

Abstract
Here we report on a novel fluorescent analog of phenytoin as a potential inhibitor of neuropathic pain with potential use as an imaging agent. Compound 2 incorporated a heptyl side chain and dansyl moiety onto the parent compound phenytoin and produced greater displacement of BTX from sodium channels and greater functional blockade with greatly reduced toxicity. Compound 2 reduced mechano-allodynia in a rat model of neuropathic pain and was visualized ex vivo in sensory neuron axons with two-photon microscopy. These results suggest a promising strategy for developing novel sodium channel inhibitors with imaging capabilities.
Keywords: Sodium channel inhibitor, Mechano-allodynia, Dansyl, Phenytoin, Neuropathic pain
1. Introduction
Human voltage-gated sodium channels (VGSCs) represent key targets for the development of anticonvulsants, anesthetics, and antiarrhythmic drugs.1–4 Despite the widespread use of sodium channel inhibitors clinically, target selectivity, side effects and toxicity have hindered drug development.
The VGSCs have nine binding sites that cause a change in their gating properties. Anesthetics bind to site 9 and cause a persistent inactivation of the channel.5,6 Site 2 binds the neurotoxin batrachotoxin (BTX) and compounds that bind to anesthetic site 9 alloster-ically displace BTX from site 2 providing a way to monitor site 9 binding.7
Diphenylhydantoin (DPH, phenytoin), a clinical VGSC inhibitor used to treat epilepsy and chronic pain, binds to site 9 of VGSCs and has served as a lead compound in the discovery of novel VGSC inhibitors.8–10 In this model, DPH binds with higher affinity to sodium channels in the open or inactive state than to channels in the resting state, resulting in decreased sodium current at high rates of stimulation.3,4,11
In a previous study, DPH analogs were designed based on a QSAR model developed from [3H]-batrachotoxin-20-α-benzoate ([3H] BTX-B) displacement in rat brain synaptoneurosomes.12 The resulting comparative molecular field analysis (COMFA) model demonstrated that replacing one of the phenyl rings of DPH with a heptyl side chain, reaching a hydrophobic region of the binding site, was preferred. The addition of a meta-chloro group on the remaining phenyl ring was optimal to binding (Scheme 1, compound 1).2 To exploit this hydrophobically receptive region of the drug pharmacophore, we added a pentyl tethered 5-(dimethylamino)-8-sulfonyl-naphthalene (Dansyl) moiety.13 Herein, we report the synthesis of phenytoin analogs and their evaluation for BTX displacement, functional blockade, toxicity, and imaging properties. A binding model of 1 and 2 is presented.
Scheme 1.

Design of DPH analogs based on a QSAR model using [3H] BTX-B displacement.
2. Results
2.1. Design and synthesis of compounds 1 and 2
The synthesis of 12 and the intermediate sulfonamide 314 have been described previously. The sulfonamide 3 was converted to the Weinreb amide 4 through an amide coupling in the presence of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI). Following an aryl Grignard addition to yield the ketone 5 the desired hydantoin scaffold 2 was prepared via the Bucherer-Berg modified Strecker reaction (Scheme 2).
Scheme 2.

Synthetic route for 2. (a) 1 M NaHCO3, dansyl chloride, TEA, H2O/acetone, room temp, 3H, 85%. (b) EDCI, N,O-dimethylhydroxylamine HCI, DMAP, DCM, room temp, 2.5 h, 75%. (c) 3-Chlorophenyl magnesium bromide, THF, 0 °C, room temp, 16 h, 60%. (d) (NH4)2CO3, KCN, H2O/EtOH/THF (3:2:1), 75 °C, 3H, microwave 35%.
2.2. Fluorescence spectrum of compound 2
The fluorescence of the hydantoin core was confirmed by analysis of the excitation-emission spectrum (Fig. 1). The fluorescence spectrum of 2 exhibited two excitation maxima with one exciting at 290 nm (S0→S2) and emitting at 510 nm (S1 →S0) and one exciting at 400 nm (S0→S1) and emitting at 513 nm (S1→S0). This equates to a Stokes shift of 113 nm.
Figure 1.

Excitation and emission spectrum of 2.
2.3. VGSC affinity, functional blockade, and acute toxicity of compounds 1 and 2
To determine if 1 and 2 bound to sodium channels like the parent compound phenytoin (DPH), they were assessed for the magnitude of displacement of [3H] BTX-B in rat forebrain membranes, where sodium channels are highly expressed. Both 1 and 2 showed greater displacement of [3H] BTX-B than DPH (Table 1).
Table 1.
