

Summary
Schizophrenia is a highly heritable disorder. Genetic risk is conferred by a large number of alleles, including common alleles of small effect that might be detected by genome-wide association studies. Here, we report a multi-stage schizophrenia genome-wide association study of up to 36,989 cases and 113,075 controls. We identify 128 independent associations spanning 108 conservatively defined loci that meet genome-wide significance, 83 of which have not been previously reported. Associations were enriched among genes expressed in brain providing biological plausibility for the findings. Many findings have the potential to provide entirely novel insights into aetiology, but associations at DRD2 and multiple genes involved in glutamatergic neurotransmission highlight molecules of known and potential therapeutic relevance to schizophrenia, and are consistent with leading pathophysiological hypotheses. Independent of genes expressed in brain, associations were enriched among genes expressed in tissues that play important roles in immunity, providing support for the hypothesized link between the immune system and schizophrenia.
Schizophrenia has a life time risk of around 1%, and is associated with substantial morbidity and mortality as well as personal and societal costs.1-3 Although pharmacological treatments are available for schizophrenia, their efficacy is poor for many patients.4 All available antipsychotic drugs are thought to exert their main therapeutic effects through blockade of the type 2 dopaminergic receptor5,6 but, since the discovery of this mechanism over 60 years ago, no new antipsychotic drug of proven efficacy has been developed based on other target molecules. Therapeutic stasis is in large part a consequence of the fact that the pathophysiology of schizophrenia is unknown. Identifying the causes of schizophrenia is therefore a critical step towards improving treatments and outcomes for those with the disorder.
High heritability points to a major role for inherited genetic variants in the aetiology of schizophrenia. 7,8 While risk variants range in frequency from common to extremely rare9, estimates10,11 suggest half to a third of the genetic risk of schizophrenia is indexed by common alleles genotyped by current genome-wide association study (GWAS) arrays. Thus, GWAS is potentially an important tool for understanding the biological underpinnings of schizophrenia.
To date, around 30 schizophrenia-associated loci10-23 have been identified through GWAS. Postulating that sample size is one of the most important limiting factors in applying GWAS to schizophrenia, we created the Schizophrenia Working Group of the Psychiatric Genomics Consortium (PGC). Our primary aim was to combine all available schizophrenia samples with published or unpublished GWAS genotypes into a single, systematic analysis.24 Here, we report the results of that analysis, including at least 108 independent genomic loci that exceed genome-wide significance. Some of the findings support leading pathophysiological hypotheses of schizophrenia or targets of therapeutic relevance, but most of the findings provide novel insights.
128 Independent Associated Loci
We obtained genome-wide genotype data from which we constructed 49 ancestry matched, non-overlapping case-control samples (46 of European and three of East Asian ancestry, 34,241 cases and 45,604 controls) and 3 family-based samples of European ancestry (1,235 parent affected-offspring trios) (Supplementary Table 1 and Supplementary text). These comprise the primary PGC GWAS dataset. Genotypes from all studies were processed by the PGC using unified quality control procedures followed by imputation of SNPs and insertion-deletions using the 1000 Genomes Project reference panel.25 In each sample, association testing was conducted using imputed marker dosages and principal components (PCs) to control for population stratification. The results were combined using an inverse-weighted fixed effects model.26 After quality control (imputation INFO score ≥ 0.6, MAF ≥ 0.01, and successfully imputed in ≥ 20 samples), we considered around 9.5 million variants. The results are summarized in Figure 1. To enable acquisition of large samples, some groups ascertained cases via clinician diagnosis rather than a research-based assessment and provided evidence of the validity of this approach (Supplementary text).11,13 Post-hoc analyses revealed the pattern of effect sizes for associated loci was similar across different assessment methods and modes of ascertainment (Extended Data Figure 1), supporting our a priori decision to include samples of this nature.
Figure 1. Manhattan plot.

Manhattan plot of the discovery genome-wide association meta-analysis of 49 case control samples (34,241 cases and 45,604 controls) and 3 family based association studies (1,235 parent affected-offspring trios). The x-axis is chromosomal position and the y-axis is the significance of association (−log10(P)).The red line shows the genome-wide significance level (5×10−8). SNPs in green are in LD with the index SNPs (diamonds) which represent independent genome-wide significant associations.
For the subset of LD (linkage disequilibrium)-independent SNPs with P < 1×10−6 in the meta-analysis, we next obtained results from deCODE genetics (1,513 cases and 66,236 controls of European ancestry). We define LD-independent SNPs as those with low LD (r2 < 0.1) to a more significantly associated SNP within a 500 kb window. Given high LD in the extended MHC region spans ~8 Mb, we conservatively include only a single MHC SNP to represent this locus. The deCODE data were then combined with those from the primary GWAS to give a dataset of 36,989 cases and 113,075 controls. In this final analysis, 128 LD-independent SNPs surpassed genome-wide significance (P ≤ 5×10−8) (Supplementary Table 2).
