

Abstract
Although prostate cancer (CaP) is the most frequently diagnosed malignant tumor in American men, the mechanisms underlying the development and progression of CaP remain largely unknown. Recent studies have shown that downregulation of miR-124 occurs in several types of human cancer, suggesting a tumor suppressive function of miR-124. Until now, however, it has been unclear whether miR-124 is associated with CaP. In the present study, we completed a series of experiments to understand the functional role of miR-124 in CaP. We detected the expression level of miR-124 in clinical CaP tissues, evaluated the influence of miR-124 on the growth of CaP cells, and investigated the mechanism underlying the dysregulation of miR-124. We found that i) miR-124 directly targets the androgen receptor (AR) and subsequently induces a upregulation of p53; ii) miR-124 is significantly down-regulated in malignant prostatic cells compared to that in benign cells and DNA methylation causes the reduced expression of miR-124; and iii) miR-124 can inhibit the growth of CaP cells in vitro and in vivo. Data from this study revealed that loss of miR-124 expression is a common event in CaP, which may contribute to pathogenesis of CaP. Our studies also suggest that miR-124 is a potential tumor suppressive gene in CaP, and restoration of miR-124 expression may represent a novel strategy for CaP therapy.
Keywords: prostate cancer, androgen, androgen receptor, miRNA, tumor suppressor
Introduction
Prostate cancer (CaP) is the most frequently diagnosed malignant tumor and was the second leading cause of cancer-related death in American men in 2011(Siegel et al 2012). Metastatic CaP can be treated effectively with androgen ablation. However, one of the most troubling aspects of this disease is that after hormone treatment, the tumor inevitably progresses from an androgen-dependent (AD) to an incurable castration-resistant (CR) form (Sadar 2011). The mechanism underlying the progression has been poorly understood. In the past decades, therefore, an important task in CaP research is to elucidate the molecular alteration occurring in the development of CR tumors. Considerable insight into the progression of CaP has been recently achieved, including the discovery of aberrantly expressed microRNAs (miRNAs).
The human genome may encode over 1000 miRNAs which negatively regulate approximately 60% of human genes (Friedman et al 2009, Lewis et al 2005). Thus, miRNAs are involved in almost all important cellular processes, not only in physiological conditions but also in diseases including cancer (Volinia et al 2006). Indeed, a number of cancer-related miRNAs have been identified recently. Some miRNAs have also been reported to be aberrantly expressed in human CaP (Shi et al 2008). These miRNAs, which act as tumor suppressor genes or oncogenes, contribute to the pathogenesis of CaP by directly targeting some proliferation-related genes or apoptotic molecules (Shenouda and Alahari 2009). In spite of these exciting findings, the role of miRNA in the development and progression of CaP has been largely unexplored. Thus, identifying CaP-associated miRNAs and investigating their roles in CaP will help us understand the mechanisms related to the development and progression of this disease.
MiR-124 is a highly conserved miRNA whose in vivo function is poorly defined. This small non-coding RNA was first reported to be highly expressed in neuronal cells (Makeyev et al 2007). Recent studies revealed that miR-124 was significantly downregulated in several types of human cancers (Ando et al 2009, Furuta et al , Lujambio et al 2007). Moreover, it regulates some proliferation-related genes such as cyclin-dependent kinase 6 (CDK6) (Agirre et al 2009), forkhead box A2 (Foxa2) (Baroukh et al 2007) and solute carrier family 16, member 1 (SLC16A1) (Agirre et al 2009). Thus, miR-124 is classified as tumor suppressor miRNA (Agirre, 2009 #11). Until now, however, the role of miR-124 in CaP has been totally unknown, although it was reported previously to be undetectable in 22Rv1 CaP cells (Mitchell et al 2008).
In the present study, we aimed to explore the role of miR-124 in CaP. We found that miR-124 directly targets the androgen receptor (AR). We also detected the expression level of miR-124 in clinical CaP tissues and found that a majority of these CaP tissues express low levels of miR-124. In addition, we observed that synthetic miR-124 mimics inhibit the proliferation of CaP cells. Since the AR plays a crucial role in the pathogenesis of CaP, our studies established a link between dysregulated miR-124, overexpressed AR and CaP cell proliferation. Our results imply that miR-124 is a potential tumor suppressor gene and downregulation of miR-124 contributes to the development and progression of CaP.
Results
MiR-124 directly targets the AR and inhibits proliferation of CaP cells
Overexpression of the AR is well known to play an important role in pathogenesis of CaP. We are very interested in determining whether certain miRNA(s) contribute to upregulation of the AR. From two algorithms, TargetScan (Release 5.2) and miRanda (August 2010 Release) that predict miRNA-binding sites in the first 436 base of the AR 3′UTR (NM_000044.2), we identified three broadly conserved miRNA-binding sites for six miRNAs (miR-130/miR-301, miR-124/miR-506 and miR-30/miR-384). In order to assess the ability of these miRNAs to regulate AR expression, AR-positive C4-2B CaP cells cultured in androgen-deprived medium were separately treated with four chemically-modified miRNA mimics (miR-124, miR-130, miR-384 or miR-506, purchased from Ambion). As shown in Fig. 1A, compared to miRNA negative control (miR-NC), treatment with miR-124 mimic resulted in a reduction of AR protein by ∼70%, while the other three miRNA mimics induced 20–40% decrease in AR expression, suggesting a potent downregulation of the AR by miR-124. To determine the influence of these miRNAs on proliferation of CaP cell, AR-positive CaP cell lines (LNCaP, C4-2B and 22Rv1) were transiently transfected with each of these miRNA mimics. Consistent with its effect on AR level, transfection of miR-124 mimic inhibited proliferation to a greater extent than the other miRNA mimics tested (SI Fig. 1). We thus focused on miR-124 in this study.
Figure 1.
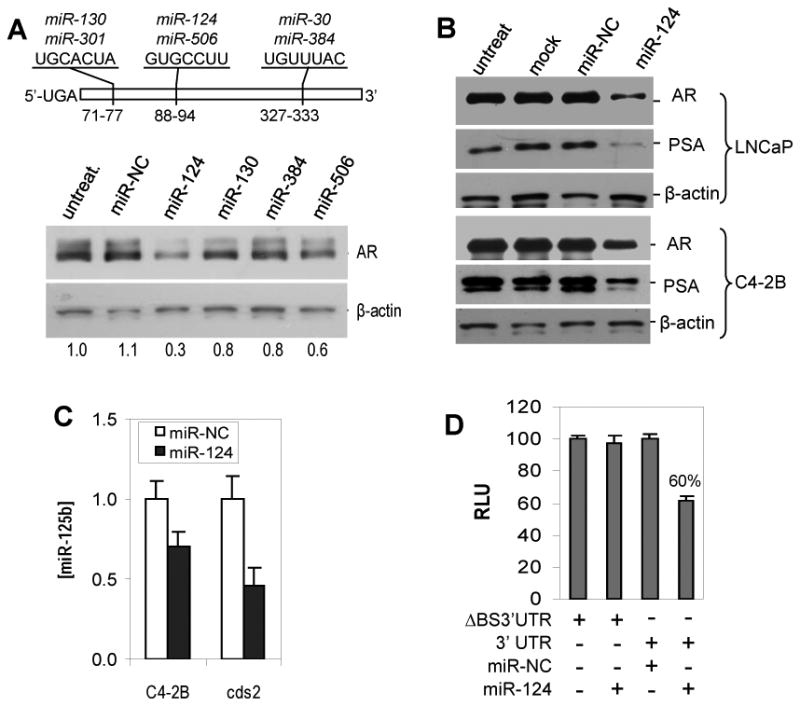
MiR-124 negatively regulates androgen receptor (AR). A) Western blot analysis of the AR expression in C4-2B CaP cells that were treated with 100 nM of synthetic miR-124, miR-130, miR-384 or miR-506. The numbers under the gels are the fold changes of AR protein relative to untreated C4-2B cells (untreat). Fold changes were calculated by scanning the AR bands and normalizing for β-actin bands. The upper portion is a schema of the first 436 bases of the AR 3′ UTR (based on the RefSeq NM_000044.2). The digital numbers indicate the predicted three miRNA-binding sites targeted by six potential AR-targeting miRNAs. B) Western blot analysis of the expression of AR and PSA in LNCaP and C4-2B cells treated with 100 nM of miR-124 mimic. Both mock and miRNA-NC were used as controls. C) Quantitative PCR (qPCR) assay of miR-125b levels in C4-2B and cds2 cells treated with 100 nM of miR-124 mimic or miR-NC. The values shown as mean ± SE (n=3) are from three independent experiments performed in triplicate. D) Luciferase analysis in C4-2B cells. The assay was repeated three times with each assay being performed in four wells, and similar results were obtained each time. The representative results are shown as mean ± SD (n = 4). The percentage represents enzyme activity in 100 nM miR-124 mimic-transfected cells relative to that in 100 nM miR-NC-transfected cells. RLU, relative luciferase unit. ΔBS3′UTR, AR 3′UTR fragment lacking the miR-124 binding site.
