

Abstract
OBJECTIVE
Determine the role of phagocytosis in the deposition of acute phase SAA protein in peripheral organs as AA amyloid.
METHODS
AA amyloidosis was induced by injection of amyloid enhancing factor (AEF) in huIL-6 transgenic mice. Clodronate liposomes were injected at different times, and the amyloid load evaluated by Congo red birefringence staining and monitoring with the amyloid specific probe 125I-labeled peptide p5R.
RESULTS
Injection of clodronate containing liposomes depleted Iba-1 positive and F4/80 positive phagocytic cells in liver and spleen for up to 5 days. Treatment prior to administration of intravenous AEF did not alter the pattern of deposition of the AEF in spleen, but inhibited the catabolism of the 125I-labeled AEF. Clodronate treatment 1 day before or 1 day after AEF administration had little effect on AA amyloid accumulation at 2 weeks; however, mice treated with clodronate liposomes 5 days after AEF induction and evaluated at 2 weeks post AEF induction showed reduced amyloid load relative to controls. At 6 weeks post-AEF there was no significant effect on amyloid load following a single clodronate treatment.
CONCLUSION
Macrophages have been shown to be instrumental in both accumulation and clearance of AA amyloid after cessation of inflammation. Our data indicate that when SAA protein is continuously present, depletion of phagocytic cells during the early course of the disease progression temporarily reduces amyloid load.
Keywords: clodronate liposomes, SAA, AA amyloidosis, macrophages, peptides, huIL-6 mice, AEF
Introduction
AA amyloidosis results from the aggregation and deposition of serum amyloid A (SAA) protein, as fibrils, in peripheral organs leading to dysfunction and death. In humans [1] and mouse models [2–5], SAA protein is elevated due to an inflammatory response. In humans, the inflammation can be due to sporadic episodes of Familial Mediterranean Fever or ongoing such as in rheumatoid arthritis. It has been shown that macrophages are involved in SAA processing and deposition [3, 6, 7] and that cell surface-expressed heparan sulfate proteoglycans play a critical role in amyloidogenesis through binding of HDL-associated SAA [6]. Additionally, Fc receptor-positive macrophages are involved in dissolution of the amyloid load once the inflammation process has been resolved [7, 8].
Two main mouse models of AA amyloidosis have been used to study the pathogenesis of the disease:
In the silver nitrate model, induction of SAA is variable and transient depending on the response of the animal to silver nitrate solution injected subcutaneously. Accumulation of AA amyloid, once induced by AEF injection, is dependent not only on the level and processing of SAA but also the loss or removal of AA once the SAA levels have diminished. A second model utilizes transgenic (huIL-6) mice that constitutively produce IL-6 resulting in ongoing inflammation. In this model, SAA serum levels are always elevated (400–4000 μg/mL) and deposition, initiated by injection of amyloid enhancing factor (AEF), is continuous resulting in an ever increasing AA load and ultimately death.
The deposition of AA amyloid in the mice is a two-phase process involving the initial seeding by AEF as well as processing of SAA for subsequent fibril growth increasing the size and extent of AA deposits. In the silver nitrate mouse model, SAA levels peak between 24 and 48 hours and clearance of the AA is affected once SAA levels are lowered [7]. It has been shown that antibody mediated resolution of the AA deposits is facilitated by Fc receptor positive phagocytes [7]. In contrast, in the huIL-6 transgenic model of AA, SAA is continuously induced by constitutive expression of the huIL-6 transgene in the transgenic mice [4, 5]. These mice can develop AA spontaneously as they age or the disease can be induced with iv AEF to produce AA deposits earlier and in a more predictable time frame [4]. In either case, AA deposition is continuous, ultimately resulting in death at approximately 6–10 weeks likely due to kidney failure [5]. As with the silver nitrate model, it is likely that phagocytic cells are involved in the AEF seeding and the subsequent growth of amyloid deposits as well as the potential clearance of these AA amyloid deposits if SAA production could be reduced.
Phagocytes are a diverse group of cells generally classified as macrophages [9]. Several subclasses of macrophage-like cells exist including monocytes, Fc receptor-positive cells and the tissue (spleen, liver and skin) antigen processing cells. General macrophage markers include F4/80 which recognizes a G-protein-coupled receptor (GPCR) adhesion protein family that is found on cells of myeloid lineage [10]. Iba-1-reactive antibodies bind allograft inflammatory-1 protein which is induced by cytokines and interferon and is mostly limited to activated macrophage type cells [11]. Phagocytic cells recognize and remove particulates, and Fc receptor cells are involved in removal of opsonized foreign bodies coated with antibody. The cytotoxic drug clodronate administered in a liposome formulation is recognized by the normal phagocyte system (reticuloendothelial system, RE) and is engulfed by these cells resulting in selective cell death and thus population depletion [12, 13]. Cells without phagocytosis activity are not exposed to the liposome-encapsulated clodronate and thus are spared. Phagocytic cell populations regenerate and in normal mice reach near normal levels by 2 weeks post-treatment [14]. Many subpopulations of macrophages have been shown to be sensitive to clodronate liposome treatment by monitoring of phagocyte function; however, other populations of macrophages are not effected [13]. It is clear that the range of phenotypes of macrophages is large and also that the role of phagocytic cells in experimental AA amyloidosis is complex. Certainly, recognition of iv injected AEF and processing of particulate deposits of AA during amyloid removal both require phagocytic functions. Production of AA from SAA-HDL likely requires receptor mediated endocytosis rather than phagocytosis, but may be mediated by similar classes of phagocytic macrophages. In the huIL-6 model of AA deposition, wherein serum SAA levels are continuously elevated, both functions could be active. We report here that depletion of phagocytic cells with clodronate liposomes does not alter the distribution of IV-injected AEF in the liver and spleen, but when treatment was initiated after AEF administration, clodronate transiently reduced the amyloid load in our mouse model of chronic AA amyloidosis.
Methods
Transgenic huIL-6 mice were bred by mating WT and hemizygous mice with subsequent genotyping by PCR analyses at ~3 weeks of age [5]. The animals were housed in the UTGSM animal facility with food and water provided ad libitum and standard 12 h light and dark schedule. All experimental protocols were conducted according to approved protocols by the UT Animal Care and Use Committee. Clodronate liposomes were purchased from VU medisch centrum (CloronateLiposomes.org, Amsterdam, the Netherlands). Liposome suspensions (0.1 mL) were injected iv at the indicated times.
