

Abstract
Over the past few decades, non-alcoholic fatty liver disease (NAFLD) has become one, if not the most common, cause of chronic liver disease affecting both adults and children. The increasing number of cases at an early age is the most worrying aspect of this pathology, since it provides more time for its evolution. The spectrum of this disease ranges from liver steatosis to steatohepatitis, fibrosis and in some cases, hepatocellular carcinoma. NAFLD may not always be considered a benign disease and hepatologists must be cautious in the presence of fatty liver. This should prompt the use of the available experimental models to understand better the pathogenesis and to develop a rational treatment of a disease that is dangerously increasing. In spite of the growing efforts, the pathogenesis of NAFLD is still poorly understood. In the present article we review the most relevant hypotheses and evidence that account for the progression of NAFLD to non-alcoholic steatohepatitis (NASH) and fibrosis. The available in vitro and in vivo experimental models of NASH are discussed and revised in terms of their validity in translational studies. These studies must be aimed at the discovery of the still unknown triggers or mediators that induce the progression of hepatic inflammation, apoptosis and fibrosis.
Keywords: Fatty Liver, Obesity, Metabolic syndrome, Inflammation, In vitro, Experimental model
Core tip: The molecular mechanism associated with the accumulation of fatty acids in the liver cells and the resulting molecular cascade leading to hepatic damage is far from being understood. Due to the development of reliable in vitro and in vivo models, we are starting to open the “black box”. This will lead to a better understanding of the active clinical condition and hopefully to a more effective treatment. This article critically reviews what is known and what has still to be discovered about the link between the accumulation of fat within the liver and the resulting damage.
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is a complex spectrum of diseases ranging from benign steatosis (usually asymptomatic) to more severe alterations like non-alcoholic steatohepatitis (NASH), cirrhosis and, in some cases, hepatocellular carcinoma (HCC). The most serious aspect of the disease is the high incidence in pediatric and adolescent populations, providing longer time for evolution[1]. Day by day, social and medical operators witness the dramatic increase in the incidence of this phenomenon. The “global society” is driving us towards a global epidemic of obesity, type 2 diabetes mellitus (T2DM) and metabolic syndrome (MS). NAFLD and NASH are strictly linked to the presence of insulin resistance (IR) and are nowadays considered the hepatic manifestation of the MS[2]. Although most typical forms of NAFLD are overwhelmingly associated with IR and MS, it cannot be said that IR and MS are invariably associated with fatty liver[3]. Interestingly, NAFLD markers have also been associated with IR in type 1 diabetes[4], which is not closely related to the MS. Hepatology and Gastroenterology communities are facing a great challenge since within few years, NAFLD will be the most important chronic liver disease worldwide (Figure 1).
Figure 1.
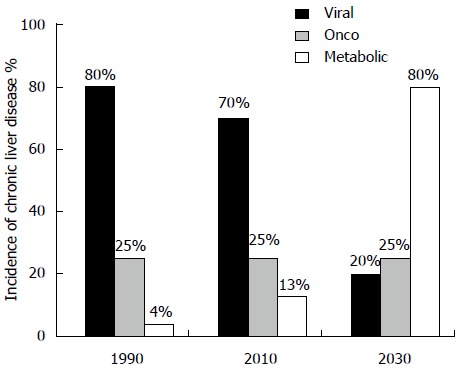
Estimation of the main etiological incidence of past, present and future chronic liver diseases according to the available data from the United States[130] and Europe[131,132].
Lifestyle changes have occurred in the industrialized societies due to the introduction of modern technologies resulting in eating more and more importantly, moving less. According to the Food and Agriculture Organization of the United Nations (FAO http://www.fao.org/docrep/x0262e/x0262e23.htm), in the next 40 years the daily caloric requirements will decrease by 350 calories. Several epidemiological studies have linked NAFLD to unhealthy diet and sedentary behaviors[5-7], and the only effective treatment for NAFLD and NASH is to guide the patient to a healthier lifestyle[8] with lifestyle coaching including personalized diet, physical activity and cognitive-behaviour therapy[9]. However, the lack of patient compliance is the main limitation of this approach. Although to a lesser extent, NAFLD can also occur in non-obese populations[10], suggesting that dietary composition is not the only cause of fatty liver. Several sets of data reviewed by Caldwell et al[3] showed that both ethnicity and genetic polymorphisms play a major role in the development and progression of the disease, and different genetic profiles might be also responsible for the variations of steatosis in the MS.
It is therefore of pivotal importance to further develop a strong translational approach to understand the pathophysiology of this new disease and to translate it into clinical practice. In the present paper, we review the most recently published data on the pathophysiology of NAFLD in an attempt to amalgamate the available information in order to contribute to the understanding of the factors involved, including a critical analysis of the in vitro and in vivo models.
PATHOGENESIS
The most accepted scheme to explain the development of NAFLD and the progression from simple steatosis to NASH is still based on theories. In 1998, Day[11] proposed the “two hits” theory. The “first hit” is characterized by the accumulation of lipids in hepatocytes due to an altered intrahepatic lipid metabolism, where insulin resistance seems to be the key pathogenic factor for the development of hepatic steatosis[12], while the “second hit” leads to hepatocyte injury, inflammation and fibrosis. Several factors were suggested to initiate the second hit such as: (1) proinflammatory cytokines and adipokines[13]; (2) mitochondrial dysfunction[13,14]; (3) oxidative stress; and (4) endoplasmic reticulum (ER) stress[15] with subsequent apoptosis. In 2010, a more complex, global and realistic model, the “multiparallel hits” hypothesis, was proposed to explain the pathogenesis of NAFLD[16]. In this model, the adipose tissue and gut-related factors play a key role in the initiation of hepatic inflammation, suggesting that simple steatosis and NASH might be two different disorders and pointing to new, non-hepatic players in the mechanisms of NAFLD and its progression.
Contributors to the development of insulin resistance
During the last few years, the interplay among gut microbiota, obesity and the metabolic consequences (liver sensitization) has become important. Some of the main factors involved in this process are summarized in Figure 2. Several clinical and experimental studies recently reviewed in detail[17] suggest that microbiotal factors may be the driving forces of IR[18], hepatic steatosis and subsequent inflammatory state. Changes in the composition of the gut microbiota might induce an increased permeability and translocation of bacterial endotoxins promoting a chronic inflammatory state. This condition can alter pathways such as insulin signaling, promoting the development of IR. The molecular basis of IR is the result of multiple genetic[19] and non-genetic mechanisms. IR can initiate a dangerous vicious circle, involving inflammation and hypercoagulability, which increases atherogenesis[20]. Data from animal models indicate that IR develops in the vasculature well before these responses are detected in muscle, liver, or adipose tissue[21]. These findings could explain the high cardiovascular risk observed in subjects with MS. Moreover, disruption in the endothelial insulin signaling can promote the development of atherosclerosis in the absence of diabetes-related risk factors including hyperglycemia and hyperinsulinemia. The development of atherosclerosis is associated with a reduced bioavailability of nitric oxide and an excessive production of reactive oxygen species[22]. The endothelial dysfunction might be mediated by FoxOs transcription factors; FoxOs inhibition in endothelial cells has been shown to have promising atheroprotective effects[23]. Altogether these findings are in agreement with previous data[24], reinforcing the idea that hepatic IR and hepatic steatosis might precede the development of T2DM. Epidemiological evidence (reviewed in detail elsewhere[25]) also suggests an association between MS and the risk of developing chronic kidney disorders beyond the contribution of hyperglycemia and high blood pressure. The increased rates of chronic kidney disease (CKD) and cardiovascular disease are the most important clinical features associated with NAFLD. To date, there is a mounting body of evidence (reviewed extensively by Targher et al[26]) suggesting that patients with NAFLD have multiple risk factors of CKD and that NAFLD is associated with an increased prevalence and incidence of CKD both in patients with and without diabetes. Renal dysfunction may be promoted by a mosaic of effects such as: (1) inflammatory cytokines released by the adipose tissue (TNF-α (tumor necrosis factor-α), IL6, adiponectin[27], leptin[28]); (2) obesity-related mechanisms such as altered renal hemodynamics; (3) excess of renal sodium reabsorption; (4) activation of renin-angiotensin and sympathetic nervous systems; and (5) physical compression of kidneys by adipose tissue.
Figure 2.
