0, 9.2 Hz, 1H), 4.45–4.55 (m, 2H), 4.58–4.63 (m, 1H), 6.56 (d, J = 6.4 Hz, 1H), 6.98–7.01 (m, 1H), 7.27 (d, J = 8.0 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 7.49–7.58 (m, 4H); addition of excess (R)-(−)-mandelic acid to a CDCl3 solution of (R)-12 gave only one signal for the acetyl methyl and one signal for the ether methyl protons; 13C NMR (DMSO-d6) δ 22.5, 41.7, 52.7, 58.2, 72.1, 120.1 (q, J = 254.9 Hz, OCF3), 121.4, 126.6, 127.7, 128.4, 137.1, 139.1, 139.3, 147.7, 169.4, 169.8; LRMS (ESI+) 433.1 [M + Na]+ (calcd for C20H21F3N2O4Na+ 433.1). Anal. (C20H21F3N2O4) C, H, F, N.
(R)-N-(3″-Acetylaminobiphenyl-4′-yl)methyl 2-Acetamido-3-methoxypropionamide ((R)-13)
Using method 1, (R)-65 (1.80 g, 4.1 mmol) and 4 M HCl (3.1 mL, 12.4 mmol) followed by Et3N (1.24 g, 12.2 mmol) and AcCl (0.38 g, 4.9 mmol) gave the desired product (R)-13 (1.05 g, 71%) as a white powder: Rf = 0.20 (CH2Cl2/CH3OH 19/1); mp 150–151 °C; [α]24.5D +11.1° (c 1.0, CH3OH); 1H NMR (CDCl3/CD3OD) δ 2.04 (s, 3H), 2.16 (s, 3H), 3.47 (s, 3H), 3.52–3.57 (m, 1H), 3.68–3.73 (m, 1H), 4.40–4.52 (m, 2H), 4.54–4.60 (m, 1H), 7.29–7.40 (m, 4H), 7.50–7.56 (m, 3H), 7.76 (s, 1H); 13C NMR (CDCl3/CD3OD) δ 22.8, 23.9, 43.1, 53.1, 59.1, 72.0, 118.6, 119.0, 122.8, 127.4, 127.9, 129.3, 137.1, 138.8, 140.3, 141.5, 170.4, 171.5; one carbonyl resonance was not detected; LRMS (ESI+) 384.2 [M + H]+ (calcd for C21H26N3O3+ 384.2); HRMS (ESI+) 384.1924 [M + H]+ (calcd for C21H26N3O3+ 384.1923).
(R)-N-(3″-Hydroxymethylbiphenyl-4′-yl)methyl 2-Acetamido-3-methoxypropionamide ((R)-14)
To a solution of (R)-73 (0.65 g, 1.6 mmol) in CH2Cl2 (16 mL) was added 4 M HCl in dioxane (1.5 mL, 6.0 mmol), and the mixture was stirred at room temperature (4 h). The reaction mixture was evaporated in vacuo and recrystallized (CH3OH/EtOAc/hexanes) to give the desired product (R)-14 (0.51 g, 90%) as a white solid: Rf = 0.48 (CH2Cl2/CH3OH 9/1); mp 152–153 °C; [α]22.5D +12.7° (c 1.0, CH3OH); 1H NMR (CDCl3/CD3OD) δ 2.00 (s, 3H), 3.34 (s, 3H), 3.45–3.49 (m, 1H), 3.67–3.72 (m, 1H), 4.42–4.46 (m, 2H), 4.51–4.55 (m, 1H), 4.69 (s, 2H), 7.27–7.31 (m, 3H), 7.35–7.41 (m, 1H), 7.44–7.46 (m, 1H), 7.50–7.54 (m, 3H); 13C NMR (CDCl3/CD3OD) δ 23.1, 43.3, 52.9, 59.3, 65.0, 72.0, 125.7, 126.1, 126.3, 127.5, 128.0, 129.1, 137.1, 140.4, 141.0, 141.8, 170.3, 171.2; LRMS (ESI+) 357.1 [M + H]+ (calcd for C20H24N2O4H+ 357.2). Anal. (C20H24N2O4) C, H, N.