For immunohistochemical staining, paraffinized tissues were subjected to antigen retrieval in pH 6.0 Na citrate buffer overnight (Dako, Carpinteria, CA) before primary antibody incubation. Staining was performed with rabbit anti Iba-1 (Wako chemicals, Richmond, VA) at 1/9000 dilution or with rat Mab anti F4/80 (AbD Serotec, Raleigh, NC; BioRad Laboratories, Hercules, CA) at 1/200 dilution coupled with horseradish peroxidase secondary antibodies (BioGenex, Fremont, CA). Peroxidase antibody presence was detected with diaminobenzidine (Vector Labs, Burlingame, CA). Citrate retrieved sections were also stained for perlecan using a 1:1000 dilution of rat MAb A7L6 (Santa Cruz Biotechnology, Inc. Santa Cruz, CA), named perlecan 1 in the text, and rat MAb HK-102 (5μg/mL, Seikagaku Biobusiness Corp, Toyko, Japan) detected with biotinylated anti-rat IgG (Biogenix, San Ramon, CA), named perlecan 2.
AEF was isolated from spleens of mice that had 3–4+ AA amyloidosis by the water flotation procedure [15]. For induction of AA deposition, lyophilized preparations of AEF were suspended in sterile phosphate buffered saline (PBS) by short probe sonication at 1 mg/mL. Mice were given iv injections (0.1 mL) of a 1/10 dilution of the material.
Radioiodination of either AEF, p5 or p5R peptide [16] was conducted as described previously [17]. Briefly, proteins were labeled in 0.1 M sodium phosphate buffer (pH 7.6) with 125I (Perkin Elmer) 10 μg freshly prepared chloramine T (Sigma, St. Louis, MO). Radioiodinated AEF was isolated from free 125I− by dialysis vs PBS (0.01 M sodium phosphate buffer, pH 7.6 with 0.15 M NaCl) for 24 h. Peptide p5R was purified by gel filtration on PD-10 resin columns equilibrated with PBS containing 0.1 mg/mL gelatin as carrier protein [17]. All radiolabeled preparations were subjected to SDS-PAGE analyses for verification of label and the p5R samples were tested for AA binding activity (16). In experiments to determine AEF deposition, 125I-labeled AEF (10 μg, ~ 30 μCi) in 0.2 mL PBS was injected iv. For analyses of AA load with p5R, radioiodinated peptide was injected iv (0.2 mL, ~5 μg, ~100 μCi) and 2 h thereafter the animals were euthanized by an overdose of inhaled isoflurane. Certain animals were analyzed by SPECT/CT imaging as previously described [17], to confirm p5R biodistribution (data not shown). A necropsy was performed on all mice with collection of muscle, liver, pancreas, spleen, left kidney, right kidney, stomach, upper intestine, lower intestine, heart and lung. A small piece of each tissue was placed in tared vials for biodistribution measurements [17] and another sample was fixed in buffered formalin for histopathology and micro-autoradiography of paraffin embedded tissue sections [17]. Slides were stained with CR for amyloid evaluation. Liver and spleen sections from all mice were graded 0–4+ by a blinded observer. The scoring system for spleen was: 1+, stain ringing follicles but not completely around; 2+, complete, sometimes double ring around follicles; 3+, double ring around follicles with some red pulp staining; 4+, stain ringing follicles with extensive red pulp staining and spotty stain in blue pulp also. Scoring for liver: 1+, light stain only in major vessel lining; 2+, heavier stain of vessel walls; 3+, staining of all vessels in portal triads with some stain extending to hepatocytes; 4+, staining extending throughout total section (>20% of total area).
Results
Experiments were done to determine the time post AEF treatment of huIL-6 mice that amyloid could first be detected and how rapid the time course of the disease occurred. Data in Table 1 show that Congo red staining could first be detected at 2 weeks post AEF injection at which time there was also an elevated level of deposition of radio-iodinated peptide p5 relative to age matched huIL-6 mice with no AEF injections. The extent of Congo red staining and the p5 deposition increased rapidly between weeks 2 to 4. Week 5 data was only slightly elevated relative to the week 4 values indicating a plateau of amyloid accumulation or our ability to quantitate it. These data were collected using radioiodinated P5 whereas P5R was used in subsequent experiments. Radioiodinated p5 has been shown to be equivalent to radioiodinated p5R in amyloid detection in these AA mice at least for early time points [16].
Table 1.
Analyses of AA amyloid in huIL-6 mice as a function of time post AEF treatment*
| Post AEF (wk) |
Congo red (0–4+) |
p5 (%ID/g) | ||
|---|---|---|---|---|
| Liver | Spleen | Liver | Spleen | |
| 0 | 0 | 0 | 0.83±0.23 | 0.70±0.15 |
| 2 | 0.59±0.33 | 1.02±0.57 | 1.9±0.52 | 3.7±1.74 |
| 3 | 1.75±0.83 | 2.62±0.65 | 6.13±3.0 | 10.2±1.8 |
| 4 | 2.5±0.93 | 3.25±0.83 | 7.7±2.7 | 12.7±1.6 |
| 5 | 3.0±0 | 4.0±0 | 10.6±2.9 | 13.2±2.9 |
huIL-6 mice were treated with 10 μg of AEF (iv) for the designated time and then sacrificed 2 h post injection of 125I P5 peptide. Liver and spleen sections were scored for Congo red stain (0–4+) and tissues analyzed for accumulation of p5 (% injected dose/gram of wet tissue) ± SD. N= 4 mice per group.
Injection of clodronate liposomes causes a selective and temporary loss of certain phagocyte populations. Images in Fig. 1 (liver) and Fig. 2 (spleen) show Iba-1 (activated macrophages) and F4/80 (general phagocyte marker) staining of tissue sections of huIL-6 mice. The cells staining in spleen for Iba-1 were perifollicular (arrows) as well as in the red pulp while cells staining for F4/80 were largely in the red pulp. Note that F4/80 positive cells in both liver and spleen are nearly completely lost 3 days after clodronate treatment, but that slight recovery can be seen by 5 days post treatment. In contrast, Iba-1-positive cells are only partially eliminated, the remaining cells possibly representing a unique non-phagocytic subset of Iba-1-positive macrophages. It should be noted that Iba-1 positive cells in kidney, heart and colon were not affected by clodronate treatment (data not shown) and these tissues do not develop detectable amyloid deposits until ~ 4 weeks post AEF. It also should be noted that huIL-6 mice have higher levels of positive cells for both markers than do wild type mice (data not shown). It is not known if the enhanced number of positive cells in huIL-6 mice represents a specific subpopulation or not.
Figure 1.