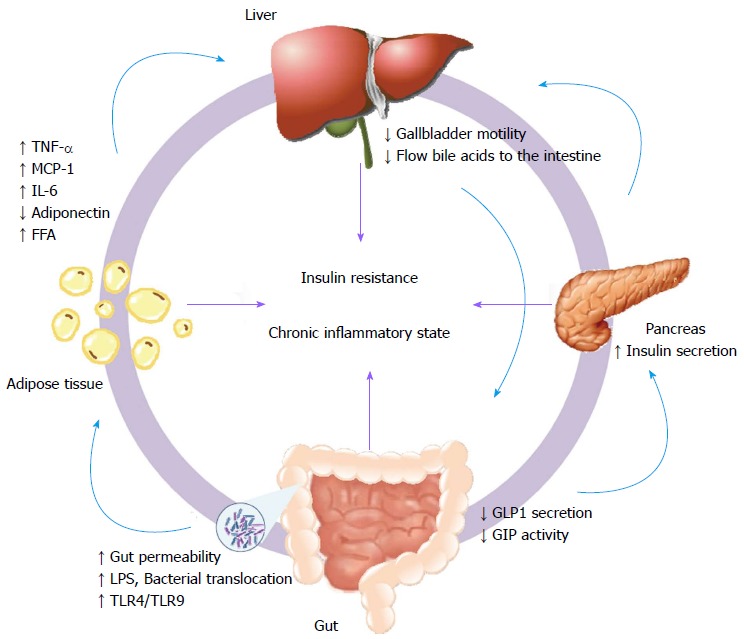
Extrahepatic factors involved in the pathogenesis of non-alcoholic fatty liver disease. The affected organs and their response are represented in a dynamic circle; in the center are indicated the main factors that contribute to the initiation/perpetuation of the hepatic injury (insulin resistance and chronic inflammatory state). The light blue arrows represent the organ-specific effects of each response. TNF-α: Tumor necrosis factor-α; MCP-1: Monocyte chemotactic protein 1; IL-6: Interleukin-6; FFA: Free fatty acids; LPS: Lipopolysaccharides; TLR: Toll-like receptor; GIP: Glucose-dependent insulinotropic peptide; GLP: Glucagon-like-peptide.
Over the last 14 years there has been a surge in the number of studies confirming that NAFLD is associated with IR (the “IR dogma”). Based on such studies, one could expect that, by correcting IR, NAFLD could be healed. Unfortunately, therapeutic studies[29] failed to confirm this expectation, suggesting a more complex interplay of factors involved in the pathogenic process.
Role of incretin hormones
Incretin hormones, such as glucose-dependent insulinotropic peptide (GIP) and glucagon-like-peptide 1 (GLP-1), are released by the gastrointestinal tract in response to nutrients that increase the glucose-mediated insulin secretion,31]. In patients with T2DM the incretin effect is severely reduced[32], due to an impaired secretion of GLP-1 and a decreased activity of GIP[33]. Recent in vitro[34] and in vivo[35] data clearly show that hepatocytes express GLP-1 receptors, and the exposure to GLP-1 agonists leads to: (1) a reduction of intracellular fat load[36,37]; (2) enhanced fat oxidation[38]; and (3) an induction of macroautophagy[39], which is a critical process for the removal of toxic fatty acids from cells. Other important regulators of glucose homeostasis are the bile acids, which through various signaling pathways regulate cholesterol, fasting and mealtime glucose, and metabolism/energy homeostasis, as well as their own synthesis and blood levels in the enterohepatic circulation[40,41]. The composition of bile acids in T2DM has been shown to be altered[42] as a consequence of a reduced gallbladder motility resulting in a reduced secretion of bile acids to the intestine. A low bile acid concentration is associated with a reduction in the secretion of GLP-1 and consequently, an impaired glucose homeostasis with a decreased insulin secretion[43]. Paradoxically, patients with NAFLD can often present a hyperinsulinemic state. However, instead of a regulation of gluconeogenesis, insulin promotes de novo lipogenesis that exacerbates hepatic lipid deposition and accelerates the development of the disease. One possible mechanism to explain this situation could be the activation of sterol regulatory transcription factor element-binding protein-1c (SREBP1c), a master transcription factor regulator of lipid synthesis, through the stimulation of the target of rapamycin complex 1 (mTORC1)[44]. The regulation of incretin hormones represents a promising strategy for NAFLD. Therapy with GLP-1 agonists (like exenatide) in T2DM patients promotes a positive effect in the liver[45], since hepatocytes express GLP-1 receptor[34]. This compound might reduce or even reverse hepatic fat accumulation and reduce the triglyceride (TG) levels, most probably as a consequence of a reduced caloric intake, which is one of the main therapeutic contributions of this kind of drug[46]. Unfortunately, the success of bile acid interventions is limited in clinical practice and the results obtained are discordant from those observed in experimental models[47]. Hyperinsulinemia in NAFLD leads to upregulation of the production of insulin-like growth factor-1 (IGF-1) and activation of insulin receptor substrate (IRS)-1. This may activate several molecules and signaling pathways including p53, mitogen-activated protein kinases (MAPK), and phosphatidylinositol-3 kinase/Akt[48]. These pathways play a significant role in carcinogenesis by inducing cell proliferation and inhibition of cell apoptosis[49,50]. Thus, NAFLD and HCC appear to be regulated by similar signaling molecules and pathways related to inflammation. This evidence is particularly interesting to support the idea that NAFLD itself could promote HCC development in earlier stages, even in the absence of cirrhosis[51,52].
Alterations in hepatic lipid metabolism
TGs are the preferred nutritional storage to buffer fluctuations in energy demand and availability. TG physical properties allow their accumulation without adverse osmotic or colloidal effects. In higher organisms, TGs are stored mainly in adipocytes, and can be accumulated in other cell types only under particular circumstances. In this regard, an interesting example was presented by Cohen et al[53] in migratory birds that store large quantities of TGs in the liver as an energy source in preparation for prolonged seasonal flights. Like migratory birds, some humans who consume excess calories deposit fat in the liver, as a maladaptive process. The moiety of the intracellular fat has distinct toxic effects. As mentioned before, hepatic accumulation of neutral cholesterol esters and TG appears not to be a threat[54,55] (though this is still an open question[56]); however, the presence of the intermediate products seems to have a more deleterious effect on liver cells. An altered lipid metabolism leads to the accumulation of intermediate products such as diacylglycerol (DG) and phospholipids (sphingolipids and ceramides)[57-59], and these compounds account for the fatty acid-induced toxicity and for the hepatic IR (Figure 3). Moreover, these metabolites promote the activation of numerous kinases, including nPKC isoforms, MAPK, ERKs and c-Jun N-terminal kinase (JNK), S6K and inhibitor kappa beta kinase beta (IKKβ), that participate in the phosphorylation of the IRS inducing positive or negative effects on the insulin pathway[60]. Recent data suggest a connection between altered cholesterol homeostasis and hepatic free cholesterol (FC) accumulation as a trigger for the pathogenesis of NASH[61,62]. Most probably, FC accumulates within the ER membrane impairing its fluidity. The resulting stiffening of the ER membrane leads to an impaired activity triggering the ER stress and eventual unfolded protein response, cell apoptosis[63,64] via JNK signaling and to the release of RE Ca2+ stores. Adjacent mitochondria readily take up the released Ca2+, and the acute Ca2+ overload results in changes in mitochondrial potential and opening of the permeability transition pores (PTPs)[65] ensuring a potent cellular cell signal[66]. Dysregulation in nuclear transcription factors SREBP-2[67], liver X-receptor (LXR)-α and farnesoid X receptor (FXR) might be the cause of cholesterol altered homoeostasis (extensively reviewed by Musso[68]). Interestingly, the incidence of NAFLD in the non-obese population has been associated with a high dietary cholesterol intake rather than intake of polyunsaturated fatty acids[69].
Figure 3.