(R)-N-(3″-Carboxybiphenyl-4′-yl)methyl 2-Acetamido-3-methoxypropionamide ((R)-15)
Using the previous procedure, (R)-74 (0.31 g, 0.7 mmol) and 4 M HCl (0.8 mL, 3.2 mmol) gave the desired product (R)-15 (0.21 g, 82%) as a white solid: Rf = 0.56 (CH2Cl2/CH3OH 9/1); mp 196–197 °C; [α]23D +9.6° (c 1.0, CH3OH); 1H NMR (CDCl3/CD3OD) δ 1.99 (s, 3H), 3.46–3.50 (m, 1H), 3.63–3.66 (m, 1H), 4.39 (dd, J = 5.8, 14.0 Hz, 1H), 4.46 (dd, J = 5.8, 14.0 Hz, 1H), 4.50–4.55 (br t, 1H), 6.78–6.84 (m, 1H), 7.16–7.21 (m, 1H), 7.28–7.56 (m, 8H); the methoxy proton resonance was not detected and believed to be overlapped with the adjacent peak near δ 3.40; 13C NMR (CDCl3/CD3OD) δ 22.8, 43.1, 52.9, 59.1, 72.1, 127.4, 128.0, 128.4, 128.8, 128.9, 131.0, 131.5, 137.5, 139.4, 141.0, 168.9, 170.4, 171.5; LRMS (ESI+) 371.2 [M + H]+ (calcd for C20H22N2O5H+ 371.2); HRMS (ESI+) 371.1607 [M + H]+ (calcd for C20H22N2O5H+ 371.1607). Anal. (C20H22N2O5·0.13H2O) C, H, N.
(R)-N-(3″-Aminobiphenyl-4′-yl)methyl 2-Acetamido-3-methoxypropionamide Hydrochloride ((R)-16)
Using the procedure for (R)-14, (R)-75 (1.18 g, 3.59 mmol) and 4 M HCl (2.7 mL, 10.8 mmol) gave the desired product (R)-16 (0.91 g, 90%) as a yellow solid: Rf = 0.00 (EtOAc/hexanes 1/1); mp 99–100 °C; [α]23.5D +10.4 ° (c 1.0, CH3OH); 1H NMR (CDCl3/CD3OD) δ 1.99 (s, 3H), 3.36 (s, 3H), 3.50 (dd, J = 4.0, 9.2 Hz, 1H), 3.78 (dd, J = 7.2, 9.2 Hz, 1H), 4.43 (dd, J = 5.8, 15.0 Hz, 1H), 4.48 (dd, J = 5.8, 15.0 Hz, 1H), 4.59–4.64 (m, 1H), 6.63–6.67 (m, 2H), 6.85 (m, 1H), 6.93–6.96 (m, 1H), 6.99–7.05 (br t, 1H), 7.20 (t, J = 7.8 Hz, 1H), 7.26 (d, J = 6.0 Hz, 2H), 7.49 (dd, J = 1.6, 6.4 Hz, 2H); 13C NMR (CD3OD) δ 22.6, 42.8, 52.8, 58.3, 72.1, 121.2, 122.0, 126.0, 126.6, 127.8, 130.4, 132.8, 137.2, 139.5, 141.4, 169.6, 169.9; LRMS (ESI+) 364.2 [M + Na – HCl]+ (calcd for C19H23NaN3O3+ 364.2); HRMS (ESI+) 342.1817 [M – Cl]+ (calcd for C19H24N3O3 342.1817).
Pharmacology
Compounds were screened under the auspices of the National Institutes of Health’s ASP. Experiments were performed in male rodents (albino Carworth Farms No. 1 mice (ip), albino Sprague–Dawley rats (ip, po)). Housing, handling, and feeding were in accordance with recommendations contained in the Guide for the Care and Use of Laboratory Animals. Anticonvulsant activity was established using the MES test,4 6 Hz,5 and the scMet test,23 according to previously reported methods.1
Catecholamine A Differentiated (CAD) Cells
CAD cells were grown at 37 °C and in 5% CO2 (Sarstedt, Newton, NC) in Ham’s F12/EMEM (GIBCO, Grand Island, NY), supplemented with 8% fetal bovine serum (Sigma, St. Louis, MO) and 1% penicillin/streptomycin (100% stocks, 10 000 U/mL penicillin G sodium and 10 000 μg/mL streptomycin sulfate).9,14 Cells were passaged every 6–7 days at a 1:25 dilution.