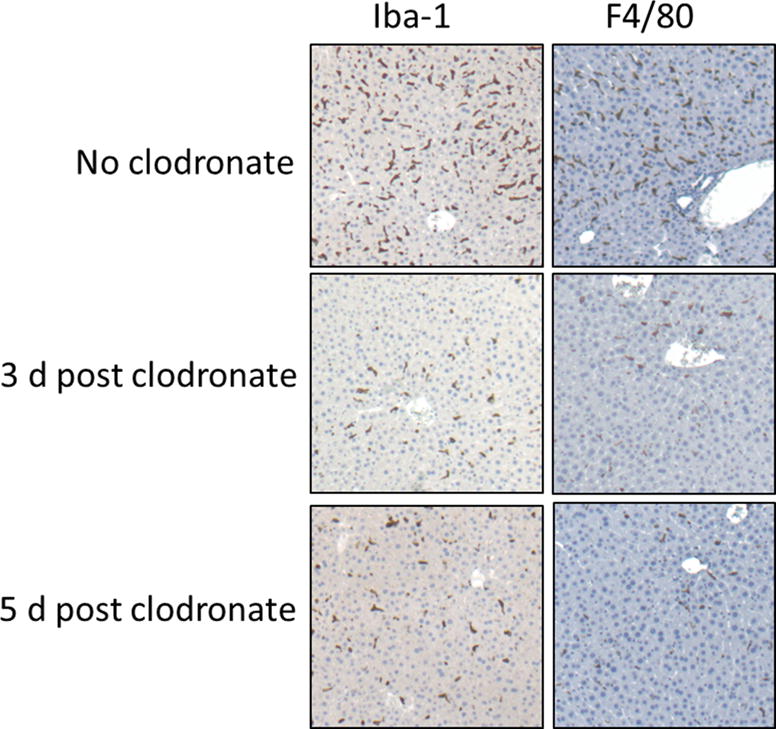
Immunohistochemical staining of liver sections from huIL-6 mice either not treated or treated with clodronate liposomes 3 or 5 d previously. Staining is shown with mAb Iba1 or mAb F4/80 antibodies both of which are markers for phagocytic cells. Original magnification 50×
Figure 2.
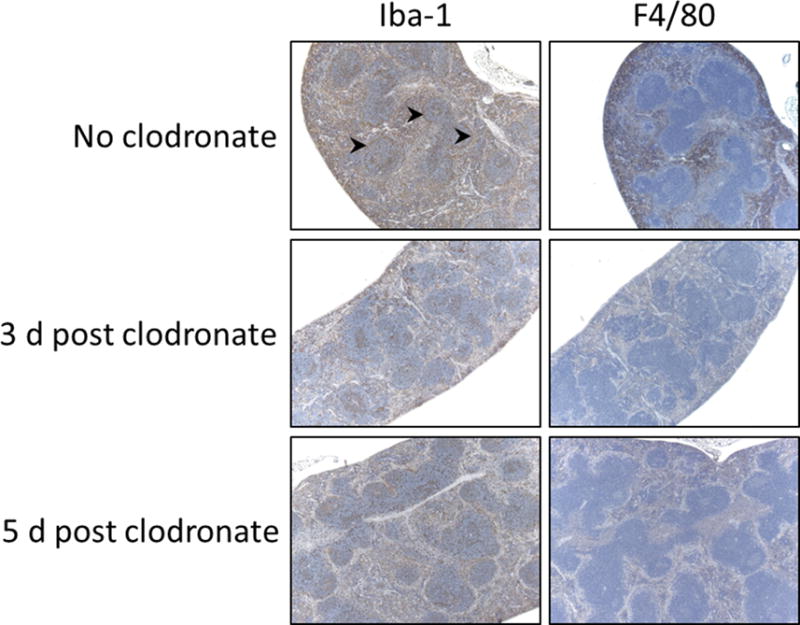
Immunohistochemical staining of spleen tissue sections as done for Fig. 1. Arrowheads point to areas of perifollicular staining in the spleen. Original magnification 50×.
To determine the effect of clodronate liposome treatment of huIL-6 mice on the deposition of AEF, preparations of AEF were radioiodinated with 125I and dialyzed to remove free iodide. When the radiolabeled AEF was dissolved in SDS-PAGE buffer and boiled before loading, the autoradiographic pattern revealed that about half of the radioactive iodide was associated with a band running with SAA mobility (Fig. 3C). There was some material of higher molecular weight and some that did not enter the gel presumably due to fibrils that were not totally dissociated before the loading. There was also about 10% of the material running at the gel front (free 125I−) that apparently was not removed by the dialysis step. Approximately 10 μg 125I AEF (the amount normally used to induce amyloid deposition) was injected into mice treated 24-h earlier with clodronate liposomes. A second group of huIL-6 mice that were not clodronate treated served as controls. Mice were euthanized at either 15 min or 4 h post-injection of 125I-labeled AEF and the biodistribution of radioactivity in the tissues analyzed by microautoradiography and radioactivity counting. Microautoradiography of liver (Fig. 3A) and spleen (Fig. 3B) harvested 15 min post injection showed little difference in the amount or pattern of 125I-labeled AEF deposition whether or not mice had been treated with clodronate liposomes. In contrast, tissues harvested 4 h post-injection showed much less 125I-labeled material in control mice than in those that had been treated with clodronate in both liver (Fig. 3A) and spleen (Fig. 3B). Furthermore, deposition of 125I in clodronate-treated mice at 4 h was similar to the patterns seen in tissues harvested at 15 min post-injection. Data for biodistribution of 125I-AEF in liver and spleen (Table 2) support the microautoradiographic analyses. These data are consistent with the hypothesis that in huIL-6 mice, AEF was internalized in RE cells of the liver and spleen and over the course of a few hours was catabolized or dehalogenated yielding the release of free 125I-iodide. This mechanism is further supported by the fact that the stomach, which contains high concentrations of iodide symporters, accumulated ~ 2x more free 125I-iodide in control mice than in clodronate treated mice (6.3%ID/g vs3.5% ID/g respectively).
Figure 3.