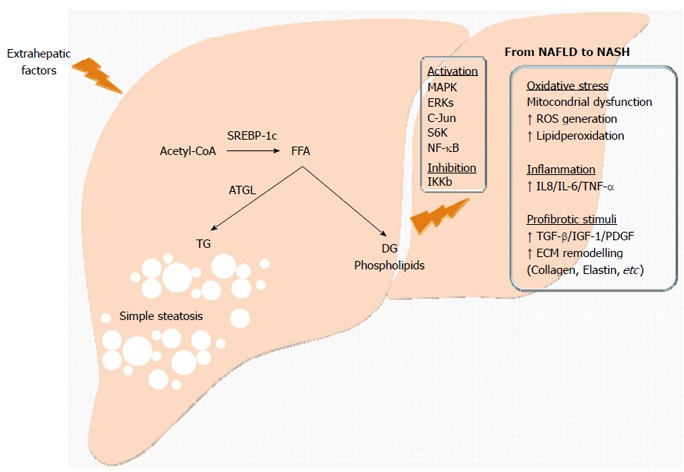
Effect of intracellular fat accumulation within the liver. Liver sensitization induces an alteration of the normal hepatic liver metabolism leading to simple steatosis with neutral triglyceride (TG) accumulation or in the more severe cases, to the production of intermediate products (DG and phospholipids) responsible for lipotoxicity. Alteration of several mediators of signaling pathways leads to the events observed during the progression from non-alcoholic fatty liver disease (NAFLD) to non-alcoholic steatohepatitis (NASH) (hepatic insulin resistance, oxidative stress, inflammation, and fibrosis). IKKb: Protein Kinase-1-mediated IB Kinase; ROS: Reactive oxygen species; IL-6: Interleukin-6; TNFα: Tumor necrosis factor-α; IGF-1: Insulin-like growth factor-1; PDGF: Platelet-derivedgrowth factor; ECM: Extracellular matrix; MAPK: Mitogen-activated protein kinases; ERKs: Extracellular signal-regulated kinases; NF-κB: Nuclear factor κB.
Chronic inflammatory state
Persistent IR associated with an excessive caloric diet and sedentary life style lead to obesity, now recognized as a chronic inflammatory disorder. Thus, inflammation is considered the major risk of obesity and is associated with white adipose tissue dysfunction. An altered adipokine profile has been suggested to play a pivotal role in the initiation and perpetuation of the pathological events[70,71]. In NAFLD, adipose tissue contributes to the systemic production of TNF-α[72], MCP1, IL6 and adiponectin; these mediators modify the hepatic inflammatory/immune system[56-59]. Furthermore, it has been reported that the adipose tissue of obese subjects presents an increased number of macrophages[73], and they might account for much of the adipose tissue inflammatory cytokine secretion. These cells presumably arise from peripheral blood monocytes that become activated by hyperinsulinemia and the abnormal levels of FFA encountered in individuals with IR. Monocytes have also been shown to be activated in poorly controlled type 1 diabetes, showing an increased ability to attach to the endothelial cells[74], one of the early stages in atherosclerosis. Moreover, it has been reported that monocytes are strongly correlated with glycated hemoglobin (HbA1c), explaining the association between monocytes and IR in type 1 diabetes[75]. Activation of these cells produces abundant quantities of cytokines such as TNF-α and IL6. Studies performed in human monocytes suggest that these cells might respond to the increased concentrations of saturated non-esterified fatty acids observed in IR conditions by producing high levels of IL6. This increased secretion of IL6 could prime these cells to generate a robust local or systemic inflammatory response contributing to the development of complications such as T2DM and atherosclerosis[76]. In the liver, fatty acid accumulation induces mainly the up-regulation of IL8, produced both by hepatocytes and non parenchymal cells[77-79]. It was reported that IL6 and TNF-α signaling via TNF-α receptor-1 are important in NASH-related development of HCC, and that hypoadiponectinemia accelerated hepatic tumor formation in the mouse model of NASH[80,81]. A detailed study of the role of the main cytokines in humans and animal models can be found in a recently published work from Braunersreuther et al[82]. Collectively, these data confirm the close relationship between lipid metabolism and liver cancer in animal experimental models, although there are still many doubts regarding human studies.
Contribution of oxidative stress
Several papers demonstrate that oxidative stress occurs during NAFLD, especially due to mitochondrial dysfunction[14,83-86]. It has been reported that activated hepatic mitochondrial metabolism[87] is a common characteristic of NAFLD in both human subjects[88] and animal models[89]. However, the regulatory connection linking FFA to altered mitochondrial function is still undefined. Currently, there are two competing views on the role of lipid beta-oxidation in the development of NAFLD[88,89]. One view holds that impaired or incomplete beta-oxidation leads to hepatic steatosis and accumulation of lipid intermediates that inhibit insulin signaling. The other view holds that increased supply of FFA to the liver results in excessive beta-oxidation that fuels reactive oxygen species (ROS) accumulation and inflammation. The loss of electrons from complexes I and III in the mitochondrial electron transport chain can combine with oxygen to generate ROS, powerful oxidizing agents that indiscriminately damage many important components of the cell including DNA, lipid membranes and proteins. ROS are known to activate pro-apoptotic pathways and initiate programmed cell death. However, it has also been reported that ROS-related lipoapoptosis appears to be cell-type dependent[90,91]. Altogether, the role of specific FFA metabolic pathways in promoting ROS accumulation and damage remain largely unclear. The oxidative stress observed in NAFLD subjects might probably be a bystander consequence of a sensitized liver, rather than the main cause of the disease.
Progression of NAFLD to NASH
The progression from simple steatosis to NASH is determined by the initiation of the fibrotic response. Understanding the regulation of the initiation, progression and perpetuation of fibrosis will be very important, particularly from a therapeutic viewpoint. Hepatic stellate cells (HSC) are the main regulators of extracellular matrix (ECM) production and play an essential role in the development of fibrosis (extensively reviewed elsewhere[92,93]). Under normal conditions, HSC have a quiescent phenotype and constitute a third of the non-parenchymal cell population; 85% of hepatic vitamin A is dissolved and stored within quiescent HSC[94]. However, these cells can be activated by noxious stimuli triggered by damaged hepatocytes. When activated, HSC undergo several phenotypic and functional changes. A decrease in the retinoid content is accompanied by a strong increase in the production of extracellular components and cell proliferation. During the initial fibrogenic process, there is a cross-talk between injured hepatocytes and HSC, which is further stimulated in a paracrine mode by the infiltrated leukocytes and activated Kupffer cells (KC). The initial process is followed by the perpetuation of the fibrogenic response. The master regulator of this process is TGF-β[95], a pro-fibrotic cytokine released by almost all the involved cells, whose effect is cell-type dependent. For instance, in mature hepatocytes TGF-β is responsible for inhibition of cell proliferation and participates in the induction of apoptosis[96], while in HSC it promotes cell activation[97] and enhanced production of ECM (collagen, elastin, proteogycans, among others) associated with a decreased degradation by inhibition of the activity of matrix metalloproteinases.
From 1980 when Ludwig et al[98] first defined the condition, great efforts have been dedicated to elucidate the underlying mechanisms involved in this multifactorial and frequent disorder. In spite of data obtained in clinical settings, animal models and in vitro systems, the molecular causes of NASH remains mostly speculative, and further investigations are needed.
In vivo and in vitro experimental models
Due to ethical considerations, mechanistic studies are difficult (or impossible) to be conducted in humans. Consequently, the development of experimental models able to mimic the human condition becomes a necessary tool in the study of the pathophysiology and progression from NAFLD to NASH. Over the last two decades, several animal models have been established and proposed as preclinical platforms for the study of NASH development and the definition of therapeutic options. Table 1 summarizes the most used animal models and their characteristics. The main advantage of this approach is the possibility to define pathogenic pathways in a cause-effect response. The goals these models need to fulfill are: (1) that the pathological pattern of liver injury reflects human steatohepatitis; and (2) that the model should reproduce the context in which human NASH develops. The most used models are genetically modified animals (see for detailed reviews[99-102]) such as the ob/ob mouse with a mutation in the leptin gene[73] or the db/db mouse which lacks the leptin receptor. However, to develop fibrosis and consequent NASH, both models require a methionine and choline deficient (MCD) diet[54]. A controversy still exists about the validity of this diet, since MCD feeding in normal animals induces weight loss and insulin sensitivity[103] despite the impairment of hepatic receptor signaling[104]. Moreover, few human diets are deficient in methionine and choline. Another genetic model consists of animals with deletions in acyl-CoA oxidase (ACOX). Although at an initial stage these animals present severe steatosis and liver inflammatory infiltration with hepatocyte apoptosis, after 6-8 mo they become resistant to steatosis with (PPAR)-α dependent liver regeneration, limiting the utility of this model for the study of steatohepatitis. Deletions in methionine adenosyltransferase (MAT)-1A (MATO mice)[105] and liver-specific pten lead to the development of steatohepatitis but without MS. Sterol regulatory element binding protein (SREBP)-1c transgenic mice, which present an overexpression of this protein in adipose tissue, show IR secondary to impaired adipose differentiation leading to severe hepatic steatosis with the histological features of steatohepatitis[106]. Conversely, these transgenic mice exhibit decreased adipose mass limiting its application to NAFLD/NASH, where adipose tissue is the storage compartment that contributes to perturbations of whole-body lipid homeostasis. An alternative genetically modified animal model is the KK-Ay mouse in which there is a heterozygous mutation of the agouti gene (KK-Ay/a). Interestingly, these animals present impaired hypothalamic appetite suppression[107] and consequently, they are hyperphagic and develop an obese phenotype. They also present hepatic steatosis in conjunction with IR. However, the main limitation of this model is that NASH does not occur spontaneously, and a MCD diet is required for the induction.