To determine whether miR-124-mediated downregulation of the AR affects the AR activity, both AR-positive LNCaP and C4-2B were treated with miR-124 mimic. Western blot analysis demonstrated that miR-124-induced down-regulation of the AR and was concomitant with a reduced PSA level (Fig. 1B). Since AR regulates miR-125b (Shi et al 2007), we used C4-2B and cds2 cells which express increased miR-125b (Shi et al 2007) to examine the effect of miR-124 mimic on endogenous miR-125b. We found that miR-124 induced downregulation of miR-125b by 30% in C4-2B and 54% in cds2 (Fig. 1C). To verify that the putative miR-124 binding site in the 3′ UTR of AR mRNA is responsible for regulation by miR-124, the 3′ UTR was cloned into a luciferase reporter vector and then cotransfected with miR-124 mimic into C4-2B cells. Luciferase activity was measured two days after transfection. As shown in Fig. 1D, transfection of miR-124 mimic resulted in 40% reduction of the enzyme activity, indicating a direct interaction between miR-124 and AR mRNA. Taken together, these data indicate that miR-124 targets the AR and affects the AR activity.
MiR-124 is significantly downregulated in prostate cancer cells
We determined the expression level of miR-124 in CaP cells. The abundance of miR-124 was first examined in seven prostate cell lines (2 benign and 5 malignant) using quantitative PCR (qPCR). We found a reduced expression of miR-124 in the malignant lines compared to the benign lines (Fig. 2A). We also performed Northern blot analysis of these cell lines and similar results were obtained (SI Fig. 2A). Next, we tested whether miR-124 is down-regulated in clinical prostate samples. We reviewed the miRNA expression profiling of our initial clinical tissue specimens and found that the abundance of miR-124 is markedly reduced in four CaP samples relative to that in one benign prostatic hyperplasia (BPH) tissue (SI Fig. 2B). We then determined whether downregulation of miR-124 is common in clinical CaP tissues. The levels of miR-124 were detected in 79 clinical prostatic tissues including 19 BPHs, 44 primary CaPs, 6 lymph node metastases, and 10 CR tumors. Although miR-124 abundances have overlap in a small number of benign and primary tumor tissues, the average miR-124 level was 2.7-fold less in primary CaPs than in BPHs (39 in CaPs vs 146 in BPHs, p<0.01) (Fig. 2B). Interestingly, 10 CR tumors and 6 metastases expressed extremely low levels of miR-124. Compared to the primary CaP samples, the averaged miR-124 level decreases by 77% in these advanced tumors (39 in CaPs vs 9 in CR & metastatic tumors, p<0.05) (Fig. 2B), suggesting a potential involvement of miR-124 in CaP progression. To confirm the downregulation of miR-124, 18 CaP samples and matched benign adjacent tissues were analyzed using qPCR. In comparison with matched benign tissues, 15 of 18 CaP samples (83%) expressed significantly reduced miR-124 (Fig. 2C). In addition, we performed Northern blot analysis in five matched prostate tissues in which sufficient quantities of RNA were available. As expected, the three CaPs (8, 11 and 16) having low miR-124 levels detected by qPCR exhibit lower signal intensity in Northern blots (SI Fig. 2C). Taken together, these data provide strong evidence that miR-124 is significantly reduced in CaP cell lines and in a majority of clinical CaP samples, and also suggest that decreased expression of miR-124 is common in human CaP tissues.
Figure 2.
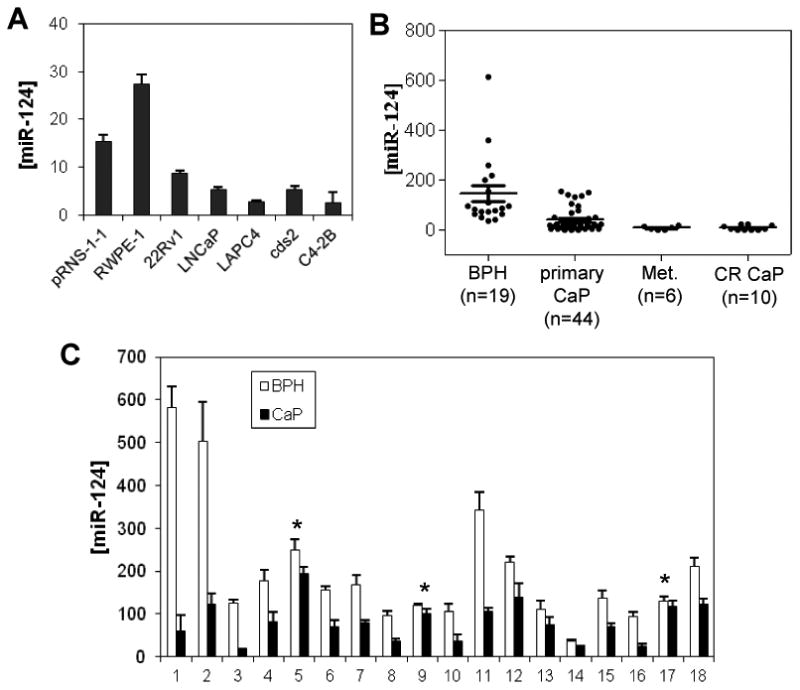
Quantitative PCR (qPCR) assays of miR-124 expression levels in CaP cells. A) MiR-124 abundance in two benign prostate cell lines (pRNS-1-1 and RWPE-1) and five CaP cell lines (22Rv1, LNCaP, LAPC-4, cds2 and C4-2B). B) MiR-124 expression levels in 19 benign prostatic hyperplasia (BPH) tissues, 44 primary CaPs, 6 lymph node metastases and 10 castration-resistant tumors. C) MiR-124 levels in 18 matched benign and malignant prostate samples. The levels of miR-124 of five pairs of prostate tissues (5, 8, 9, 11 & 16) were assayed using Northern blot analysis and similar results were obtained. The Northern blot results are shown in SI Figure 2B. “*”, no statistical difference (p>0.05) between BPH and CaP tissues. In A, B and C, qPCR assays were repeated three times with each assay being performed in triplicate, and similar results were obtained each time. The values are shown as mean ± SE (n=3).
MiR-124 and AR are negatively correlated in human CaP tissues
Having demonstrated that miR-124 targets the AR and is significantly downregulated in CaP cells, we therefore asked whether clinical CaP samples having low level of miR-124 overexpressed the AR. To address the issue, eight additional matched pairs of CaP and BPH were examined for expression of AR protein using immunohistochemical (IHC) analysis. In these samples, miR-124 level detected by qPCR was significantly decreased in CaP samples compared to that in BPH. Conversely, IHC results demonstrated that AR immunostaining was more intense in 7 of 8 CaP samples than that in BPH matches. These results reveal an inverse relationship between miR-124 and AR expression levels in prostatic tissues. Fig. 3 shows the representative results from four matches. In CaP samples AR protein staining is much higher than that in matched BPH tissues (Fig. 3A), while the expression of miR-124 is significantly decreased in CaP tissue compared to that in BPH tissues (Fig. 3B). Therefore, our data suggest that down-regulation of miR-124 results in increased expression of the AR in clinical CaP tissues.
Figure 3.