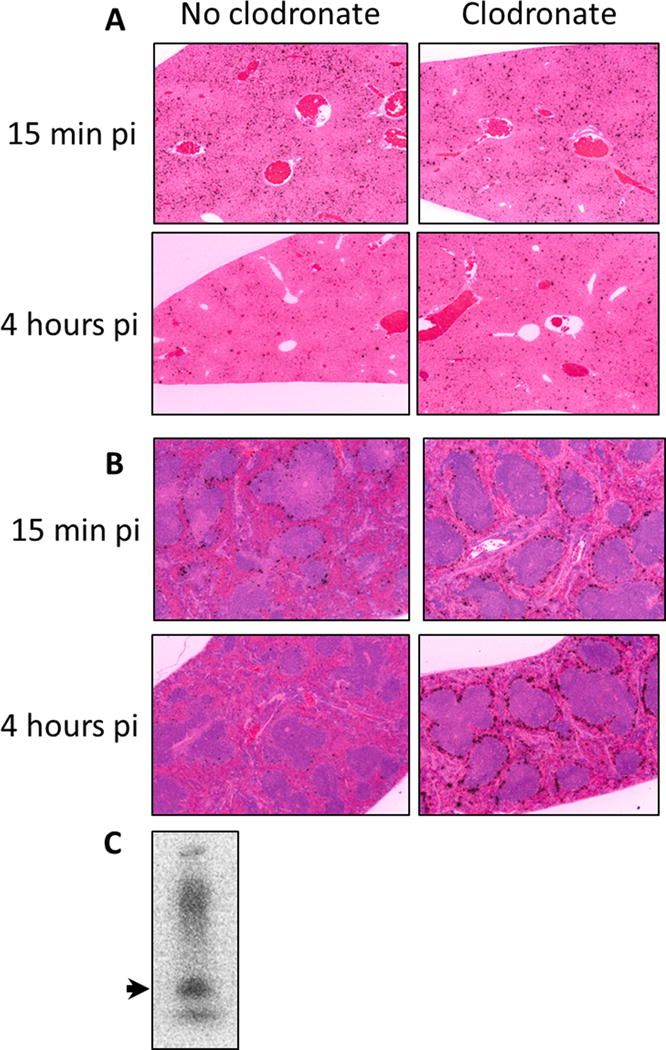
Microautoradiography of A) liver or B) spleen sections from mice injected with 125I-AEF. Tissues harvested either 15 min or 4 h post-injection were fixed, paraffin embedded and sectioned before dipping slides in autoradiographic emulsion. Slides were developed and counter stained with H&E for tissue visualization (Original magnification 50×). Black silver grains designate sites of 125I decays. Panel C shows the autoradiographic pattern of radioiodinated AEF profile on SDS-PAGE. The arrow points to the mobility of SAA.
Table 2.
* Biodistribution (%ID/g) of 125I-AEF in huIL6 mice with or without clodronate liposome treatment
| 15 min post injection | |||||
|---|---|---|---|---|---|
| Spleen | Liver | ||||
| Clodronate + | Control | t-test | Clodronate + | Control | t-test |
| 8.3 ± 3.1 | 6.4 ± 0.6 | p = 0.51 | 17.8 ± 3.5 | 19.5 ± 1.4 | p = 0.48 |
| 4 h post injection | |||||
| Spleen | Liver | ||||
| Clodronate + | Control | t-test | Clodronate + | Control | t-test |
| 18.1 ± 11.5 | 2.4 ± 0.5 | p = 0.014 | 11.4 ± 2.9 | 6.3 ± 1.1 | p = 0.006 |
huIL6 mice, either clodronate treated or controls (day −1), were injected with 125I AEF (see Figure 3) and at the designated times, animals (n=3/group for 15 min and N=5/group for 4 h) were euthanized and analyzed for % injected dose/gram of wet tissue of 125I content ± standard deviation.
For accurate analyses of amyloid load in mice that had been treated or not treated with clodronate and to determine the effect of clodronate on the amount of perlecan detected in amyloid deposits, huIL6 mice that had been injected with AEF for 4 weeks prior were treated with clodronate liposomes or control vehicle. Mice were sacrificed three days later and tissues stained for expression of Iba positive cells, perlecan (two different antibodies), Congo red staining and P5R deposition. A panel of images for spleen, liver and kidney for a mouse from each group, chosen so that they could be readily compared due to their similar Congo red staining (Fig. 4). The column 1 panels indicate that P5R deposition was similar in liver, spleen and kidney papilla whether or not animals had been treated with clodronate liposomes. The second column of panels documents that Congo red staining for these tissues was also similar. This is to be expected since mice had extensive amyloid at the time of clodronate treatment and the mice were sacrificed only 3 days post treatment. These data indicate that amyloid detection by PR5 was not significantly affected by prior clodronate treatment. The third column of panels shows that Iba-1 positive cells were greatly reduced in liver and spleen in the clodronate treated mice. Very few Iba-1 positive cells were noted in kidney papilla of either treated or untreated animals. Panels in columns 4 and 5 show staining for perlecan using 2 different anti-perlecan antibodies. Perlecan staining as detected by either antibody was much reduced in liver and spleen of clodronate treated animals while that in kidney papilla was relatively unaffected. These data support the contention that perlecan in the amyloid deposits is associated with Iba-1 positive cells.
Figure 4.
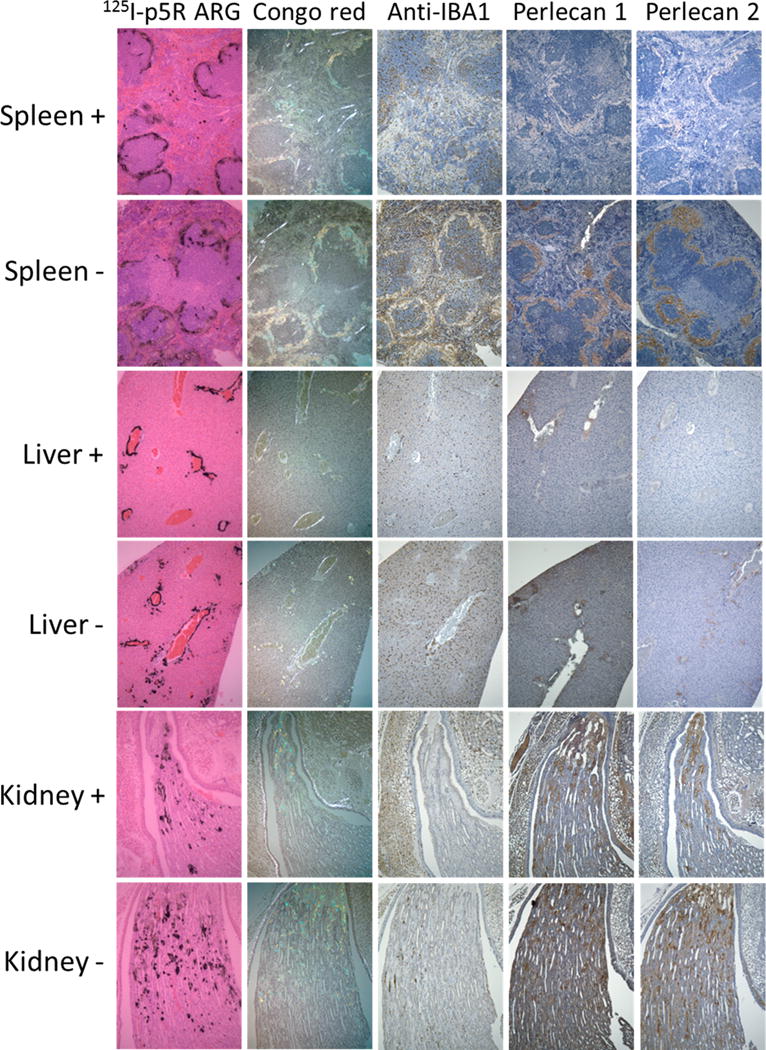
Stained sections from huIL-6 mice that had been given AEF 4 weeks before sacrifice and were either treated with clodronate (+) or not treated (−) 3 days prior to sacrifice. Sections from liver, spleen and kidney papilla are included. Column 1 images are autoradiography of sections harvested 2 h post iv injection with 125I p5R. Column 2 figures show sections stained with Congo red in birefringence microscopy. Column 3 shows sections stained with iba-1 antibody. Column 4 shows sections stained with rat anti perlecan Mab A7L6 and column 5 shows sections stained with rat Mab HK-102. Original magnification 100×.