Table 1.
Summary of the major findings obtained among the most widespread in vivo models
| Model | Genetic manipulation | Diet modifications | Obesity | Metabolic syndrome (IR) | Hepatosteatosis | Steatohepatitis | Fibrosis |
| ob/ob | Leptin Deficient | No | Yes | Yes | Yes | Yes (in males) | No (protected) |
| mice | Yes | Variable | Yes | Yes | Yes | Yes | |
| MCD | (loss weight in some) | ||||||
| db/db | Mutation on leptin receptor | No | Yes | Yes | Yes | No | No |
| mice | Yes | Variable | No | Yes | Yes | Yes | |
| MCD | (age-related weight gain) | ||||||
| AOX | Nullizygous for acycil - CoA oxidase | No | No | No | Yes | Yes | No |
| null mice | [before 6-8 mo | (before 6-8 mo) | |||||
| Resistant | Resistant | ||||||
| (after 8 mo)] | (after 8 mo) | ||||||
| MATO | Nullizygous for (MAT)-1A | No | No | No | Yes | Yes | Yes |
| null mice | |||||||
| pten | Liver specific pten deletion | No | No | No | Yes | Yes | Yes |
| null mice | |||||||
| (SREBP)-1c | SREBP-1c overexpressed in adipose tissue | No | No | Yes | Yes | Yes | Yes |
| transgenic mice | |||||||
| KK-Ay | Heterozygous mutation on agouti gene (KK-Ay/a) | No | Yes | Yes | Yes | No | No |
| mice | Yes | Yes | Yes | Yes | Yes | No | |
| MCD | |||||||
| LIRKO | Liver-specific Leptin receptor KO | No | No | Hepatic IR | No | - | - |
| mice | |||||||
| C57Bl/6J | No | Yes | Yes | Yes | Yes | Yes | Yes |
| HFHC | (mild) | ||||||
| HF | |||||||
| Cholesterol-Cholate | No | Yes | No | No | Yes | Yes | Yes |
| (Atherogenic diet) | Cholesterol | (only hepatic IR) | (over 1-6 mo) | (over 1-6 mo) | (over 1-6 mo) | ||
| Cholate |
MCD: Methionine choline deficient diet; HFHC: High fat-high carbohydrate diet; HF: High fructose diet; KO: Knock-out; IR: Insulin resistance.
The use of diet-induced models, extensively reviewed elsewhere[102], is another strategy in the study of NASH development. Different diets for small animals have been characterized[108-110] with good results in the development of steatosis and inflammation, but marginal results in generating fibrosis. Different effects depending on the composition of the diet have been reported. High-carbohydrate diets stimulate moderate hepatic lipogenesis in rats, whereas animals fed with high-fat diets present a strong inhibition of this anabolic pathway. The plasma TG levels are higher in the high-carbohydrates diets, whereas the high-fat diet determines an accumulation of TG in the liver. However, both diets induce an increase of plasmatic levels of glucose and insulin[111]. Regarding the generation of fibrosis, promising evidence has emerged from mice fed with an atherogenic diet containing 1.25% cholesterol and 0.5% cholate[112]. Under these dietary conditions, a progressive formation of steatosis is observed associated with an evident inflammatory response, induction of oxidative stress and development of fibrosis in 6-24 wk. However, these animals are systematically insulin-sensitive, albeit they develop hepatic IR and surprisingly, they show a weight loss. This makes the cholesterol-cholate model substantially different from human NASH, severely limiting its application. A valid tool for the study of hepatic IR and the effect of insulin on leptin homeostasis is represented by LIRKO mice, a liver-specific insulin receptor knock-out[113]. These animals present abnormal glucose metabolism and progressive liver dysfunction, and display focal dysplasia and hyperplastic nodules. However, serum TG levels are decreased, most probably by the inability of insulin to promote TG synthesis in the liver and by reduced lipolysis in adipose tissue. In spite of the hyperinsulinemia and IR, these animals are not obese[114]. A promising approach is the administration of a high-fat diet associated with high fructose to male C57Bl/6J mice, which induces results similar to those observed in human NASH[115]. In spite of the promising results, substantial objections remain: (1) the long term exposure required for observing the pathological phenotype[116]; (2) the inclusion of only male animals excluding the application of this approach to the female population; and (3) rodents might adapt to high-fat feeding and become resistant to the development of obesity[117].
Worthy of attention is the fact that under specific experimental settings, animals can develop NASH from simple steatosis. However, the data fail to explain why in humans only some individuals develop NASH while others can live with NAFLD with no complications[118]. This crucial issue is still an open question, and most probably may be related to a different response of the cell to fat storage[119,120].
Contrary to other liver diseases in which in vitro models are important tools in research, convincing data are still missing in NAFLD and NASH. One of the reasons may be related to the use of a rather simplistic set-up to tackle the multistep process of the development of NASH. The use of an in vitro approach presents several advantages and disadvantages, as recently reviewed in detail[121]. A broad spectrum of in vitro validated possibilities is available, such as the use of primary cell culture, immortalized cell lines, or an even more sophisticated system such as precision-cut slices of perfused liver. The main obstacle of the in vitro system is the extrapolation of the results to the much more complex human environment. A good example of this limitation is the choice of free fatty acids (FFA), since it has been reported that individual FFA have distinct inherent steatotic and toxic activities, the saturated FFA presenting the highest toxicity[122]. In normal and in NAFLD subjects, the most abundant FFAs in liver triglycerides are oleic acid (18:1) and palmitoleic acid (16:1) for unsaturated, and palmitic acid (16:0) and stearic acid (18:0) for saturated FFAs[123]. The relative concentration has been demonstrated to be a determinant in their hepatic accumulation and toxicity[124]. For instance, different effects of oleic and palmitic acid were reported on lipid accumulation and on the induction of apoptosis. Oleic acid was shown in several hepatic cell lines to be more steatogenic than palmitic acid[125] but less toxic that the latter. Long chain FFAs are highly insoluble in the aqueous phase, and for this reason are carried in blood associated with serum albumin. Whereas under physiological conditions the FFA: albumin ratio is around 2:1[78] under pathological states, the ratio can be as high as 7.5:1[126]. This simple, but fundamental, detail is often disregarded in several studies. In addition, since the development of NAFLD and the progression to NASH involve several cell types, another crucial point is the cell type used in the experimental system. The vast majority of the published data has been obtained in hepatocyte cultures, but for the study of the progression to fibrosis, other cell types such as HSC and KC must be considered. The crucial role played by the interaction among the different cell types points to the need of much more controlled experimental setups to provide a comprehensive approach to the molecular mechanisms involved. For this reason, the establishment of co-culture systems has been acknowledged to be promising in the last few years[77,127-129] with regard to the study of the different intracellular mechanisms.
In any case, in spite of the progress in the molecular biology of NAFLD/NASH, the main limitation of these in vitro approaches remains the different models and experimental variables used in the different laboratories. This makes each study somewhat unique and independent from the others. A better definition of the experimental conditions and standardized models would greatly contribute to improving the possibility of achieving solid results.
CONCLUSION
A translational approach to NAFLD and NASH is just at the beginning. The disease is rather new and is still based on a negative definition, but we now know that it is linked to a metabolic dysfunction of the glucose and/or lipid hepatic pathways. The number of patients affected by this disorder is exponentially growing worldwide, and NAFLD diagnosis must be performed early to prevent the progression to NASH, cirrhosis and HCC or to cardiovascular diseases, and to adopt effective preventive strategies. We now have the experimental models to investigate the still unknown reasons why only some types of sugars and lipids induce progressive hepatic inflammation, apoptosis and fibrosis. We hope they will help us to understand the inner mechanism of the damage and design better drugs that will combine with a much healthier lifestyle to fight this plague.