As in meta-analyses of other complex traits which identified large numbers of common risk variants, 27,28 the test statistic distribution from our GWAS deviates from the null (Extended Data Figure 2). This is consistent with the previously documented polygenic contribution to schizophrenia. 10,11 The deviation in the test statistics from the null (λGC = 1.47, λ1000 = 1.01) is only slightly less than expected (λGC = 1.56) under a polygenic model given fully informative genotypes, the current sample size, and the lifetime risk and heritability of schizophrenia.29
Multiple additional lines of evidence allow us to conclude the deviation between the observed and null distributions in our primary GWAS indicates a true polygenic contribution to schizophrenia. First, applying a novel method 30 that uses LD information to distinguish between the major potential sources of test statistic inflation, we found our results are consistent with polygenic architecture but not population stratification (Extended Data Figure 3). Second, for 78% of 263 LD-independent SNPs with P <1×10−6 in the case-control GWAS, the risk alleles from were overrepresented in cases in the independent samples from deCODE (P = 5.6 ×10−21). This degree of consistency between the primary GWAS and the replication data is highly unlikely to occur by chance (P = 1×10−9). (Note the tested alleles surpassed the P < 10−6 threshold in our GWAS before we added either the trios or deCODE data to the meta-analysis. This trend test is therefore independent of the primary case-control GWAS). Third, analysing the 1,235 parent-proband trios, we again found excess transmission of the schizophrenia-associated allele at 69% of the LD-independent SNPs with P <1×10−6 in the case-control GWAS. This is again unlikely to occur by chance (P = 1×10−9) and additionally excludes population stratification as fully explaining the associations that surpassed our threshold for seeking replication. Fourth, we used the trios trend data to estimate the expected proportion of true associations at P <1×10−6 in the discovery GWAS, allowing for the fact that half of the index SNPs are expected to show the same allelic trend in the trios by chance, and that some true associations will show opposite trends given the limited number of trio samples (Supplementary Material). Given the observed trend test results, around 67% (95% CI 64%-73%) or N = 176 of the associations in the scan at P <1×10−6 are expected to be true, and therefore the number of associations that will ultimately be validated from this set of SNPs will be considerably more than those that now meet genome-wide significance. Taken together, these analyses indicate that the observed deviation of test statistics from the null primarily represents polygenic association signal, and that the considerable excess of associations at the tail of extreme significance largely correspond to true associations.
Independently associated SNPs do not translate to well-bounded chromosomal regions. Nevertheless, it is useful to define physical boundaries for the SNP associations to identify candidate risk genes. We defined an associated locus as the physical region containing all SNPs correlated at r2 > 0.6 with each of the 128 index SNPs. Associated loci within 250 kb of each other were merged. This resulted in 108 physically distinct associated loci, 83 of which have not been previously implicated in schizophrenia and therefore harbour potential novel biological insights into disease aetiology (Supplementary Table 3; regional plots in Supplementary Figure 1). The significant regions include all but 5 loci previously reported as genome-wide significant in large samples (Supplementary Table 3).
Characterization of Associated Loci
Of the 108 loci, 75% include protein-coding genes (40% a single gene) and a further 8% are within 20 kb of a gene (Supplementary Table 3). Notable associations relevant to major hypotheses of the aetiology and treatment of schizophrenia include DRD2 (the target of all effective antipsychotic drugs) and multiple genes (e.g. GRM3, GRIN2A, SRR, GRIA1) involved in glutamatergic neurotransmission and synaptic plasticity. In addition, associations at CACNA1C, CACNB2, and CACNA1I, which encode voltage-gated calcium channel subunits, extend previous findings implicating members of this family of proteins in schizophrenia and other psychiatric disorders. 11,13,31,32 Genes encoding calcium channels, and proteins involved in glutamatergic neurotransmission and synaptic plasticity have been independently implicated in schizophrenia by studies of rare genetic variation, 33-35 suggesting convergence at a broad functional level between studies of common and rare genetic variation. We highlight in the supplementary text genes of particular interest within associated loci with respect to current hypotheses of schizophrenia aetiology or treatment (although we do not imply these genes are necessarily the causal elements).
For each of the schizophrenia associated loci, we identified a credible causal set of SNPs (for definition see Supplementary text). 36 In only 10 instances (Supplementary Table 4) was the association signal credibly attributable to a known nonsynonymous exonic polymorphism. The apparently limited role of protein coding variants is consistent both with exome sequencing findings 33 and with the hypothesis that most associated variants detected by GWAS exert their effects through altering gene expression rather than protein structure37,38 and with the observation that schizophrenia risk loci are enriched for expression quantitative trait loci (eQTL).39
To try to identify eQTLs that could explain associations with schizophrenia, we merged the credible causal set of SNPs defined above with eQTLs from a meta-analysis of human brain cortex eQTL studies (N=550) and an eQTL study of peripheral venous blood (N=3754)40 (Supplementary Text). Multiple schizophrenia loci contained at least one eQTL for a gene within 1 Mb of the locus (Supplementary Table 4). However, in only 12 instances was the eQTL plausibly causal (two in brain, and nine in peripheral blood, one in both). This low proportion suggests that if most risk variants are regulatory, available eQTL catalogs do not yet provide power, cellular specificity, or developmental diversity to provide clear mechanistic hypotheses for follow-up experiments.
Brain and Immunity
To further explore the regulatory nature of the schizophrenia associations, we mapped the credible sets (N=108) of causal variants onto sequences with epigenetic markers characteristic of active enhancers in 56 different tissues and cell lines (Supplementary Text). Schizophrenia associations were significantly enriched at enhancers active in brain (Figure 2) but not in tissues unlikely to be relevant to schizophrenia (e.g., bone, cartilage, kidney, and fibroblasts). Brain tissues used to define enhancers consist of heterogeneous populations of cells. Seeking greater specificity, we contrasted genes enriched for expression in neurons and glia using mouse ribotagged lines41. Genes with strong expression in multiple cortical and striatal neuronal lineages were were enriched for associations, providing support for an important neuronal pathology in schizophrenia (Extended Data Figure 4) but this is not statistically more significant than, or exclusionary of, contributions from other lineages42.
