

Abstract
Insulin and adrenergic stimulation are two divergent regulatory systems that may interact under certain pathophysiological circumstances. Here, we characterized a complex consisting of insulin receptor (IR) and β2-adrenergic receptor (β2AR) in the heart. The IR/β2AR complex undergoes dynamic dissociation under diverse conditions such as Langendorff perfusions of hearts with insulin or after euglycemic-hyperinsulinemic clamps in vivo. Activation of IR with insulin induces protein kinase A (PKA) and G-protein receptor kinase 2 (GRK2) phosphorylation of the β2AR, which promotes β2AR coupling to the inhibitory G-protein, Gi. The insulin-induced phosphorylation of β2AR is dependent on IRS1 and IRS2. After insulin pretreatment, the activated β2AR-Gi signaling effectively attenuates cAMP/PKA activity after β-adrenergic stimulation in cardiomyocytes and consequently inhibits PKA phosphorylation of phospholamban and contractile responses in myocytes in vitro and in Langendorff perfused hearts. These data indicate that increased IR signaling, as occurs in hyperinsulinemic states, may directly impair βAR-regulated cardiac contractility. This β2AR-dependent IR and βAR signaling cross-talk offers a molecular basis for the broad interaction between these signaling cascades in the heart and other tissues or organs that may contribute to the pathophysiology of metabolic and cardiovascular dysfunction in insulin-resistant states.
Introduction
Insulin and adrenergic stimulation represent two divergent regulatory systems that interact with overlapping signaling pathways in adipocytes, liver, and skeletal and cardiac muscle. Hyperinsulinemia is a uniform characteristic of obesity and type 2 diabetes (1), which increases insulin receptor (IR) signaling in the myocardium (2). It was recently demonstrated that hyperactivation of insulin signaling in the myocardium contributes to adverse left ventricular (LV) remodeling in pressure overload cardiac hypertrophy (induced by transverse aortic constriction) (3). Heart failure, which is associated with elevated sympathetic adrenergic activity, is characterized by generalized insulin resistance, hyperinsulinemia (4), and impaired insulin-mediated glucose uptake in the myocardium (2). Diabetes and obesity increase the risk of heart failure and induce cardiac dysfunction, which has been termed diabetic cardiomyopathy (5,6). Given that dysfunction of these regulatory systems commonly occurs in cardiovascular diseases (7–9), it is likely that molecular cross-talk between insulin and adrenergic receptor regulatory systems exists within the cardiovascular system.
Stimulation of β-adrenergic receptors (βARs), which are prototypical members of the G-protein–coupled receptor superfamily, is best known for its regulation of contractile function in the heart. Ligand binding to βARs induces cAMP-dependent protein kinase A (PKA) activation (10) leading to phosphorylation of various substrates including phospholamban (PLB) (10–12) to increase myocyte contractility, stroke volume, and cardiac output (13). Among the cardiac βARs, β1AR is the major subtype that couples to the stimulatory G protein, Gs, to stimulate contractile function, whereas β2AR is able to couple to both Gs and Gi but with minimal effect on contractile function (10–12). Conversely, activation of IRs, which are receptor tyrosine kinases, promotes phosphorylation of IR substrates (IRS-1 and -2) leading to Akt activation, which promotes glucose uptake, glucose metabolism, and insulin-mediated cardiac and skeletal muscle growth (14). Stimulation of βARs also increases glucose uptake in cardiac and skeletal muscle cells (15,16). Insulin and adrenergic stimulation share common downstream signaling components including Gi (17), arrestin (18), and G-protein receptor kinase (GRK)2 (16,19,20). Stimulation with either insulin or adrenergic receptors antagonizes the ability of the other to activate glucose transport (8) and to modulate myocyte survival (21).
An earlier study suggested that insulin augmented adrenergic stimulation of contractility in isolated papillary muscles (22). However, in an ischemia-reperfusion study, insulin inhibited β-adrenergic responses in the heart(s) (23). Also, phosphatidylinositol 3-kinase, a downstream kinase in the insulin signaling pathway, inhibits β-adrenergic–induced contractile responses in isolated cardiomyocytes (24). Earlier studies revealed that insulin induced β2AR phosphorylation and internalization in HEK293 cells and adipocytes (25–27); however, a comprehensive understanding of the molecular mechanisms underlying insulin’s effects on β2AR signaling in the heart remains to be achieved.
