

Abstract
Objective
Spinal muscular atrophy (SMA) is one of the most common severe hereditary diseases of infancy and early childhood in North America, Europe, and Asia. SMA is usually caused by deletions of the survival motor neuron 1 (SMN1) gene. A closely related gene, SMN2, modifies the disease severity. SMA carriers have only 1 copy of SMN1 and are relatively common (1 in 30–50) in populations of European and Asian descent. SMN copy numbers and SMA carrier frequencies have not been reliably estimated in Malians and other sub‐Saharan Africans.
Methods
We used a quantitative polymerase chain reaction assay to determine SMN1 and SMN2 copy numbers in 628 Malians, 120 Nigerians, and 120 Kenyans. We also explored possible mechanisms for SMN1 and SMN2 copy number differences in Malians, and investigated their effects on SMN mRNA and protein levels.
Results
The SMA carrier frequency in Malians is 1 in 209, lower than in Eurasians. Malians and other sub‐Saharan Africans are more likely to have ≥3 copies of SMN1 than Eurasians, and more likely to lack SMN2 than Europeans. There was no evidence of gene conversion, gene locus duplication, or natural selection from malaria resistance to account for the higher SMN1 copy numbers in Malians. High SMN1 copy numbers were not associated with increased SMN mRNA or protein levels in human cell lines.
Interpretation
SMA carrier frequencies are much lower in sub‐Saharan Africans than in Eurasians. This finding is important to consider in SMA genetic counseling in individuals with black African ancestry. Ann Neurol 2014;75:525–532
Spinal muscular atrophy (SMA) is caused by deletions and other mutations in the survival motor neuron 1 (SMN1) gene at chromosome 5q13.1 These genetic lesions result in loss of α‐motor neurons, leading to muscle weakness and atrophy. SMN2, which is highly homologous to SMN1, modifies the severity of disease.4 About 95 to 98% of SMA patients are homozygous for deletion of SMN1, and the remaining 2 to 5% are compound heterozygotes for deletion and other mutation in SMN1. SMA is a leading genetic cause of infant mortality, with an estimated incidence of 1 in 8,000 to 10,000 live births and a carrier frequency of 1 in 30–50 in populations of European ancestry.6 Recent data suggest that the SMA carrier frequency is lower in persons of black African ancestry in Cuba,7 South Africa,8 and the United States.9
Despite the abundance of other autosomal recessive neuromuscular diseases due to relatively high consanguinity in Mali,10 only 1 case of SMA has been clinically diagnosed in the country (unpublished data). Given the low incidence of SMA in Mali, we hypothesized that the SMA carrier frequency in this country is low. Here we show that in Malian and other sub‐Saharan African populations, SMN1 copy numbers are higher than in Europeans and Asians. We also explore possible explanations for SMN copy number variation in Mali, and investigate relationships between SMN copy number and SMN mRNA and protein levels.
Subjects and Methods
Sampling of Healthy Controls and DNA Extraction
Protocols were approved by the neurosciences institutional review board (IRB) at the National Institutes of Health (NIH), and the Ethical Committee at the Faculty of Medicine and Odontostomatology (FMOS), University of Bamako. All participants provided written informed consent.
Healthy adult FMOS students of Malian descent and nationality were eligible for the study. In a pilot study we collected blood samples from 40 donors at the NIH Blood Bank and 30 students at the FMOS. In addition, we used 15 samples collected from healthy controls for a previous NIH study.11 We subsequently recruited another 671 students at the FMOS. Genomic DNA was extracted from whole blood at the NIH and from buffy coats at the FMOS using the Gentra Puregene blood kit (Qiagen, Gaithersburg, MD). DNA samples were shipped to the NIH, and aliquots were shipped to Integrated Genetics (Westborough, MA) for SMN1 copy number determination.
We also obtained DNA samples from healthy controls of Luhya (Kenya, n = 120) and Yoruba (Nigeria, n = 120) ethnicities from the Coriell Institute (Camden, NJ).