Figure 2. Enrichment in enhancers.

Cell and tissue type specific enhancers were identified using ChIP-seq datasets (H3K27ac signal) from 56 cell line and tissue samples (Y-axis). We defined cell and tissue type enhancers as the top 10% of enhancers with the highest ratio of reads in that cell or tissue type divided by the total number of reads. Enrichment of credible causal associated SNPs from the schizophrenia GWAS was compared with frequency matched sets of 1000 Genomes SNPs (Supplementary Text). The X-axis is the −log10(P) for enrichment. P-values are uncorrected for the number of tissues/cells tested. A −log10(P) of roughly 3 can be considered significant after Bonferroni correction. Descriptions of cell and tissue types at the Roadmap Epigenome website (http://www.roadmapepigenomics.org).
Schizophrenia associations were also strongly enriched at enhancers that are active in tissues with important immune functions, particularly B-lymphocyte lineages involved in acquired immunity (CD19 and CD20 lines, Figure 2). These enrichments remain significant even after excluding the extended MHC region and regions containing brain enhancers (enrichment P for CD20 <10−6), demonstrating that this finding is not an artefact of correlation between enhancer elements in different tissues and not driven by the strong and diffuse association at the extended MHC. Epidemiological studies have long hinted at a role for immune dysregulation in schizophrenia, the present findings provide genetic support for this hypothesis. 43
Aiming to develop additional biological hypotheses beyond those that emerge from inspection of the individual loci, we further undertook a limited mining of the data through gene-set analysis. However, since there is no consensus methodology by which such analyses should be conducted, nor an established optimal significance threshold for including loci, we sought to be conservative, using only two of the many available approaches44,45 and restricting analyses to genes within genome-wide significant loci. Neither approach identified gene-sets that were significantly enriched for associations after correction for the number of pathways tested (Supplementary Table 5) although nominally significantly enrichments were observed among several predefined candidate pathways (Extended Data Table 1). A fuller exploratory analysis of the data will be presented elsewhere.
Overlap with rare mutations
CNVs associated with schizophrenia overlap with those associated with ASD and intellectual disability9 as do genes with deleterious de novo mutations. 34 Here, we find significant overlap between genes in the schizophrenia GWAS associated intervals and those with de novo nonsynonymous mutations in schizophrenia (P=0.0061) (Extended Data Table 2), suggesting that mechanistic studies of rare genetic variation in schizophrenia will be informative for schizophrenia more widely. We also find evidence for overlap between genes in schizophrenia GWAS regions and those with de novo nonsynonymous mutations in intellectual disability (P=0.00024) and ASD (P=0.035), providing further support for the hypothesis that these disorders have partly overlapping pathophysiologies.9,34
Polygenic Risk Score Profiling
Previous studies have shown that risk profile scores (RPS) constructed from alleles showing modest association with schizophrenia in a discovery GWAS can predict case-control status in independent samples, albeit with low sensitivity and specificity.10,11,16 This finding was robustly confirmed in the present study. The estimate of Nagelkerke R2 (a measure of variance in case-control status explained) depends on the specific target dataset and threshold (PT) for selecting risk alleles for RPS analysis (Extended Data Figure 5 & 6a). However, using the same target sample as earlier studies and PT =0.05, R2 is now increased from 0.03 (reference10) to 0.184 (Extended Data Figure 5). Assuming a liability-threshold model, a lifetime risk of 1%, independent SNP effects, and adjusting for case-control ascertainment, RPS now explains about 7% of variation on the liability scale46 to schizophrenia across the samples (Extended Data Figure 6b), about half of which (3.4%) is explained by genome-wide significant loci.
We also evaluated the capacity of RPS to predict case-control status using a standard epidemiological approach to a continuous risk factor. We illustrate this in three samples, each with different ascertainment schemes (Figure 3). The Danish sample is population-based (i.e. inpatient and outpatient facilities), the Sweden sample is based on all cases hospitalized for schizophrenia in Sweden, and the MGS study was ascertained specially for genetic studies from clinical sources in the US and Australia. We grouped individuals into RPS deciles and estimated the odds ratios for affected status for each decile with reference to the lowest risk decile. The odds ratios increased with greater number of schizophrenia risk alleles in each sample, maximizing for the tenth decile in all samples: Denmark 7.8 (95% CI 4.4-13.9), Sweden 15.0 (95% CI 12.1-18.7), and MGS 20.3 (95% CI 14.7-28.2). Given the need for measures that index liability to schizophrenia,47,48 the ability to stratify individuals by RPS offers new opportunities for clinical and epidemiological research. Nevertheless, we stress that the sensitivity and specificity of RPS do not support its use as a predictive test. For example in the Danish epidemiological sample, the area under the receiver operating curve is only 0.62 (Extended Data Figure 6c, Supplementary Table 6).
Figure 3. Odds ratio by risk score profile.