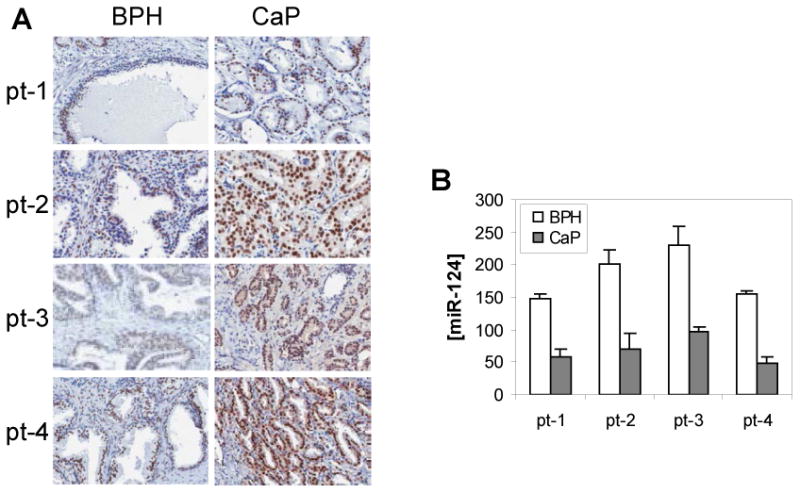
The expression levels of androgen receptor (AR) and miR-124 in four matched prostate tissues (pt-1 to pt-4). A) Immunohistochemical analysis of the AR protein in four human CaP samples (right) and their benign prostate tissue (left). B) qPCR detection of the abundance of miR-124 in both BPH and CaP tissues. The values are shown as mean ± SE from three independent experiments.
MiR-124 is methylated in CaP cells
The question remained: why is miR-124 downregulated in CaP cells? We first confirmed that androgens do not regulate miR-124, based on our observation that miR-124 levels were similar in androgen R1881-treated and untreated CaP cell lines (data not shown). Previous study has shown that DNA methylation regulates the expression of miRNAs (Sinkkonen et al 2008). Moreover, computer analysis demonstrated that the miR-124-1 at 8p23.1 and the miR-124-3 at 20q13.33 are embedded within CpG islands and the miR-124-2 at 8q12.3 is 760 bp downstream of a CpG island (Agirre et al 2009). In order to determine that DNA methylation downregulates miR-124 in CaP cells, we performed a demethylation experiment in three androgen-independent cell lines (22Rv1, C4-2B and cds2). Cells were treated with 50 μM of 5′-Aza-2′-Deoxycytidine (Aza) for 4 days and miR-124 was detected by qPCR. It was found that Aza treatment induced 2- to 5-fold increase in the miR-124 level compared to that in the untreated cells (Fig. 4A). Since miR-124 targets the AR as shown in Figure 1, we tested whether Aza downregulates expression of the AR. It was found that treatment with Aza reduced the AR level by ∼40% in LNCaP and ∼ 60% in C4-2B cells (Fig. 4B). We then assessed the methylation status of two benign prostate cell lines (pRNS-1-1 and RWPE-1) and five CaP cell lines using methylation-specific PCR (MSP). The 5′ DNA fragments of three miR-124 genes were strongly amplified by MSP primers in all cell lines except RWPE-1 whose miR-124-2 fragment was not amplified by the MSP primers (Fig. 4C). The fragments of miR-124-1 and miR-124-2 were also amplified weakly by unmethylated DNA primers in some of these cell lines (Fig. 4C). MSP data suggest different extents of DNA methylation at the 5′ regions of miR-124 genes. In order to validate DNA methylation, the 5′ DNA fragments of three miR-124 genes in six cell lines were amplified using COBRA (combined bisulfite restriction analysis) primers, cloned and sequenced. It was found that 40-80% of CpG sites were methylated in miR-124-1, 20-57% in miR-124-2 and 15-31% in miR-124-3, in five CaP cell lines, and the CpG methylation are 18%, 13% and 8%, respectively, in benign RWPE-1 cells (Fig. 4D).
Figure 4.
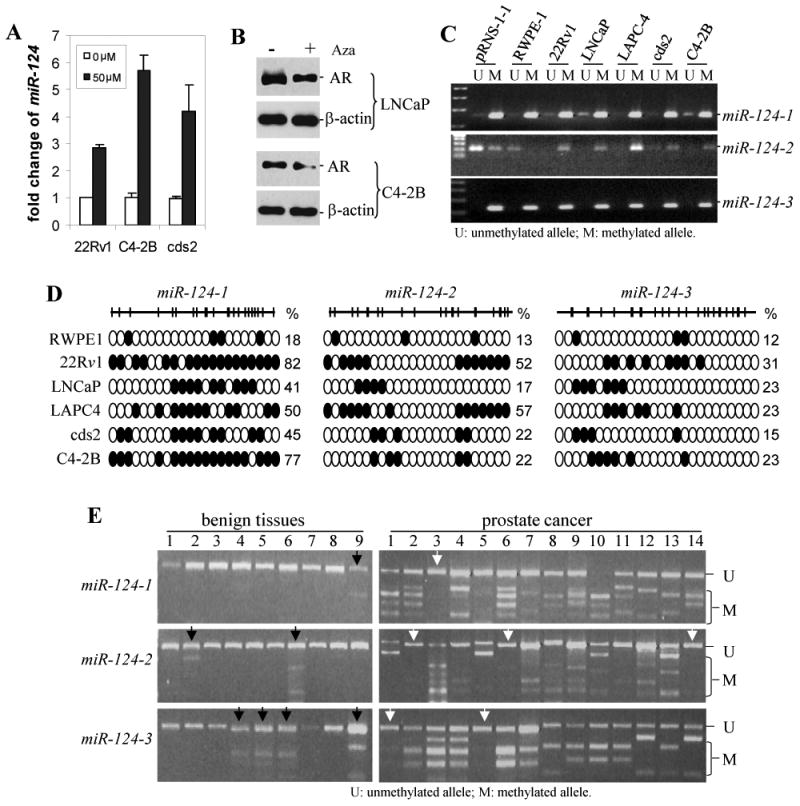
Methylation of miR-124 in CaP cells. A) Expression levels of miR-124 in 22Rv1, C4-2B and cds2 CaP cell lines before and after treatment with 50 μM of Aza. Results are expressed as fold change of miR-124 relative to the untreated control. The assay was repeated three times with each assay being performed in three wells, and similar results were obtained each time. The representative results are shown as mean ± SD (n = 3). B) Western blot analysis of the AR expression in LNCaP and C4-2B cells treated with 50 μM of Aza. C) Methylation-specific PCR (MSP) assay of the 5′ CpG islands of miR-124-1, miR-124-2 and miR-124-3 in seven prostatic cell lines: two benign lines (pRNS-1-1 and RWPE-1) and 5 malignant lines (22Rv1, LNCaP, LAPC-4, cds2 and C4-2B). D) Bisulfite sequencing analysis of miR-124 CpG island methylation in RWPE-1 cell line and five CaP cell lines. The top schematic view represents individual amplified 5′ DNA fragments of three miR-124 genes, which are located at -1895 to -1655 upstream of pre-miR-124-1, -1456 to -1188 of pre-miR-124-2 and -1465 to -1246 of pre-miR-124-3. The vertical bars denote individual CpG dinucleotides. Methylation profiles of miR-124 CpG island fragments in six cell lines tested were demonstrated in the bottom of the schema. CpGs are represented by open circles if unmethylated and by black circles if methylated. Each row exhibits methylated CpGs from at least three clones. The numbers on the right of each row are the percentage of methylated CpG dinucleotides. E) COBRA analysis of methylation of miR-124-1, miR-124-2 and miR-124-3 in 9 BPH tissues and 14 CaP samples. Purified PCR product was digested with BstU1 that cuts methylated CGCG. The black arrows indicate the BPH samples having detectable methylation, and the white arrows indicate the CaP samples without methylation.
We next determined the methylation status in 9 BPH tissues and 14 CaP tissues using COBRA. The 5′ DNA fragments of three miR-124 genes that contain 2, 5 and 6 CGCG sites, respectively, were amplified with COBRA primers and digested with the restriction enzyme BstU1 which only cleaves methylated CGCG sites. It was found that BstU1 was able to cut the miR-124-1 PCR products in 13 of 14 (93%) CaP samples, miR-124-2 in 11 of 14 (79%) samples and miR-124-3 in 12 of 14 (86%) samples, while the BstU1-digested BPH DNA fragments of miR-124 genes are 11% (1/9), 22% (2/9) and 44% (4/9), respectively (Fig. 4E), indicating that significant miR-124 methylation occurs in clinical CaP specimens. Taken together, our data suggest that DNA methylation occurring at the 5′ regions of miR-124 genes causes, at least in part, the reduced expression of miR-124 in CaP cells.