A series of experiments was performed to assess the effect of clodronate liposome treatment on the accumulation of AA amyloid in huIL-6 mice. The burden of AA amyloid was measured by visual scoring of Congo red stained tissues, by visual scoring of 125I-p5R peptide deposition in autoradiography tissue sections, and finally by the quantitative evaluation of biodistribution of 125I-p5R in tissue 2 h post-injection of peptide.
The first set of experiments examined amyloid deposition in three groups of mice. Group A mice were treated with clodronate 1 day after AEF injection, group B were treated with clodronate 1 day before AEF treatment and group C received no clodronate. Two weeks after AEF administration, mice were euthanized for analyses of AA deposition as described above. Previous work with this model has indicated that at 2 weeks post-AEF, mice are in a rapid phase of amyloid accumulation. Generally, at this time point >90% of the mice have developed amyloid in the spleen and liver; however the extent of the deposits can be quite variable. Two weeks post-AEF is also the most sensitive time point to assess the efficacy of anti-AA amyloid treatments (see Table 1). Two independent experiments with 4 or 5 mice per group were performed ~ 3 months apart and the data pooled for analyses. Data for Congo red scores and 125I-p5R visual scores are shown in Table 3 and biodistribution data in presented in Fig. 5. ANOVA analyses of these data determined there was no significance of main effect for P5R biodistribution data (Fig 5), liver, p = 0.93; spleen p = 0.54 or p5R scoring data (Table 3), liver. p = 0.66; spleen, p = 0.60. For the Congo red scoring (Table 3), both data sets for liver and spleen violated the test of homogeneity and Kruskal Wallis Tests were done. For liver, this test gave a non-significant main effect p = 0.225; however, for spleen the Kruskal Wallis test was significant, p = 0.03. A follow up Mann-Whitney U test revealed that the data for B versus C groups was significant p = 0.011. Thus, splenic amyloid as assessed by Congo red staining showed a significant difference between groups B and C, but the result was not supported either by 125I-p5R scoring or biodistribution analyses (Fig. 5).
Table 3.
* Effect of clodronate liposome treatment day −1 or +1 or not treated on amyloid accumulation at 2 wk post AEF.
| Congo red quantitation (0–4) | |||
|---|---|---|---|
| AEF after clodronate (A) | AEF before clodronate (B) | No clodronate (C) | |
| Liver | 0.67 ± 0.81 | 0.59 ± 0.59 | 0.24 ± 0.19 |
| Spleen | 0.39 ± 0.63 | 0.32 ± 0.18** | 0.12 ± 0.05** |
| 125I-p5R autoradiograph scoring (0–4) | |||
| Liver | 1.13 ± 0.78 | 1.14 ± 0.56 | 0.87 ± 0.78 |
| Spleen | 0.72 ± 0.62 | 1.08 ± 0.56 | 0.73 ± 0.69 |
huIL-6 mice (n=9/ group) were injected with 10 μg of AEF (day 0) and also were treated with clodronate liposomes either A) the day before AEF (day −1) or B) the day after AEF (day +1) or C) no clodronate treatment.
At 2 weeks post AEF injection, mice were injected with 125I P5R and tissues were harvested 2 h post injection. Congo red scoring or p5R scoring of autoradiographs were done. In addition animal tissues were evaluated for 125I P5R (see Figure 5).
B vs C, p= 0.011
Figure 5.
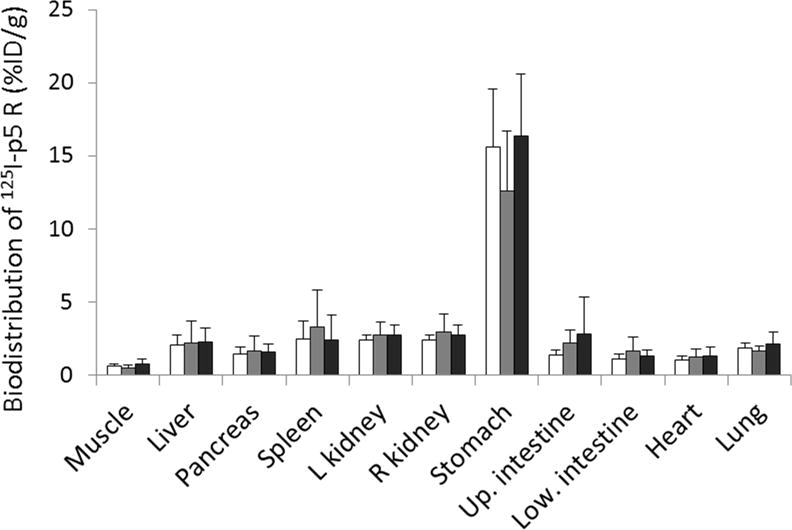
Biodistribution of amyloid detection peptide p5R radiolabeled with 125I. Animals (n=9 per group pooled from 2 separate experiments) were injected with clodronate liposomes at day −1 (white bars), day +1 (gray bars) or no clodronate injected (black bars) relative to AEF injection at day 0. At 2 weeks post-AEF, mice were injected with 125I-p5R and euthanized 2 h later. Data are shown as % injected dose/gram of wet tissue ± SD bars. There were no significant differences in biodistribution (ANOVA analyses) among the 3 groups for liver or spleen.
The second set of experiments was performed to compare amyloid load in mice treated with clodronate 5 days post-AEF. This time was chosen to assess the role of phagocytic cells on the rapid amyloid deposition that occurred early in the disease course (see Table 1). The same measures of AA deposition were employed as were used in the first experiments. Mice were euthanized either 2 weeks or 6 weeks post-AEF. The experiment with 2 week sacrifice was repeated ~3 months later and the data for the two experiments pooled. The 6 week data was for a single experiment with n=5. Data for Congo red scoring and 125I-p5R scoring are shown in Table 4. Data for both of these methods of evaluation showed that there were statistically significant differences for amyloid deposition in the liver and spleens of mice euthanized 2 weeks post-AEF. By 6 weeks post-AEF, the Congo red scoring showed significant differences, but these values were not supported by 125I-p5R scoring or 125I-p5R biodistribution values (see Fig 6). To support these analyses, biodistibution measurements for 125I-p5R were compared (Fig 6). The biodistribution data support the Congo red and 125I-p5R scoring data in that significant reduction of amyloid detected at 2 weeks post-AEF was noted in the 2 week analyses; however by 6 weeks, there was no significant difference in amyloid load detected by 125I-p5R in the clodronate-treated versus untreated groups.
