

Antimicrobial peptides (AMPs) are naturally occurring antibiotics found in essentially all living organisms [1]. In the past two decades, AMPs have attracted considerable interest because of their potential therapeutic use as anti-infective agents [1–2]. There exists, however, a number of serious challenges preventing AMP from reaching a pharmaceutical market including their rapid in vivo degradation, high production costs, and reduced activity in physiological conditions [2]. Efforts to overcome these problems while retaining the peptides’ natural anti-infective properties resulted in the emergence of a rapidly expanding field of non-natural mimics of antimicrobial peptides.
Good understanding of the structure-activity relationships in AMPs is essential in the effort to create a successful peptidomimetic compound. It has been proposed that AMPs kill pathogens by disrupting the cell membrane, or by invading the cytoplasm and inhibiting core metabolic functions [1a, 3]. In both cases the pathogen’s membrane plays a crucial role either as an immediate target or as a barrier that must be traversed. Despite the immense diversity of AMPs discovered, they commonly have a net cationic charge and tend to adopt highly amphiphilic topologies in which the hydrophilic and hydrophobic side chains segregate into opposing regions of the molecule [1b]. The overall positive charge ensures the initial attraction of AMPs to anionic bacterial membranes and imparts some measure of selectivity, since mammalian cell membranes are largely zwitterionic. An amphipathic organization is supposedly essential at latter stages of membrane permeabilization [1]. Although the antimicrobial activity of AMPs is often attributed to their ability to fold upon contact with microbial surfaces [1b], it has been recently shown that conformational preorganization is not obligatory [4]. The question remains, then: what is the effect of structural flexibility on the antimicrobials’ mode of action? Herein, we show that the flexibility of antimicrobial compounds may aid in the penetration of bacterial cell wall barrier which poses a serious challenge for conformationally rigid antimicrobials. As a result, the mode of interaction of conformationally flexible and restrained antimicrobials with bacterial membrane lipids is different. In addition, it is demonstrated that conformational rigidity is not required for the membrane disruptive activity of compounds tested.
In order to understand the role of this design variable better, we have undertaken a detailed structural study of the interactions of two non-natural antimicrobials, acyl-lysyl octamer C12K-7α8 [4b] and arylamide foldamer [5] (hereafter termed OAK-1 and AA-1 respectively) with model bacterial membranes. We used lipid A and 1,2-dipalmitoyl-sn-glycero-3-phosphoglycerol (DPPG) (Scheme 1) monolayers (LMs) at the air-liquid interface while utilizing highly sensitive synchrotron X-ray scattering methods. OAK-1 has been previously shown to remain unstructured both in buffer and in the presence of charged liposomes [4b]. In contrast, the structure of AA-1 contains two 1,3-phenylene diamine units connected by a 4,6-dialkoxy-substituted isophthalic acid that ensures the formation of a hydrogen bond network and consequently compact, rigid molecular geometry [5].
Scheme 1.

Structures of the bacterial membrane components DPPG and lipid A, antimicrobials OAK-1 and AA-1 (dotted lines denote hydrogen bonds).
In this approach, LMs represent the external leaflet of the bacterial membranes, while OAK-1 or AA-1 are introduced into the subphase of a Langmuir trough to comprise the extracellular fluid and thus mimic the approach of the antimicrobials toward the bacterial surface. The rationale behind this choice of lipids is following: lipid A is the hydrophobic anchor of lipopolysaccharide (LPS) that makes up the external leaflet of the outer membrane of most Gram-negative bacteria, while PG is the dominant anionic phospholipid of the bacterial cytoplasmic membranes.
Results for OAK-1 and AA-1 were compared with previous X-ray studies of the interactions of natural AMPs LL-37 [6] and Protegrin-1 (PG-1) [7] with DPPG and lipid A monolayers. These peptides are conformationally rigid and adopt α-helical and β-sheet conformations respectively. It should be emphasized here that we did not compare activities of these antimicrobials against either DPPG or lipid A directly, since the concentrations of the antimicrobials used were different. Instead, we aimed to establish whether diverse antimicrobials exert similar membrane disruptive and penetration activities against diffferent anionic bacterial lipids.