VGSC affinity, functional blockade, and acute toxicity15
| Compound | % [3H] BTX-B displacement (40 µM) |
% Nav1.2 functional block (10 µM) |
Acute toxicity (i.p.) LD50 (mg/kg) |
|---|---|---|---|
| DPH | 28 | 11±4 [139–178] |
15615 |
| Compound 1 | 70 | 31 ±5 [55–175] |
93 |
| Compound 2 | 86 | 59 ±4 [646–2650] |
2000 |
% BTX displacement: In vitro displacement of [3H] BTX-B by DPH, 1, and 2 in rat forebrain membranes. Compounds were applied at 40 µm. % Navl.2 block was measured in vitro in HEK cells expressing Nav 1.2 compounds were applied at 10 µM. Compound toxicity was calculated using the OECD up-and-down test guideline 425 (LD50). Values are statistical estimates based on the long-term fate of 7–10 mice per dose. Beneath the LD50 values are 95% confidence intervals where the assumed sigma is 0.5 mg/kg. The DPH acute toxicity data was previously published.
To test whether the compounds were functionally active as sodium channel blockers, the effects of 1 and 2 on human embryonic kidney cells (HEK 293) stably expressing hNav1.2 sodium channels was assessed using the whole-cell patch-clamp method. Inhibition of sodium currents represents functional blockade of VGSCs. 1 and 2 inhibited sodium currents by 30% and 59%, respectively (Table 1).
Acute toxicity experiments were performed using the OECD Up-and-Down Test to reduce the number of animals required. The toxicity of DPH and 1 were comparable. Notably, the toxicity of 2 was reduced in mice by up to 13-fold.
2.4. Effect of compounds 1 and 2 on mechano-allodynia
Phenytoin has been used clinically to reduce symptoms associated with neuropathic pain. To determine if 1 and 2 reduce neuropathic pain, they were tested in a rat model of sciatic nerve constriction. After nerve constriction, rats developed a significant reduction in threshold to von Frey filaments applied to the affected hindpaw, as expected with this model (Fig. 2, Presurgery vs Base-line-1). Both 1 and 2 increased withdrawal threshold, with a significant increase in threshold exhibited for 2. The effect of gabapentin (an anti-seizure drug and calcium channel blocker) on mechano-allodynia is shown in a separate cohort of rats to provide an approximate comparison with a drug that is used clinically to treat neuropathic pain.
Figure 2.

Compound 2 and mechano-allodynia in rats. Withdrawal threshold to von Frey filaments was measured 2 h after a subcutaneous injection of 1 (C1) or 2 (C2) in rats with sciatic nerve ligation and mechano-allodynia. N = 5 rats per group. Data represent the mean and SEM; 1-way ANOVA with Tukey’s post-hoc tests. C1; compound 1, C2; compound 2. Drug dose is in mg of drug per kg body weight of rat. *p <0.05, **p <0.01, ***p <0.001.
2.5. Effect of compound 2 on motor control and seizure induction
Because of its reduced toxicity and efficacy in a rodent model of neuropathic pain, 2 was examined for its effects on motor control and balance. A rotarod test was performed to test 2 at three different doses. None of the mice demonstrated neurological impairment at any of the drug doses tested at 0.5 and 4 h after drug administration (Table 2).
Table 2.
Compound 2 and the rotarod test in mice
| Compound 2 (mg/kg) | Motor impairment/total tested |
|
|---|---|---|
| 0.5 (h) | 4.0 (h) | |
| 30 | 0/4 | 0/2 |
| 100 | 0/8 | 0/4 |
| 300 | 0/4 | 0/2 |
Mice were injected intraperitoneally with 2 and tested at 0.5 and 4.0 h on the rotarod test. Normal controls maintained equilibrium on a rod rotating at 6 rpm for one minute. Numbers show number of mice that did not complete each test out of the number of mice tested.
The parent compound phenytoin is used clinically as an anticonvulsant. To assess whether 2 reduced seizures, it was tested in rodent models of seizure induction. Compound 2 was ineffective in both the maximal electroshock test (MES) and subcutaneous metrazol seizure threshold (scMET) models of epilepsy, unlike the parent compound DPH, which increased seizure thresholds.
2.6. Visualization of compound 2 ex vivo
Incorporation of a dansyl group into the drug pharmacophore of 2 suggested it might be useful as a fluorescent ligand. To determine if 2 could be visualized ex vivo in neuronal tissue, sections from dorsal root ganglia (DRG) (the nerve root and neuronal cell bodies of the constricted sciatic nerve), were treated with 2, and visualized with two-photon confocal microscopy. Differential staining of 2 was observed in DRG tissue and showed heaviest localization in axons and nerve fibers compared to neuronal cell bodies (Fig. 3).