Footnotes
Supported by European Union Seventh Framework Program (FP7/2007-2013) under grant agreement, No. Health-F2-2009-241762, for the project FLIP; Italian National Grant MIUR (Art.13 D.LGS 297/99 - Progetto Nutrizione e Salute); and an in house grant from Fondazione Italiana Fegato, ONLUS
P- Reviewers: Bulum T, Kayadibi H, Lonardo A, Montecucco F, Perazzo H, Takahashi Y, Zara V S- Editor: Ma YJ L- Editor: Logan S E- Editor: Ma S
References
- 1.Sartorio A, Del Col A, Agosti F, Mazzilli G, Bellentani S, Tiribelli C, Bedogni G. Predictors of non-alcoholic fatty liver disease in obese children. Eur J Clin Nutr. 2007;61:877–883. doi: 10.1038/sj.ejcn.1602588. [DOI] [PubMed] [Google Scholar]
- 2.Chen SH, He F, Zhou HL, Wu HR, Xia C, Li YM. Relationship between nonalcoholic fatty liver disease and metabolic syndrome. J Dig Dis. 2011;12:125–130. doi: 10.1111/j.1751-2980.2011.00487.x. [DOI] [PubMed] [Google Scholar]
- 3.Caldwell SH, Ikura Y, Iezzoni JC, Liu Z. Has natural selection in human populations produced two types of metabolic syndrome (with and without fatty liver)? J Gastroenterol Hepatol. 2007;22 Suppl 1:S11–S19. doi: 10.1111/j.1440-1746.2006.04639.x. [DOI] [PubMed] [Google Scholar]
- 4.Bulum T, Kolarić B, Duvnjak L, Duvnjak M. Nonalcoholic fatty liver disease markers are associated with insulin resistance in type 1 diabetes. Dig Dis Sci. 2011;56:3655–3663. doi: 10.1007/s10620-011-1807-7. [DOI] [PubMed] [Google Scholar]
- 5.Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, Natale S, Vanni E, Villanova N, Melchionda N, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917–923. doi: 10.1053/jhep.2003.50161. [DOI] [PubMed] [Google Scholar]
- 6.Zelber-Sagi S, Nitzan-Kaluski D, Goldsmith R, Webb M, Blendis L, Halpern Z, Oren R. Long term nutritional intake and the risk for non-alcoholic fatty liver disease (NAFLD): a population based study. J Hepatol. 2007;47:711–717. doi: 10.1016/j.jhep.2007.06.020. [DOI] [PubMed] [Google Scholar]
- 7.Zelber-Sagi S, Nitzan-Kaluski D, Goldsmith R, Webb M, Zvibel I, Goldiner I, Blendis L, Halpern Z, Oren R. Role of leisure-time physical activity in nonalcoholic fatty liver disease: a population-based study. Hepatology. 2008;48:1791–1798. doi: 10.1002/hep.22525. [DOI] [PubMed] [Google Scholar]
- 8.Centis E, Moscatiello S, Bugianesi E, Bellentani S, Fracanzani AL, Calugi S, Petta S, Dalle Grave R, Marchesini G. Stage of change and motivation to healthier lifestyle in non-alcoholic fatty liver disease. J Hepatol. 2013;58:771–777. doi: 10.1016/j.jhep.2012.11.031. [DOI] [PubMed] [Google Scholar]
- 9.Scaglioni F, Marino M, Ciccia S, Procaccini A, Busacchi M, Loria P, Lonardo A, Malavolti M, Battistini NC, Pellegrini M, et al. Short-term multidisciplinary non-pharmacological intervention is effective in reducing liver fat content assessed non-invasively in patients with nonalcoholic fatty liver disease (NAFLD) Clin Res Hepatol Gastroenterol. 2013;37:353–358. doi: 10.1016/j.clinre.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 10.Liu CJ. Prevalence and risk factors for non-alcoholic fatty liver disease in Asian people who are not obese. J Gastroenterol Hepatol. 2012;27:1555–1560. doi: 10.1111/j.1440-1746.2012.07222.x. [DOI] [PubMed] [Google Scholar]
- 11.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 12.Bugianesi E, Moscatiello S, Ciaravella MF, Marchesini G. Insulin resistance in nonalcoholic fatty liver disease. Curr Pharm Des. 2010;16:1941–1951. doi: 10.2174/138161210791208875. [DOI] [PubMed] [Google Scholar]
- 13.Diehl AM, Li ZP, Lin HZ, Yang SQ. Cytokines and the pathogenesis of non-alcoholic steatohepatitis. Gut. 2005;54:303–306. doi: 10.1136/gut.2003.024935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–28. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 15.Bachar E, Ariav Y, Ketzinel-Gilad M, Cerasi E, Kaiser N, Leibowitz G. Glucose amplifies fatty acid-induced endoplasmic reticulum stress in pancreatic beta-cells via activation of mTORC1. PLoS One. 2009;4:e4954. doi: 10.1371/journal.pone.0004954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836–1846. doi: 10.1002/hep.24001. [DOI] [PubMed] [Google Scholar]
- 17.Moschen AR, Kaser S, Tilg H. Non-alcoholic steatohepatitis: a microbiota-driven disease. Trends Endocrinol Metab. 2013;24:537–545. doi: 10.1016/j.tem.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 18.Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143:913–916.e7. doi: 10.1053/j.gastro.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 19.Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology. 2011;53:1883–1894. doi: 10.1002/hep.24283. [DOI] [PubMed] [Google Scholar]
- 20.Montecucco F, Bertolotto M, Vuilleumier N, Franciosi U, Puddu A, Minetti S, Delrio A, Quercioli A, Bergamini E, Ottonello L, et al. Acipimox reduces circulating levels of insulin and associated neutrophilic inflammation in metabolic syndrome. Am J Physiol Endocrinol Metab. 2011;300:E681–E690. doi: 10.1152/ajpendo.00527.2010. [DOI] [PubMed] [Google Scholar]
- 21.Kim F, Pham M, Maloney E, Rizzo NO, Morton GJ, Wisse BE, Kirk EA, Chait A, Schwartz MW. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arterioscler Thromb Vasc Biol. 2008;28:1982–1988. doi: 10.1161/ATVBAHA.108.169722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gage MC, Yuldasheva NY, Viswambharan H, Sukumar P, Cubbon RM, Galloway S, Imrie H, Skromna A, Smith J, Jackson CL, et al. Endothelium-specific insulin resistance leads to accelerated atherosclerosis in areas with disturbed flow patterns: a role for reactive oxygen species. Atherosclerosis. 2013;230:131–139. doi: 10.1016/j.atherosclerosis.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 23.Tsuchiya K, Tanaka J, Shuiqing Y, Welch CL, DePinho RA, Tabas I, Tall AR, Goldberg IJ, Accili D. FoxOs integrate pleiotropic actions of insulin in vascular endothelium to protect mice from atherosclerosis. Cell Metab. 2012;15:372–381. doi: 10.1016/j.cmet.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis RC, Castellani LW, Hosseini M, Ben-Zeev O, Mao HZ, Weinstein MM, Jung DY, Jun JY, Kim JK, Lusis AJ, et al. Early hepatic insulin resistance precedes the onset of diabetes in obese C57BLKS-db/db mice. Diabetes. 2010;59:1616–1625. doi: 10.2337/db09-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carbone F, Montecucco F, Mach F, Pontremoli R, Viazzi F. The liver and the kidney: two critical organs influencing the atherothrombotic risk in metabolic syndrome. Thromb Haemost. 2013;110:940–958. doi: 10.1160/TH13-06-0499. [DOI] [PubMed] [Google Scholar]
- 26.Targher G, Chonchol M, Pichiri I, Zoppini G. Risk of cardiovascular disease and chronic kidney disease in diabetic patients with non-alcoholic fatty liver disease: just a coincidence? J Endocrinol Invest. 2011;34:544–551. doi: 10.3275/7614. [DOI] [PubMed] [Google Scholar]
- 27.Wisse BE. The inflammatory syndrome: the role of adipose tissue cytokines in metabolic disorders linked to obesity. J Am Soc Nephrol. 2004;15:2792–2800. doi: 10.1097/01.ASN.0000141966.69934.21. [DOI] [PubMed] [Google Scholar]
- 28.Wolf G, Chen S, Han DC, Ziyadeh FN. Leptin and renal disease. Am J Kidney Dis. 2002;39:1–11. doi: 10.1053/ajkd.2002.29865. [DOI] [PubMed] [Google Scholar]
- 29.Lonardo A, Bellentani S, Ratziu V, Loria P. Insulin resistance in nonalcoholic steatohepatitis: necessary but not sufficient - death of a dogma from analysis of therapeutic studies? Expert Rev Gastroenterol Hepatol. 2011;5:279–289. doi: 10.1586/egh.11.19. [DOI] [PubMed] [Google Scholar]