Membrane penetration activities of OAK-1 (1.3 μM) and AA-1 (9 μM) were probed by measuring the relative change in area per lipid molecule, ΔA/A, at a constant surface pressure of 30 mN/m (see Supporting Information). Figure 1a shows that OAK-1 and AA-1 readily incorporate into the anionic lipid monolayers inducing ΔA/A increase of 38±2% and 60±4% in both films, respectively. These insertion trends of OAK-1 and AA-1 are in line with membrane penetration activities of LL-37 and PG-1 [6a, 6b, 7a].
Figure 1.

Membrane-penetration activity of OAK-1 and AA-1. a) Relative change in area per molecule of DPPG or lipid A after injection of OAK-1 (1.3 μM) and AA-1 (9μM). b) and c) X-ray reflectivity data (symbols) and corresponding fits (lines) normalized by Fresnel reflectivity plotted against scattering vector qZ. b) DPPG monolayer before (squares) and after OAK-1 (inverted triangles) or AA-1 (rhombs) injection; c) lipid A monolayer before (squares) and after OAK-1 (inverted triangles) or AA-1 (rhombs) injection. d) and e) the electron density distribution in the anionic lipid monolayers perpendicular to the aqueous interface following OAK-1 or AA-1 injection. For clarity b) and c) data are shifted vertically.
The impact of OAK-1 and AA-1 on the molecular structure of the LMs was assessed with grazing incidence X-ray diffraction (GIXD) [8]. GIXD measurements provide a direct structural information on the lateral molecular organization in the LMs at the sub-nanometer scale. Our diffraction data reveal that both OAK-1 and AA-1 exert strong membrane disruptive activity, albeit in a different manner. The diffraction pattern of the DPPG monolayer yields two first-order Bragg peaks (Figure 2a), indicative of the centered rectangular unit cell with dimensions a = 5.53 Å, b = 8.57 Å, γ = 90° and area Auc = 47.4 Å2, which is in good agreement with previous reports [6a, 7a]. The GIXD diffraction pattern of the lipid A monolayer at 30 mN/m yields a single Bragg peak (Figure 2b), indicative of the hexagonal lattice with parameters a = b = 5.06 Å, γ = 120° and area Auc = 22.2 Å2. A hexagonal packing mode was observed, as anticipated for a molecule with a truncated cone shape like lipid A [9] (Scheme 1), and agrees well with previously published data [6b].
Figure 2.
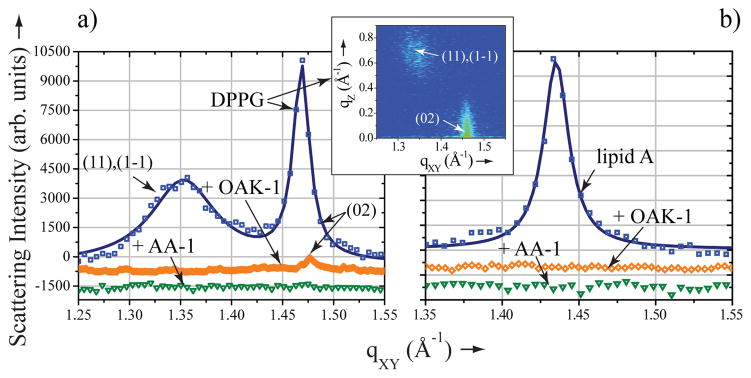
Membrane disruptive activity of OAK-1 and AA-1. Grazing incidence X-ray diffraction data (symbols) and corresponding fits (lines):scattering intensity, integrated over qZ range, against scattering vector qXY of a) DPPG monolayer before (squares) and after OAK-1 (rhombs) or AA-1 (inverted triangles) injection; insert) 2D contour map of Bragg peaks (qXY) against Bragg rods (qZ) of DPPG; b) lipid A monolayer before (squares) and after OAK-1 (rhombs) or AA-1 (inverted triangles) injection.