Quantification of SMN Copy Number
SMN1 and SMN2 copy numbers were quantified in 628 and 613 of the 671 Malian samples, respectively. SMN1 copy number was quantified in 542 of the samples at Integrated Genetics and an additional 86 samples at the NIH. SMN2 copy numbers were quantified in all 613 samples at the NIH. The copy number determinations were done by quantitative real‐time polymerase chain reaction (qPCR) technique based on Taqman technology (Life Technologies, Carlsbad, CA; Roche Molecular Systems, Pleasanton, CA).9 The same methods were used at the NIH to quantify SMN1 and SMN2 copy numbers in the Nigerian and Kenyan samples. Primers and methods for the SMN copy number estimation were previously published.11
Identification of SMN Hybrid Genes
We amplified SMN from intron 6 to exon 8 by PCR from genomic DNA using previously reported primers and conditions13 (12 NIH samples and 20 Malian samples with ≥3 SMN1 copies). We then used the TA cloning kit (Life Technologies) to subclone 15 to 20 colonies per sample. PCR products from each clone were subsequently digested by DdeI and EcoRV. Tris‐borate‐EDTA gel electrophoresis was done as published.13
PCR products for each clone were also sequenced at the National Institute of Neurologic Disorders and Stroke DNA sequencing facility. Sequences were analyzed based on the 5 known nucleotide differences between SMN1 and SMN2 intron 6 to exon 8.14 SMN hybrids were identified by the association of exon 7 from SMN1 and exon 8 from SMN2, or vice versa. To validate results, we amplified and sequenced SMN intron 6 to exon 8 in 18 Nigerian samples (11 with 3–4 copies of SMN1 and 7 with 2 copies of SMN1) and 10 NIH and 10 Malian samples known to lack SMN2 (16 colonies per sample).
SMN1 Copy Number and Malaria Susceptibility
We obtained 1,204 genomic DNA samples from a cohort of children aged 6 months to 17 years in the village of Kenieroba, Mali. This cohort was followed through 4 complete transmission seasons (2008–2011) to record the frequency and severity of all Plasmodium falciparum malaria episodes (Lopera‐Mesa et al, in preparation). We quantified SMN1 copy number in the samples by qPCR using SYBR Green I dye (Life Technologies). We performed a Poisson regression on malaria episodes using SMN1 copy number and other variables known or suspected to influence malaria incidence and parasite density. The cohort study was also approved by the Ethical Committee at FMOS and by the IRB at the National Institute of Allergy and Infectious Diseases. Parents or guardians of the children provided written informed consent.
SMN1 Copy Number and Gene Duplication at the SMA Locus
We performed a real‐time SYBR Green I dye–based qPCR assay using primers15 to quantify the copy numbers of SMN1 exon 7, SMN2 exon 7, and the SMN1‐neighboring genes NAIP (neuronal apoptosis inhibitory protein) exon 5 and H4F5t exon 2 in 200 Malian DNA samples. Three Tunisian DNA samples with known copy number were used as controls.15 We compared the copy numbers of SMN1 exon 7 and the other genes to assess duplication at the SMA locus, using the Pearson chi‐square test and p < 0.05 to indicate a significant difference.
SMN Gene Localization by Fluorescent In Situ Hybridization Analysis
Metaphase preparations from Epstein–Barr virus–transformed lymphoblastoid cells were done by standard air‐drying technique, and fluorescent in situ hybridization (FISH) was done with labeled DNA by nick‐translation technique, essentially as described.16 Fifty nanograms of labeled probe (P13996, a 28kb genomic SMN clone kindly provided by Arthur Burghes, Ohio State University, Columbus, OH) was applied to each slide. Blocking, hybridization, and counterstaining were done as previously described.16
SMN Expression Analysis by qPCR
We obtained 24 lymphoblastoid cell lines with 1 to 4 copies of SMN1 from Nigerian and Kenyan donors (Coriell Institute). RNA and cDNA were prepared as described.11
We used Taqman qPCR to determine SMN1 copy numbers in 39 DNA samples from unrelated individuals in the Centre d'Etude du Polymorphisme Humain (CEPH) collection (Paris, France). We then used SMN mRNA expression data for the corresponding CEPH lymphoblasts from GeneNetwork at the University of Tennessee (http://www.genenetwork.org/ webqtl/WebQTL.py?cmd=sch&refseq=NM_000344&species= human; record ID ILMN_1665022) and compared the mean SMN mRNA level to the SMN1 copy number.
We determined SMN1 and SMN2 copy numbers and SMN mRNA expression levels in 15 human induced pluripotent stem (iPS) cell lines from non‐SMA individuals (7 healthy controls, 6 patients with spinal and bulbar muscular atrophy, and 2 patients with α‐sarcoglycan mutations). The iPS cells were made by lentiviral transduction or mRNA transfection with the reprogramming factors Oct4, Klf4, Sox2, and c‐Myc (Millipore, Billerica, MA, and Stemgent, Cambridge, MA, respectively).