MiR-124 induces apoptosis
Previous studies demonstrated that the AR upregulates miR-125b, while miR-125b represses expression of p53 (Le et al 2009) and facilitates proliferation of CaP cells (Shi et al, Shi et al 2007). Since miR-124 targets the AR and downregulates miR-125b, we asked whether increased miR-124 level increases apoptotic cell death. To address the issue, LNCaP and C4-2B cells were transfected with miR-124 mimic and then cultured for 4 days. Apoptotic death cells were quantitatively detected by Annexin V binding assay. We observed that miR-124 mimic induced 10.4% of LNCaP cells and 9.7% of C4-2B cells to undergo apoptotic cell death (Fig. 5A). The comparative figure for the miR-NC cells was 0.7% and 1.1%, respectively (p<0.01). A representative Annexin V assay is shown in SI Fig. 3 in which miR-124 mimic-induced apoptosis (upper-right and lower-right squares in quadrant gates) were 11.4% of LNCaP cells and 8.4%) of C4-2B cells. To provide biochemical evidence for the occurrence of apoptosis, we determined whether miR-124 mimic increases the expression of p53 and caspase 3. As expected, treatment of C4-2B cells with miR-124 mimic induced a ∼9.0-fold enhanced expression of p53 (Fig. 5B) and ∼2.0-fold increase in caspase 3 (Fig. 5C). These data indicate that miR-124, at least in part, induces apoptosis by activating the p53 signaling pathway.
Figure 5.
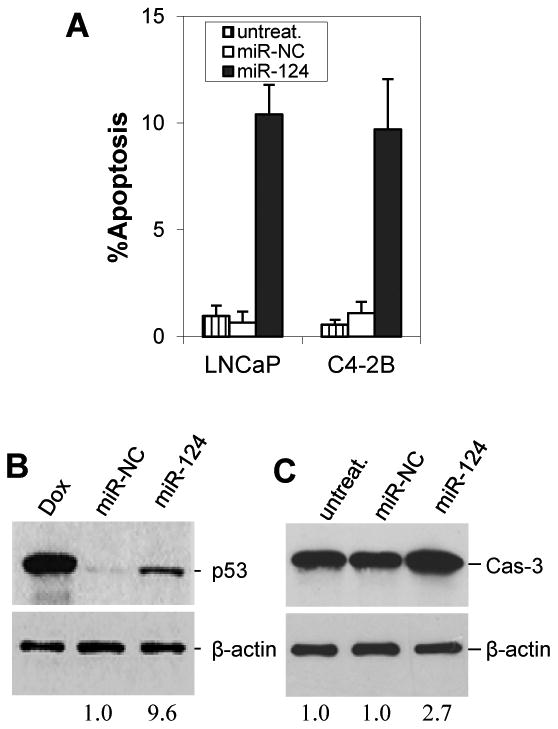
MiR-124 induces apoptosis and upregulates p53. A) Annexin V assay of apoptosis. LNCaP and C4-2B cells were treated with 100 nM miR-124 mimic or miR-NC for 4 days and stained with Annexin V and propidium iodide. Both early and late apoptotic cells are combined. The values are shown as mean ± SE from three independent experiments. B) Western blotting analysis of p53 expression in 100 nM miR-124 mimic-transfectd C4-2B cells. Doxorubicin (Dox)-treated cells were used as positive control. C) Western blotting analysis of caspase 3 (Cas-3) in 100 nM miR-124 mimic-transfectd C4-2B cells. In both B and C, the numbers under the gels are the fold changes of p53 and Cas-3 in miR-124 mimic-treated C4-2B cells relative to miR-NC-treated cells.
MiR-124 inhibits xenograft growth of CaP cells
Having demonstrated that miR-124 inhibits proliferation of AR-positive CaP cells and induces apoptosis, we tested whether this miRNA exerts an inhibitory effect on xenograft growth of CaP cells in vivo. We employed 22Rv1 CaP cells to address this question due to their expression of low (Figure 2A) or undetectable (Mitchell et al 2008) level of miR-124, as well as their ability to efficiently form tumors in nude mice. Overexpression of miR-124 in 22Rv1 was established using lentiviral vector system that expresses 28-fold elevation of miR-124 relative to controls (data not shown). Xenograft tumors were generated by subcutaneously inoculating intact male nude mice with 22Rv1-lenti-miR-124 cells or 22Rv1-lenti-vector cells. Tumors in miR-124 group and control groups became palpable 18 days after inoculation, suggesting that miR-124 did not affect the onset of 22Rv1 tumor. However, overexpression of miR-124 significantly inhibited the growth of 22Rv1 tumors after three weeks when compared to controls (Fig. 6A). At the end of the experiments, qPCR analysisof miR-124 level was performed in 3 lenti-miR-124 tumors. It was found that miR-124 levels were 23-fold increase in the miR-124 tumors compared to the control tumors (Fig. 6B), similar to that in cultured 22Rv1-lenti-miR-124 cells, revealing a stable expression of miR-124 in vivo. In addition, one lenti-vector tumor and two lenti-miR-124 tumors were examined by Western blot analysis for their AR expression. As shown in Fig. 6C, lenti-miR-124 tumors express moderate reduction of the full-length AR. Interestingly, overexpression of miR-124 in 22Rv1 cells induced a downregulation of the truncated AR that may contribute to the aggressive features of 22Rv1 cells (Tepper et al 2002). Collectively, our results suggest that miR-124 inhibits growth of 22Rv1 xenograft tumors by repressing AR expression.
Figure 6.
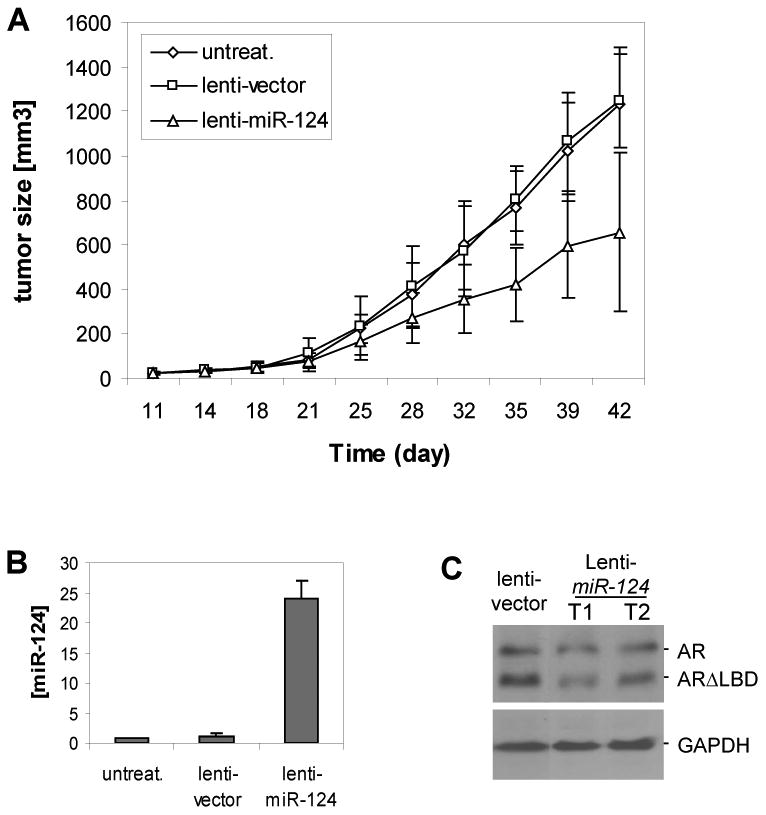
Inhibition of xenograftic tumor growth by miR-124. A). Five nude mice per group were injected subcutaneously with 2× 106 22Rv1-miR-124 cells, 22Rv1-vector cells, or untreated 22Rv1 cells. The growth of xenograft tumors were measured twice per week. Each time point represents mean ± SD of five independent values (mm3). B) qPCR assay of miR-124 level in xenograft tumors. The assay was repeated twice and similar results were obtained each time. The representative results are shown as mean ± SD (n = 3). C) Western blot assay of the AR expression in two 22Rv1-miR-124 tumors that express the full-length AR and a truncated AR (Tepper et al 2002).
Discussion
Although miR-124 has been reported to be involved in several other cancer types (Ando et al 2009, Furuta et al , Lujambio et al 2007), its role in CaP is largely unknown. To date, only one study has reported that miR-124 was undetectable in 22Rv1 CaP cells (Mitchell et al 2008). The present study explored the role of miR-124 in CaP. We found that miR-124 directly targets the AR, suggesting a potential tumor suppressor gene in CaP. In addition, our expression analysis revealed that miR-124 was significantly downregulated in a majority of clinical CaP specimens tested, which suggests that dysregulation of miR-124 may be a common event in patients with CaP and loss of miR-124 may contribute to the progression of CaP.