In this study, we characterized signaling cross-talk in which IRs and β2ARs form a novel complex in the heart. This complex directly exerts a β2AR-dependent impact on intracellular transduction of βAR signaling pathways that regulate cardiac contractility in cardiomyocytes and the myocardium. Stimulation of IR promotes IRS-dependent and GRK2-mediated phosphorylation of β2AR in isolated cardiomyocytes and in ex vivo Langendorff perfused hearts or after euglycemic-hyperinsulinemic clamps in vivo. Stimulation of the IR also promotes dissociation of the β2AR-IR complex and promotes β2AR internalization. Internalization of β2AR selectively promotes Gi coupling to attenuate cAMP/PKA signaling (28), which inhibits contractile response in isolated neonatal and adult cardiomyocytes and in Langendorff perfused hearts. Our results not only underscore the critical role of signaling cross-talk and integration between IR and βARs for contractile regulation in the myocardium but also provide a potential general mechanism to understand cross-talk between IR and βAR regulatory systems in other metabolic disorders and cardiac diseases.
Research Design and Methods
Langendorff Perfusion Heart Preparation
Animal experiments were performed following the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All procedures were approved by the Institutional Animal Care and Use Committees at the University of California, Davis; the University of Utah; Temple University; and the Carver College of Medicine of the University of Iowa. The isolated heart perfusion technique was described previously (29). Hearts were excised from mice under anesthesia (120 mg/kg body wt i.p. sodium pentobarbital) and were rapidly placed on a Langendorff apparatus. Hearts were perfused at a constant pressure of 80 mmHg with a solution containing 113.8 mmol/L NaCl, 22 mmol/L NaHCO3, 4.7 mmol/L KCl, 1.2 mmol/L KH2PO4, 1.1 mmol/L MgSO4, 11 mmol/L glucose, 2 mmol/L CaCl2, and 2 mmol/L Na-pyruvate and aerated with 95% oxygen and 5% carbon dioxide, pH 7.35–7.4, to which was added isoproterenol (ISO) at concentrations ranging from 10-14 to 10-6 (mol/L). A water-filled balloon was inserted into the left ventricle and adjusted to achieve a LV end diastolic pressure of 10 mmHg. The balloon was connected to a Millar pressure system (Millar Instruments, Houston, TX), and the pressure was measured with a pressure catheter (SPR-671; Millar Instruments) connected to an ADInstruments PowerLab 16/30 with LabChart Pro-6.0 (ADInstruments, Boston, MA). The heart rate was maintained at 480 bpm by pacing the right ventricle with a Grass SD9 Stimulator. Once a stable effect of the previous dose (∼5 min) was obtained, the next dose was applied. LV pressure, LV end diastolic pressure, and the maximum rate of positive and negative change (dP/dt) in LV pressure were recorded. Data were analyzed offline with LabChart Pro-6.0.
Euglycemic-Hyperinsulinemic Clamps
Euglycemic-hyperinsulinemic clamps were performed in nonsedated mice as previously described with minor changes (30). In summary, the mouse jugular vein was catheterized under tribromoethanol anesthesia (250 mg/kg body wt by single intraperitoneal injection). Mice were allowed to recover for 5 days with one heparin flush on day 3 before the clamp procedure. All mice were fasted overnight before the clamp procedure day to synchronize the metabolic state. On the day of the procedure, mice were single housed in a standard housing cage with a tether arm attached to the catheter. A dual infusion pump (Harvard Apparatus, Boston, MA) was used to infuse insulin at a constant flow rate (10 mU/kg/min). A glucose solution was infused at a variable rate to maintain plasma glucose at a target value of 75–110 mg/dL and held at that level for 60 min. A comparable rate of saline infusion was used as control. Glucose was monitored using tail vein blood at 5-min intervals with a glucometer (Glucometer Elite; Bayer, Tarrytown, NY).
Isolated Working Heart Perfusions and Intraperitoneal Injection
Mouse hearts were isolated from anesthetized mice and perfused in the working mode in Krebs-Henseleit buffer supplemented with 5 mmol/L glucose and 0.4 mmol/L palmitate in the presence or absence of 1 nmol/L insulin as previously described by our group (31). Cardiac-restricted IRS1-KO and IRS2-KO mice were injected with isoproterenol (ISO) (2 mg/kg) for 10 min. The hearts were harvested for Western blot.