Figure 3.

Two-photon confocal microscopy image of compound-2 in rat dorsal root ganglia paraffin sections. Rat DRG sections (5 µM) were deparaffinized and treated with 2 at 100 µM for 5 min before washing and imaging. Samples were excited at 725 nm and monitored at 510 nm. Scale bar, 100 µm B. Inset from panel A.
2.7. SAR of novel phenytoin derivatives
The relationship between modification of the phenyl groups of phenytoin and [3H] BTX-B displacement was assessed (Table 3). Replacing a phenyl ring with a heptyl side chain resulted in improved BTX displacement (1). Substitution of aromatic groups was favorable (3a) although binding was diminished when the methylene linker was replaced with an ethylene linker (3c) possibly due to steric hindrance. Modification of the heptyl side chain to increase hydrophobicity was favorable (2b, 11a) whereas activity was lost when side chain hydrophobicity was reduced (11b). Addition of a hydrophobic dansyl group resulted in increased BTX displacement (2).
Table 3.
Structure–activity relationships of novel phenytoin derivatives
| Compound name | %BTX | Structure |
|---|---|---|
| Phenytoin (DPH) | 28 | 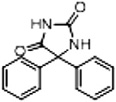 |
| Compound 1 (2a in Supplementary Scheme 1) | 70 | 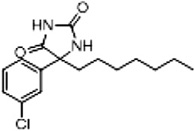 |
| 3a (Supplementary Scheme 1) | 72 | 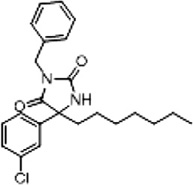 |
| 3d (Supplementary Scheme 1) | 83 | 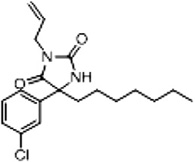 |
| 3c (Supplementary Scheme 1) | 17 | 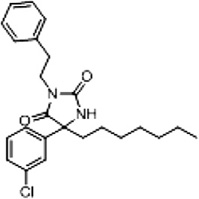 |
| 2b (Supplementary Scheme 1) | 79 | 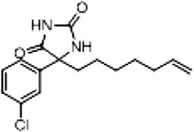 |
| 11a (Supplementary Scheme 2) | 87 | 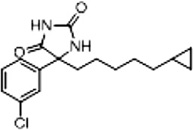 |
| 11b (Supplementary Scheme 2) | 28 | 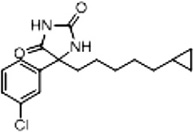 |
| Compound 2 (Scheme 2 and Supplementary Scheme 5) | 86 | 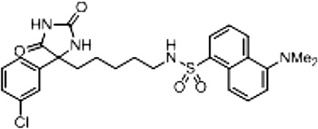 |
%BTX is the percent displacement of [3H] batrachotoxin-20-α-benzoate (BTX) from the saturated site 2 of VGSCs in rat brain synaptoneurosomes at 40 µM of test compound. Compounds that displaced greater than 50% of the [H] BTX-B were considered active analogs for further study.
2.8. Model of compound 1 and 2 at the BTX binding site of sodium channels
The sodium channel homology model is described in a previous report.16 Since 1 and 2 displace BTX, we hypothesized that they bind to the open state of the sodium channel. Compounds 1 and 2 were docked into the homology model using the program Sur-FlexDock incorporated in Sybyl X.17 The starting structure was the unminimized constructed 3-D structure. A preliminary conformation search was performed using systemic search/analysis in Sybyl X. To be consistent with the mutation studies18–20 and previous known interactions of BTX analogs, the docked positions of 1 and 2 were remodeled using step-by-step manual docking with restrained Molecular Dynamics simulations followed by energy minimization. In the restrained MD simulations, the optimal H-bond and hydrophobic distance restraints were set between the sodium channel pore forming residues and the compounds.
A structural model of 1 and 2 docked to the sodium channel is shown in Figure 4. Although, some uncertainly remains for several residues, our binding model predicted residues F1283, F1579, L1582, V1583, Y1586 in IVS6, and T1279, L1280 in IIIS6, and L788, F791, L792, in IIS6 and F430, I433, L437 in IS6 contributed to the tight binding interaction of 1 (Fig. 4A) and 2 (Fig. 4B). The shape and size of 1 and 2 (shown in green) are complimentary to the model of the BTX binding site (shown in cyan), where the imi-dazolidine-2,4-dione fits into the sodium channel sub-cavity formed by T1279 (IIIS6), L1280 (IIIS6), F791(II) and L792 (II).