- 30.Rask E, Olsson T, Söderberg S, Johnson O, Seckl J, Holst JJ, Ahrén B. Impaired incretin response after a mixed meal is associated with insulin resistance in nondiabetic men. Diabetes Care. 2001;24:1640–1645. doi: 10.2337/diacare.24.9.1640. [DOI] [PubMed] [Google Scholar]
- 31.Holst JJ, Gromada J. Role of incretin hormones in the regulation of insulin secretion in diabetic and nondiabetic humans. Am J Physiol Endocrinol Metab. 2004;287:E199–E206. doi: 10.1152/ajpendo.00545.2003. [DOI] [PubMed] [Google Scholar]
- 32.Nauck M, Stöckmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia. 1986;29:46–52. doi: 10.1007/BF02427280. [DOI] [PubMed] [Google Scholar]
- 33.Gautier JF, Choukem SP, Girard J. Physiology of incretins (GIP and GLP-1) and abnormalities in type 2 diabetes. Diabetes Metab. 2008;34 Suppl 2:S65–S72. doi: 10.1016/S1262-3636(08)73397-4. [DOI] [PubMed] [Google Scholar]
- 34.Gupta NA, Mells J, Dunham RM, Grakoui A, Handy J, Saxena NK, Anania FA. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology. 2010;51:1584–1592. doi: 10.1002/hep.23569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ding X, Saxena NK, Lin S, Gupta NA, Anania FA. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology. 2006;43:173–181. doi: 10.1002/hep.21006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trevaskis JL, Griffin PS, Wittmer C, Neuschwander-Tetri BA, Brunt EM, Dolman CS, Erickson MR, Napora J, Parkes DG, Roth JD. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am J Physiol Gastrointest Liver Physiol. 2012;302:G762–G772. doi: 10.1152/ajpgi.00476.2011. [DOI] [PubMed] [Google Scholar]
- 37.Ben-Shlomo S, Zvibel I, Shnell M, Shlomai A, Chepurko E, Halpern Z, Barzilai N, Oren R, Fishman S. Glucagon-like peptide-1 reduces hepatic lipogenesis via activation of AMP-activated protein kinase. J Hepatol. 2011;54:1214–1223. doi: 10.1016/j.jhep.2010.09.032. [DOI] [PubMed] [Google Scholar]
- 38.Svegliati-Baroni G, Saccomanno S, Rychlicki C, Agostinelli L, De Minicis S, Candelaresi C, Faraci G, Pacetti D, Vivarelli M, Nicolini D, et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011;31:1285–1297. doi: 10.1111/j.1478-3231.2011.02462.x. [DOI] [PubMed] [Google Scholar]
- 39.Sharma S, Mells JE, Fu PP, Saxena NK, Anania FA. GLP-1 analogs reduce hepatocyte steatosis and improve survival by enhancing the unfolded protein response and promoting macroautophagy. PLoS One. 2011;6:e25269. doi: 10.1371/journal.pone.0025269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Staels B, Fonseca VA. Bile acids and metabolic regulation: mechanisms and clinical responses to bile acid sequestration. Diabetes Care. 2009;32 Suppl 2:S237–S245. doi: 10.2337/dc09-S355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 42.Andersén E, Karlaganis G, Sjövall J. Altered bile acid profiles in duodenal bile and urine in diabetic subjects. Eur J Clin Invest. 1988;18:166–172. doi: 10.1111/j.1365-2362.1988.tb02408.x. [DOI] [PubMed] [Google Scholar]
- 43.Knop FK. Bile-induced secretion of glucagon-like peptide-1: pathophysiological implications in type 2 diabetes? Am J Physiol Endocrinol Metab. 2010;299:E10–E13. doi: 10.1152/ajpendo.00137.2010. [DOI] [PubMed] [Google Scholar]
- 44.Laplante M, Sabatini DM. mTORC1 activates SREBP-1c and uncouples lipogenesis from gluconeogenesis. Proc Natl Acad Sci USA. 2010;107:3281–3282. doi: 10.1073/pnas.1000323107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blaslov K, Zibar K, Bulum T, Duvnjak L. Effect of exenatide therapy on hepatic fat quantity and hepatic biomarkers in type 2 diabetic patients. Clin Res Hepatol Gastroenterol. 2013:Epub ahead of print. doi: 10.1016/j.clinre.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 46.Holst JJ, Vilsbøll T, Deacon CF. The incretin system and its role in type 2 diabetes mellitus. Mol Cell Endocrinol. 2009;297:127–136. doi: 10.1016/j.mce.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 47.Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, Adorini L, Sciacca CI, Clopton P, Castelloe E, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013;145:574–582.e1. doi: 10.1053/j.gastro.2013.05.042. [DOI] [PubMed] [Google Scholar]
- 48.Tanaka S, Wands JR. Insulin receptor substrate 1 overexpression in human hepatocellular carcinoma cells prevents transforming growth factor beta1-induced apoptosis. Cancer Res. 1996;56:3391–3394. [PubMed] [Google Scholar]
- 49.Ish-Shalom D, Christoffersen CT, Vorwerk P, Sacerdoti-Sierra N, Shymko RM, Naor D, De Meyts P. Mitogenic properties of insulin and insulin analogues mediated by the insulin receptor. Diabetologia. 1997;40 Suppl 2:S25–S31. doi: 10.1007/s001250051393. [DOI] [PubMed] [Google Scholar]
- 50.Page JM, Harrison SA. NASH and HCC. Clin Liver Dis. 2009;13:631–647. doi: 10.1016/j.cld.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 51.Stickel F, Hellerbrand C. Non-alcoholic fatty liver disease as a risk factor for hepatocellular carcinoma: mechanisms and implications. Gut. 2010;59:1303–1307. doi: 10.1136/gut.2009.199661. [DOI] [PubMed] [Google Scholar]
- 52.Yu J, Shen J, Sun TT, Zhang X, Wong N. Obesity, insulin resistance, NASH and hepatocellular carcinoma. Semin Cancer Biol. 2013;23:483–491. doi: 10.1016/j.semcancer.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 53.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, Bhanot S, Monia BP, Li YX, Diehl AM. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–1374. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 55.Malhi H, Gores GJ. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin Liver Dis. 2008;28:360–369. doi: 10.1055/s-0028-1091980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bass NM. Lipidomic dissection of nonalcoholic steatohepatitis: moving beyond foie gras to fat traffic. Hepatology. 2010;51:4–7. doi: 10.1002/hep.23458. [DOI] [PubMed] [Google Scholar]
- 57.Choi SS, Diehl AM. Hepatic triglyceride synthesis and nonalcoholic fatty liver disease. Curr Opin Lipidol. 2008;19:295–300. doi: 10.1097/MOL.0b013e3282ff5e55. [DOI] [PubMed] [Google Scholar]
- 58.Bikman BT, Summers SA. Ceramides as modulators of cellular and whole-body metabolism. J Clin Invest. 2011;121:4222–4230. doi: 10.1172/JCI57144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774–788. doi: 10.1002/hep.23719. [DOI] [PubMed] [Google Scholar]
- 60.Tanti JF, Jager J. Cellular mechanisms of insulin resistance: role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr Opin Pharmacol. 2009;9:753–762. doi: 10.1016/j.coph.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 61.Wouters K, van Bilsen M, van Gorp PJ, Bieghs V, Lütjohann D, Kerksiek A, Staels B, Hofker MH, Shiri-Sverdlov R. Intrahepatic cholesterol influences progression, inhibition and reversal of non-alcoholic steatohepatitis in hyperlipidemic mice. FEBS Lett. 2010;584:1001–1005. doi: 10.1016/j.febslet.2010.01.046. [DOI] [PubMed] [Google Scholar]
- 62.Zhao L, Chen Y, Tang R, Chen Y, Li Q, Gong J, Huang A, Varghese Z, Moorhead JF, Ruan XZ. Inflammatory stress exacerbates hepatic cholesterol accumulation via increasing cholesterol uptake and de novo synthesis. J Gastroenterol Hepatol. 2011;26:875–883. doi: 10.1111/j.1440-1746.2010.06560.x. [DOI] [PubMed] [Google Scholar]