Cell Culture
Neonatal cardiomyocytes were isolated from 1- to 2-day-old wild-type, β1AR-KO, and β2AR-KO and cardiac-restricted IRS1-KO and IRS2-KO mouse pups. Adult mouse cardiomyocytes were isolated from wild-type and mutant (mut) mice as indicated and cultured as described previously (32). Adult rat cardiomyocytes were provided by Dr. Donald Bers (University of California, Davis). In a subset of experiments, H9c2 cardiomyoblasts were cultured in DMEM plus 10% FBS for experiments.
Adenovirus Infection and Plasmid Transfection
Neonatal and adult cardiomyocytes were infected with adenoviruses (100 multiplicity of infection) as previously described to express the PKA activity biosensor (A-kinase activity reporter 3 [AKAR3] [33]) or the cAMP biosensor (indicator of cAMP using epac 3 [ICUE3] [32]) or C terminal of inhibitory G protein [Gi-ct], GFP-βARKct, Flag-β2AR, Flag-GRKmut β2AR, or Flag-PKAmut β2AR (28) as indicated for 24 h. Small interfering RNA oligos targeting the mouse IR (IDT, Coralville, IA) were transfected into wild-type neonatal cardiomyocytes, and experiments were conducted after 48 h expression. GRK2, IRS1, and IRS2 mouse small hairpin RNA (shRNA) plasmids (Sigma-Aldrich, St. Louis, MO) were used to create recombinant lentiviruses. Neonatal cardiomyocytes were infected with GRK2 shRNA, IRS1 shRNA, or IRS2 shRNA lentivirus for 24 h and cultured for an additional 48 h.
Fluorescent Resonance Energy Transfer Measurement
Myocytes expressing PKA or cAMP biosensors were rinsed and maintained in PBS for fluorescent resonance energy transfer (FRET) recordings (32). Cells were imaged on a Zeiss Axiovert 200M microscope with a 40×/1.3NA oil-immersion objective lens and a cooled charge-coupled device (CCD) camera. Dual emission ratio imaging was acquired with a 420DF20 excitation filter, a 450DRLP diachronic mirror, and two emission filters (475DF40 for cyan and 535DF25 for yellow). The acquisition was set with 200-ms exposure in both channels and 20-s elapses. Images in both channels were subjected to background subtraction, and ratios of yellow-to-cyan color were calculated at different time points.
Adult Myocyte-Shortening Assay
Cells were stimulated with ISO at indicated concentrations after treatment with or without insulin (100 nmol/L) for 30 min. Adult myocytes were placed in a dish with HEPES buffer (34) and electrically stimulated at 30 V/cm at 0.5 Hz at room temperature. Cell length was recorded with a charge coupled device camera. Cell contraction shortening was analyzed by IonOptix software (IonOptix, Boston, MA) and normalized as the increase over the basal levels after being fitted to a sigmoidal curve. The maximal shortening was normalized to the baseline value.
Western Blot Analysis
Whole-cell and heart tissue lysates were prepared in lysis buffer (50 mmol/L Tris, pH 7.4; 2.5 mmol/L EDTA; 150 mmol/L NaCl, 25 mmol/L sodium pyrophosphate, and 1% (v/v) NonidetP40, 1% Na-deoxycholate, 0.1% SDS, and protease inhibitor cocktail tablets (Thermo Scientific, Chicago, IL) after washing twice with ice-cold PBS. The lysates without boiling were resolved by SDS-PAGE. Proteins were transferred to a Nitrocellulose membrane (Millipore, Billerica, MA), and incubated with the primary antibody followed by IRDye 680CW goat-anti mouse or with IRDye 800CW goat-anti rabbit secondary antibodies. Specific proteins were detected by an Odyssey scanner (LI-COR, Lincoln, NE). The primary antibodies used for Western blotting were as follows: total and phosphorylated PLB at Ser16 and Thr17 (Bradilla, Leeds, U.K.), IR (SCBT, Santa Cruz, CA), total and phosphorylated β2AR at Ser261/262 (28) and Ser355/356 (SCBT), total and phosphorylated pAkt Ser473 (Cell Signaling, Danvers, MA), γ-tubulin, GFP, and GRK2 (SCBT).