Figure 4.

Model showing 1 and 2 docked into the BTX binding site of the hNaV channel. The ball and stick model shows BTX binding site (white), and the feasibility of binding of 1 (panel A), 2 (panel B) (green) and [3H] BTX-B (cyan).
3. Discussion
Blockers of human voltage-gated sodium channels are used clinically as anticonvulsants, antiarrhythmics, anesthetics, and more recently, to treat neuropathic pain. While widely used, issues of channel selectivity, off-target effects, and toxicity continue to hinder further drug development. In this study we synthesized compounds with increased affinity for the BTX binding site of VGSCs using deductions derived from our 3-D QSAR developed from phenytoin (DPH).21 Incorporation of a heptyl side chain conferred additional displacement of [3H] BTX-B and further functional blockade of VGSCs in patch-clamp electrophysiology experiments. The addition of a dansyl moiety (2) resulted in further BTX displacement and functional blockade compared to DPH or l.
Incorporation of a dansyl group into the pharmacophore of phenytoin resulted in a fluorescent analog of phenytoin. Compound 2 was visualized in the sciatic nerve and dorsal root ganglia (DRG) ex vivo with two-photon microscopy. The drug bound primarily to axons, consistent with structural studies on sodium channel density which indicate higher protein levels in the axon than the soma.22,23
Compound 2 significantly reduced mechano-allodynia associated with neuropathic pain after sciatic nerve compression comparable to that observed with gabapentin, a drug used clinically to treat chronic pain.24,25 Notably, 2 was less toxic than the parent compound DPH suggesting it may hold improvements over other sodium channel blockers used to treat neuropathic pain, such as carbamazepine and lamotrigine.26–29
Structural modeling of 1 and 2 binding to the open state of VGSCs suggests that the increased displacement of [3H] BTX-B by 2 versus 1 likely stems from the following favorable additional interactions within hydrophobic regions of the sodium channel: (1) the amide group of imidazolidine-2,4-dione may form hydrogen bonds with 1279(IIIS6); (2) hydrogen bond interactions may occur between the dansyl sulphamide with N434(I) and Y1586(IV); (3) the dansyl group may form favorable hydrophobic contacts with F1283 (IIIS6), L437(I), L788(II), L1280(IIIS6) in a hydrophobic region of the binding site.
Somewhat surprisingly, 2 did not suppress seizure induction, suggesting differences between parent drug and 2 in pharmacokinetics or dynamics. Possible explanations include changes in sodium channel isoform selectivity, poor brain-penetrance relative to the parent compound, changes in selectivity for state-dependent inhibition allowing for improved selectivity for depolarized neurons, or blocking at lower frequencies of stimulation and producing a tonic block.30
In conclusion, compound 2 may represent a novel approach for the development of safer sodium channel inhibitors to treat neuropathic pain. The enhanced hydrophobic groups in the receptive region of the anesthetic binding region improved BTX displacement and increased functional blockade of sodium channels while reducing compound toxicity. By using drug design to incorporate a dansyl fluorophore into the drug pharmacophore, we synthesized an analog of a sodium channel inhibitor with the potential to be used as an imaging agent that may be useful in biodistribution studies both in vivo and ex vivo.
4. Experimental
4.1. Chemistry general procedures
Commercially available reagents were purchased from Sigma-Aldrich. 1H and 13C NMR spectra were recorded on a Varian 400-NMR magnet. Flash chromatographic purifications were done on Biotage SP1 chromatography system monitoring at 254 nm.
4.2. Synthesis
4.2.1. 6-(5-(Dimethylamino)naphthalene-1-sulfonamido)-N-methoxy-N-methylhexanamide (4)
EDCI (2.93 g, 20.6 mmol), N,O-dimethylhydroxylamine HCI (2.02 g, 20.6 mmol), and 4-dimethylaminopyridine (DMAP) (2.54 g, 20.6 mmol) were added to a solution of acid 3 (3.02 g, 8.23 mmol) in DCM. The reaction was stirred for 2.5 h before being quenched with brine (20 mL). The phases were separated and the organic phase was washed with 2 M HCI (10 mL) and brine (10 mL) before being dried over Na2SO4. The solution was concentrated and purified by flash chromatography (1:20 MeOH/DCM) to yield a yellow oil (2.54 g, 75%). 1H NMR (400 MHz, CDC13): δ 1.21 (m, 2H), 1.42 (m, 4H), 2.27 (t, J = 7.3 Hz, 2H), 2.87 (m, 8H), 3.12 (s, 3H), 3.61 (s, 3H), 5.17 (t, J = 6.1 Hz, 1H), 7.16 (d, J = 7.5 Hz, 1H), 7.51 (m, 1H), 8.21 (dd,J = 1.2,7.3 Hz, 1H,), 8.31 (d,J = 8.7 Hz, 1H,), 8.52 (d, J = 8.5 Hz, 1H,). 13C NMR (100 MHz. CDC13): δ 134.9, 130.1, 129.7, 129.5, 129.3, 128.1, 123.1, 118.8, 115.0, 61.0, 45.3, 42.9, 31.3,29.1,25.9,23.6.