Odds ratio for schizophrenia by risk score profile (RPS) decile in the Sweden (Sw1-6), Denmark (Aarhus), and Molecular Genetics of Schizophrenia studies (Supplementary text). Risk alleles and weights were derived from ‘leave one out’ analyses in which those samples were excluded from the GWAS meta-analysis (Supplementary text). The threshold for selecting risk alleles was PT < 0.05. The RPS were converted to deciles (1=lowest, 10=highest RPS), and nine dummy variables created to contrast deciles 2-10 to decile 1 as the reference. Odds ratios and 95% confidence intervals (bars) were estimated using logistic regression with PCs to control for population stratification.
Finally, seeking evidence for non-additive effects on risk, we tested for statistical interaction between all pairs of 125 autosomal SNPs that reached genome-wide significance. P-values for the interaction terms were distributed according to the null, and no interaction was significant after correction for multiple comparisons. Thus, we find no evidence for epistatic or non-additive effects between the significant loci (Extended Data Figure 7). It is possible that such effects could be present between other loci, or occur in the form of higher order interactions.
Discussion
In the largest molecular genetic study of schizophrenia ever conducted, or indeed of any neuropsychiatric disorder, we demonstrate the power of GWAS to identify large numbers of risk loci. We also show that the use of alternative ascertainment and diagnostic schemes designed to rapidly increase sample size does not inevitably introduce a crippling degree of heterogeneity. That this is true for a phenotype like schizophrenia where there are no biomarkers or supportive diagnostic tests provides grounds to be optimistic about applying GWAS to other disorders where diagnosis is similarly clinician-based, and recruitment traditionally constrained by the need for expensive and time consuming assessments.
We further show that the associations are not randomly distributed across genes of all classes and function; rather they converge upon genes that are expressed in certain tissues and cellular types. The findings include molecules that are the current, or the most promising, targets for therapeutics, and point to systems that align with the predominant aetiological hypotheses of the disorder. This suggests that the many novel findings we report also provide a aetiologically relevant foundation for mechanistic and treatment development studies. We additionally find overlap between genes affected by rare variants in schizophrenia and those within GWAS loci, and broad convergence in the functions of some of the clusters of genes implicated by both sets of genetic variants, particularly genes related to abnormal glutamatergic synaptic and calcium channel function. How variation in these genes impact function to increase risk for schizophrenia cannot be answered by genetics, but the overlap strongly suggests that common and rare variant studies are complementary rather than antagonistic, and that mechanistic studies driven by rare genetic variation will be informative for schizophrenia.
Extended Data
Extended Data Figure 1. Homogeneity of Effects Across Studies.
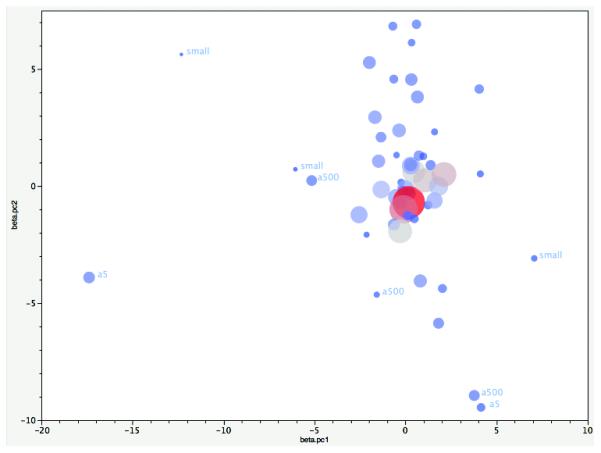
Plot of the first two principal components (PC) from principal components analysis (PCA) of the logistic regression β coefficients for autosomal genome-wide significant associations. The input data were the β coefficients from 52 samples for 112 independent SNP associations (excluding 3 chrX SNPs and 13 SNPs with missing values in Asian samples). PCAs were weighted by the number of cases. Each circle shows the location of a study on PC1 and PC2. Circle size and colour are proportional to the number of cases in each sample (larger and redder circles correspond to more cases). Most samples cluster. Outliers had either small numbers of cases (“small”) or were genotyped on older arrays. Abbreviations. a500 (Affymetrix 500K); a5 (Affymetrix 5.0). Studies that did not use conventional research interviews are in the central cluster (CLOZUK, Sweden, and Denmark-Aarhus studies).
Extended Data Figure 2. Quantile-quantile plot.
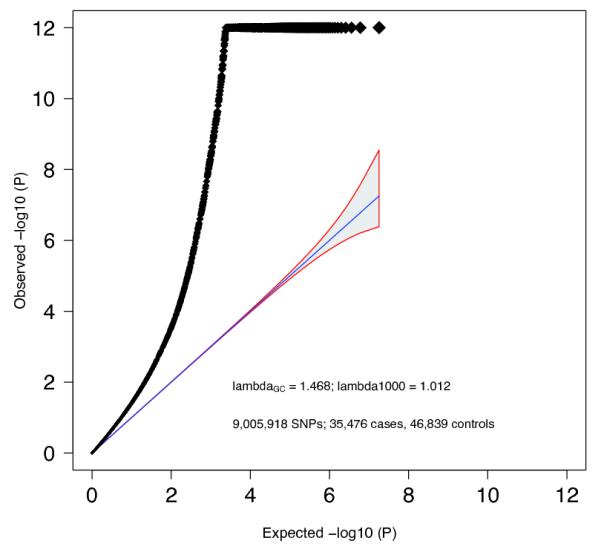
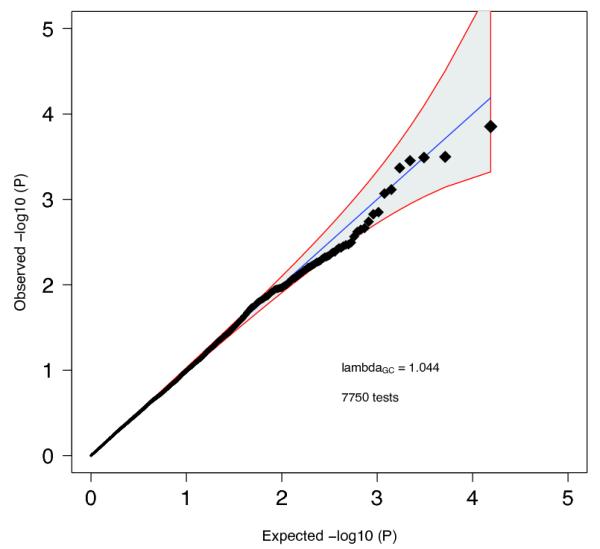
Quantile-quantile plot of GWAS meta-analysis. Expected −log10(P) -values are those expected under the null hypothesis. For clarity, we avoided expansion of the Y-axis by setting association P-values < 10−12 to 10−12. The shaded area surrounded by a red line indicates the 95% confidence interval under the null. Lambda is the observed median χ2 test statistic divided by the median expected χ2 test statistic under the null.