Coimmunoprecipitation
Heart tissues were used to detect endogenous protein interactions. Heart tissues were lysed with a FastPrep-24 homogenizer for 20 s in immunoprecipitation assay buffer (50 mmol/L Tris-HCl [pH 7.5], 150 mmol/L NaCl, 1%NP-40, 0.25% deoxycholate, 9.4 mg/50 mL sodium orthovanadate, and 1% sodium dodecyl sulfate). Lysates were cleared by centrifugation (40,000 rpm for 30 min at 4°C) and subjected to immunoprecipitation with Protein A beads (Repligen, Waltham, MA). The immunoprecipitates were resolved via SDS-PAGE and blotted with antibodies against IR (1:500) or β2AR (1:500). Primary antibodies were visualized with IRDye 680CW goat anti-mouse or with IRDye 800CW goat anti-rabbit secondary antibodies using an Odyssey scanner (LI-COR).
Statistical Analysis
One or two-way ANOVA followed by post hoc Turkey test or Student t test were performed using Prism (GraphPad Software, San Diego, CA). P < 0.05 was considered statistically significant.
Results
Insulin Impairs β-Adrenergic Stimulation of Contractility in Mouse Hearts
We hypothesized that insulin could impair adrenergic signal transduction in the myocardium and impair adrenergic-induced contractile responses. In Langendorff perfused mouse hearts, insulin alone (1 nmol/L) did not significantly affect cardiac contractility in mouse hearts as evidenced by LV developed pressure (LVDP) (57.5 ± 13.6 mmHg without insulin vs. 53.3 ± 4.6 mmHg with insulin), maximal +dP/dt (rate of rise of ventricular pressure) (1,944.8 ± 508.9 mmHg/s without insulin vs. 1,766.0 ± 202.9 mmHg/s with insulin), and minimum –dP/dt (rate of fall of ventricular pressure) (−1,520.5 ± 318.2 mmHg/s without insulin vs. −1,344.7 ± 115.8 mmHg/s with insulin). β-Adrenergic stimulation with ISO induced a dose-dependent increase in cardiac contractility (Fig. 1A–C) with half-maximal effective concentration (EC50) of ∼0.01 nmol/L and maximal response at concentrations >1 nmol/L. ISO increased peak LVDP from 57.5 ± 13.6 to 132.7 ± 5.6 mmHg (130.7%), maximal +dP/dt from 1,944.8 ± 508.9 to 5,868.3 ± 981.5 mmHg/s (201.7%), and minimum −dP/dt from −1,520.5 ± 318.2 to −4,351.8 ± 358.3 mmHg/s (186.2%). In contrast, pretreatment with 1 nmol/L of insulin significantly attenuated the ISO-induced contractile responses, changing the EC50 by 5- to 10-fold (∼0.1 nmol/L). In comparison with hearts stimulated with ISO alone, the maximal responses of LVDP (from 53.3 ± 4.6 to 78.9 ± 10.0 mmHg; 48.0%), +dP/dt (from 1,766.0 ± 202.9 to 2,584.5 ± 156.2 mmHg/s; 46.4%), and –dP/dt (from −1,344.7 ± 115.8 to −2,077.7 ± 164.8 mmHg/s; 54.5%) were all substantially decreased (Fig. 1A–C). Consistent with contractility data, adrenergic stimulation, but not insulin stimulation, significantly induced PKA phosphorylation of Ser16 and Ca2+/calmodulin-dependent protein kinase (CaMK)II phosphorylation of Thr17 of PLB, a critical regulator of calcium cycling that mediates cardiac contractility. In comparison, neither stimulation altered the expression of SERCA and the ratio between PLB and SERCA (Supplementary Fig. 1). However, pretreatment with insulin significantly reduced PKA and (CaMK)II phosphorylation of PLB induced by ISO (Fig. 1D). These data suggest that insulin blunts β-adrenergic responsiveness of cardiomyocytes, which may contribute to impaired cardiac function in hyperinsulinemic states.
Figure 1.