4.2.2. N-(6-(3-Chlorophenyl)-6-oxohexyl)-5-(dimethylamino) naphthalene-1-sulfonamide (5)
A flame dried round bottom flask was charged with amide 4 (1.56 g, 3.82 mmol) which was dissolved in THF (30 mL). The flask was cooled to 0 °C and 3-chlorophenyl-magnesium bromide solution was added dropwise (0.50 M, 39.0 mL, 19.1 mmol). This was warmed to room temperature and stirred overnight. The reaction was quenched with saturated ammonium chloride solution (20 mL). The organic phase was extracted with EtOAc (3 × 25 mL), washed with brine (25 mL), and dried under Na2SO4. It was then concentrated to dryness and purified by flash chromo-tography (1:1 EtOAc/hex) to yield a yellow oil (1.04 g, 60%). 1H NMR (400 MHz, CDCI3):δ 1.21 (m, 2H), 1.43 (m, 4H), 2.71 (t, J = 7.2 Hz, 2H), 2.83 (s, 6H), 2.91 (dd, J = 6.8 Hz, 13.2 Hz, 2H), 5.30 (t, J = 6.2 Hz, 1H), 7.12 (d, J =7.0 Hz, 1H), 7.34 (t, J = 7.9 Hz, 1H), 7.49 (m, 3H), 7.72 (d, J = 7.8 Hz, 1H), 7.83 (s, 1H), 8.23 (d, J = 6.1 Hz, 1H), 8.34 (d, J = 8.7 Hz, 1H), 8.50 (d, J = 8.5 Hz, 1H). 13C NMR NMR (100 MHz. CDCI3): δ 198.6, 151.7, 138.1, 134.7, 134.6, 132.7, 130.1, 129.7, 129.6, 129.4, 129.3, 128.1, 127.8, 125.9, 123.0, 118.6, 114.9, 45.2, 45.2, 42.8, 38.0, 29.0, 25.7, 22.9.
4.2.3. N-(5-(4-(3-Chlorophenyl)-2,5-dioxoimidazolidin-4-yl)pentyl)-5-(dimethylamino) naphthalene-1-sulfonamide (2)
The ketone 4 (300 mg, 0.65 mmol) was dissolved in THF (1 mL). EtOH (2 mL) was added followed by KCN (213 mg, 3.3 mmol) and ammonium carbonate (628 mg, 6.5 mmol). Water (3 mL) was added and the vessel was sealed. The tube was placed in a CEM Discover microwave (temperature = 75 °C, power = 150 W) for 3 h. The product was extracted with DCM (3 × 25 mL), washed with brine (3 × 35 mL) and the organic layer was dried over Na2SO4 and evaporated to dryness. It was purified by flash chromo-tography (1:1 EtOAc/Hex) to yield a light green crystalline solid (301 mg, 35%). 1H NMR (400 MHz, CDCI3): δ 1.12 (m, 4H), 1.25 (m, 2H) 1.96 (m, 2H), 2.86 (m, 8H), 5.72 (t, J = 6.0 Hz, 1H), 7.13 (d, J = 7.5 Hz, 1H), 7.28 (m, 2H), 7.38 (m, 1H), 7.49 (dd, J = 8.6, 17.0 Hz, 3H), 7.85 (s, 1H), 8.19 (d, J = 7.3 Hz, 1H), 8.29 (d, J = 8.6 Hz, 1H), 8.51 (d, J = 8.4 Hz, 1H), 9.21 (s, 1H). 13C NMR (100 MHz. CDCI3): δ 175.0, 157.9, 139.8, 139.4, 134.7, 134.6, 130.3, 130.0, 129.5, 129.4, 128.7, 128.4, 128.2, 125.6, 125.2, 123.6, 123.2, 115.1, 68.4, 45.3, 42.5, 38.3, 28.8, 25.7, 22.9, 14.1. LC–MS (ESI): m/z 530 (M+H)+; HRMS (TOF): C26H29C1N4O4S. Calcd (M+1) 529.1598. Found 529.1688.