Table 4.
* Accumulation of amyloid at 2 weeks or 6 weeks post-AEF in mice treated with clodronate liposomes.
| Congo red quantitation (0–4) | ||||||
|---|---|---|---|---|---|---|
| 2 wk post AEF | 6 wk post AEF | |||||
| Clodronate | Control | t-test | Clodronate | Control | t-test | |
| Liver | 0.24 ± 0.29 | 1.13 ± 0.77 | p = 0.007 | 2.1 ± 0.22 | 2.6 ± 0.25 | p=0.013 |
| Spleen | 0.38 ± 0.59 | 1.43 ± 0.88 | p = 0.006 | 2.5 ± 0.35 | 3.0 ± 0.0 | p = 0.027 |
| 125I-p5R autoradiograph scoring (0–4) | ||||||
| Liver | 0.59 ± 0.49 | 1.61 ± 0.98 | p = 0.16 | 3.2 ± 0.27 | 3.5±0.0 | p=0.074 |
| Spleen | 0.53 ± 0.43 | 1.77 ± 0.78 | p = 0.002 | 1.75 ± 0.66 | 1.62 ± 0.25 | p = 0.73 |
huIL-6 mice were injected with 10μg AEF and treated with clodronate liposomes (day 5) or not treated. After 2 weeks, mice were injected iv with 125I p5R and tissues were harvested 2 h later. Tissues were scored for Congo red stain or for 125I p5R autoradiography. Tissues were also evaluated for biodistribution (see figure 6). Values are 0–4+ scoring ± standard deviation. N=10 for 2 week and n=5 for 6 week groups.
Figure 6.
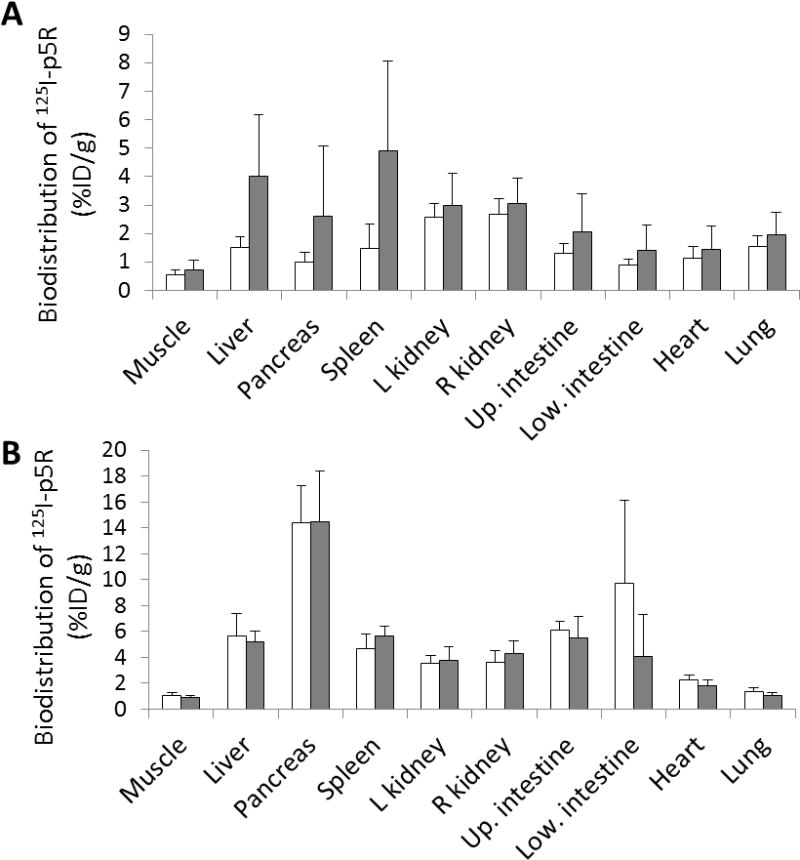
Biodistribution of 125I-p5R as for Figure 5 except that mice were not treated (white bars) or treated (gray bars) with clodronate liposomes 5 d after AEF and euthanized 2 weeks (n=10, top panel) or 6 weeks (n=5, bottom panel) post-AEF. Data are shown as % injected dose/gram of wet tissue ± SD For the 2 week data (top panel) there was a significant difference in 125I-p5R accumulation in liver and spleen (p=0.007 and 0.006, respectively). In contrast at 6 weeks post-AEF injection there was no significant difference in 125I-p5R accumulation in either liver or spleen (p= 0.646 and 0.201, respectively).
Discussion
Macrophages have many functions, and their involvement in immune functions has been reviewed recently [9]. There have also been many reports documenting the effect of clodronate liposomes on various macrophage functions [13]. The effect of clodronate liposomes is likely greatest in those phagocytic cells exposed to the circulation or in areas with relatively permeable endothelium. The effect is also thought to be directed to cells with phagocytic activity since the liposomes do not release the toxic clodronate unless they are engulfed and lysed within a cell. We and others have shown that toxic effects of the clodronate are transient, disappearing 2 weeks after treatment [14]. It is possible that this time frame varies with different phagocyte functions, but we have not tested this hypothesis. Our data indicate that some Iba-1-positive, and the majority of F4/80-positive cells in the spleen and liver were absent after clodronate liposome treatment and that the loss of staining persisted for 3–5 days at which time some recovery was observed. With the loss of these phagocytic cells we expected a difference in the capturing and deposition of AEF which is particulate. Analyses at 15 min post injection, when most particulate material has been cleared from the circulation showed no difference in amount or pattern of 125I AEF (Fig 3, Table 1). The experiment was repeated and mice analyzed at 4 h post injection. At this time point the clodronate-treated mice retained the original pattern of 125I microautoradiography. The loss of radiohalides, such as iodide from radiotracers has been documented previously [18]. Our work with peptide-bound 125I indicated that dehalogenation occurs relatively rapidly and is nearly complete by 4 h post injection [17]. The apparent loss of 125I-AEF in the untreated mice is thus likely due to dehalogenation and subsequent loss of 125I from the deposited AEF. The fact that 125I-AEF is sequestered in the marginal zone of the spleen but is protected from dehalogenation in the clodronate treated animals suggests that the material binds to extracellular receptors of cells or debris in the spleen but the material is not internalized (Note that dehalogenation is deemed to occur only intracellularly where the enzyme that mediates this effect resides). Whether there was a difference in AEF processing in the clodronate treated vs non-treated mice, the AEF remained active and induced disease in both groups.