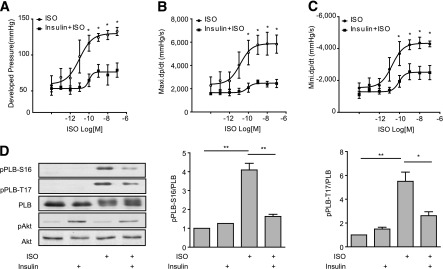
Insulin inhibits βAR-induced phosphorylation of PLB and cardiac contractility in murine hearts. Mouse hearts were cannulated for Langendorff perfusion with different concentrations of ISO in the absence or presence of pretreatment with insulin (1 nmol/L for 30 min). The LVDP (A), maximal +dP/dt (B), and minimal −dP/dt (C) were analyzed and plotted against doses of ISO. D: Mouse hearts were cannulated for Langendorff perfusion with ISO (100 nmol/L for 10 min) with or without pretreatment with insulin (1 nmol/L for 30 min). Heart lysates were used to detect phosphorylation of Akt (Ser473) and PLB at the Ser16 site (PKA site) and Thr17 site ([CaMK]II site). The signals were normalized against their respective total proteins. n = 5. *P < 0.05; **P < 0.01, by one-way ANOVA between groups. Maxi., maximal; Mini., minimal.
Activation of IR by Insulin Impairs β-Adrenergic Signaling in Cardiomyocytes in a β2AR-Dependent Manner
We then used FRET-based biosensors AKAR3 (33) for PKA activities to directly determine the impact of insulin-adrenergic signaling cross-talk on cAMP and PKA activities in neonatal cardiomyocytes. Stimulation of βAR with ISO induced a dose-dependent increase in the PKA AKAR3-FRET ratio (EC50 0.43 nmol/L) (Fig. 2A and Supplementary Fig. 2). Insulin dose-dependently impaired ISO-induced increase(s) in the PKA AKAR3-FRET ratio by right shifting the ISO-induced dose response curves (EC50 3.78 nmol/L at 10 nmol/L and EC50 38.2 nmol/L at 100 nmol/L of insulin treatment) (Fig. 2A). Pretreatment with increasing doses of insulin almost completely inhibited the increase(s) in PKA AKAR3-FRET ratio induced at 1 nmol/L of ISO (Fig. 2A). ISO (100 nmol/L) induced a robust increase in sarcomere shortening in adult cardiomyocytes (Fig. 2B). While insulin alone minimally affected the baseline shortening, pretreatment with insulin significantly attenuated the ISO-induced myocyte shortening (Fig. 2B). In comparison, insulin did not affect the forskolin-induced cAMP FRET response and myocyte contractile shortening response (Supplementary Fig. 3). To further assess which βAR subtype signaling was modulated by insulin, we applied the β1AR selective agonist dobutamine or the β2AR selective agonist clenbuterol. Dobutamine robustly increased the PKA AKAR3-FRET ratio, which was significantly attenuated after pretreatment with insulin (Fig. 2C). In comparison, clenbuterol induced a modest increase in PKA AKAR3-FRET ratio, which was also reduced by pretreatment with insulin (Fig. 2C). In agreement, dobutamine, but not clenbuterol, significantly increased myocyte contractile shortening, which is consistent with the predominant role of β1AR in promoting cardiac contractility in hearts (10–12). In contrast, β2AR-induced cAMP and PKA activities are compartmentalized along the plasma membrane and play a minimal role in inducing phosphorylation of PLB and the contractile response (10–12). Thus, whereas pretreatment with insulin significantly attenuated the dobutamine-induced contractile response (Fig. 2D), insulin had little impact on contractility when cells were exposed to clenbuterol. Consistent with the contractile shortening data, dobutamine but not clenbuterol induced a strong phosphorylation of Ser16 and Thr17 of PLB, which was significantly attenuated by insulin pretreatment (Fig. 2E–H).
Figure 2.

Insulin attenuates βAR-induced PKA activities and contractile responses in adult cardiomyocytes. A and C: Adult rat cardiomyocytes expressing the PKA biosensor AKAR3 were stimulated with different concentrations of insulin and ISO as indicated (A) or with βAR agonists (dobutamine, 1 μmol/L; or clenbuterol, 10 nmol/L) after incubation with insulin (100 nmol/L for 30 min) (C). The changes in the PKA FRET ratio were recorded, and the maximal increases in PKA FRET ratio were plotted. B and D: Adult rat cardiomyocytes were stimulated with βAR agonists (dobutamine, 1 μmol/L; or clenbuterol, 10 nmol/L) after incubation with insulin (100 nmol/L for 30 min) as indicated. Contractile shortening was recorded and plotted. n indicates the number of cells tested. E–H: The cells were lysed to detect phosphorylation of Akt (Ser473) and PLB (Ser16 or Thr17); the phosphorylation levels were normalized against their respective total protein. n = 4. *P < 0.05, **P < 0.01, ***P < 0.001 by one-way ANOVA between groups as indicated. AU, arbitrary units; Ins, insulin.