4.3. Modeling and docking of compounds 1 and 2 to the sodium channel
The modeling of the sodium channel is described in our previous paper.16 Docking studies between ligand and the sodium channel were carried out using the program SurFlexDock (Sybyl X. Tripos Inc., St. Louis, USA) and AUTODOCK 4.017 with all the parameters set to default. Molecular dynamics simulations were carried out using AMBER 8.031 using the default parameters. The structure of the Nav-compound 2 complex was then refined by molecular dynamics simulation using the Amber 9.0 program suite. The charge and force field parameters of compounds were obtained using the most recent Antechamber module where 2 was minimized at the MP2/6-31G* level using Gaussian 03. The SHAKE algorithm was used to fix the bonds involving hydrogen.32 The protocol for the molecular dynamics simulation is described below: The total charge of the system was neutralized by addition of a chloride ion. The system was solvated in a 12 Å cubic box of water where the TIP3P model 8 was used. 2000 steps of minimization of the system were performed in which the sodium channel was constrained by a force constant of 75 kcal/mol/Å2. After minimization, a 10 ps simulation was used to gradually raise the temperature of the system to 298 K while the complex was constrained by a force constant of 15 kcal/mol/Å. Another 25 ps of equilibrium run was used where only the backbone atoms of the complex were constrained by a force constant of 5 kcal/mol/Å. A final production run of 200 ps was performed with no constraints. When applying constraints, the initial complex structure was used as a reference structure. All the molecular dynamics simulations were at constant temperature and pressure. The PME method10 was used and the non-bonded cutoff distance was set at 12 Å. The time step was 5 fs, and the neighboring pairs list was updated in every 25 steps.
4.4. [3H] BTX-B displacement assay
The [3H] BTX-B binding assays were performed by Novascreen Biosciences (Hanover, MD). Rat forebrain membranes (10 mg tissue/well) were incubated with [3H] BTX-B (30–60 Ci/mmol) in 50 mM HEPES (pH 7.4) containing 130 mM choline chloride, at 37 °C for 60 min. The reaction was terminated by rapid vacuum filtration of reaction contents onto glass fiber filters. Radioactivity trapped on the filters was determined and compared to controls to measure binding of [3H] BTX-B with the Na+ channel site 2 binding site. Reactions were pre-incubated with test compounds (40 µM) to measure percent [3H] BTX-B displacement. Aconitine (1 µM) was used as a positive control (Sigma-Aldrich, St. Louis, MO). Each experiment was replicated at least conce for each compound.
4.5. Sodium hNav1.2 channel electrophysiology
Sodium currents were recorded using the whole-cell configuration of the patch-clamp recording technique with a Axopatch 200 amplifier (molecular devices) as described.16 Compounds were prepared as 100 mM stock solutions in dimethyl sulfoxide (DMSO) and diluted to desired concentration in perfusion solution. The maximum DMSO concentration used was 0.1% and had no effect on current amplitude. All experiments were performed at room temperature (20–22 °C). After establishing whole-cell, a minimum series resistance compensation of 75% was applied. Sodium currents were elicited by a depolarizing step from a holding potential of −100 mV to +10 mV for 25 ms at 15 s intervals. Compounds were applied after a 3 min control period and continued until steady state current amplitude was observed. All data represent percentage mean block ± standard error of the mean (SEM).
4.6. Biological evaluation
4.6.1. Animals
Adult female Sprague-Dawley rats were used for peripheral nerve injury and evaluation of tactile sensitivity (Taconic Farms, Germantown, NY: 150 g at the start of the experiment). Rats were housed in the Georgetown University Research Resource Facility, maintained on a 12 h light/dark cycle and allowed free access to food and water. For acute toxicity testing, motor impairment, and anticonvulsant screening, male albino mice (CF-1) 18–25 g were used. All experiments were approved by the Georgetown University Animal Care and Use Committee and performed in accordance with the National Institutes of Health guidelines on animal care.
4.6.2. Acute toxicity testing
Toxicity was calculated using the OECD Up-and-Down Test Guideline 425.33 This protocol significantly reduces the number of animals required to estimate the acute toxicity of a chemical when compared to previous methods for LD50.