Western Blot Analysis
We extracted protein lysates from the fibroblast and lymphoblastoid cell lines and performed a semiquantitative Western blot using 40μg of protein lysate and human monoclonal anti‐SMN antibody as described previously.17
Statistical Methods
Confidence intervals (CIs) on proportions were calculated by exact (Clopper–Pearson) method, and comparisons between proportions used the central Fisher exact test and the associated CIs.18 To test for a relationship to malaria, we performed 2 models: a Poisson regression on the malaria episodes adjusted for linear overdispersion and a generalized estimating equation model on the log10 parasite (P. falciparum) density values. As in Lopera‐Mesa et al (in preparation), the models adjusted for age, hemoglobin type, α‐thalassemia, glucose‐6‐phosphate dehydrogenase deficiency, ABO blood group, village, sex, and ethnicity. SMN1 copy number was included as a continuous variable not rounded to the nearest integer.
We used Spearman rank correlation with correction for ties to examine the association between SMN mRNA expression and SMN1 copy number, and used Fisher exact test to examine the association between gender and SMN1 copy number.
Results
High SMN1 and Low SMN2 Copy Numbers in Sub‐Saharan Africans
We determined SMN1 copy number in individuals of black African ancestry (628 Malians of mixed ethnicity, 120 Nigerians of Yoruba ethnicity, and 120 Kenyans of Luhya ethnicity). Malian adult participants were 70% male and had a mean ± standard deviation age of 22 ± 2.2 years. The frequency of SMN1 copy numbers in these 3 populations and from other healthy populations reported in the literature9 are listed (Table 1). In our study, samples with 4 or 5 copies of SMN1 could not be reliably distinguished from each another and were combined. The frequency of SMA carriers with just 1 copy of SMN1 was 1 in 209 (3 in 628; 95% CI = 1 in 74–1,014) in the Malian population. Overall, the SMN1 copy number distribution was significantly different between the Malians and other sub‐Saharan Africans compared to previously reported European and Asian populations (p < 0.0001). The distribution of SMN1 copy number for the Malians was also significantly different from the Nigerians (p = 0.006) and Kenyans (p = 0.002), likely due to 53% of the Malians having ≥3 copies of SMN1, compared to 38% for both the Nigerian and Kenyan samples. There was significant association between SMN1 copy number and gender in the Malians (p = 0.002), likely due to the males having a higher proportion (57%) of ≥3 copies of SMN1 compared to 41% for females.
Table 1. Frequency of SMN1 Copy Number in Different Geographical Regions.
| Sub‐Saharan Africa | North America | Europe | Asia | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SMN1 Copy # | Mali, n = 628 | Nigeria, n = 120 | Kenya, n = 120 | African American, n = 1,015 | Caucasian, n = 1,028 | Germany,n = 140 | France, n = 621 | Sweden, n = 502 | South Korea, n = 326 | Iran, n = 200 |
| 1 | 3 (0.5%) | 2 (2%) | 1 (1%) | 11 (1%) | 28 (2.7%) | 4 (4%) | 13 (2%) | 9 (2%) | 7 (2%) | 10 (5%) |
| 2 | 295 (47%) | 72 (60%) | 73 (61%) | 529 (52%) | 935 (91%) | 132 (94%) | 593 (96%) | 479 (95%) | 254 (78%) | 150 (75%) |
| 3 | 249 (40%) | 30 (25%) | 27 (22%) | 475 (47%)a | 65 (6.3%) | 3 (2%) | 15 (2%) | 14 (3%) | 62 (19%) | 30 (15%) |
| 4 | 81 (12.5%)b | 16 (13%) | 19 (16%) | – | – | 1 (1%) | – | – | 3 (1%) | 10 (5%) |
| Study | ‐‐‐‐‐‐‐‐Present study‐‐‐‐‐‐‐‐ | Hendrickson 20099 | Feldkötter 200219 | Corcia 201220 | Lee 200421 | Hasanzad 200922 | ||||
Three or more SMN1 copies.
Four or more SMN1 copies.