Cortical Neurons
Rat cortical neuron cultures were prepared from cortices dissected from embryonic day 19 brains exactly as described.48,49
Culturing HEK293 Cells Expressing NaV1.1, NaV1.3, NaV1.5, and NaV1.7
The cDNA gene encoding NaV1.1 was codon-optimized and synthesized using the open reading frame (accession no. NC_000002.11) and subcloned into the vector pTarget. The cDNA genes encoding NaV1.3 from rat and NaV1.7 from human were subcloned into the vector pcDNA3.1-mod. The cDNA encoding NaV1.5 from human50 was introduced into pcDNA3.1(+) with the CMV promoter. The constructs were then transfected into HEK293 cells using the calcium phosphate precipitation technique. After 48 h, the cells were passaged into 100 mm dishes and treated with G418 (Geneticin, Life Technologies) at 800 μg/mL to select for neomycin resistant cells. After 2 weeks, colonies were picked and split. The colonies were then tested for channel expression with whole-cell patch-clamp technique. The cell line was then maintained with 500 μg/mL G418. NaV1.1,34 NaV1.3,51 NaV1.5,52 and NaV1.753 stable cells were grown under standard tissue culture conditions (5% CO2 at 37 °C) in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum.
Electrophysiology
Whole-cell voltage clamp recordings were performed at room temperature on HEK293 cells, CAD cells, and cortical neurons using an EPC 10 amplifier (HEKA Electronics, Lambrecht/Pfalz Germany). Electrodes were pulled from thin-walled borosilicate glass capillaries (Warner Instruments, Hamden, CT) with a P-97 electrode puller (Sutter Instrument, Novato, CA) such that final electrode resistances were 1–2 MΩ when filled with internal solutions. The internal solution for recording Na+ currents contained the following (in mM): 110 CsCl, 5 MgSO4, 10 EGTA, 4 ATP Na2-ATP, 25 HEPES (pH 7.2, 290–310 mOsm/L). The external solution contained the following (in mM): 100 NaCl, 10 tetraethylammonium chloride (TEA-Cl), 1 CaCl2, 1 CdCl2, 1 MgCl2, 10 d-glucose, 4 4-AP, 0.1 NiCl2, 10 HEPES (pH 7.3, 310–315 mOsm/L). Whole-cell capacitance and series resistance were compensated with the amplifier. Series resistance error was always compensated to be less than ±3 mV. Cells were considered only when the seal resistance was less than 3 MΩ. Linear leak currents were digitally subtracted by P/4.
Data Acquisition and Analysis
Signals were filtered at 10 kHz and digitized at 10–20 kHz. Analysis was performed using Fitmaster and Origin8.1 (OriginLab Corporation, MA, USA). For activation curves, conductance (G) through Na+ channels was calculated using the equation G = I/(Vm – Vrev), where Vrev is the reversal potential, Vm is the membrane potential at which the current was recorded, and I is the peak current. Activation and inactivation curves were fitted to a single-phase Boltzmann function G/Gmax = 1/{1 + exp[(V – V50)/k]}, where G is the peak conductance, Gmax is the fitted maximal G, V50 is the half activation voltage, and k is the slope factor. Additional details of specific pulse protocols are described in the results text or figure legends.
Statistical Analyses
Differences between mean values were compared by either paired or unpaired two-tailed Student’s t-tests or an analysis of variance (ANOVA), when comparing multiple groups (repeated measures whenever possible). If a significant difference was determined by ANOVA, then a Dunnett’s or Tukey’s post hoc test was performed. Data are expressed as the mean ± SEM, with p < 0.05 considered as the level of significance.