Extended Data Figure 3. LD Score regression consistent with polygenic inheritance.
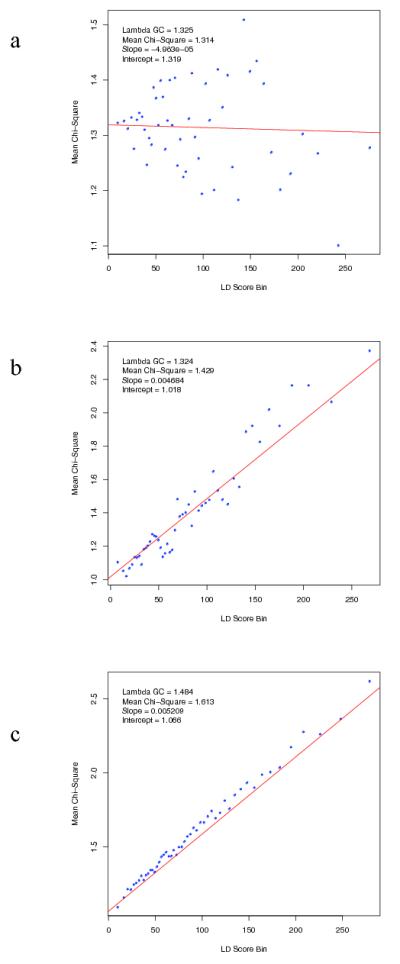
The relationship between marker χ2 association statistics and linkage disequilibrium (LD) as measured by the LD Score. LD Score is the sum of the r2 values between a variant and all other known variants within a 1 cM window, and quantifies the amount of genetic variation tagged by that variant. Variants were grouped into 50 equal-sized bins based on LD Score rank. LD Score Bin and Mean χ2 denotes mean LD score and test statistic for markers each bin. We simulated (Supplementary Text) test statistics under two scenarios: (a) no true association, inflation due to population stratification (b) polygenic inheritance (λGC =1.32), in which we assigned independent and identically distributed per-normalized-genotype effects to a randomly selected subset of variants. Panel (c) present results from the PGC schizophrenia GWAS (λGC =1.48). The real data are strikingly similar to the simulated data summarized in (b) but not (a). The intercept estimates the inflation in the mean χ2 that results from confounding biases, such as cryptic relatedness or population stratification. Thus, the intercept of 1.066 for the schizophrenia GWAS suggests that ~90% of the inflation in the mean χ2 results from polygenic signal. The results of the simulations are also consistent with theoretical expectation (see Supplementary Text).
Extended Data Figure 4. Enrichment of Associations in Tissues and Cells.
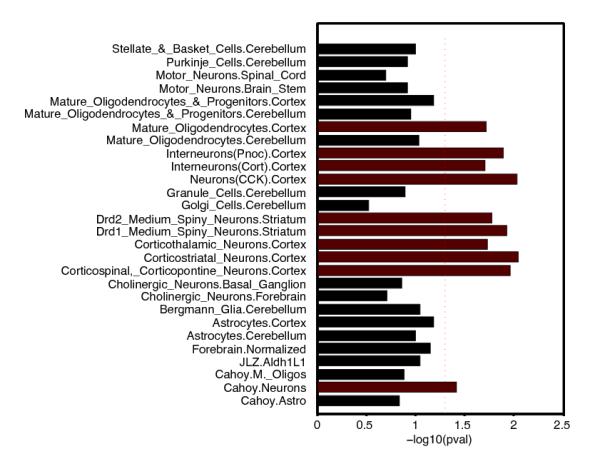
Genes whose transcriptional start is nearest to the most associated SNP at each schizophrenia-associated locus were tested for enriched expression in a) purified brain cell subsets obtained from mouse ribotagged lines41. The red dotted line indicates P=0.05.
Extended Data Figure 5. MGS Risk Profile Score Analysis.