We also determined SMN2 copy number in 613 Malian, 120 Nigerian, and 120 Kenyan samples, and compared the copy number distribution to those of healthy subjects reported in the literature (Table 2). The SMN2 copy number distribution in Malians was significantly different from Nigerians (p = 0.02) but not Kenyans (p = 0.26). When we compare the results in sub‐Saharan Africans from our study with previously reported results in Europeans (see Table 2), the percentage of Africans with no SMN2 (19–27%) is significantly higher (odds ratio = 3.7; 95% CI = 2.8–4.8; p < 0.0001) than in individuals of European ancestry (8–9%).19
Table 2. Frequency of SMN2 Copy Number in Different Geographical Regions.
| Sub‐Saharan Africa | Europe | |||||
|---|---|---|---|---|---|---|
| SMN2 Copy# | Mali, n = 613 | Nigeria, n = 120 | Kenya, n = 120 | Germany, n = 100 | France, n = 621 | Sweden, n = 502 |
| 0 | 150 (24%) | 33 (27%) | 23 (19%) | 9 (9%) | 52 (8%) | 37 (8%) |
| 1 | 276 (45%) | 66 (55%) | 53 (44%) | 38 (38%) | 239 (38%) | 212 (42%) |
| 2 | 172 (28%) | 21 (18%) | 38 (32%) | 48 (48%) | 321 (52%) | 247 (49% |
| 3 | 15 (3%)a | — | 6 (5%) | 5 (5%) | 9 (2%) | 6 (1%) |
| Study | ‐‐‐‐‐‐‐‐Present study‐‐‐‐‐‐‐ | Anhuf 200323 | Corcia 201220 | |||
Three or 4 SMN2 copies.
Causes of SMN Copy Number Differences
The differences in SMN1 and SMN2 copy numbers between sub‐Saharan Africans and Eurasians could be due to increased SMN2 to SMN1 gene conversion, natural selection of high SMN1 copy number (e.g., for protection against P. falciparum malaria), or SMN1 duplication at the SMA locus or elsewhere in the African genome.
To investigate a role for gene conversion, we used standard genotyping to identify 20 Malian and 12 NIH samples with ≥3 SMN1 copies. We found that 14% (49 of 343) and 9% (15 of 173) of SMN clones were SMN1/2 hybrids by restriction digestion in the Malian and NIH samples, respectively (Table 3). Identification of hybrid clones by sequencing gave similar results (see Table 3). The restriction digestion results were also similar in 11 Nigerian samples, particularly those with ≥3 SMN1 copies, where 4% of clones (7 of 165) were SMN hybrids. The frequency of SMN hybrid clones increased with SMN1 copy number; we found no SMN hybrids in 10 Malian and 10 NIH samples with a 2SMN1/0SMN2 genotype. Thus, SMN hybrid genes may account for part but not all of the differences in SMN1 and SMN2 copy numbers we observed.
Table 3. Frequency of SMN1, SMN2, and SMN Hybrid Genes Identified by Restriction Endonuclease Digestion and Sequence Analysis.
| Number of Clones Identified by Restriction Digestion | |||||
|---|---|---|---|---|---|
| Origin of Samples | SMN1 | SMN2 | SMN Hybrid | Total | |
| Mali, n = 20 | 264 | 40 | 49 (14%) | 343 | |
| NIH, n = 12 | 136 | 22 | 15 (9%) | 173 | |
| Number of Clones Identified by Sequencing | |||||
| SMN1 | SMN2 | SMN1>2 Hybrid | SMN2>1 Hybrid | Total | |
| Mali, n = 20 | 195 | 2 | 3 | 46 (19%) | 246 |
| NIH, n = 12 | 111 | 3 | 4 | 9 (7%) | 127 |
To explore whether high SMN1 copy numbers may have been naturally selected for resistance to P. falciparum malaria or high parasite densities, we included our SMN1 copy data in an analysis of genetic resistance to these outcomes (Lopera‐Mesa et al, in preparation). We saw no difference in average number of malaria episodes by type of episode or whether SMN1 copy number is ≥3 (Table 4). In an analysis that accounts for age, sickle‐cell trait, and other malaria‐protective host factors, we found no association between SMN1 copy number and malaria incidence. Specifically, the relative risk (RR) of a malaria episode (severe or mild) for each additional SMN1 copy is not significantly different from 1 (RR = 0.97, 95% CI = 0.92–1.03, p = 0.3). For comparison, these data are sufficient to show clearly the protective effect of sickle‐cell trait (RR = 0.67, 95% CI = 0.58–0.76, p < 0.0001). Also, the change in average log10 parasitemia for each additional SMN1 copy number is not significantly different from 0 (−0.004; 95% CI = −0.046 to 0.039, p = 0.9).