The introduction of AA-1 into the subphase underneath either anionic lipid monolayer leads to complete disappearance of the diffraction peaks, thus inducing a disordered phase (Figure 2a,b). When OAK-1 is introduced underneath the lipid A monolayer, the diffraction peak also dissapears (Figure 2b), implying a complete disruption of the molecular order within the film. However, the X-ray diffraction pattern of DPPG after the injection of OAK-1 still indicates presence of the ordered lipid structure yielding a single weak Bragg peak with intensity 5 to 10 fold lower than that obtained prior to peptide injection (Figure 2a). Such decrease in intensity of the scattered X-rays could be observed when liquid-ordered domains occupy notably smaller portion of the DPPG monolayer surface area. In addition, a significant Bragg peak widening, indicative of the coherence length decrease from 298 Å to less than 100 Å, implies that the size of the liquid-ordered domains decrease considerably.
Interestingly, neither LL-37 nor PG-1 natural AMPs can disrupt the molecular order of lipid A [6a, 6b, 7a]. Nevertheless, all structured antimicrobials tested induced complete deterioration of the ordered structure in DPPG monolayer [6a, 6b, 7a]. Thus, GIXD results provide evidence that at least two out of three conformationally rigid antimicrobials exert stronger membrane disruptive activity against DPPG as compared to lipid A. Remarkably, OAK-1 exhibits the opposite trend disintegrating the structure of lipid A more efficiently.
X-ray reflectivity (XR) was used to determine the ability of peptide mimics to penetrate into the hydrophobic core of the lipid film. [8]. Analysis of the XR data yields the electron density profile ρ(z) across the film perpendicular to the interface. The measurements were conducted before and after introduction of antimicrobial into the system. Pure lipids measurements showed the acyl chains region of 16.9 Å and 15.2 Å thick for the DPPG and lipid A, respectively, while the corresponding average electron densities ρ were 0.318 e−/Å3 and 0.317 e−/Å3.
Figures 1b and 1c reveal that introduction of either OAK-1 or AA-1 into the system leads to profound changes in the reflectivity profiles of the anionic lipids. Results of lipid/antimicrobial systems XR analysis are summarized in Table 1 and detailed in Supporting Information. After injection of AA-1, the electron density of the acyl chain region (ρac) of DPPG is considerably higher than that of lipid A (Figure 1e). Our XR results allow us to estimate a contribution from AA-1 to ρac, which comprise 2.95 e−/Å2 for DPPG and no contribution for lipid A. Thus, AA-1 can efficiently penetrate into the hydrocarbon region of DPPG, but resides in the headgroup region of lipid A. Neville et al have previously shown using XR that neither LL-37 nor PG-1 can penetrate into the hydrophobic core of the lipid A, but both AMPs span the DPPG monolayer completely [6a, 6b, 7a].
Table 1.
X-ray reflectivity data fitting parameters for the hydrocarbon region of DPPG and lipid A monolayers after injection of the antimicrobials.
| Experiment | Lac, Å[a] | ρac, e−/Å3[a] | Laa, Å[b] | ρaa, e−/Å3[b] |
|---|---|---|---|---|
| DPPG/OAK-1 | 6.3 | 0.272 | 8.4 | 0.352 |
| lipid A/OAK-1 | 8.1 | 0.254 | 6.7 | 0.357 |
| DPPG/AA-1 | 8.9 | 0.217 | 9.3 | 0.471 |
| lipid A/AA-1 | 9.9 | 0.268 | NA | NA |
Lac and ρac are thickness and electron density of the box containing lipid acyl chains only;
Laa and ρaa are thickness and electron density of the box composed of both lipid acyl chains and antimicrobials.
Strikingly, unstructured OAK-1 inserts into the hydrocarbon region of lipid A with similar propensity as into that of DPPG (Figure 1d), contributing to corresponding ρac 0.96 e−/Å2 and 0.78 e−/Å2. This unique membrane penetration behavior of OAK-1 cannot be simply explained by its high positive charge or hydrophobicity, because LL-37 is more hydrophobic and carries 3 additional positively charged residues [4b]. Instead, we propose that OAK-1’s structural flexibility plays the decisive role in its membrane insertion propensity.