In this study, we address the potential mechanisms underlying miR-124 down-regulation in CaP cells. Since methylation of tumor suppressor genes (TSGs) is very common in both early and advanced stages of CaP (Graff et al 1995, Kang et al 2004, Lou et al 1999, Woodson et al 2004), miR-124 acting as a potential TSG in CaP may be susceptible to epigenetic modification. Indeed, we observed DNA methylation of three miR-124 genes in CaP cell lines and clinical CaP samples, an explanation for the reduction of miR-124 levels. This is supported by the fact that demethylation treatment with Aza increased the abundance of miR-124 in CaP cells. These data provide strong evidence that miR-124 is downregulated in CaP in part as a result of DNA methylation. Our results are consistent with those reported in other cancer types in which miR-124 loci are aberrantly methylated (Agirre et al 2009, Furuta et al , Lujambio et al 2007). However, methylation of miR-124 loci was not detected in a recent report (Rauhala et al 2010). In that study, Rauhala et al. treated CaP cells with 1 μM of Aza for two days followed by 300 nM of trichostatin A (TSA, a histone deacetylase inhibitor) for 24 hours. They identified 38 methylated miRNAs without miR-124. Mazar et al reported that Aza upregulates miR-34b expression in a dose-dependent manner and 1 μM of Aza did not induce elevation of miR-34b in melanoma cells (Mazar et al 2011). Since up to 100 μM of Aza failed to induce cytotoxicity in CaP cell lines (Walton et al 2008), we used 50 μM of Aza to treat CaP cells for four days, which induced significant elevation of miR-124. It is thus likely that our ability to detect the increased level of miR-124 is due to the higher concentration of Aze used. Besides the epigenetic regulation, the expression level of miR-124 can be affected by genetic factors, such as mutation and heterozygosity (LOH). Indeed, we detected a G→C point mutation (or SNP) in the 5′ region of miR-124-2 (SI Fig. 4). Mutation (or SNP) in 5′ region can reduce expression of mature miRNA by altered miRNA processing (Davis and Hata 2009, Jazdzewski et al 2008). In addition, a previous study revealed that tumor suppressive miRNAs are frequently located in LOH regions (Calin et al 2004). We know that miR-124-1 is located at 8p23.1 and miR-124-2 at 8q12.3. LOH in these two regions were reported to occur in approximately 50% of CaPs (Chang et al 2007, Matsuyama et al 2003, Perinchery et al 1999). Thus, LOH may affect the levels of miR-124-1 & miR-124-2. In order to elucidate the mechanism underlying downregulation of miR-124, further study is needed to detect the frequencies of DNA mutation (or SNP) and LOH in three miR-124 loci.
An interesting and clinically relevant discovery is that miR-124 directly targets the AR. The AR contributes to the initiation and progression of CaP. A study has demonstrated that a moderately altered AR expression is sufficient to convert AD CaP cells to a CR form (Chen et al 2004). The AR has been reported to be upregulated in clinical CaPs, particularly those that are CR. Why AR is upregulated in CaP remains poorly understood. AR gene amplification appears to account for the AR upregulation in ∼30% CR CaPs (Koivisto et al 1997); however, it is unknown how the AR is upregulated in the other CR tumors. Since the 3′UTR of the AR mRNA contains a conserved miR-124 binding site, we performed several experiments to validate the regulation of the AR by miR-124. We also observed an inverse correlation between miR-124 abundance and the AR expression level in clinical CaP samples tested. Therefore, our study demonstrates that dysregulation of miR-124 may elevate the expression of the AR, which is a new mechanistic explanation for overexpression of the AR in CaP. In this present study, identification of miR-124 as AR-targeting miRNA was based on the TargetScan and miRanda prediction programs. We noted that miR-124, as well as other three predicted miRNAs as shown in Figure 1A, were not selected as AR-targeting miRNAs in a recent study (Ostling et al 2011). In that study, the authors transfected five AR-positive CaP cell lines with 20 nM of human miRNA library and selected 71 miRNA candidates that affect the level of AR in all five cell lines. However, miRNA repression of target molecular expression is dose-dependent (Yamakuchi et al 2008). The dose of miR-124 mimic used in this study is 100 nM. In addition, miRNA functions in a cell type- and context-dependent manner (Olive et al 2010), and some AR-targeting miRNAs may not equally affect the AR level in all five CaP cell lines. Therefore, different treatment conditions and different selection criteria may account for the differential results.
Although we focus on the AR in this study, we realize that miR-124 targets hundreds of human genes, and other potential targets may also be relevant for the tumor suppressive function of miR-124 in CaP. We have identified a putative miR-124 binding site in the 3′UTR of high mobility group AT-hook 1 gene (HMGA1). In a pilot study, transfection of C4-2B cells with miR-124 mimic resulted in reduction of HMGA1 expression by 60%; furthermore, two clinical CaP specimens having low level of miR-124 did overexpress cytoplasmic HMGA1 (see SI Fig. 5), suggesting a link between low miR-124 and overexpression of HMGA1 in CaP. Previous studies revealed that HMGA1 was highly expressed in advanced CaPs (Takaha et al 2002, Tamimi et al 1993), and cytoplasmic HMGA1 directly interacts with p53, leading to p53 inactivation (Frasca et al 2006, Pierantoni et al 2006). Therefore, functional characterization of this miR-124 target will provide new insight into elucidating the molecular alteration occurring in advanced CaP. Additionally, cyclin-dependent kinase 6 (CDK6) and forkhead box A2 (Foxa2) have been identified as promising targets of miR-124 (Agirre et al 2009, Baroukh et al 2007). Both CDK6 and Foxa2 were previously reported to be elevated in CaP cells and correlate with tumor grade (Chen et al 1996, Mirosevich et al 2006, Qi et al). Moreover, these two proteins interact with the AR and enhance the AR activity (Lim et al 2005, Yu et al 2005). Further studies are required to understand whether upregulation of these two molecules in CaP cells are the result of decreased expression of miR-124.
Consistent with a previous report in nerve cells (Cao et al 2007), restoration of miR-124 level by transfecting CaP cells with synthetic miR-124 induces increased apoptosis. We also found that treatment of CaP cells with miR-124 upregulated the expression of p53, which is due at least partially to miR-124-mediated inhibition of AR/miR-125b signaling (Shi et al 2007). Thus, upregulation of p53 signaling may play a key role in miR-124-induced apoptosis. Since miR-124 induces apoptosis, we evaluated its effect on proliferation of CaP cells. Restoration of miR-124 level significantly inhibited the growth of CaP cells. More interestingly, when lentiviral-transduced 22Rv1 CaP cells that stably express miR-124 were injected subcutaneously into intact male nude mice, xenograft tumor growth also was significantly inhibited. Therefore, further testing of miR-124 in pre-clinical models of CaP will help define its ultimate therapeutic potential for treatment of CaP.
In conclusion, this study has shown that loss of miR-124 expression may be a common event in CaP. Since miR-124 negatively regulates the AR, downregulation of miR-124, as found in CaP, increases expression of the receptor. Therefore, deregulation of miR-124 as well as other AR-targeting miRNA candidates (Ostling et al 2011) contributes to high levels of AR expression in clinical CaP. Additionally, our data suggest that downregulation of miR-124 may be involved in the pathogenesis of CaP. Our evidence supporting the ability of miR-124 to induce apoptosis in CaP cells encourages the hope that restoring miR-124 function in CaP cells, either by itself or in conjunction with other therapies, will offer improved survival, which will be through delayed development or treatment of CR prostate cancer.
Materials and Methods
Cell lines
Two immortalized benign prostatic epithelial cell lines (RWPE-1 and pRNS-1-1) and five CaP cell lines (LNCaP, C4-2B, cds2, 22Rv1 and LAPC-4) were employed in this study. RWPE-1 (provided by Dr. Mukta. Webber, Michigan State University, East Lansing, MI) and pRNS-1-1 (provided by Dr. Johng Rhim, University of the Health Sciences, Bethesda, MD) were maintained in keratinocyte serum-free medium supplemented with 50 mg/ml bovine pituitary extract and 5 ng/ml epidermal growth factor. CaP cell lines were maintained in RPMI 1640 medium containing 10% fetal bovine serum. Both C4-2B and cds2 cell lines are derivatives of LNCaP (Lin et al 2001, Shi et al 2004).