- 63.Li L, Hossain MA, Sadat S, Hager L, Liu L, Tam L, Schroer S, Huogen L, Fantus IG, Connelly PW, et al. Lecithin cholesterol acyltransferase null mice are protected from diet-induced obesity and insulin resistance in a gender-specific manner through multiple pathways. J Biol Chem. 2011;286:17809–17820. doi: 10.1074/jbc.M110.180893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hager L, Li L, Pun H, Liu L, Hossain MA, Maguire GF, Naples M, Baker C, Magomedova L, Tam J, et al. Lecithin: cholesterol acyltransferase deficiency protects against cholesterol-induced hepatic endoplasmic reticulum stress in mice. J Biol Chem. 2012;287:20755–20768. doi: 10.1074/jbc.M112.340919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 66.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 67.Musso G, Cassader M, Bo S, De Michieli F, Gambino R. Sterol regulatory element-binding factor 2 (SREBF-2) predicts 7-year NAFLD incidence and severity of liver disease and lipoprotein and glucose dysmetabolism. Diabetes. 2013;62:1109–1120. doi: 10.2337/db12-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Musso G, Gambino R, Cassader M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog Lipid Res. 2013;52:175–191. doi: 10.1016/j.plipres.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 69.Yasutake K, Nakamuta M, Shima Y, Ohyama A, Masuda K, Haruta N, Fujino T, Aoyagi Y, Fukuizumi K, Yoshimoto T, et al. Nutritional investigation of non-obese patients with non-alcoholic fatty liver disease: the significance of dietary cholesterol. Scand J Gastroenterol. 2009;44:471–477. doi: 10.1080/00365520802588133. [DOI] [PubMed] [Google Scholar]
- 70.Asrih M, Jornayvaz FR. Inflammation as a potential link between nonalcoholic fatty liver disease and insulin resistance. J Endocrinol. 2013;218:R25–R36. doi: 10.1530/JOE-13-0201. [DOI] [PubMed] [Google Scholar]
- 71.Marra F, Bertolani C. Adipokines in liver diseases. Hepatology. 2009;50:957–969. doi: 10.1002/hep.23046. [DOI] [PubMed] [Google Scholar]
- 72.Tilg H, Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. N Engl J Med. 2000;343:1467–1476. doi: 10.1056/NEJM200011163432007. [DOI] [PubMed] [Google Scholar]
- 73.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kunt T, Forst T, Früh B, Flohr T, Schneider S, Harzer O, Pfützner A, Engelbach M, Löbig M, Beyer J. Binding of monocytes from normolipidemic hyperglycemic patients with type 1 diabetes to endothelial cells is increased in vitro. Exp Clin Endocrinol Diabetes. 1999;107:252–256. doi: 10.1055/s-0029-1212108. [DOI] [PubMed] [Google Scholar]
- 75.Bulum T, KolariÄ B, Duvnjak L. Decreased serum monocytes and elevated neutrophils as additional markers of insulin resistance in type 1 diabetes. Int J Diabetes Dev Ctries. 2013:1–6. [Google Scholar]
- 76.Bunn RC, Cockrell GE, Ou Y, Thrailkill KM, Lumpkin CK, Fowlkes JL. Palmitate and insulin synergistically induce IL-6 expression in human monocytes. Cardiovasc Diabetol. 2010;9:73. doi: 10.1186/1475-2840-9-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chavez-Tapia NC, Rosso N, Tiribelli C. Effect of intracellular lipid accumulation in a new model of non-alcoholic fatty liver disease. BMC Gastroenterol. 2012;12:20. doi: 10.1186/1471-230X-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Joshi-Barve S, Barve SS, Amancherla K, Gobejishvili L, Hill D, Cave M, Hote P, McClain CJ. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology. 2007;46:823–830. doi: 10.1002/hep.21752. [DOI] [PubMed] [Google Scholar]
- 79.Chávez-Tapia NC, Rosso N, Uribe M, Bojalil R, Tiribelli C. Kinetics of the inflammatory response induced by free fatty acid accumulation in hepatocytes. Ann Hepatol. 2013;13:113–120. [PubMed] [Google Scholar]
- 80.Bråkenhielm E, Veitonmäki N, Cao R, Kihara S, Matsuzawa Y, Zhivotovsky B, Funahashi T, Cao Y. Adiponectin-induced antiangiogenesis and antitumor activity involve caspase-mediated endothelial cell apoptosis. Proc Natl Acad Sci USA. 2004;101:2476–2481. doi: 10.1073/pnas.0308671100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kamada Y, Matsumoto H, Tamura S, Fukushima J, Kiso S, Fukui K, Igura T, Maeda N, Kihara S, Funahashi T, et al. Hypoadiponectinemia accelerates hepatic tumor formation in a nonalcoholic steatohepatitis mouse model. J Hepatol. 2007;47:556–564. doi: 10.1016/j.jhep.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 82.Braunersreuther V, Viviani GL, Mach F, Montecucco F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J Gastroenterol. 2012;18:727–735. doi: 10.3748/wjg.v18.i8.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 84.Cortez-Pinto H, Chatham J, Chacko VP, Arnold C, Rashid A, Diehl AM. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: a pilot study. JAMA. 1999;282:1659–1664. doi: 10.1001/jama.282.17.1659. [DOI] [PubMed] [Google Scholar]
- 85.Serviddio G, Sastre J, Bellanti F, Viña J, Vendemiale G, Altomare E. Mitochondrial involvement in non-alcoholic steatohepatitis. Mol Aspects Med. 2008;29:22–35. doi: 10.1016/j.mam.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 86.Teodoro JS, Rolo AP, Duarte FV, Simões AM, Palmeira CM. Differential alterations in mitochondrial function induced by a choline-deficient diet: understanding fatty liver disease progression. Mitochondrion. 2008;8:367–376. doi: 10.1016/j.mito.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 87.Leamy AK, Egnatchik RA, Young JD. Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog Lipid Res. 2013;52:165–174. doi: 10.1016/j.plipres.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011;14:804–810. doi: 10.1016/j.cmet.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Satapati S, Sunny NE, Kucejova B, Fu X, He TT, Méndez-Lucas A, Shelton JM, Perales JC, Browning JD, Burgess SC. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J Lipid Res. 2012;53:1080–1092. doi: 10.1194/jlr.M023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res. 2006;47:2726–2737. doi: 10.1194/jlr.M600299-JLR200. [DOI] [PubMed] [Google Scholar]
- 91.Hickson-Bick DL, Sparagna GC, Buja LM, McMillin JB. Palmitate-induced apoptosis in neonatal cardiomyocytes is not dependent on the generation of ROS. Am J Physiol Heart Circ Physiol. 2002;282:H656–H664. doi: 10.1152/ajpheart.00726.2001. [DOI] [PubMed] [Google Scholar]
- 92.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gressner OA, Weiskirchen R, Gressner AM. Evolving concepts of liver fibrogenesis provide new diagnostic and therapeutic options. Comp Hepatol. 2007;6:7. doi: 10.1186/1476-5926-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dooley S, Delvoux B, Streckert M, Bonzel L, Stopa M, ten Dijke P, Gressner AM. Transforming growth factor beta signal transduction in hepatic stellate cells via Smad2/3 phosphorylation, a pathway that is abrogated during in vitro progression to myofibroblasts. TGFbeta signal transduction during transdifferentiation of hepatic stellate cells. FEBS Lett. 2001;502:4–10. doi: 10.1016/s0014-5793(01)02656-4. [DOI] [PubMed] [Google Scholar]
- 96.Cavin LG, Romieu-Mourez R, Panta GR, Sun J, Factor VM, Thorgeirsson SS, Sonenshein GE, Arsura M. Inhibition of CK2 activity by TGF-beta1 promotes IkappaB-alpha protein stabilization and apoptosis of immortalized hepatocytes. Hepatology. 2003;38:1540–1551. doi: 10.1016/j.hep.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 97.Proell V, Carmona-Cuenca I, Murillo MM, Huber H, Fabregat I, Mikulits W. TGF-beta dependent regulation of oxygen radicals during transdifferentiation of activated hepatic stellate cells to myofibroblastoid cells. Comp Hepatol. 2007;6:1. doi: 10.1186/1476-5926-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55:434–438. [PubMed] [Google Scholar]
- 99.Anstee QM, Goldin RD. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol. 2006;87:1–16. doi: 10.1111/j.0959-9673.2006.00465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koteish A, Mae Diehl A. Animal models of steatohepatitis. Best Pract Res Clin Gastroenterol. 2002;16:679–690. doi: 10.1053/bega.2002.0332. [DOI] [PubMed] [Google Scholar]
- 101.Larter CZ, Yeh MM. Animal models of NASH: getting both pathology and metabolic context right. J Gastroenterol Hepatol. 2008;23:1635–1648. doi: 10.1111/j.1440-1746.2008.05543.x. [DOI] [PubMed] [Google Scholar]