Acknowledgments
This work is supported by grants, in part, from the NINDS (Grant 1 R41 NS080278) and the North Carolina Biotechnology Center (Technology Enhancement Grant) administered by the University of North Carolina—Chapel Hill and a National Scientist Development Award from the American Heart Association (Grant SDG5280023 to R.K.). We thank the NINDS and the ASP at the National Institutes of Health with Drs. Tracy Chen, Jeffrey Jiang, and John Kehne for kindly performing the pharmacological studies via the ASP’s contract site at the University of Utah with Drs. H. Wolfe, H. S. White, and K. Wilcox. T.R.C., C.B., and Y.X. are supported by NIH Grant NS053422. The content is solely the responsibility of the authors and does not represent the official views of the National Center for Research Resources, and National Institutes of Health.
Glossary
Abbreviations Used
- AAA
α-aminoamide
- ASD
antiseizure drug
- ASP
Anticonvulsant Screening Program
- CAD
catecholamine A differentiated
- CF3O
trifluoromethoxy
- CNS
central nervous system
- ED50
effective dose (50%)
- FAA
fuctionalized amino acid
- IBCF
isobutyl chloroformate
- HEK
human embryonic kidney
- IC50
concentration at which half of the channels have transitioned to a slow-inactivated state
- ip
intraperitoneally
- MAC
mixed anhydride coupling
- MES
maximal electroshock seizure
- NINDS
National Institutes of Neurological Disorders and Stroke
- NMM
N-methylmorpholine
- PI
protective index
- po
orally
- RMP
resting membrane potential
- scMet
scMetrazol
- TD50
neurological impairment (toxicity, 50%)
- TEA-Cl
tetraethylammonium chloride
- V1/2
voltage of half-maximal (in)activation
- VGSC
voltage-gated Na+ channel
Supporting Information Available
Figures S1–S5, synthetic procedures, experimental and spectral data for the intermediates and final products evaluated in this study, tables of elemental analysis results and high-resolution MS data, and 1H NMR and 13C NMR spectra for (R)-4–(R)-16. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∞ H.L.: Cell Dynamics Research Center, Gwangju Institute of Science and Technology, Gwangju, Korea.
Author Present Address
○ K.D.P.: Center for Neuro-Medicine, Brain Science Institute, Korea Institute of Science and Technology, Seoul, Korea.
Author Contributions
# H.L. and K.D.P. contributed equally to this study.
The authors declare the following competing financial interest(s): H.K. has a royalty-stake position in (R)-1 and is the founder of NeuroGate Therapeutics, Inc.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Choi D.; Stables J. P.; Kohn H. Synthesis and anticonvulsant activities of N-benzyl-2-acetamidopropionamide derivatives. J. Med. Chem. 1996, 39, 1907–1916. [DOI] [PubMed] [Google Scholar]
- Perucca E.; Yasothan U.; Clincke G.; Kirkpatrick P. Lacosamide. Nat. Rev. Drug Discovery 2008, 7, 973–974. [DOI] [PubMed] [Google Scholar]
- Stoehr T.; Kupferberg H. J.; Stables J. P.; Choi D.; Harris R. H.; Kohn H.; Walton N.; White H. S. Lacosamide, a novel anticonvulsant drug, shows efficacy with a wide safety margin in rodent models for epilepsy. Epilepsy Res. 2007, 74, 147–154. [DOI] [PubMed] [Google Scholar]
- White H. S.; Woodhead J. H.; Franklin M. R.; Swinyard E. A.; Wolf H. H.. General principles: experimental selection, quantification, and evaluation of antiepileptic drugs. In Antiepileptic Drugs, 4th ed.; Levy R. H., Mattson R. H., Meldrum B. S., Eds.; Raven Press: New York, 1995; pp 99–110. [Google Scholar]
- Barton M. E.; Klein B. D.; Wolff H. H.; White H. S. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001, 47, 217–227. [DOI] [PubMed] [Google Scholar]