Clinical samples
Collection and use of CaP patient specimens were approved by the Institutional Review Board (UCD IRB#: 200312072-6). Primary CaP samples from radical prostatectomy, metastatic pelvic lymph nodes, CR tumor specimens and BPH tissues were obtained fresh from surgical excision by the Department of Urology, University of California, Davis. These CR tumors were described in our previous publication (de Vere White et al 1997): six from stage D patients and four from patients receiving combined androgen blockade therapy prior to radical prostatectomy. To ensure that portions of CaP tissues were enriched for tumor cells, a surgeon and a pathologist together sectioned the excised tissue, and a fresh piece was taken from the palpated cancer and rapidly frozen to -80°C until use. A permanent section was then cut from the immediately adjacent area for Hematoxylin and Eosin (H&E) staining. Based upon histological review of H&E section, it was confirmed that these CaP samples used in this study were tumor cell-enriched.
Reporter plasmid construction and luciferase assay
To construct reporter plasmids, a 0.45-kb DNA fragment of the AR 3′UTR containing the putative miR-124 binding site was prepared by PCR. The AR 3′UTR fragment lacking the miR-124 binding site was used as negative control. DNA fragments were cloned into the pMIR-REPORT Luciferase vector (Ambion) downstream of the reporter gene. The sequences and cloning direction of these PCR products were validated by DNA sequencing. For luciferase assay, cells (4×104 per well) were seeded into 24-well plates and cultured for 24 hours. The cells were then co-transfected with reporter plasmids and 100 nM chemically-modified miR-124 mimic or miRNA negative control (miR-NC). The pRL-SV40 Renilla luciferase plasmid (Promega) was used as an internal control. Two days later, cells were harvested and lysed with passive lysis buffer (Promega). Luciferase activity was measured using a dual luciferase reporter assay (Promega). Luciferase activity was normalized by Renilla luciferase activity.
Western blot analysis
Cells were grown to 70–80% confluence and lysed in lysis buffer consisting of 25 mmol/L Tris-HCl (pH7.5), 150 mmol/LNaCl, 1 mmol/L EDTA, 1% Triton X-100, 0.1% SDS and 1× proteinase inhibitor cocktail, and equal concentrations of denatured protein samples were loaded on a 10% SDS–polyacrylamide gel. After electrophoresis, proteins were transferred to an Immobilon PVDF membrane, and immunoblot analysis was conducted by the standard method.
qPCR
For quantitative expression analysis of miRNA, total RNA was isolated from fresh-frozen prostatic tissues, cultured cells or xenograft tumors using TRIzol reagent (Life Technologies, Inc.). The miRNA level was measured using a TaqMan miRNA assay kit (Applied Biosystems, Foster City, CA) following the manufacturer's instruction and PCR amplification was carried out in a 7900HT Fast Real-time PCR system (Applied Biosystems). Real-time PCR was performed in triplicate for each sample. The abundance of miRNA was measured using Ct (threshold cycle) following the approach described previously (Schmittgen and Livak 2008). ΔCt was calculated by subtracting the Ct of U6 RNA from the Ct of the miRNA tested. ΔΔCt was calculated by subtracting the ΔCt of a reference sample from the ΔCt of each sample. For clinical tissues, the reference sample is one CaP specimen tested that expresses the lowest level of miRNA. Fold change was generated using the equation 2-ΔΔCt.
Methylation assay
Genomic DNA was isolated from prostate cell lines using standard procedures, and from CaP cell-enriched tumor tissues following our previous method (Shi et al 1996). Bisulfite modification of DNA (1.0 μg) was carried out by using an EZ DNA methylation-direct kit (Zymo Research, Irvine, CA). For the prostate cell lines, the methylation of 5′ CpG island regions was detected by MSP using the primers specific for either methylated or unmethylated DNA. The methylated CpG dinucleotides were validated using bisulfite DNA sequencing. Amplified CpG islands were separately cloned into pCR 2.1 vector using TOPO TA Cloning Kit (Invitrogen) and at least three clones per PCR product were picked up for DNA sequencing. In clinical prostate tissues, the methylation status was assessed by COBRA assay. COBRA primer-amplified PCR products were purified using Amicon 30 filter (Millipore), digested by BstUI and separated on a 3% agarose gel (1% agarose plus 2% NuSieve GTG low-melting temperature agaros). The primers used for MSP and COBRA were designed based on “The Li Lab” program (http://www.urogene.org/methprimer/indexl.html). The primer sequences and amplified size are listed in SI Table 1.
Annexin V binding assay
MiR-124-induced apoptosis was analyzed using a FACS Annexin V assay kit (Trevigen, Inc., Gaithersburg, MD) following the protocol provided by the manufacturer. Briefly, cells were transfected with miR-124 mimic and incubated for 4 days. The cells were harvested, washed once with PBS, re-suspended in 100 μl of 1× Annexin V binding buffer containing 1 μl of Annexin V-FITC conjugate and 10 μl of propidium iodide solution and then incubated for 30 min at room temperature in the dark. Subsequently, 400 μl 1× Annexin V binding buffer was added and samples analyzed on a FACScan flow cytometer. Data analysis was performed using FACScan software (Becton Dickinson).
Animal experiment
A lentiviral miR-124 expression vector that expresses a ∼500-base human pre-miR-124-1 and the empty lentiviral vector were purchased from System Biosciences (SBI, Mountain View, CA). Pseudovirus production and cell transduction were performed following the manufacturer's protocol. The resulting 22Rv1-miR-124 and 22Rv1-vector cells were selected through fluorescence-activated cell sorting (FACS) and were maintained in medium described above. Overexpression of miR-124 in infected 22Rv1 cells was confirmed by qPCR. Animal studies were performed in male athymic nu/nu mice (4- to 6-week-old; Harlan Laboratories). Mice were injected subcutaneously with suspensions of 2×106 22Rv1-miR-124 cells or 22Rv1-vector cells in a mixture (1:1 vol/vol) of culture medium and Matrigel (Becton Dickinson). Tumor growth was monitored and tumor dimensions were measured twice per week. Tumor volumes were calculated according to the formula: ½ (length×width×height). Animal studies were performed according to protocols approved by the Animal Care and Use Committee at University of California, Davis.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software 5.0. Student's t-test was used to analyze the difference in miR-124 levels between different groups. Statistical significance was set up to p<0.05 in each test.
Supplementary Material
SI Figure 1. miR-124 inhibits the proliferation of CaP cells. WST-1 cell proliferation analysis of AR-positive CaP cell lines. Cells (4×103 per well) were plated in 96-well plates for 24 hours, and then transfected with 100 nM synthetic miRNA mimics (miR-124m, miR-130m, miR-384m or miR-506m) or miRNA negative control (miR-NC) using lipofectamine 2000 (Invitrogen). The transfection protocol was optimized using a fluorescent pEGFP-N1 vector (Clontech) to ensure a transfection efficiency >90%. WST-1 cell proliferation assay was carried out at Day 5 after transfection. The growth changes were demonstrated as M ± SD (n = 3). AR-negative DU-145 cells were used as control that shows a slight inhibition of proliferation caused by miRNA mimics. “*” indicates a significant difference (p<0.05) relative to miR-NC.
SI Figure 2. Downregulation of miR-124 in CaP cell lines and in clinical specimens. A) To validate the qPCR results shown in Figure 2A, Northern blot assay was performed for miR-124 expression in seven prostate cell lines (two benign and five malignant). The miR-124 expression pattern is similar to that observed in qPCR. The numbers under the gel are the fold changes of miR-124 in CaP cells (22Rv1, LNCaP, LAPC4, cds2 and C4-2B) relative to in benign cells (pRNS-1-1 and RWPE-1). B) The expression levels of miR-124 in one BPH and four CaP tissues (including a lymph node metastasis, Met) was summarized from a previous microarray profiling assay (left). The miR-124 levels detected in the microarray profiling were validated by qPCR (right). C) Northern blot assay for miR-124 in five CaP specimens that have sufficient amount of RNA. Three CaP samples (8, 11 and 16) having low miR-124 levels detected by qPCR exhibit lower signal intensity in North blots, compared to their BPH matches. Two CaP samples (5 and 9) and their BPH matches exhibit similar signal intensity in North blots. The numbers under the gel are the fold changes of miR-124 in CaP cells relative to in benign cells. B, BPH; C, CaP. U6 was used as loading control.
SI Figure 3. LNCaP (top) and C4-2B (bottom) cells transfected with 100 nM of miR-124m or miR-NC were grown in 10% FBS medium for four days. Both early apoptosis and later apoptosis were analyzed using Annexin V apoptosis assay protocol.
SI Figure 4. A point mutation or single nucleotide polymorphism (SNP) was detected at the 5′CpG region (1267 upstream of pre-miR-124-2) in a clinical CaP specimen. The wild-type “G” changed to “C” and was methylated (pointed out by an arrow).