Table 4. Incidence of Plasmodium falciparum Malaria Episodes, Stratified by SMN1 Copy Number.
| Categorization | Average No. of Malaria Episodes | |||
|---|---|---|---|---|
| SMN1 Copy# | No. | Mild and Severe | Mild | Severe |
| ≤2 (low) | 999 | 2.85 | 2.63 | 0.21 |
| ≥3 (high) | 205 | 2.89 | 2.63 | 0.26 |
Regarding the possibility of locus duplication, we did not find increased copy numbers of other genes at the SMA locus (NAIPt and H4F5t), excluding the possibility of segmental duplication (data available on request). FISH analysis showed that all SMN1 copies are localized to the SMA locus at chromosome 5q13.1 (Fig 1).
Figure 1.
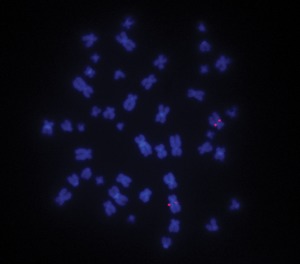
Fluorescence in situ hybridization (FISH) analysis at the SMN locus. FISH analysis in lymphoblastoid cell line #19441 with a 4 SMN1/0 SMN2 genotype shows signal amplification of both copies on chromosome 5 and not elsewhere.
Effects of SMN Copy Number Differences
We did not find significant increases in SMN mRNA level with increasing SMN1 copy number in 24 lymphoblast samples of Nigerian and Kenyan origin (p = 0.14; Fig 2A), in 15 fibroblast samples of unknown ethnic origin from the Coriell Institute (not shown), or in 39 lymphoblast samples of unknown ethnic origin from CEPH (p = 0.88; see Fig 2B). SMN protein levels measured by Western blot analysis also did not vary with SMN1 copy number in lymphoblasts or fibroblasts (not shown). iPS cells showed a trend toward increasing SMN mRNA levels with increasing SMN1 copy number, but this was not significant (p = 0.32; see Fig 2C).
Figure 2.
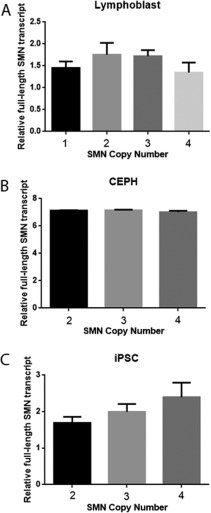
Total SMN mRNA levels in different cells. (A) Coriell lymphoblasts (n = 3 with SMN1 copy number 1, 7 with copy number 2, 11 with copy number 3, and 3 with copy number 4). (B) Centre d'Etude du Polymorphisme Humain (CEPH) lymphoblasts (n = 33 with SMN1 copy number 2, 3 with copy number 3, and 3 with copy number 4). (C) Induced pluripotent stem cells (iPSC; n = 8 with SMN1 copy number 2, 4 with copy number 3, and 3 with copy number 4). Mean and standard error of the mean are shown.
Discussion
Until recently SMA was described as a “panethnic” condition, implying similar carrier frequencies and disease incidence worldwide. However, most SMA carrier frequency data have been derived from populations in the United States, Europe, and Asia. Most population‐based studies of SMA carrier frequency have provided few or no data on ethnicity, and few studies have been performed in African populations. Studies on SMA patients in North Africa24 show similar results to those in the United States and Europe. However, inherited neurological diseases in general and SMA in particular have been understudied in sub‐Saharan Africa, in part because of the lack of genetic diagnostics. Although SMA type 1 is reported to be rare in black Africans,7 it is not known whether genetic modifiers of SMA are present in this population. To address this possibility, we investigated SMN copy number variation in healthy Malian adults. We sampled medical students at the University of Bamako because they are representative of the general Malian population in ethnic background and geographic origin, and research facilities are available for rapid onsite processing of blood samples. The low SMA carrier frequency we found in this population is consistent with the apparent rarity of SMA in Mali.10
The cause of high SMN1 copy number in sub‐Saharan Africans relative to Eurasians remains to be determined, although our findings exclude such possibilities as gene conversion, selective pressure due to malaria resistance, and locus duplication. SMN1 to SMN2 gene conversion has been reported in SMA patients with late onset disease, milder phenotype, or both.28 Conversely, SMN2 to SMN1 conversion is suggested by the finding that individuals with higher SMN1 copies tend to have lower SMN2 copies.31 Although we were able to identify previously reported13 SMN1/2 hybrids in Malians and Nigerians, their frequency is too low to explain the high SMN1 and low SMN2 copy numbers in these populations. The relative prevalence of high SMN1 (and low SMN2) copy number in sub‐Saharan Africa, or low SMN1 (and high SMN2) copy number elsewhere, suggests that diseases with significant morbidity or mortality in these regions select for SMN variation. Although we found no overall association with malaria susceptibility, other infectious diseases or environmental hazards may be selecting for SMN copy number variants. The finding that SMN1 copy number can vary from 0 to 4 per chromosome in whites14 suggested to us that locus duplication may have increased SMN1 copy number to ≥4 in Malians. Because pseudogenes may arise from retrotranscription33 and be located at genomic loci distinct from their origin, we used FISH to analyze this possibility in our Malian samples. FISH analysis showed SMN signal only at 5q13.1, not at 6p21.3 (a paralogous locus containing NAIP exon 9,34 which has a high degree of nucleotide similarity to SMN) or elsewhere.