- Lothman E. W.; Williamson J. M. Closely spaced recurrent hippocampal seizures elicit two types of heightened epileptogenesis: a rapidly developing, transient kindling and a slowly developing, enduring kindling. Brain Res. 1994, 649, 71–84. [DOI] [PubMed] [Google Scholar]
- Errington A. C.; Stohr T.; Heers C.; Lees G. The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol. Pharmacol. 2008, 73, 157–169. [DOI] [PubMed] [Google Scholar]
- Sheets P. L.; Heers C.; Stoehr T.; Cummins T. R. Differential block of sensory neuronal voltage-gated sodium channels by lacosamide, lidocaine and carbamazepine. J. Pharmacol. Exp. Ther. 2008, 326, 89–99. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Park K. D.; Salome C.; Wilson S. M.; Stables J. P.; Liu R.; Khanna R.; Kohn H. Development and characterization of novel derivatives of the antiepileptic drug lacosamide that exhibit far greater enhancement in slow inactivation of voltage-gated sodium channels. ACS Chem. Neurosci. 2011, 2, 90–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salome C.; Salome-Grosjean E.; Park K. D.; Morieux P.; Swendiman R.; DeMarco E.; Stables J. P.; Kohn H. Synthesis and anticonvulsant activities of (R)-N-(4′-substituted)benzyl 2-acetamido-3-methoxypropionamides. J. Med. Chem. 2010, 53, 1288–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salome C.; Salome-Grosjean E.; Stables J. P.; Kohn H. Merging the structural motifs of functionalized amino acids and α-aminoamides: compounds with significant anticonvulsant activities. J. Med. Chem. 2010, 53, 3756–3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morieux P.; Stables J. P.; Kohn H. Synthesis and anticonvulsant activities of N-benzyl (2R)-2-acetamido-3-oxysubstituted propionamide derivatives. Bioorg. Med. Chem. 2008, 16, 8968–8975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morieux P.; Salome C.; Park K. D.; Stables J. P.; Kohn H. The structure–activity of the 3-oxy site in the anticonvulsant N-benzyl (2R)-2-acetamido-3-methoxypropionamide. J. Med. Chem. 2010, 53, 5716–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Oxford G. S. Voltage-dependent ion channels in CAD cells: a catecholaminergic neuronal line that exhibits inducible differentiation. J. Neurophysiol. 2000, 84, 2888–2895. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wilson S. M.; Brittain J. M.; Ripsch M. S.; Salome C.; Park K. D.; White F. A.; Khanna R.; Kohn H. Merging structural motifs of functionalized amino acids and α-aminoamides results in novel anticonvulsant compounds with significant effects on slow and fast inactivation of voltage-gated sodium channels and in the treatment of neuropathic pain. ACS Chem. Neurosci. 2011, 2, 317–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson G. W.; Zimmerman J. E.; Callahan F. M. A reinvestigation of the mixed anhydride method of peptide synthesis. J. Am. Chem. Soc. 1967, 89, 5012–5017. [DOI] [PubMed] [Google Scholar]
- Park K. D.; Yang X.-F.; Lee H.; Dustrude E.; Wang Y.; Khanna R.; Kohn H. Discovery of lacosamide affinity bait agents that exhibit potent voltage-gated sodium channel blocking properties. ACS Chem. Neurosci. 2013, 4, 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaura N.; Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar]
- For a comparable procedure for resolving stereoisomers, see the following:Weisman G. R.Nuclear magnetic resonance analysis using chiral solvating agents. In Asymmetric Synthesis—Analytical Methods; Morrison J. D., Ed.; Academic Press: New York, 1983; Vol. 1, pp 153–172. [Google Scholar]
- Stables J. P.; Kupferberg H. G.. The NIH Anticonvulsant Drug Development (ADD) program: preclinical anticonvulsant screening project. In Molecular and Cellular Targets for Antiepileptic Drugs; Avanzini G., Regesta G., Tanganelli P., Avoli M., Eds.; John Libbey: London, 1997; pp 191–198. [Google Scholar]