In order to understand this phenomenon let’s try to fill up 2D space with balls of a particular diameter and then introduce a rod into this pattern. A larger diameter of the balls results in a higher volume of unoccupied space. Applying this logic to lipids and antimicrobials, incorporation of conformationally rigid antimicrobials into lipids with larger cross-sectional diameter (CSD) should lead to more profound packing perturbations and loss of hydrophobic contacts between the lipid acyl chains. This explains why none of the structured antimicrobials inserted into the hydrophobic core of lipid A with CSD of 13 Å, but penetrated into the hydrocarbon chains of DPPG with CSD of 8 Å (Scheme 1).
Unstructured antimicrobials can adopt their conformation in such a way as to minimize induced perturbations and to fill up the space between lipids. Therefore, the CSD of lipids is a somewhat less crucial parameter in activity of flexible antimicrobials. This notion is corroborated by a similar propensity of OAK-1 to insert into lipid A and DPPG.
Based on the limited data presented here and in previous reports, we suggest that the mode of interaction of conformationally flexible and restrained antimicrobials with bacterial membrane lipids is different. The flexible OAK-1 penetrates deep into the DPPG and lipid A monolayers effectively disrupting and hence presumably increasing permeability or enhancing breakdown of the bacterial cytoplasmic and outer membranes. The AA-1 and natural structural AMPs employ similar mechanism of disruption of the bacterial cytoplasmic membrane, but, strikingly, bind with high affinity and mask lipid A of the bacterial outer membrane projecting from the outside. In addition, these two binding modes of structured antimicrobials could coexist in the membrane regions containing lipids of similar negative surface charge density, but sufficiently different CSD.
In conclusion, we report results of the first X-ray study of interactions between non-natural AMP mimics and model bacterial membranes aimed at understanding the role of structural flexibility on the activity of antimicrobials. Our results show that conformational flexibility does not prevent antimicrobials from exerting a strong membrane disruptive activity. In addition, we demonstrate that while penetration of lipid A represents a serious challenge for the conformationally rigid antimicrobials, the flexible OAK-1 incorporates into lipid A with the same propensity as into DPPG. This implies that the structured and flexible antimicrobials act on the bacterial outer membrane differently. Over the last years, a number of novel synthetic oligomers [10] and linear polymers [11] with favorable antimicrobial efficacy, yet no specific or regular conformation, have been identified. Our results will aid in the rational design and optimization of emerging and future non-natural antimicrobial agents.
Experimental Section
A detailed explanation of the XR data analysis and additional experimental details can be found in Supporting Information.
Supplementary Material
Footnotes
We thank A. Antipova for her help with the monolayer experiments. DG is supported by NIH, R01 AI073892 grant. AM is supported by the Israel Science Foundation (grant 283/08). ChemMatCARS Sector 15 is principally supported by the National Science Foundation/Department of Energy (NSF/CHE-0822838). Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Andrey Ivankin, Center for Molecular Study of Condensed Soft Matter (μCoSM), and Division of Physics, BCPS Department, Illinois Institute of Technology, 3440 S Dearborn Street, Chicago, IL 60616 (USA).
Liran Livne, Department of Biotechnology & Food Engineering, Technion-Israel Institute of Technology, Haifa 32000, (Israel).
Prof. Amram Mor, Department of Biotechnology & Food Engineering, Technion-Israel Institute of Technology, Haifa 32000, (Israel)
Prof. Gregory A. Caputo, Department of Chemistry and Biochemistry, Rowan University, 201 Mullica Hill Road, Glassboro, NJ 08028 (USA)
Prof. William F. DeGrado, Department of Biochemistry and Biophysics, University of Pennsylvania, School of Medicine, 36th & Hamilton Walk, Philadelphia, PA 19104-6059 (USA)
Dr. Mati Meron, CARS, University of Chicago Chicago, IL 60637 (USA)
Dr. Binhua Lin, CARS, University of Chicago Chicago, IL 60637 (USA)
Prof. David Gidalevitz, Center for Molecular Study of Condensed Soft Matter (μCoSM), and Division of Physics, BCPS Department, Illinois Institute of Technology, 3440 S Dearborn Street, Chicago, IL 60616 (USA).