4.6.3. Motor impairment and anticonvulsant screening
Compounds were tested at the anticonvulsant screening program at NINDS for their effect on motor coordination and for their efficacy in reducing seizures in rodents.34 A rotarod test was used for motor coordination. Mice were placed on a rod that rotates at 6 rpm and animals were considered impaired if they fell off the rod three times during a 1-min period. For anticonvulsant screening, mice received 30, 100, or 300 mg/kg of test compound intra-peritoneally, rats received 30 mg/kg of test compound peroral, or rats received varying doses intraperitoneally before seizure induction with maximal electric shock (MES). For the subcutaneous Metrazol test (scMET), rats were pretreated with 50 mg/kg peroral test compound before seizure induction with MET injection.
4.6.4. Peripheral nerve injury and evaluation of tactile sensitivity
Rats underwent a unilateral ligation of the sciatic nerve35 with modifications.36 Animals that developed significant allodynia to von Frey filaments by the up and down method37,38 were selected for drug testing. Drug was dissolved in DMSO and diluted in PEG400 to 200 mg/ml stock solution. Aliquots of drug stock were diluted in vehicle (PEG400/phosphate buffered saline) prior to injection. Rats (n = 5 per group) were injected subcutaneously at 50 or 100 mg/kg body weight. Response to von Frey filaments was tested at 2 h after injection. Controls received vehicle without drug. Group differences were analyzed with a one-way analysis of variance with Tukey’s post hoc tests, with significance set at p <0.05.
Supplementary Material
Acknowledgments
We thank Charles Bauer (Novascreen, Caliper Life Sciences, Hanover, MD) for performing the [3H] BTX-B assays and the James P. Stables at NINDS for help in the evaluation of compound 2 in the Anticonvulsant Drug Development Program. Financial assistance was provided by NIH Grants CA105435-04 and R21NS061069 in addition to the Georgetown Drug Discovery Program and a new investigator grant from R24HD050845 (LHM). This work was supported by the Lombardi Cancer Center shared resources.
Abbreviations
- VGSC
voltage-gated sodium channel
- hNav1.2
human sodium channel 1.2
- BTX
batrachotoxin
- DPH
diphenylhydantoin
Footnotes
Competing financial interests
A patent application has been filed by Georgetown University on the behalf of the inventors that are listed as authors in this article.
Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2012.06.042.
References and notes
- 1.Dib-Hajj SD, Black JA, Waxman SG. Pain Med. 2009;10:1260. doi: 10.1111/j.1526-4637.2009.00719.x. [DOI] [PubMed] [Google Scholar]
- 2.Lenkowski PW, Batts TW, Smith MD, Ko SH, Jones PJ, Taylor CH, McCusker AK, Davis GC, Hartmann HA, White HS, Brown ML, Patel MK. Neuropharmacology. 2007;52:1044. doi: 10.1016/j.neuropharm.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rogawski MA, Loscher W. Nat. Med. 2004;10:685. doi: 10.1038/nm1074. [DOI] [PubMed] [Google Scholar]
- 4.Anger T, Madge DJ, Mulla M, Riddall D. J. Med. Chem. 2001;44:115. doi: 10.1021/jm000155h. [DOI] [PubMed] [Google Scholar]
- 5.Scheib H, McLay I, Guex N, Clare JJ, Blaney FE, Dale TJ, Tate SN, Robertson GM. J. Mol. Model. 2006;12:813. doi: 10.1007/s00894-005-0066-y. [DOI] [PubMed] [Google Scholar]
- 6.Lipkind GM, Fozzard HA. Mol. Pharmacol. 2005;68:1611. doi: 10.1124/mol.105.014803. [DOI] [PubMed] [Google Scholar]
- 7.Linford NJ, Cantrell AR, Cm Y, Scheuer T, Catterall WA. Proc. Natl. Acad. Sci. U.S.A. 1998;95:13947. doi: 10.1073/pnas.95.23.13947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ko SH, Jochnowitz N, Lenkowski PW, Batts TW, Davis GC, Martin WJ, Brown ML, Patel MK. Neuropharmacology. 2006;50:865. doi: 10.1016/j.neuropharm.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 9.Lenkowski PW, Ko SH, Anderson JD, Brown ML, Patel MK. Eur. J. Pharm. Sci. 2004;21:635. doi: 10.1016/j.ejps.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Wang GK, Russell C, Wang SY. Pain. 2004;110:166. doi: 10.1016/j.pain.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 11.Mula M. Cent. Nerv. Syst. Agents Med. Chem. 2009;9:79. doi: 10.2174/187152409788452063. [DOI] [PubMed] [Google Scholar]