Given that gene conversion, natural selection by malaria, and locus duplication did not explain the relatively high SMN1 copy numbers in Malians, we considered potential roles for the high degree of consanguinity and the “bottleneck phenomenon.” Data on genetic disorders in Arab populations extracted from the Catalogue of Transmission Genetics in Arabs database (http://www.cags.org.ae) indicate a relative abundance of recessive disorders associated with the practice of consanguinity, with a rate of first cousin marriage of 25 to 30%.35 Consanguinity was reported by 27% of our Malian participants, and parental first cousin marriage was reported by 17%. If consanguinity is causal, then we would expect similar SMA carrier frequencies and SMN copy number distributions in other populations with high consanguinity, but this has not been reported. For example, studies in Saudi Arabia and Egypt found an SMA carrier frequency of 1 in 20.36 Finally, high SMN1 copy numbers in sub‐Saharan Africans may be due to the bottleneck phenomenon, meaning that by chance the population that migrated out of Africa to Asia and Europe had lower SMN1 copy number or randomly drifted in this direction after the outmigration.39 This may be the most likely explanation for the different SMN1 distributions in these areas. Regardless of the cause, our assessment of SMN copy number in different ethnic groups helps to appropriately target SMA carrier testing and provide accurate risk assessment to individuals from different populations.
A fundamental question is how SMN1 copy number variation affects gene expression and phenotypic traits. It has been reported that African populations have a higher frequency of NAIP duplication than Eurasians, with a relative increase in transcription of this gene.40 Although we detected no such correlation between SMN1 copy number and expression levels in lymphoblasts and fibroblasts, there was a trend toward correlation in iPS cells. The lack of correlation in lymphoblasts and fibroblasts may indicate feedback regulation to maintain constant SMN mRNA and protein levels in these cells, or reflect a limit on gene transcription due to epigenetic factors. Further study of this phenomenon in different cell types under different conditions could help to identify factors responsible for regulating SMN1 expression, which could be targets for therapeutics development.
Authorship
H.A.S., M.T., A.G., F.D., F.N.Y., K.Ba., N.B., G.L., Y.I.C., and Y.M. contributed to the writing and approval of the protocol, organized and participated in the study design and sampling in Mali, and edited the manuscript. A.A., M.G., and A.S. provided control DNA samples for the gene duplication analysis and contributed to the experimental design and statistical analysis. They also participated in the writing of the manuscript. B.H. and T.S. generated data and edited the manuscript. R.M.F., M.D., and M.P.F. provided DNA samples, helped design the study, did statistical analysis, and participated in the manuscript writing. C.G., G.C., M.B., and K.Z. provided key cellular reagents, helped with the study design and data analysis, and provided critical feedback on the manuscript. H.‐S.L., A.D. and E.P. helped with the study design, generated data, and helped in the writing of the manuscript. S.A. did statistical analysis for the protocol design and the manuscript and gave critical feedback. M.S., K.C., J.N., A.B.Sc., A.B.Si., G.H., K.Br., K.G.M., B.G.B., and K.H.F. wrote the protocol, participated in the study design, generated data, and wrote the manuscript.
Potential Conflicts of Interest
B.H.: Employer Integrated Genetics is a commercial testing laboratory that performs SMN1 copy number analysis.
Acknowledgment
This work was supported by intramural research funding from the NIH National Institute of Neurologic Disorders and Stroke (NINDS) and National Institute of Allergy and Infectious Diseases.
We thank Dr J. E. Bailey‐Wilson, Dr A. F. Wilson, L. Fernandez‐Rhodes, C. Watts, M. J. White, A. Kokkinis, and Dr S. Parodi for their help with study design; Drs S. Doumbia, S. Ceryak, E. Hoffman, and H. Kaminski for their valuable input; Drs A. Burghes and V. McGovern for providing us with the P13996 clone; Dr C. Sumner for providing us with genomic DNA samples and leukocyte concentrates from SMA patients and families; Drs A. Tounkara at the University of Bamako and L. Goldfarb at NINDS for the use of their laboratories for DNA extraction and analysis; J. Nagel for DNA sequencing; Dr G. Tullo for help with the malaria susceptibility analysis; and the study participants for their time and effort in contributing to the success of this project.