- Porter R. J.; Cereghino J. J.; Gladding G. D.; Hessie B. J.; Kupferberg H. J.; Scoville B.; White B. G. Antiepileptic drug development program. Cleveland Clin. Q. 1984, 51, 293–305. [DOI] [PubMed] [Google Scholar]
- Dunham N. W.; Miya T.-S. A note on a simple apparatus for detecting neurological deficit in rats and mice. J. Am. Pharm. Assoc. 1957, 46, 208–209. [DOI] [PubMed] [Google Scholar]
- White H. S.; Woodhead J. H.; Wilcox K. S.; Stables J. P.; Kupferberg H. J.; Wolf H. H.. General principles: discovery and preclinical development of antiepileptic drugs. In Antiepileptic Drugs, 5th ed.; Levy R. H., Mattson R. H., Meldrum B. S., Perruca E., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, 2002; pp 36–48. [Google Scholar]
- Swinyard E. A. Laboratory evaluation of antiepileptic drugs: review of laboratory methods. Epilepsia 1969, 10, 107–119. [DOI] [PubMed] [Google Scholar]
- Morimoto K.; Fahnestock M.; Racine R. J. Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog. Neurobiol. 2004, 73, 1–60. [DOI] [PubMed] [Google Scholar]
- McNamara J. O.; Byrne M. C.; Dasheiff R. M.; Fitz J. G. The kindling model of epilepsy: a review. Prog. Neurobiol. 1980, 15, 139–159. [DOI] [PubMed] [Google Scholar]
- Loscher W.; Schmidt D. Which animal models should be used in the search for new antiepileptic drugs? A proposal based on experimental and clinical considerations. Epilepsy Res. 1988, 2, 145–181. [DOI] [PubMed] [Google Scholar]
- Hodgkin A. L.; Huxley A. F. The dual effect of membrane potential on sodium conductance in the giant axon of Loligo. J. Physiol. 1952, 116, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterall W. A. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium currents. Neuron 2000, 26, 13–25. [DOI] [PubMed] [Google Scholar]
- Kallen R. G.; Cohen S. A.; Barchi R. L. Structure, function and expression of voltage-dependent sodium channels. Mol. Neurobiol. 1993, 7, 383–428. [DOI] [PubMed] [Google Scholar]
- Ulbricht W. Voltage clamp studies of veratinized frog nodes. J. Cell. Comp. Physiol. 1965, 66Suppl. 291–98. [Google Scholar]
- Perucca P.; Mula M. Antiepilpetic drug effects on mood and behavior: molecular targets. Epilepsy Behav. 2013, 26, 440–449. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Brittain J. M.; Jarecki B. W.; Park K. D.; Wilson S. M.; Wang B.; Hale R.; Meroueh S. O.; Cummins T. R.; Khanna R. In silico docking and electrophysiological characterization of lacosamide binding sites on collapsin response mediator protein 2 (CRMP-2) identifies a pocket important in modulating sodium channel slow inactivation. J. Biol. Chem. 2010, 285, 25296–25307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King A. M.; Yang X.-F.; Wang Y.; Dustrude E. T.; Barbosa C.; Due M. R.; Piekarz A. D.; Wilson S. M.; White F. A.; Salome C.; Cummins T. R.; Khanna R.; Kohn H. Identification of the benzyloxyphenyl pharmacophore: a structural unit that promotes sodium channel slow inactivation. ACS Chem. Neurosci. 2012, 3, 1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B. Slow inactivation of the sodium conductance in squid giant axons. Pronase resistance. J. Physiol. 1978, 283, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean B. P. The action potential in mammalian central neurons. Nat. Rev. Neurosci. 2007, 8, 451–465. [DOI] [PubMed] [Google Scholar]
- Do M. T. H.; Bean B. P. Subthreshold sodium currents and pacemaking of subthalamic neurons: modulation by slow inactivation. Neuron 2003, 39, 109–120. [DOI] [PubMed] [Google Scholar]