- 12.Anderson JD, Hansen TP, Lenkowski PW, Walls AM, Choudhury IM, Schenck HA, Friehling M, Holl GM, Patel MK, Sikes RA, Brown ML. Mol. Cancer Ther. 2003;2:1149. [PubMed] [Google Scholar]
- 13.Weber G, Farris FJ. Biochemistry. 1979;18:3075. doi: 10.1021/bi00581a025. [DOI] [PubMed] [Google Scholar]
- 14.Smith AB, 3rd, Rucker PV, Brouard I, Freeze BS, Xia S, Horwitz SB. Org Lett. 2005;7:5199. doi: 10.1021/ol0520166. [DOI] [PubMed] [Google Scholar]
- 15.Biton V. Clin. Neuropharmacol. 2007;30:230. doi: 10.1097/wnf.0b013e3180413d7d. [DOI] [PubMed] [Google Scholar]
- 16.De Oliveira EO, Graf KM, Patel MK, Baheti A, Kong HS, MacArthur LH, Dakshanamurthy S, Wang K, Brown ML, Paige M. Bioorg. Med. Chem. 2011;19:4322. doi: 10.1016/j.bmc.2011.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morris G, Goodsell D, Halliday R, Huey R, Hart W, Belew R, Olson AJ. Comput. Chem. 1998;19:1639. [Google Scholar]
- 18.Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Science. 1994;265:1724. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- 19.Yarov-Yarovoy V, Brown J, Sharp EM, Clare JJ, Scheuer T, Catterall W. A. J. Biol. Chem. 2001;276:20. doi: 10.1074/jbc.M006992200. [DOI] [PubMed] [Google Scholar]
- 20.Yarov-Yarovoy V, McPhee JC, Idsvoog D, Pate C, Scheuer T, Catterall W. A. J. Biol. Chem. 2002;277:35393. doi: 10.1074/jbc.M206126200. [DOI] [PubMed] [Google Scholar]
- 21.Brown ML, Zha CC, Van Dyke CC, Brown GB, Brouillette WJ. J. Med. Chem. 1999;42:1537. doi: 10.1021/jm980556l. [DOI] [PubMed] [Google Scholar]
- 22.Van Wart A, Trimmer JS, Matthews G. J. Comp. Neurol. 2007;500:339. doi: 10.1002/cne.21173. [DOI] [PubMed] [Google Scholar]
- 23.Kole MH, Ilschner SU, Kampa BM, Williams SR, Ruben PC, Stuart GJ. Nat. Neurosci. 2008;11:178. doi: 10.1038/nn2040. [DOI] [PubMed] [Google Scholar]
- 24.Moore RA, Wiffen PJ, Deny S, McCmay HJ. Cochrane Database Syst. Rev. 2011 CD007938. [Google Scholar]
- 25.Finnerup NB, Sindrup SH, Jensen TS. Pain. 2010;150:573. doi: 10.1016/j.pain.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 26.Besag FM, Berry DJ, Pool F, Newbery JE, Subel B. Epilepsia. 1998;39:183. doi: 10.1111/j.1528-1157.1998.tb01356.x. [DOI] [PubMed] [Google Scholar]
- 27.Benes J, Parada A, Figueiredo AA, Alves PC, Freitas AP, Learmonth DA, Cunha RA, Garrett J, Soares-da-Silva P. J. Med. Chem. 1999;42:2582. doi: 10.1021/jm980627g. [DOI] [PubMed] [Google Scholar]
- 28.Rogers M, Tang L, Madge DJ, Stevens EB. Semin. Cell Dev. Biol. 2006;17:571. doi: 10.1016/j.semcdb.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 29.Devor M. J. Pain. 2006;7:S3. doi: 10.1016/j.jpain.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 30.Theile JW, Cummins TR. Front. Pharmacol. 2011;2:54. doi: 10.3389/fphar.2011.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. J. Comput. Chem. 2005;26:1668. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanson RN, Lee CY, Friel CJ, Dilis R, Hughes A, DeSombre ER. J. Med. Chem. 2003;46:2865. doi: 10.1021/jm0205806. [DOI] [PubMed] [Google Scholar]
- 33.OECD. Vol. 2001. Paris: 1998. [Google Scholar]
- 34.NINDS. Bethesda, MD: 2012. [Google Scholar]
- 35.Bennett GJ, Xie YK. Pain. 1988;33:87. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 36.Mosconi T, Kruger L. Pain. 1996;64:37. doi: 10.1016/0304-3959(95)00077-1. [DOI] [PubMed] [Google Scholar]
- 37.Dixon WJ. Annu. Rev. Pharmacol. Toxicol. 1980;20:441. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- 38.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. J. Neurosci. Methods. 1994;53:55. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.