The patterns of AEF deposition in liver and spleen correspond with the known distribution of phagocytic cells in these organs, notably Kupfer cells in liver and perifollicular (marginal zone) phagocytes in spleen. It is interesting that amyloid deposition in spleen coincides with the distribution of the marginal zone phagocytes while that in the liver differs significantly (see Figs. 1 and 3, and ref 16). It is possible that there are two different kinds of phagocytes in liver and only those near and within portal triads actually support amyloid accumulation. In the silver nitrate model, liver deposition was noted first within the central veins and throughout the parenchyma. In addition, AA deposits (and HSPGs) were found in some but not all of Kupfer cell areas of the liver [19]. Another possibility is that in the IL-6 model AEF is only active in spleen and that depositions in liver and other organs is the result of release of amyloidogenic fragments from the spleen which seeded other organs. This latter hypothesis is supported by the fact that in the IL-6 model, only mice with splenic AA develop hepatic amyloid and that disease in the liver is always temporally secondary to the appearance of splenic AA in this model. However, it has been shown that in the silver nitrate model, even splenectomized mice develop liver AA deposits [3].
A series of control experiments were conducted to demonstrate that the earliest time post-AEF injection that amyloid could be detected and quantitated was about 2 weeks post AEF (Table 1). These experiments were conducted previously when we were using the radioiodinated peptide P5 for amyloid detection. For subsequent experiments we have used radioiodinated p5R peptide; however, published data [16] indicate that at least for early time points with light amyloid load, the two peptides yield similar results. Although AA amyloid likely begins to accumulate shortly after injection of AEF, in the huIL-6 model, quantitation of the amounts of AA were most consistent when done 2 weeks post injection and the earliest times examined were at 1 week post injection (data not shown). A second control experiment was done to show that in mice bearing amyloid deposits, Congo red staining and P5R deposition gave similar result regardless of the clodronate treatment. In these control experiments, we also demonstrated that perlecan staining in liver and spleen correlated with iba-1 (macrophage) staining in these tissues.
To investigate the effect of clodronate treatment on amyloid deposition in animals injected 2 weeks earlier with AEF, mice were treated one day before or after AEF injection. The amount of amyloid deposited in the liver and spleen evaluated 2 weeks after AEF showed no significant differences among the groups (Fig. 5 and table 3). This result was unexpected since we anticipated that the clodronate effect would be persistent for a large fraction of the 2 week period. These data suggest that both the sequestration of circulating AEF (as a seed) and the initial stages of AA amyloidogenesis do not require the presence of phagocytic macrophages. It is possible that there is significant circulating processed SAA that is capable of fibril formation when AEF is available. Alternatively, in the huIL-6 mice it is possible that circulating HDL-free SAA is available for fibril elongation. Arai et al. have shown, by using anti-SAA and anti-AA antibodies that deposits of murine AA contain full-length SAA at the surface of the phagocytic cells [20], but it is not known whether this mechanism represents the dominant method of fibril deposition/elongation. It is also possible that clodronate treatment was largely reversed by the time the fast phase of amyloid deposition was occurring, or that the particular function of macrophages required for amyloid accumulation has a different time frame of expression.
We have observed that AEF-induced huIL-6 mice are in a very fast phase of amyloid accumulation for the first few weeks (see Table 1). We chose 5 d for clodronate treatment to induce macrophage “paralysis” at early periods of this critical time. Data in Fig. 6 and Table 4 show that amyloid deposition in day 5 clodronate-treated mice was significantly reduced in the liver and spleen at 2 weeks post-AEF induction. Since the clodronate effect lasts for between 5 d and 2 weeks, it is likely that animals had little phagocyte function during this period. There could be two competing processes occurring: amyloid accumulation and amyloid removal. Both processes are thought to involve macrophages [6–8]. Experience with the huIL-6 model indicates that accumulation dominates in this competition, since once induced by AEF injection, huIL-6 mice never survive. Thus, these data suggest that the clodronate effect is mediated by inhibition of the deposition process of the disease. This is consistent with other work demonstrating that SAA is internalized and processed by phagocytic cells becoming more fibrillogenic [6] and we hypothesize that this is the main function inhibited by clodronate treatment during this time of AA amyloidogenesis. Since heparan sulfate has been implicated in the deposition process, it may be that the SAA HDL particulate associated with heparan sulfate forms a structure stimulating opsonization of the particle for SAA processing [6]. This could also be a point at which clodronate liposomes inhibit amyloid deposition. Indeed, our data in Fig 3 indicate that perlecan is much reduced in established amyloid (4 weeks post AEF) of clodronate treated mice. Regardless of the details of the mechanism, the implication is that if one could intervene in macrophage function at this stage of the disease in humans, significant reduction in amyloid deposition might occur and recovery from the disease may be faster and more complete.
Mice treated 5 d post-AEF induction and analyzed 6 weeks later showed no significant difference in amyloid load as detected by 125I-p5R peptide (Fig 6 and Table 4); however, there was some significant difference in Congo red scoring (Table 4). Although there is some suggestion of significant difference in amyloid load, it is likely that the relentless amyloid deposition process in these mice is so rapid that most of the difference that existed at 2 weeks post-AEF was overwhelmed. It is possible that multiple clodronate treatments given at the correct times during the disease process might maintain the animals in a reduced state of amyloid accumulation; however, in the huIL-6 model, the production of IL-6 and therefore SAA is incessant and progression of the disease is auto-catalytic such that the mice will ultimately succumb. It may be possible, in human AA disease such as associated with Familial Mediterranean Fever [21, 22], strategically timed treatments could totally abrogate amyloid deposition during the critical phase of the disease. It should be noted that clodronate liposomes have been used to treat human disease and thus translation to human treatment is a possibility. During the review of this paper, a study was published examining the effect of clodronate liposome injection on AA deposition and different splenic macrophage populations using the silver nitrate model [23]. This work demonstrated that indeed different macrophage populations in the spleen respond differently to clodronate treatment and that clodronate treatment in the silver nitrate model retarded AA development, but did not eliminate it.
Conclusion
Timely treatment of progressive AA amyloid deposition in huIL-6 mice with clodronate liposomes resulted in inhibition of amyloid deposition by inducing temporary paralysis of a subset of phagocytic macrophages. The effect was likely do to inhibition of SAA processing to AA in these cells. The effect was transient and any curative treatment must be well timed and likely delivered in multiple doses.