- Vilin Y. Y.; Ruben P. C. Slow inactivation in voltage-gated sodium channels: molecular substrates and contributions to channelopathies. Cell Biochem. Biophys. 2001, 35, 171–190. [DOI] [PubMed] [Google Scholar]
- Errington A. C.; Stohr T.; Lees G. Voltage gated ion channels: targets for anticonvulsant drugs. Curr. Top. Med. Chem. 2005, 5, 15–30. [DOI] [PubMed] [Google Scholar]
- Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug–receptor reaction. J. Gen. Physiol. 1977, 69, 497–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin A. L. Resurgence of sodium channel research. Annu. Rev. Physiol. 2001, 63, 871–894. [DOI] [PubMed] [Google Scholar]
- Lee H.; Park K. D.; Yang X.-F.; Dustrude E. T.; Wilson S. M.; Khanna R.; Kohn H. (Biphenyl-4-yl)methylammonium chlorides: potent anticonvulsants that modulate Na+ currents. J. Med. Chem. 2013, 56, 5931–5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevarello P.; Bonsignori A.; Dostert P.; Heidempergher F.; Pinciroli V.; Colombo M.; McArthur R. A.; Salvati P.; Post C.; Fariello R. G.; Varasi M. Synthesis and anticonvulsant activity of a new class of 2-[(arylalkyl)amino]alkanamide derivatives. J. Med. Chem. 1998, 41, 579–590. [DOI] [PubMed] [Google Scholar]
- Salvati P.; Maj R.; Caccia C.; Cervini M. A.; Fornaretto M. G.; Lamberti E.; Pevarello P.; Skeen G. A.; White H. S.; Wolf H. H.; Faravelli L.; Mazzanti M.; Mancinelli M.; Varasi M.; Fariello R. G. Biochemical and electrophysiological studies on the mechanism of action of PNU-151774E, a novel antiepileptic compound. J. Pharmacol. Exp. Ther. 1999, 288, 1151–1159. [PubMed] [Google Scholar]
- Ben-Menachem E.; Biton V.; Jatuzis D.; Abou-Khalil B.; Doty P.; Rudd G. D. Efficacy and safety of oral lacosamide as adjunctive therapy in adults with partial-onset seizures. Epilepsia 2007, 48, 1308–1317. [DOI] [PubMed] [Google Scholar]
- Cummins T. R.; Sheets P. L.; Waxman S. G. The roles of sodium channels in nociception: implications for mechanisms of pain. Pain 2007, 131, 243–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagal S. K.; Brown A. D.; Cox P. J.; Omoto K.; Owen R. M.; Pryde D. C.; Sidders B.; Skerratt S. E.; Stevens E. B.; Storer R. I.; Swain N. A. Ion channels as therapeutic targets: a drug discovery perspective. J. Med. Chem. 2013, 56, 593–624. [DOI] [PubMed] [Google Scholar]
- Brittain J. M.; Piekarz A. D.; Wang Y.; Kondo T.; Cummins T. R.; Khanna R. An atypical role for collapsing response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated Ca2+ channels. J. Biol. Chem. 2009, 284, 31375–31390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittain J. M.; Wang Y.; Eruvwetere O.; Khanna R. Cdk5-mediated phosphorylation of CRMP-2 enhances its interaction with CaV2.2. FEBS Lett. 2012, 586, 3813–3818. [DOI] [PubMed] [Google Scholar]
- Gellens M. E.; George A. L.; Chen L. Q.; Chahine M.; Hom R.; Barchi R. L.; Kallen R. G. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins T. R.; Aglieco F.; Renganathan M.; Herzog R. I.; Dib-Hajj S. D.; Waxman S. G. Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J. Neurosci. 2001, 21, 5952–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W.; Xiao Y.; Chen H.; Ashpole N. M.; Pekarz A. D.; Ma P.; Hudmon A.; Cummins T. R.; Shou W. The human NaV1.5 F1486 deletion associated with long QT syndrome leads to impaired sodium channel inactivation and reduced lidocaine sensitivity. J. Physiol. 2012, 590, 5123–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theile J. W.; Cummins T. R. Inhibition of Navbeta4 peptide-mediated resurgent sodium currents in Nav1.7 channels by carbamazepine, riluzole, and anandamide. Mol. Pharmacol. 2011, 80, 724–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.