We further used myocytes from mice lacking individual βAR genes to determine which βAR subtype is involved in insulin-induced signaling cross-talk. In wild-type neonatal myocytes, insulin pretreatment reduced the ISO-induced changes in cAMP ICUE3 and PKA AKAR3-FRET ratios (Fig. 3A and B). In myocytes lacking β2ARs (β2AR-KO), we examined the insulin effect on β1AR, the major β1AR subtype that is responsible for cardiac contractile response to catecholamines in the heart. Interestingly, insulin did not influence ISO-induced and β1AR-mediated increases in cAMP and PKA FRET ratio (Fig. 3A and B), suggesting that expression and activation of β2ARs is necessary for the insulin effect. As controls, in myocytes lacking β1ARs (β1AR-KO), activation of β2AR induced small increases in cAMP and PKA FRET ratios, which were sensitive to insulin pretreatment (Fig. 3A and B). These data indicate that despite the minor expression of β2AR in the heart, the cross-talk between IR and β2AR is sufficient to attenuate the cAMP signal induced by activation of both β1AR and β2AR in wild-type cells.
Figure 3.

The effects of insulin on βAR subtype signaling–induced cAMP-PKA activities and contractile responses in β1AR-KO and β2AR-KO cardiomyocytes. Wild-type, β1AR-KO, and β2AR-KO neonatal cardiomyocytes expressing the cAMP biosensor ICUE3 (A) or PKA biosensor AKAR3 (B) were treated with or without insulin (100 nmol/L for 30 min) prior to stimulation with ISO (100 nmol/L) or as indicated. The changes in the cAMP FRET ratio and PKA FRET ratio were recorded, and the maximal responses were plotted. ***P < 0.001 by Student t test relative to ISO group; n indicates the number of cells tested. Adult β1AR-KO (C) or β2AR-KO (E) myocytes paced at 0.5 Hz were pretreated with or without insulin (100 nmol/L for 30 min) before βAR agonists (ISO, 100 nmol/L) as indicated. Contractile shortening was recorded and plotted. ***P < 0.001 by one-way ANOVA between groups; n indicates the number of cells tested. D and F: Cells were lysed to detect phosphorylation of Akt (Ser473) and PLB (Ser16 or Thr17); n = 4. AU, arbitrary units; WT, wild-type.
We then validated the cross-talk between IR and βAR subtype signaling cascades in contractile shortening in adult cardiomyocytes. ISO failed to increase contractile shortening or phosphorylation of PLB in β1AR-KO myocytes (Fig. 3C and D). In contrast, in β2AR-KO myocytes, ISO induced a robust response in contractile shortening, which was not affected by insulin pretreatment (Fig. 3E). Consistently, ISO also promoted a strong phosphorylation of Ser16 and Thr17 of PLB, which was not affected by insulin pretreatment (Fig. 3F).
β2AR Is Necessary for the Inhibitory Effect of Insulin on Cardiac Contractility in Mouse Hearts
We further validated the necessary role of β2AR in cross-talk between IR and βAR signaling cascades in animal hearts. In Langendorff perfused β2AR-KO mouse hearts, β-adrenergic stimulation with ISO induced a strong increase in cardiac contractility, including increased peak LVDP, maximal +dP/dt, and minimum −dP/dt (Fig. 4A–C). Unlike the case of wild-type hearts, pretreatment with 1 nmol/L of insulin did not significantly attenuate the ISO-induced contractile responses, with no reduction in the maximal responses of LVDP, maximal +dP/dt, or minimal −dP/dt (Fig. 4A–C). Consistent with contractility data, adrenergic stimulation, but not insulin stimulation, significantly induced PKA phosphorylation of Ser16 and (CaMK)II phosphorylation of Thr17 of PLB (Fig. 4D). Pretreatment with insulin did not significantly reduce ISO-induced PKA and (CaMK)II phosphorylation of PLB (Fig. 4D). These data confirm the essential role of β2AR in mediating the cross-talk between IR and βARs that impairs cardiac contractility in animal hearts.
Figure 4.