References
- 1.Brzustowicz LM, Lehner T, Castilla LH, et al. Genetic mapping of chronic childhood‐onset spinal muscular atrophy to chromosome 5q11.2‐13.3. Nature 1990;344:540–541 [DOI] [PubMed] [Google Scholar]
- 2.Melki J, Abdelhak S, Sheth P, et al. Gene for chronic proximal spinal muscular atrophies maps to chromosome 5q. Nature 1990;344:767–768 [DOI] [PubMed] [Google Scholar]
- 3.Zerres K, Rudnik‐Schoneborn S. Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol 1995;52:518–523 [DOI] [PubMed] [Google Scholar]
- 4.Lefebvre S, Burlet P, Liu Q, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 1997;16:265–269 [DOI] [PubMed] [Google Scholar]
- 5.Wirth B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy. Hum Mutat 2000;15:228–237 [DOI] [PubMed] [Google Scholar]
- 6.Ogino S, Wilson RB, Gold B. New insights on the evolution of the SMN1 and SMN2 region: simulation and meta‐analysis for allele and haplotype frequency calculations. Eur J Hum Genet 2004;12:1015–1023 [DOI] [PubMed] [Google Scholar]
- 7.Zaldivar T, Montejo Y, Acevedo AM, et al. Evidence of reduced frequency of spinal muscular atrophy type I in the Cuban population. Neurology 2005;65:636–638 [DOI] [PubMed] [Google Scholar]
- 8.Labrum R, Rodda J, Krause A. The molecular basis of spinal muscular atrophy (SMA) in South African black patients. Neuromusc Disord 2007;17:684–692 [DOI] [PubMed] [Google Scholar]
- 9.Hendrickson BC, Donohoe C, Akmaev VR, et al. SMN1 allele frequencies in the major ethnic groups within North America. J Med Genet 2009;46:641–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meilleur KG, Coulibaly S, Traoré M, et al. Genetic testing and counseling for hereditary neurological diseases in Mali, West Africa. J Community Genet 2011;2:33–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sumner CJ, Kolb SJ, Harmison GG, et al. SMN mRNA and protein levels in the peripheral blood: biomarkers for SMA clinical trials. Neurology 2006;66:1067–1073 [DOI] [PubMed] [Google Scholar]
- 12.Chan V, Yip B, Yam I, et al. Carrier incidence for spinal muscular atrophy in Southern Chinese. J Neurol 2004;251:1089–1093 [DOI] [PubMed] [Google Scholar]
- 13.Van der Steege G, Grootscholten PM, Cobben JM, et al. Apparent gene conversions involving the SMN gene in the region of the spinal muscular atrophy locus on chromosome 5. Am J Hum Genet 1996;59:834–838 [PMC free article] [PubMed] [Google Scholar]
- 14.Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet 1999;8:1177–1183 [DOI] [PubMed] [Google Scholar]
- 15.Amara A, Adala L, Ben Charfeddine I, et al. Correlation of SMN2, NAIP, p44, H4F5 and occludin genes copy number with spinal muscular atrophy phenotype in Tunisian patients. Eur J Paediatr Neurol 2012;16:167–174 [DOI] [PubMed] [Google Scholar]
- 16.Dutra AS, Mignot E, Puck JM. Gene localization and syntenic mapping by FISH in the dog. Cytogenet Cell Genet 1996;74:113–117 [DOI] [PubMed] [Google Scholar]
- 17.Burnett BG, Muñoz E, Tandon A, et al. Regulation of SMN protein stability. Mol Cell Biol 2009,29:1107–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fay MP. Confidence intervals that match Fisher's exact or Blaker's exact tests. Biostatistics 2010;11:373–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feldkötter M, Schwarzer V, Wirth R, et al. Quantitative analyses of SMN1 and SMN2 based on real‐time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002;70:358–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Corcia P, Ingre C, Blasco H, et al. Homozygous SMN2 deletion is a protective factor in the Swedish ALS population. Eur J Hum Genet 2012;20:588–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee T‐M, Kim S‐W, Jung S‐C. Quantitative analysis of SMN1 gene and estimation of SMN1 deletion carrier frequency in Korean population based on real‐time PCR. J Korean Med Sci 2004;19:870–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hasanzad M, Azad M, Kahrizi K, et al. Deletions in the survival motor neuron gene in Iranian patients with spinal muscular atrophy. Ann Acad Med 2009;38:139–141 [PubMed] [Google Scholar]