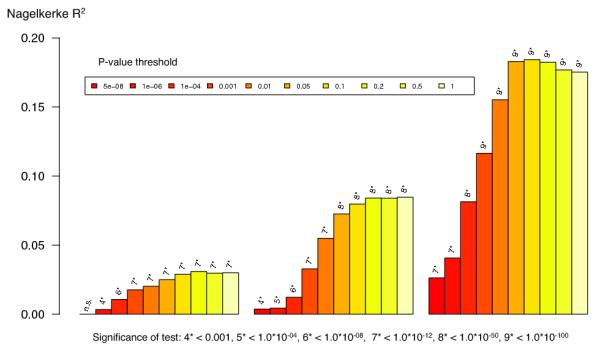
Polygenic risk profile score (RPS) analyses using the MGS18 sample as target, and deriving risk alleles from three published schizophrenia datasets (X-axis): ISC (2615 cases and 3338 controls) 10, PGC1 (excluding MGS, 9320 cases and 10,228 controls) 16, and the current meta analysis (excluding MGS) with 32,838 cases and 44,357 controls. Samples sizes differ slightly from original publication due to different analytical procedures. This shows the increasing RPS prediction with increasing training dataset size reflecting improved precision of estimates of the SNP effect sizes. The proportion of variance explained (Y-axis; Nagelkerke’s R2) was computed by comparison of a full model (covariates + RPS) score to a reduced model (covariates only). Ten different P-value thresholds (PT) for selecting risk alleles are denoted by the colour of each bar (legend above plot). For significance testing, see the bottom legend which denotes the P-value for the test that R2 is different from zero. All numerical data and methods used to generate these plots are available in Supplementary Tables 6, 7, and Supplementary Text.
Extended Data Figure 6. Risk Profile Score Analysis.

We defined 40 target subgroups of the primary GWAS dataset and performed 40 leave-one-out GWAS analyses (see Supplementary Material) from which we derived risk alleles for RPS analysis (X-axis) for each target subgroup. a) The proportion of variance explained (Y-axis; Nagelkerke’s R2) was computed for each target by comparison of a full model (covariates + RPS) score to a reduced model (covariates only). For clarity, 3 different P-value thresholds (PT) are presented denoted by the colour of each bar (legend above plot) as for Extended Data Figure 5 but for clarity we restrict to fewer P-value thresholds (PT of 5×10−8, 1×10−4, and 0.05) and removed the significance values. (b) The proportion of variance on the liability scale from risk scores calculated at the PT 0.05 with 95% CI bar assuming baseline population disease risk of 1%. (C) Area under the receiver operating curve (AUC). All numerical data and methods used to generate these plots are available in Supplementary Tables 6, 7, and Supplementary Text.
Extended Data Figure 7. Epistasis.
Extended Data Table 1. ALIGATOR and INRICH.
Gene sets that have been reported to be enriched for schizophrenia associations and or rare mutations were tested for enrichment for genome wide significant associations using ALIGATOR44 and INRICH45. Specifically, we tested the glutamatergic postsynaptic proteins comprising activity-regulated cytoskeleton-associated protein (ARC) and N-methyl-d-aspartate receptor (NMDAR) complexes33-35, other curated synaptic gene-sets14,51, targets of fragile X mental retardation protein (FMRP) 33-35, calcium channels 11,33, and TargetScan predicted MIR137 sets11,16. The MIR137 TargetScan sets contain computationally predicted conserved miRNA target sites in 3′UTRs of human genes52. The current version is v6, but the version used in the prior PGC SCZ report16 was based on v5 (filtered for a probability of conserved targeting > 0.9). We report the results of both analyses for consistency with previous work. The association at the extended MHC complex was not included given the extensive linkage disequilibrium at this region spans large numbers of genes. NA means that the pathway in question contained fewer than 2 significant genes (for ALIGATOR) or regions (INRICH).
| SET | ALIGATOR | INRICH |
|---|---|---|
| Postsynaptic sets | ||
| ARC | NA | 1 |
| NMDAR | NA | 0.458 |
|
| ||
| Curated pre- and postsynaptic sets | ||
| Cell adhesion and trans8 synaptic signalling | 0.902 | 0.44 |
| Structural plasticity | NA | NA |
| Excitability | NA | NA |
|
| ||
| FMRP sets | ||
| FMRP | 0.0066 | 5.00E-05 |
|
| ||
| MIR137 sets | ||
| Targetscan v5 with PCT > 0.9 | 0.0371 | 0.0103 |
| Targetscan v6.2 | 0.059 | 0.0024 |
|
| ||
| Calcium signalling sets | ||
| CACN* channel subunits | 0.0338 | 0.022 |
Extended Data Table 2. de novo overlap.
Test of overlap between genes mapping to schizophrenia-associated loci in the present study and genes affected by nonsynonymous (NS) de novo mutations. Enrichment was calculated using the dnenrich permutation framework as described34. Genes within the GWS loci were weighted by 1/N, where N is the number of coding genes within each associated locus. The observed test statistic (stat) is the sum of weights of genes impacted by de novo mutations. The expected test statistics are calculated by averaging over 50,000 permuted de novo mutation sets. Genes within schizophrenia-associated loci affected by de novo mutations are listed (multiple hits listed in parentheses). Cohorts: SCZ = schizophrenia, ID = intellectual disability, ASD = autism spectrum disorder. All mutations analysed annotated according to a unified system (see supplementary tables 1 and 2 of ref34). Genes with LoF de novo mutations are underlined and in italics.