β2AR is necessary for the inhibitory effects of insulin on cardiac contractility. β2AR-KO mouse hearts were cannulated for Langendorff perfusion with ISO in the absence or presence of pretreatment with insulin (1 nmol/L for 30 min). The LVDP (A), maximal +dP/dt (B), and minimal −dP/dt (C) were analyzed and plotted. D: Mouse hearts were cannulated for Langendorff perfusion with ISO (100 nmol/L for 10 min) with or without pretreatment with insulin (1 nmol/L for 30 min). Heart lysates were used to detect phosphorylation of Akt (Ser473), PLB at Ser16 (PKA site), and Thr17 ([CaMK]II site), respectively. The signals were normalized against their respective total proteins, respectively. n = 5. *P < 0.05 by one-way ANOVA between groups.
IR and β2AR Exist in a Complex, and Insulin Induces PKA- and GRK-Mediated Phosphorylation of β2AR in Murine Hearts
We then sought to determine the molecular mechanism for the IR/β2AR cross-talk in animal hearts. Isolated mouse hearts were perfused in the Langendorff mode with or without insulin (1 nmol/L) for 5 min. We observed significant increase(s) in phosphorylation of β2AR at Ser261/262 (PKA sites) and Ser355/356 (GRK sites) relative to saline-perfused controls (Fig. 5A). Meanwhile, in the hearts of mice subjected to euglycemic-hyperinsulinemic clamps for 60 min, we also observed significant increase(s) in phosphorylation of β2AR at both PKA and GRK sites in hearts obtained from hyperinsulinemic animals (Fig. 5B). Activation of IR signaling was supported by increased phosphorylation of Akt in hearts exposed to insulin ex vivo and in vivo (Fig. 5A and B). In comparison, the protein levels of Gi were not changed by insulin perfusion (Supplementary Fig. 4). Moreover, we found that IRs and β2ARs form membrane complexes that may facilitate signaling cross-talk. Coimmunoprecipitation with anti-IR antibody showed that both receptors form complexes in mouse hearts, but the association was significantly reduced in hearts after exposure to insulin for 10 min ex vivo (Fig. 5C) or in mouse hearts after euglycemic-hyperinsulinemic clamps for 60 min in vivo (Fig. 5D).
Figure 5.
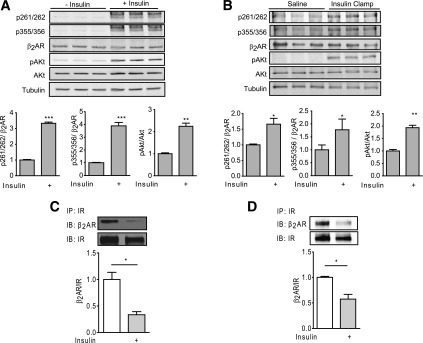
Insulin induces β2AR phosphorylation in mouse hearts. Isolated working mouse hearts were perfused with or without insulin (1 nmol/L) prior to harvest (A). Mice were subjected to euglycemic-hyperinsulinemic clamps or to sham saline infusions for 60 min, and the hearts were harvested (B). PKA and GRK-mediated phosphorylation of the β2AR at Ser261/262 and Ser355/356 were detected by Western blot. The levels of phosphorylation were normalized against total β2AR. n = 3. *P < 0.05; **P < 0.01 or ***P < 0.001 by Student t test relative to controls. Heart lysates obtained from Langendorff perfused mouse hearts with insulin (100 nmol/L) for 10 min (C) or from animals after sham or euglycemic-hyperinsulinemic clamps (60 min) (D) were immunoprecipitated (IP) with anti-IR antibody. The pulled-down proteins were detected by Western blot with antibodies as indicated and normalized against their respective IP proteins. n > 3. *P < 0.05 by t test between indicated groups. IB, immunoblot.