- 102.Takahashi Y, Soejima Y, Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol. 2012;18:2300–2308. doi: 10.3748/wjg.v18.i19.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rinella ME, Green RM. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol. 2004;40:47–51. doi: 10.1016/j.jhep.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 104.Schattenberg JM, Wang Y, Singh R, Rigoli RM, Czaja MJ. Hepatocyte CYP2E1 overexpression and steatohepatitis lead to impaired hepatic insulin signaling. J Biol Chem. 2005;280:9887–9894. doi: 10.1074/jbc.M410310200. [DOI] [PubMed] [Google Scholar]
- 105.Martínez-Chantar ML, Corrales FJ, Martínez-Cruz LA, García-Trevijano ER, Huang ZZ, Chen L, Kanel G, Avila MA, Mato JM, Lu SC. Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase 1A. FASEB J. 2002;16:1292–1294. doi: 10.1096/fj.02-0078fje. [DOI] [PubMed] [Google Scholar]
- 106.Nakayama H, Otabe S, Ueno T, Hirota N, Yuan X, Fukutani T, Hashinaga T, Wada N, Yamada K. Transgenic mice expressing nuclear sterol regulatory element-binding protein 1c in adipose tissue exhibit liver histology similar to nonalcoholic steatohepatitis. Metabolism. 2007;56:470–475. doi: 10.1016/j.metabol.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 107.Schattenberg JM, Galle PR. Animal models of non-alcoholic steatohepatitis: of mice and man. Dig Dis. 2010;28:247–254. doi: 10.1159/000282097. [DOI] [PubMed] [Google Scholar]
- 108.Tetri LH, Basaranoglu M, Brunt EM, Yerian LM, Neuschwander-Tetri BA. Severe NAFLD with hepatic necroinflammatory changes in mice fed trans fats and a high-fructose corn syrup equivalent. Am J Physiol Gastrointest Liver Physiol. 2008;295:G987–G995. doi: 10.1152/ajpgi.90272.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kawasaki T, Igarashi K, Koeda T, Sugimoto K, Nakagawa K, Hayashi S, Yamaji R, Inui H, Fukusato T, Yamanouchi T. Rats fed fructose-enriched diets have characteristics of nonalcoholic hepatic steatosis. J Nutr. 2009;139:2067–2071. doi: 10.3945/jn.109.105858. [DOI] [PubMed] [Google Scholar]
- 110.Lieber CS, Leo MA, Mak KM, Xu Y, Cao Q, Ren C, Ponomarenko A, DeCarli LM. Model of nonalcoholic steatohepatitis. Am J Clin Nutr. 2004;79:502–509. doi: 10.1093/ajcn/79.3.502. [DOI] [PubMed] [Google Scholar]
- 111.Ferramosca A, Conte A, Damiano F, Siculella L, Zara V. Differential effects of high-carbohydrate and high-fat diets on hepatic lipogenesis in rats. Eur J Nutr. 2014;53:1103–1114. doi: 10.1007/s00394-013-0613-8. [DOI] [PubMed] [Google Scholar]
- 112.Matsuzawa N, Takamura T, Kurita S, Misu H, Ota T, Ando H, Yokoyama M, Honda M, Zen Y, Nakanuma Y, et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology. 2007;46:1392–1403. doi: 10.1002/hep.21874. [DOI] [PubMed] [Google Scholar]
- 113.Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, Kahn CR. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000;6:87–97. [PubMed] [Google Scholar]
- 114.Cohen SE, Kokkotou E, Biddinger SB, Kondo T, Gebhardt R, Kratzsch J, Mantzoros CS, Kahn CR. High circulating leptin receptors with normal leptin sensitivity in liver-specific insulin receptor knock-out (LIRKO) mice. J Biol Chem. 2007;282:23672–23678. doi: 10.1074/jbc.M704053200. [DOI] [PubMed] [Google Scholar]
- 115.Kohli R, Kirby M, Xanthakos SA, Softic S, Feldstein AE, Saxena V, Tang PH, Miles L, Miles MV, Balistreri WF, et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology. 2010;52:934–944. doi: 10.1002/hep.23797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ito M, Suzuki J, Tsujioka S, Sasaki M, Gomori A, Shirakura T, Hirose H, Ito M, Ishihara A, Iwaasa H, et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol Res. 2007;37:50–57. doi: 10.1111/j.1872-034X.2007.00008.x. [DOI] [PubMed] [Google Scholar]
- 117.Romestaing C, Piquet MA, Bedu E, Rouleau V, Dautresme M, Hourmand-Ollivier I, Filippi C, Duchamp C, Sibille B. Long term highly saturated fat diet does not induce NASH in Wistar rats. Nutr Metab (Lond) 2007;4:4. doi: 10.1186/1743-7075-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yilmaz Y. Review article: is non-alcoholic fatty liver disease a spectrum, or are steatosis and non-alcoholic steatohepatitis distinct conditions? Aliment Pharmacol Ther. 2012;36:815–823. doi: 10.1111/apt.12046. [DOI] [PubMed] [Google Scholar]
- 119.Lonardo A, Loria P, Argo C, Caldwell S. Perspectives on cellular dysfunction in nonalcoholic steatohepatitis: a case of ‘multiorganelle failure’? Proceedings of a virtual workshop on nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol. 2011;5:135–139. doi: 10.1586/egh.11.24. [DOI] [PubMed] [Google Scholar]
- 120.Arrese M. Burning hepatic fat: therapeutic potential for liver-specific thyromimetics in the treatment of nonalcoholic fatty liver disease. Hepatology. 2009;49:348–351. doi: 10.1002/hep.22783. [DOI] [PubMed] [Google Scholar]
- 121.Chavez-Tapia NC, Rosso N, Tiribelli C. In vitro models for the study of non-alcoholic fatty liver disease. Curr Med Chem. 2011;18:1079–1084. doi: 10.2174/092986711794940842. [DOI] [PubMed] [Google Scholar]
- 122.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 123.Araya J, Rodrigo R, Videla LA, Thielemann L, Orellana M, Pettinelli P, Poniachik J. Increase in long-chain polyunsaturated fatty acid n - 6/n - 3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin Sci (Lond) 2004;106:635–643. doi: 10.1042/CS20030326. [DOI] [PubMed] [Google Scholar]
- 124.Gómez-Lechón MJ, Donato MT, Martínez-Romero A, Jiménez N, Castell JV, O’Connor JE. A human hepatocellular in vitro model to investigate steatosis. Chem Biol Interact. 2007;165:106–116. doi: 10.1016/j.cbi.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 125.Ricchi M, Odoardi MR, Carulli L, Anzivino C, Ballestri S, Pinetti A, Fantoni LI, Marra F, Bertolotti M, Banni S, et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J Gastroenterol Hepatol. 2009;24:830–840. doi: 10.1111/j.1440-1746.2008.05733.x. [DOI] [PubMed] [Google Scholar]
- 126.Listenberger LL, Ory DS, Schaffer JE. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J Biol Chem. 2001;276:14890–14895. doi: 10.1074/jbc.M010286200. [DOI] [PubMed] [Google Scholar]
- 127.Krause P, Saghatolislam F, Koenig S, Unthan-Fechner K, Probst I. Maintaining hepatocyte differentiation in vitro through co-culture with hepatic stellate cells. In Vitro Cell Dev Biol Anim. 2009;45:205–212. doi: 10.1007/s11626-008-9166-1. [DOI] [PubMed] [Google Scholar]
- 128.Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, Jang MK, Guenther ND, Mederacke I, Friedman R, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58:1461–1473. doi: 10.1002/hep.26429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Coulouarn C, Corlu A, Glaise D, Guénon I, Thorgeirsson SS, Clément B. Hepatocyte-stellate cell cross-talk in the liver engenders a permissive inflammatory microenvironment that drives progression in hepatocellular carcinoma. Cancer Res. 2012;72:2533–2542. doi: 10.1158/0008-5472.CAN-11-3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kim WR, Brown RS, Terrault NA, El-Serag H. Burden of liver disease in the United States: summary of a workshop. Hepatology. 2002;36:227–242. doi: 10.1053/jhep.2002.34734. [DOI] [PubMed] [Google Scholar]
- 131.Blachier M, Leleu H, Peck-Radosavljevic M, Valla DC, Roudot-Thoraval F. The burden of liver disease in Europe: a review of available epidemiological data. J Hepatol. 2013;58:593–608. doi: 10.1016/j.jhep.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 132.Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis. 2010;28:155–161. doi: 10.1159/000282080. [DOI] [PubMed] [Google Scholar]