| Disease group | NS (N) | P | 0 NS in PGC2 loci | Observed (stat) | Expected (stat) | Genes |
|---|---|---|---|---|---|---|
| SCZ | 702 | 0.0061 | 25 | 10.97 | 5.27 | CACNACI(x2) CCDC39 CDCH(x2) CRCL CUL3 DPEP2 DPYD(x2) EP300 ESAM GRIN2A LRP1 NCAN PDCDCC PTPRF RIMSC SBNOC SGSM2 SLC7A6 STAGC TMEM2C9 ZDHHC5 ZNF536 |
| ID | 141 | 0.00002 | 11 | 6.87 | 1.05 | GRIAC GRIN2A(x2) LRPC NEKC NGEF SATB2 SREBF2 STAGC TCFH(x2) |
| ASD | 789 | 0.035 | 19 | 9.99 | 5.93 | APH1A CNOTC CSMDC CUL3 CYPC7AC CYP26BC EPHX2 LRPC MAPK3 MEF2C MPP6 MYOC5A NISCH PBRMC PRKDC RIMS1 TSNAREC WDR55 ZNF80HA |
| Controls | 434 | 0.15 | 16 | 4.88 | 3.28 | ANKRDHH CCCorf87 CCDC39 CDK2APC CHRMH DPEP2 EP300 LRPC LRRCH8 MAN2AC MYOCA OSBPL3 RAIC SF3BC SREBF2 TLE3 |
Supplementary Material
Acknowledgements and Consortia
Core funding for the Psychiatric Genomics Consortium is from the US National Institute of Mental Health (U01 MH094421). We thank Dr Thomas Lehner (NIMH). The work of the contributing groups was supported by numerous grants from governmental and charitable bodies as well as philanthropic donation. Details are provided in the supplementary material. Membership of the Wellcome Trust Case Control Consortium and of the Psychosis Endophenotype International Consortium are provided in the Supplementary Text.
Footnotes
URLs: Results can be downloaded from the Psychiatric Genomics Consortium website (http://pgc.unc.edu) and visualized using Ricopili (http://www.broadinstitute.org/mpg/ricopili). Genotype data for the samples where the ethics permit deposition are available upon application from the NIMH Genetics Repository (https://www.nimhgenetics.org).
Competing Financial Interests: Several of the authors are employees of the following pharmaceutical companies; Pfizer (C.R.S., J.R.W., H.S.X.), F. Hoffman-La Roche (E.D., L.E.), Eli Lilly (D.A.C., Y.M., L.N.) and Janssen (S.G., D.W., Q.S.L.; also N.C. an ex-employee). Others are employees of deCODE genetics (S.S., H.S., K.S.). None of these companies influenced the design of the study, the interpretation of the data, the amount of data reported, or financially profit by publication of the results which are pre-competitive. The other authors declare no competing interests.
References
- 1.Saha S, Chant D, McGrath J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Archives of General Psychiatry. 2007;64:1123–31. doi: 10.1001/archpsyc.64.10.1123. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization . The Global Burden of Disease: 2004 Update. WHO Press; Geneva: 2008. [Google Scholar]
- 3.Knapp M, Mangalore R, Simon J. The global costs of schizophrenia. Schizophrenia Bulletin. 2004;30:279–93. doi: 10.1093/oxfordjournals.schbul.a007078. [DOI] [PubMed] [Google Scholar]
- 4.Lieberman JA, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–23. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- 5.Carlsson A, Lindqvist M. Effect of Chlorpromazine or Haloperidol on Formation of 3methoxytyramine and Normetanephrine in Mouse Brain. Acta Pharmacologica et Toxicologica. 1963;20:140–4. doi: 10.1111/j.1600-0773.1963.tb01730.x. [DOI] [PubMed] [Google Scholar]
- 6.van Rossum JM. The significance of dopamine-receptor blockade for the mechanism of action of neuroleptic drugs. Archives Internationales de Pharmacodynamie et de Therapie. 1966;160:492–4. [PubMed] [Google Scholar]
- 7.Lichtenstein P, et al. Recurrence risks for schizophrenia in a Swedish national cohort. Psychological Medicine. 2006;36:1417–26. doi: 10.1017/S0033291706008385. [DOI] [PubMed] [Google Scholar]
- 8.Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Archives of General Psychiatry. 2003;60:1187–92. doi: 10.1001/archpsyc.60.12.1187. [DOI] [PubMed] [Google Scholar]
- 9.Sullivan PF, Daly MJ, O’Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nature Reviews Genetics. 2012;13:537–51. doi: 10.1038/nrg3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.International Schizophrenia Consortium Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–52. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ripke S, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nature Genetics. 2013;45:1150–9. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikeda M, et al. Genome-wide association study of schizophrenia in a Japanese population. Biological Psychiatry. 2011;69:472–8. doi: 10.1016/j.biopsych.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 13.Hamshere ML, et al. Genome-wide significant associations in schizophrenia to ITIH3/4, CACNA1C and SDCCAG8, and extensive replication of associations reported by the Schizophrenia PGC. Molecular Psychiatry. 2012 doi: 10.1038/mp.2012.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Donovan M, et al. Identification of novel schizophrenia loci by genome-wide association and follow-up. Nature Genetics. 2008;40:1053–5. doi: 10.1038/ng.201. [DOI] [PubMed] [Google Scholar]
- 15.Rietschel M, et al. Association between genetic variation in a region on chromosome 11 and schizophrenia in large samples from Europe. Molecular Psychiatry. 2011 doi: 10.1038/mp.2011.80. [DOI] [PubMed] [Google Scholar]
- 16.Schizophrenia Psychiatric Genome-Wide Association Study Consortium Genome-wide association study identifies five new schizophrenia loci. Nature Genetics. 2011;43:969–76. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irish Schizophrenia Genomics Consortium. Wellcome Trust Case Control Consortium Genome-wide association study implicates HLA-C*01:02 as a risk factor at the MHC locus in schizophrenia. Biological Psychiatry. 2012 doi: 10.1016/j.biopsych.2012.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi J, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–7. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi Y, et al. Common variants on 8p12 and 1q24.2 confer risk of schizophrenia. Nature Genetics. 2011;43:1224–7. doi: 10.1038/ng.980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stefansson H, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–7. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steinberg S, et al. Common variants at VRK2 and TCF4 conferring risk of schizophrenia. Human Molecular Genetics. 2011;20:4076–81. doi: 10.1093/hmg/ddr325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yue WH, et al. Genome-wide association study identifies a susceptibility locus for schizophrenia in Han Chinese at 11p11. Nature Genetics. 2011;43:1228–31. doi: 10.1038/ng.979. [DOI] [PubMed] [Google Scholar]