- 23.Anhuf D, Eggermann T, Rudnik‐Schoneborn S, et al. Determination of SMN1 and SMN2 copy number using TaqMan technology. Hum Mutat 2003;22:74–78 [DOI] [PubMed] [Google Scholar]
- 24.Radhakrishnan K, Thacker AK, Maloo JC, et al. Descriptive epidemiology of some rare neurological diseases in Benghazi, Libya. Neuroepidemiology 1988;7:159–164 [DOI] [PubMed] [Google Scholar]
- 25.Bouhouche A, Benomar A, Birouk N, et al. High incidence of SMN1 gene deletion in Moroccan adult‐onset spinal muscular atrophy patients. J Neurol 2003;250:1209–1213 [DOI] [PubMed] [Google Scholar]
- 26.Essawi ML, Effat LK, Shanab GML, et al. Molecular analysis of SMN1 and NAIP genes in Egyptian patients with spinal muscular atrophy. Bratisl Lek Listy 2007;108:133–137 [PubMed] [Google Scholar]
- 27.Lumaka A, Bone D, Lukoo R, et al. Werdnig‐Hoffmann disease: report of the first case clinically identified and genetically confirmed in central Africa (Kinshasa‐Congo). Genet Counsel 2009;20:349–358 [PubMed] [Google Scholar]
- 28.Velasco E, Valero C, Valero A, et al. Molecular analysis of the SMN and NAIP genes in Spanish spinal muscular atrophy (SMA) families and correlation between number of copies of cBCD541 and SMA phenotype. Hum Mol Genet 1996;5:257–263 [DOI] [PubMed] [Google Scholar]
- 29.Campbell L, Potter A, Ignatius J, et al. Genomic variation and gene conversion in spinal muscular atrophy: implications for disease process and clinical phenotype. Am J Hum Genet 1997;61:40–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazzei R, Gambardella A, Conforti FL, et al. Gene conversion events in adult onset spinal muscular atrophy. Acta Neurol Scand 2003;109:151–154 [DOI] [PubMed] [Google Scholar]
- 31.Ogino S, Gao S, Leonard DGB, et al. Inverse correlation between SMN1 and SMN2 copy numbers: evidence for gene conversion from SMN2 to SMN1. Eur J Hum Genet 2003;11:275–277 [DOI] [PubMed] [Google Scholar]
- 32.Chen T‐H, Tzeng C‐C, Wang C‐C, et al. Identification of bidirectional gene conversion between SMN1 and SMN2 by simultaneous analysis of SMN dosage and hybrid genes in a Chinese population. J Neurol Sci 2011;308:83–87 [DOI] [PubMed] [Google Scholar]
- 33.Hurles M. Gene duplication: the genomic trade in spare parts. PLoS Biol 2004;2:900–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banyer JL, Goldwurm S, Cullen L, et al. The spinal muscular atrophy gene region at 5q13.1 has a paralogous chromosomal region at 6p2l.3. Mamm Genome 1998;9:235–239 [DOI] [PubMed] [Google Scholar]
- 35.Tadmouri GO, Nair P, Obeid T, et al. Consanguinity and reproductive health among Arabs. Reprod Health 2009;6:17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shawky RM, El‐Sayed NS. Clinico‐epidemiologic characteristics of spinal muscular atrophy among Egyptians. Egyptian J Med Human Genet 2011;12:25–30 [Google Scholar]
- 37.El‐Rajeh S, Bademosi O, Ismail H, et al. A community survey of neurological disorders in Saudi Arabia. The Thugbah Study. Neuroepidemiology 1993;12:164–178 [DOI] [PubMed] [Google Scholar]
- 38.Al‐Jumah M, Majumdar R, Rehana Z, et al. A pilot study of spinal muscular atrophy carrier screening in Saudi Arabia. J Pediatr Neurol 2007;5:221–224 [Google Scholar]
- 39.Reed FA, Tishkoff S. African human diversity, origins and migrations. Curr Opin Genet Dev 2006;16:597–605 [DOI] [PubMed] [Google Scholar]
- 40.Boniotto M, Tailleux L, Lomma M, et al. Population variation in NAIP functional copy number confers increased cell death upon Legionella pneumophila infection. Hum Immunol 2011;73:196–200 [DOI] [PubMed] [Google Scholar]