Acknowledgments
This work was supported by the University of Tennessee Graduate School of Medicine through the Molecular Imaging and Translational Research Program and by Award number R01DK079984 from the National Institute of Diabetes and Digestive and Kidney Diseases.
Abbreviations
- SAA
serum amyloid A protein
- AEF
amyloid enhancing factor
- PBS
phosphate buffered saline
Footnotes
Declaration of Interest section
Authors SJK and JSW are inventors on a pending patent application associated with the use of the peptide P5R for amyloid detection and imaging.
References
- 1.Blancas-Mejia LM, Ramirez-Alvarado M. Systemic Amyloidoses. Ann Rev Biochem. 2013;82:745–774. doi: 10.1146/annurev-biochem-072611-130030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Axelrad MA, Kisilevsky R, Willmer J, Chen SJ, Skinner M. Kinetics of amyloid deposition. 1. The effects of amyloid-enhancing factor. Lab Invest. 1982;47:139–146. [PubMed] [Google Scholar]
- 3.Kisilevsky R, Boudreau L. Kinetics of amyloid deposition. 1. The effects of amyloid-enhancing factor and splenectomy. Lab Invest. 1983;48:53–59. [PubMed] [Google Scholar]
- 4.Wall JS, Kennel SJ, Paulus MJ, Gleason S, Gregor J, Baba J, Schell M, Richey T, O’Nullian B, Donnell R, Hawkins PN, Weiss DT, Solomon A. Quantitative high-resolution microradiographic imaging of amyloid deposits in a novel murine model of AA amyloidosis. Amyloid. 2005;12(3):149–56. doi: 10.1080/13506120500222359. [DOI] [PubMed] [Google Scholar]
- 5.Solomon A, Weiss DT, Schell M, Hrncic R, Murphy CL, et al. Transgenic mouse model of AA amyloidosis. Am J Pathol. 1999;154:1267–1272. doi: 10.1016/S0002-9440(10)65378-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elimova E, Kisilevsky R, Ancin JB. Heparan sulfate promotes the aggregation of HDL-associated serum amyloid A: evidence for a proamyloidogenic histidine molecular switch. FASEB J. 2009;23:3436–3448. doi: 10.1096/fj.09-134981. [DOI] [PubMed] [Google Scholar]
- 7.Nystrom SN, Westermark GT. AA-amyloid is cleared by endogenous immunological mechanisms. Amyloid. 2012;19:138–145. doi: 10.3109/13506129.2012.711391. [DOI] [PubMed] [Google Scholar]
- 8.Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, Hutchinson WL, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature. 2010;469:93–97. doi: 10.1038/nature09494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wynn TA, Chawla A, Pollard JW. Marcophage biology in development, homeostasis and disease. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol. 1981;10:805–15. doi: 10.1002/eji.1830111013. [DOI] [PubMed] [Google Scholar]
- 11.Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S. Biochem biophys Res Comm. 1996;224:855. doi: 10.1006/bbrc.1996.1112. [DOI] [PubMed] [Google Scholar]
- 12.Pinto AJ, Stewart D, van Rooijen N, Morahan PS. Selective depletion of liver and splenic macrophages using liposomes encapsulating the drug dichloromethylene diphosphonate: effects on antimicrobial resistance. J Leukoc Biol. 1991;49:579–86. doi: 10.1002/jlb.49.6.579. [DOI] [PubMed] [Google Scholar]
- 13.Van Rooijen N, van Kesteren-Hendrikx E. Clodronate liposomes: perspectives in research and therapeutics. J Liposome Res. 2002;12:81–94. doi: 10.1081/lpr-120004780. [DOI] [PubMed] [Google Scholar]
- 14.Kennel SJ, Woodward JD, Rondinone AJ, Wall J, Huang Y, Mirzadeh S. The fate of MAB-targeted Cd125mTe/ZnS nanoparticles in vivo. Nuc Med Biol. 2008;35:501–514. doi: 10.1016/j.nucmedbio.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Pras M, Schubert M, Zucker-Franklin D, Rimon A, Franklin EC. J Clin Invest. 1968;47:924–933. doi: 10.1172/JCI105784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wall JS, Williams A, Richey T, Stuckey A, Huang Y, Wooliver C, Macy S, Heidel E, et al. A binding-site barrier affects imaging efficiency of high affinity amyloid-reactive peptide radiotracers in vivo. PLOSone. 2013;8:e66181. doi: 10.1371/journal.pone.0066181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wall JS, Richey T, Stuckey A, Donnell R, Macy S, Martin EB, Williams A, Higuchi K, Kennel SJ. In vivo molecular imaging of peripheral amyloidosis using heparin-binding peptides. PNAS. 2011;108:E586–E594. doi: 10.1073/pnas.1103247108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hagan PL, Halpern SE, Chen A, Krishnan L, Frinke J, Bartholomew RM, David GS, Carlo D. In vivo kinetics of radiolabeled monoclonal anti-CEA antibodies in animal models. J Nuc Med. 1985;26:1418–23. [PubMed] [Google Scholar]
- 19.Snow AD, Bramson R, Mar H, Wight TN, Kisilevsky R. A temporal and ultrastructural relationship between heparin sulfate proteoglycans and AA amyloid in experimental amyloidosis. J Histochem Cytochem. 1991;39:1321–1330. doi: 10.1177/39.10.1940305. [DOI] [PubMed] [Google Scholar]
- 20.Arai K, Miura K, Baba S, Shirasawa H. Transformation from SAA2-fibrils to AA-fibrils in amyloid fibrillogenesis: in vivo observations in murine spleen using anti-SAA and anti-AA antibodies. J Pathol. 1994;173(2):127–134. doi: 10.1002/path.1711730209. [DOI] [PubMed] [Google Scholar]
- 21.Akar S, Yuksel F, Tunca M, Soysal O, Solmaz D, Gerdan V, Celik A, Sen G, Onen F, Akkoc N. Famalial Mediterranean fever:risk factors, causes of death and prognosis in the cholchicine era. Medicine (Baltimore) 2012;91:131–6. doi: 10.1097/MD.0b013e3182561a45. [DOI] [PubMed] [Google Scholar]
- 22.Ozturk MA, Kanbay M, Kasapoglu B, Onat AM, Guz G, Furst DE, Ben-Chetrit E. Therapeutic approach to familial Mediterranean fever: a review update. Clin Exp Rheumatol. 2011;29:s77–86. [PubMed] [Google Scholar]
- 23.Lundmark K, Shariapanahi AV, Westermark GT. Depletion of spleen macrophages delays AA amyloid development: A study performed in the rapid mouse model of AA amyloidosis. PLoS ONE. 2013:8e79104. doi: 10.1371/journal.pone.0079104. doi: 10.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]