- 23.Lencz T, et al. Genome-wide association study implicates NDST3 in schizophrenia and bipolar disorder. Nature Communications. 2013;4:2739. doi: 10.1038/ncomms3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Psychiatric GWAS Consortium A framework for interpreting genomewide association studies of psychiatric disorders. Molecular Psychiatry. 2009;14:10–7. doi: 10.1038/mp.2008.126. [DOI] [PubMed] [Google Scholar]
- 25.Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Begum F, Ghosh D, Tseng GC, Feingold E. Comprehensive literature review and statistical considerations for GWAS meta-analysis. Nucleic Acids Research. 2012;40:3777–84. doi: 10.1093/nar/gkr1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lango Allen H, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–8. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jostins L, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, et al. Genomic inflation factors under polygenic inheritance. European Journal of Human Genetics. 2011;19:807–12. doi: 10.1038/ejhg.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bulik-Sullivan BK, et al. LD Score Regression Distinguishes Confounding from Polygenicity in Genome-Wide Association Studies. 2014 doi: 10.1038/ng.3211. http://dx.doi.org/10.1101/002931. [DOI] [PMC free article] [PubMed]
- 31.Ferreira M, et al. Collaborative genome-wide association analysis of 10,596 individuals supports a role for Ankyrin-G (ANK3) and the alpha-1C subunit of the L-type voltage-gated calcium channel (CACNA1C) in bipolar disorder. Nature Genetics. 2008;40:1056–8. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cross-Disorder Group of the Psychiatric Genomics Consortium Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–9. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Purcell SM, et al. A burden of ultra-rare disruptive mutations concentrated in synaptic gene networks increases risk of schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fromer M, et al. De novo mutations in schizophrenia identify pathogenic gene networks regulating synaptic strength and overlap with autism and intellectual disability. Nature. 2014;506:179–84. [Google Scholar]
- 35.Kirov G, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Molecular Psychiatry. 2012;17:142–53. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wellcome Trust Case Control, C. et al. Bayesian refinement of association signals for 14 loci in 3 common diseases. Nature Genetics. 2012;44:1294–301. doi: 10.1038/ng.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicolae DL, et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genetics. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maurano MT, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;237:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richards AL, et al. Schizophrenia susceptibility alleles are enriched for alleles that affect gene expression in adult human brain. Molecular Psychiatry. 2012;17:193–201. doi: 10.1038/mp.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wright F, et al. Heritability and genomics of gene expression in peripheral blood. Nature Genetics. 2014;46:430–7. doi: 10.1038/ng.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doyle JP, et al. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749–62. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tkachev D, et al. Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet. 2003;362:798–805. doi: 10.1016/S0140-6736(03)14289-4. [DOI] [PubMed] [Google Scholar]
- 43.Benros ME, Mortensen PB, Eaton WW. Autoimmune diseases and infections as risk factors for schizophrenia. Annals of the New York Academy of Sciences. 2012;1262:56–66. doi: 10.1111/j.1749-6632.2012.06638.x. [DOI] [PubMed] [Google Scholar]
- 44.Holmans P, et al. Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. American Journal of Human Genetics. 2009;85:13–24. doi: 10.1016/j.ajhg.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee P, O’Dushlaine C, Thomas B, Purcell S. InRich: Interval-based enrichment analysis for genome-wide association studies. Bioinformatics. 2012;28:1797–9. doi: 10.1093/bioinformatics/bts191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee SH, Goddard ME, Wray NR, Visscher PM. A better coefficient of determination for genetic profile analysis. Genetic Epidemiology. 2012;36:214–24. doi: 10.1002/gepi.21614. [DOI] [PubMed] [Google Scholar]
- 47.Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. American Journal of Psychiatry. 2003;160:636–45. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- 48.Insel T, et al. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. American Journal of Psychiatry. 2010;167:748–51. doi: 10.1176/appi.ajp.2010.09091379. [DOI] [PubMed] [Google Scholar]
- 49.Howie B, Marchini J, Stephens M. Genotype imputation with thousands of genomes. G3. 2011;1:457–70. doi: 10.1534/g3.111.001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nature Methods. 2012;9:179–81. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- 51.Lips ES, et al. Functional gene group analysis identifies synaptic gene groups as risk factor for schizophrenia. Molecular Psychiatry. 2012;17:996–1006. doi: 10.1038/mp.2011.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.