References
- 1.a) Jenssen H, Hamill P, Hancock REW. Clin Microbiol Rev. 2006;19:491. doi: 10.1128/CMR.00056-05. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zasloff M. Nature. 2002;415:389. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 2.Hancock REW, Sahl HG. Nat Biotech. 2006;24:1551. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 3.Brogden KA. Nat Rev Microbiol. 2005;3:238. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 4.a) Mowery BP, Lee SE, Kissounko DA, Epand RF, Epand RM, Weisblum B, Stahl SS, Gellman SH. J Am Chem Soc. 2007;129:15474. doi: 10.1021/ja077288d. [DOI] [PubMed] [Google Scholar]; b) Radzishevsky IS, Rotem S, Bourdetsky D, Navon-Venezia S, Carmeli Y, Mor A. Nat Biotech. 2007;25:657. doi: 10.1038/nbt1309. [DOI] [PubMed] [Google Scholar]
- 5.Choi S, Isaacs A, Clements D, Liu DH, Kim H, Scott RW, Winkler JD, DeGrado WF. Proc Natl Acad Sci USA. 2009;106:6968. doi: 10.1073/pnas.0811818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Neville F, Cahuzac M, Konovalov O, Ishitsuka Y, Lee KYC, Kuzmenko I, Kale GM, Gidalevitz D. Biophys J. 2006;90:1275. doi: 10.1529/biophysj.105.067595. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Neville F, Hodges CS, Liu C, Konovalov O, Gidalevitz D. Biochim Biophys Acta Biomembr. 2006;1758:232. doi: 10.1016/j.bbamem.2006.01.025. [DOI] [PubMed] [Google Scholar]; c) Neville F, Ivankin A, Konovalov O, Gidalevitz D. Biochim Biophys Acta Biomembr. 2010;1798:851. doi: 10.1016/j.bbamem.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Neville F, Cahuzac M, Nelson A, Gidalevitz D. J Phys Condens Matter. 2004;16:S2413. [Google Scholar]; e) Neville F, Gidalevitz D, Kale G, Nelson A. Bioelectrochemistry. 2007;70:205. doi: 10.1016/j.bioelechem.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 7.a) Neville F, Ishitsuka Y, Hodges CS, Konovalov O, Waring AJ, Lehrer R, Lee KYC, Gidalevitz D. Soft Matter. 2008;4:1665. doi: 10.1039/b718295c. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gidalevitz D, Ishitsuka YJ, Muresan AS, Konovalov O, Waring AJ, Lehrer RI, Lee KYC. Proc Natl Acad Sci USA. 2003;100:6302. doi: 10.1073/pnas.0934731100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Als-Nielsen J, Jacquemain D, Kjaer K, Leveiller F, Lahav M, Leiserowitz L. Phys Rep. 1994;246:252. doi: 10.1126/science.252.5012.1532. [DOI] [PubMed] [Google Scholar]
- 9.a) Netea MG, van Deuren M, Kullberg BJ, Cavaillon JM, Van der Meer JWM. Trends Immunol. 2002;23:135. doi: 10.1016/s1471-4906(01)02169-x. [DOI] [PubMed] [Google Scholar]; b) Schromm AB, Brandenburg K, Loppnow H, Zahringer U, Rietschel ET, Carroll SF, Koch MHJ, Kusumoto S, Seydel U. J Immunol. 1998;161:5464. [PubMed] [Google Scholar]
- 10.a) Som A, Tew GN. J Phys Chem B. 2008;112:3495. doi: 10.1021/jp077487j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Som A, Yang LH, Wong GCL, Tew GN. J Am Chem Soc. 2009;131:15102. doi: 10.1021/ja9067063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mowery BP, Lindner AH, Weisblum B, Stahl SS, Gellman SH. J Am Chem Soc. 2009;131:9735. doi: 10.1021/ja901613g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.