SI Figure 5. Analyses of the expression of HMGA1 in CaP cells. A) Western blotting analysis of HMGA1 expression in 100 nM miR-124m-transfectd C4-2B cells. The numbers under the gels are the fold changes of HMGA1 in miR-124m-treated C4-2B cells relative to miR-NC-treated cells. Our pilot results indicate that miR-124m treatment induced the downregulation of HMGA1 by ∼60%. B) Immunohistochemical analysis of the expression of HMGA1 in two matched BPH and CaP tissues. CaP samples (right) and their BPHs (left) were immunostained using anti-HMGA1 antibody (sc-26348, Santa Cruz Biotechnology, Inc.). The abundance of miR-124 in these tissues was measured using qPCR, and the values are shown on the top of each image. CaP tissues express lower level of miR-124 than BPHs. Immunostaining for HMGA1 is more intense in two CaP samples than that in BPH matches. Moreover, localization of HMGA1 in the cytoplasm of CaP cells was obvious compared to that in BPH calls.
Acknowledgments
This work was supported in part by NIH 1RO1CA92068 (R. deVere White), Department of Defense PCRP PC080488 (R. deVere White) and The LANIE Foundation. We thank Dr. Mukta. Webber for providing RWPE-1 cell line and Dr. Johng Rhim for pRNS-1-1 cell line. We also gratefully thank Dr. Melanie C. Bradnam for her editorial assistance and Stephanie Soares for critically reading the manuscript.
Supported by NCI grant (CA92069) and DOD Grant (PC080488).
Footnotes
Conflict of interest: The authors declare no conflict of interest.
References
- Agirre X, Vilas-Zornoza A, Jimenez-Velasco A, Martin-Subero JI, Cordeu L, Garate L, et al. Epigenetic silencing of the tumor suppressor microRNA Hsa-miR-124a regulates CDK6 expression and confers a poor prognosis in acute lymphoblastic leukemia. Cancer Res. 2009;69:4443–4453. doi: 10.1158/0008-5472.CAN-08-4025. [DOI] [PubMed] [Google Scholar]
- Ando T, Yoshida T, Enomoto S, Asada K, Tatematsu M, Ichinose M, et al. DNA methylation of microRNA genes in gastric mucosae of gastric cancer patients: its possible involvement in the formation of epigenetic field defect. Int J Cancer. 2009;124:2367–2374. doi: 10.1002/ijc.24219. [DOI] [PubMed] [Google Scholar]
- Baroukh N, Ravier MA, Loder MK, Hill EV, Bounacer A, Scharfmann R, et al. MicroRNA-124a regulates Foxa2 expression and intracellular signaling in pancreatic beta-cell lines. J Biol Chem. 2007;282:19575–19588. doi: 10.1074/jbc.M611841200. [DOI] [PubMed] [Google Scholar]
- Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Pfaff SL, Gage FH. A functional study of miR-124 in the developing neural tube. Genes Dev. 2007;21:531–536. doi: 10.1101/gad.1519207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang BL, Liu W, Sun J, Dimitrov L, Li T, Turner AR, et al. Integration of somatic deletion analysis of prostate cancers and germline linkage analysis of prostate cancer families reveals two small consensus regions for prostate cancer genes at 8p. Cancer Res. 2007;67:4098–4103. doi: 10.1158/0008-5472.CAN-06-4570. [DOI] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Chen Y, Robles AI, Martinez LA, Liu F, Gimenez-Conti IB, Conti CJ. Expression of G1 cyclins, cyclin-dependent kinases, and cyclin-dependent kinase inhibitors in androgen-induced prostate proliferation in castrated rats. Cell Growth Differ. 1996;7:1571–1578. [PubMed] [Google Scholar]
- Davis BN, Hata A. Regulation of MicroRNA Biogenesis: A miRiad of mechanisms. Cell Commun Signal. 2009;7:18. doi: 10.1186/1478-811X-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vere White R, Meyers F, Chi SG, Chamberlain S, Siders D, Lee F, et al. Human androgen receptor expression in prostate cancer following androgen ablation. European urology. 1997;31:1–6. doi: 10.1159/000474409. [DOI] [PubMed] [Google Scholar]
- Frasca F, Rustighi A, Malaguarnera R, Altamura S, Vigneri P, Del Sal G, et al. HMGA1 inhibits the function of p53 family members in thyroid cancer cells. Cancer Res. 2006;66:2980–2989. doi: 10.1158/0008-5472.CAN-05-2637. [DOI] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta M, Kozaki KI, Tanaka S, Arii S, Imoto I, Inazawa J. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis. 31:766–776. doi: 10.1093/carcin/bgp250. [DOI] [PubMed] [Google Scholar]
- Graff JR, Herman JG, Lapidus RG, Chopra H, Xu R, Jarrard DF, et al. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res. 1995;55:5195–5199. [PubMed] [Google Scholar]
- Jazdzewski K, Murray EL, Franssila K, Jarzab B, Schoenberg DR, de la Chapelle A. Common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proc Natl Acad Sci U S A. 2008;105:7269–7274. doi: 10.1073/pnas.0802682105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang GH, Lee S, Lee HJ, Hwang KS. Aberrant CpG island hypermethylation of multiple genes in prostate cancer and prostatic intraepithelial neoplasia. J Pathol. 2004;202:233–240. doi: 10.1002/path.1503. [DOI] [PubMed] [Google Scholar]
- Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, et al. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997;57:314–319. [PubMed] [Google Scholar]
- Le MT, Teh C, Shyh-Chang N, Xie H, Zhou B, Korzh V, et al. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009;23:862–876. doi: 10.1101/gad.1767609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Lim JT, Mansukhani M, Weinstein IB. Cyclin-dependent kinase 6 associates with the androgen receptor and enhances its transcriptional activity in prostate cancer cells. Proc Natl Acad Sci U S A. 2005;102:5156–5161. doi: 10.1073/pnas.0501203102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DL, Tarnowski CP, Zhang J, Dai J, Rohn E, Patel AH, et al. Bone metastatic LNCaP-derivative C4-2B prostate cancer cell line mineralizes in vitro. Prostate. 2001;47:212–221. doi: 10.1002/pros.1065. [DOI] [PubMed] [Google Scholar]
- Lou W, Krill D, Dhir R, Becich MJ, Dong JT, Frierson HF, Jr, et al. Methylation of the CD44 metastasis suppressor gene in human prostate cancer. Cancer Res. 1999;59:2329–2331. [PubMed] [Google Scholar]
- Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setien F, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67:1424–1429. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- Makeyev EV, Zhang J, Carrasco MA, Maniatis T. The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell. 2007;27:435–448. doi: 10.1016/j.molcel.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama H, Pan Y, Yoshihiro S, Kudren D, Naito K, Bergerheim US, et al. Clinical significance of chromosome 8p, 10q, and 16q deletions in prostate cancer. Prostate. 2003;54:103–111. doi: 10.1002/pros.10173. [DOI] [PubMed] [Google Scholar]
- Mazar J, Khaitan D, DeBlasio D, Zhong C, Govindarajan SS, Kopanathi S, et al. Epigenetic regulation of microRNA genes and the role of miR-34b in cell invasion and motility in human melanoma. PloS one. 2011;6:e24922. doi: 10.1371/journal.pone.0024922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirosevich J, Gao N, Gupta A, Shappell SB, Jove R, Matusik RJ. Expression and role of Foxa proteins in prostate cancer. Prostate. 2006;66:1013–1028. doi: 10.1002/pros.20299. [DOI] [PubMed] [Google Scholar]
- Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105:10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive V, Jiang I, He L. mir-17-92, a cluster of miRNAs in the midst of the cancer network. The international journal of biochemistry & cell biology. 2010;42:1348–1354. doi: 10.1016/j.biocel.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostling P, Leivonen SK, Aakula A, Kohonen P, Makela R, Hagman Z, et al. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res. 2011;71:1956–1967. doi: 10.1158/0008-5472.CAN-10-2421. [DOI] [PubMed] [Google Scholar]
- Perinchery G, Bukurov N, Nakajima K, Chang J, Hooda M, Oh BR, et al. Loss of two new loci on chromosome 8 (8p23 and 8q12-13) in human prostate cancer. Int J Oncol. 1999;14:495–500. doi: 10.3892/ijo.14.3.495. [DOI] [PubMed] [Google Scholar]
- Pierantoni GM, Rinaldo C, Esposito F, Mottolese M, Soddu S, Fusco A. High Mobility Group A1 (HMGA1) proteins interact with p53 and inhibit its apoptotic activity. Cell Death Differ. 2006;13:1554–1563. doi: 10.1038/sj.cdd.4401839. [DOI] [PubMed] [Google Scholar]
- Qi J, Nakayama K, Cardiff RD, Borowsky AD, Kaul K, Williams R, et al. Siah2-dependent concerted activity of HIF and FoxA2 regulates formation of neuroendocrine phenotype and neuroendocrine prostate tumors. Cancer Cell. 18:23–38. doi: 10.1016/j.ccr.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauhala HE, Jalava SE, Isotalo J, Bracken H, Lehmusvaara S, Tammela TL, et al. miR-193b is an epigenetically regulated putative tumor suppressor in prostate cancer. Int J Cancer. 2010;127:1363–1372. doi: 10.1002/ijc.25162. [DOI] [PubMed] [Google Scholar]
- Sadar MD. Small molecule inhibitors targeting the “achilles' heel” of androgen receptor activity. Cancer Res. 2011;71:1208–1213. doi: 10.1158/0008-5472.CAN_10-3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Shenouda SK, Alahari SK. MicroRNA function in cancer: oncogene or a tumor suppressor? Cancer Metastasis Rev. 2009;28:369–378. doi: 10.1007/s10555-009-9188-5. [DOI] [PubMed] [Google Scholar]
- Shi XB, Xue L, Ma AH, Tepper CG, Kung HJ, White RW. miR-125b promotes growth of prostate cancer xenograft tumor through targeting pro-apoptotic genes. Prostate. 71:538–549. doi: 10.1002/pros.21270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi XB, Bodner SM, deVere White RW, Gumerlock PH. Identification of p53 mutations in archival prostate tumors. Sensitivity of an optimized single-strand conformational polymorphism (SSCP) assay. Diagn Mol Pathol. 1996;5:271–278. doi: 10.1097/00019606-199612000-00008. [DOI] [PubMed] [Google Scholar]
- Shi XB, Ma AH, Tepper CG, Xia L, Gregg JP, Gandour-Edwards R, et al. Molecular alterations associated with LNCaP cell progression to androgen independence. Prostate. 2004;60:257–271. doi: 10.1002/pros.20039. [DOI] [PubMed] [Google Scholar]
- Shi XB, Xue L, Yang J, Ma AH, Zhao J, Xu M, et al. An androgen-regulated miRNA suppresses Bak1 expression and induces androgen-independent growth of prostate cancer cells. Proc Natl Acad Sci U S A. 2007;104:19983–19988. doi: 10.1073/pnas.0706641104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi XB, Tepper CG, White RW. MicroRNAs and prostate cancer. J Cell Mol Med. 2008;12:1456–1465. doi: 10.1111/j.1582-4934.2008.00420.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2012;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- Sinkkonen L, Hugenschmidt T, Berninger P, Gaidatzis D, Mohn F, Artus-Revel CG, et al. MicroRNAs control de novo DNA methylation through regulation of transcriptional repressors in mouse embryonic stem cells. Nat Struct Mol Biol. 2008;15:259–267. doi: 10.1038/nsmb.1391. [DOI] [PubMed] [Google Scholar]
- Takaha N, Hawkins AL, Griffin CA, Isaacs WB, Coffey DS. High mobility group protein l(Y): a candidate architectural protein for chromosomal rearrangements in prostate cancer cells. Cancer Res. 2002;62:647–651. [PubMed] [Google Scholar]
- Tamimi Y, van der Poel HG, Denyn MM, Umbas R, Karthaus HF, Debruyne FM, et al. Increased expression of high mobility group protein l(Y) in high grade prostatic cancer determined by in situ hybridization. Cancer Res. 1993;53:5512–5516. [PubMed] [Google Scholar]
- Tepper CG, Boucher DL, Ryan PE, Ma AH, Xia L, Lee LF, et al. Characterization of a novel androgen receptor mutation in a relapsed CWR22 prostate cancer xenograft and cell line. Cancer Res. 2002;62:6606–6614. [PubMed] [Google Scholar]
- Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton TJ, Li G, Seth R, McArdle SE, Bishop MC, Rees RC. DNA demethylation and histone deacetylation inhibition co-operate to re-express estrogen receptor beta and induce apoptosis in prostate cancer cell-lines. Prostate. 2008;68:210–222. doi: 10.1002/pros.20673. [DOI] [PubMed] [Google Scholar]
- Woodson K, Hanson J, Tangrea J. A survey of gene-specific methylation in human prostate cancer among black and white men. Cancer Lett. 2004;205:181–188. doi: 10.1016/j.canlet.2003.11.027. [DOI] [PubMed] [Google Scholar]
- Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008;105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Gupta A, Wang Y, Suzuki K, Mirosevich J, Orgebin-Crist MC, et al. Foxa1 and Foxa2 interact with the androgen receptor to regulate prostate and epididymal genes differentially. Ann N Y Acad Sci. 2005;1061:77–93. doi: 10.1196/annals.1336.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SI Figure 1. miR-124 inhibits the proliferation of CaP cells. WST-1 cell proliferation analysis of AR-positive CaP cell lines. Cells (4×103 per well) were plated in 96-well plates for 24 hours, and then transfected with 100 nM synthetic miRNA mimics (miR-124m, miR-130m, miR-384m or miR-506m) or miRNA negative control (miR-NC) using lipofectamine 2000 (Invitrogen). The transfection protocol was optimized using a fluorescent pEGFP-N1 vector (Clontech) to ensure a transfection efficiency >90%. WST-1 cell proliferation assay was carried out at Day 5 after transfection. The growth changes were demonstrated as M ± SD (n = 3). AR-negative DU-145 cells were used as control that shows a slight inhibition of proliferation caused by miRNA mimics. “*” indicates a significant difference (p<0.05) relative to miR-NC.
SI Figure 2. Downregulation of miR-124 in CaP cell lines and in clinical specimens. A) To validate the qPCR results shown in Figure 2A, Northern blot assay was performed for miR-124 expression in seven prostate cell lines (two benign and five malignant). The miR-124 expression pattern is similar to that observed in qPCR. The numbers under the gel are the fold changes of miR-124 in CaP cells (22Rv1, LNCaP, LAPC4, cds2 and C4-2B) relative to in benign cells (pRNS-1-1 and RWPE-1). B) The expression levels of miR-124 in one BPH and four CaP tissues (including a lymph node metastasis, Met) was summarized from a previous microarray profiling assay (left). The miR-124 levels detected in the microarray profiling were validated by qPCR (right). C) Northern blot assay for miR-124 in five CaP specimens that have sufficient amount of RNA. Three CaP samples (8, 11 and 16) having low miR-124 levels detected by qPCR exhibit lower signal intensity in North blots, compared to their BPH matches. Two CaP samples (5 and 9) and their BPH matches exhibit similar signal intensity in North blots. The numbers under the gel are the fold changes of miR-124 in CaP cells relative to in benign cells. B, BPH; C, CaP. U6 was used as loading control.
SI Figure 3. LNCaP (top) and C4-2B (bottom) cells transfected with 100 nM of miR-124m or miR-NC were grown in 10% FBS medium for four days. Both early apoptosis and later apoptosis were analyzed using Annexin V apoptosis assay protocol.
SI Figure 4. A point mutation or single nucleotide polymorphism (SNP) was detected at the 5′CpG region (1267 upstream of pre-miR-124-2) in a clinical CaP specimen. The wild-type “G” changed to “C” and was methylated (pointed out by an arrow).
SI Figure 5. Analyses of the expression of HMGA1 in CaP cells. A) Western blotting analysis of HMGA1 expression in 100 nM miR-124m-transfectd C4-2B cells. The numbers under the gels are the fold changes of HMGA1 in miR-124m-treated C4-2B cells relative to miR-NC-treated cells. Our pilot results indicate that miR-124m treatment induced the downregulation of HMGA1 by ∼60%. B) Immunohistochemical analysis of the expression of HMGA1 in two matched BPH and CaP tissues. CaP samples (right) and their BPHs (left) were immunostained using anti-HMGA1 antibody (sc-26348, Santa Cruz Biotechnology, Inc.). The abundance of miR-124 in these tissues was measured using qPCR, and the values are shown on the top of each image. CaP tissues express lower level of miR-124 than BPHs. Immunostaining for HMGA1 is more intense in two CaP samples than that in BPH matches. Moreover, localization of HMGA1 in the cytoplasm of CaP cells was obvious compared to that in BPH calls.