Insulin Induces IRS-Dependent PKA and GRK Phosphorylation of β2AR for Biased Activation of Gi in Cardiomyocytes
Activation of IR induces downstream signaling via recruitment of the adaptor signaling proteins, insulin receptor substrates (IRSs). Deletion of either IRS1 orIRS2 in mouse hearts (35) abolished insulin-induced phosphorylation of β2AR and the inhibitory effect of insulin (100 nmol/L) on the ISO-induced cAMP FRET responses (Fig. 6A and B). Inhibition of IRS autophosphorylation with PQ401 also abolished the inhibitory effect of insulin (100 nmol/L) on the ISO-induced cAMP FRET responses (Fig. 6C). Accordingly, silencing of the IR with an IR-specific small interfering RNA abolished the insulin effects on the βAR-induced cAMP signal (Fig. 6D). These data suggest that cross-talk between IR and βARs are dependent on the interaction between the IR and IRS proteins. A recent study indicates that insulin can promote formation of IRS-GRK2 complexes in animal hearts (16). While different GRKs are implicated in agonist-induced phosphorylation of β2ARs in a cell type–specific manner (28,36), we have previously shown that GRK2 is necessary for βAR agonist–induced GRK phosphorylation of β2ARs at Ser355/356 in cardiomyocytes (28). Inhibition of GRK2 also abolished the insulin-induced phosphorylation of β2ARs at GRK sites Ser355/356 in H9C2 cardiomyoblasts (Supplementary Fig. 5A) and the insulin-mediated impairment of β-adrenergic stimulation of cAMP signaling in neonatal cardiomyocytes (Fig. 7A). Meanwhile, the PKA inhibitor H89 abolished the insulin-induced phosphorylation of β2ARs at PKA sites Ser261/262 in H9C2 cardiomyoblasts (Supplementary Fig. 5B) and partially rescued the insulin-mediated impairment of ISO-induced cAMP FRET response in neonatal cardiomyocytes (Fig. 7B). As a control, H89 minimally affected the maximal cAMP signaling induced by ISO alone (Fig. 7B). We further examined the role of β2AR phosphorylation in IR-β2AR cross-talk by introducing either wild-type or mutant β2ARs into β2AR-KO neonatal cardiomyocytes. Insulin impaired cAMP generation induced by ISO in cells expressing the wild-type β2AR, but the effect of insulin was absent in the cells expressing either PKAmut β2AR that lacks the PKA phosphorylation sites or GRKmut β2AR that lacks the GRK phosphorylation sites (Fig. 7C). In agreement, inhibition of GRK2 by overexpressing βARKct, a dominant negative inhibitor of GRK2, abolished the inhibitory effect of insulin on the ISO-induced PKA FRET response in neonatal myocytes and fractional shortening in adult rat cardiomyocytes (Fig. 7D and E).
Figure 6.
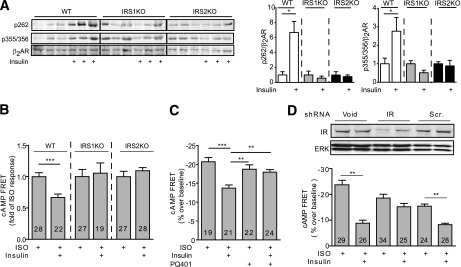
IR and IRS proteins are necessary for insulin-induced phosphorylation of β2AR and inhibition of adrenergic signaling. A: Cardiac-restricted IRS1-KO and IRS2-KO mice were injected with ISO, and the changes in phosphorylation of β2AR at the PKA site Ser262 and GRK site Ser355/356 were detected with antibodies, respectively, and normalized against the total β2AR. *P < 0.05, by one-way ANOVA relative to wild-type (WT) controls; n = 4. IRS1-KO and IRS2-KO (B) and wild-type (C and D) neonatal cardiomyocytes that expressed the cAMP biosensor ICUE3 together with scrambled, IR-specific shRNA were treated with PQ401 (5 μmol/L for 2 h) and insulin (100 nmol/L for 30 min) before stimulation with ISO (100 nmol/L) as indicated. The changes in the cAMP FRET ratio were recorded, and the maximal increases in cAMP FRET ratio were plotted. **P < 0.01, ***P < 0.001 by Student t test relative to ISO group; n indicates the number of cells tested. ERK, extracellular signal–related kinase; Scr., scramble; shRNA, small hairpin RNA.
Figure 7.

Insulin induces GRK2-dependent inhibition of βAR-induced cAMP activities and contractile shortening in cardiomyocytes. A and B: The ISO (100 nmol/L)-induced changes in the cAMP FRET ratio were recorded in neonatal cardiomyocytes with GRK2 shRNA or PKA inhibitor H89 after incubation with insulin (100 nmol/L for 30 min) as indicated. The maximal increases in cAMP FRET ratio were plotted. C: β2AR-KO neonatal cardiomyocytes expressing wild-type or mutant β2ARs harboring either the PKA (PKAmut) or GRK (GRKmut) phosphorylation sites. The ISO (100 nmol/L)-induced changes in the cAMP FRET ratio were recorded after treatment with insulin (100 nmol/L) for 30 min. The maximal increases in cAMP FRET ratio were plotted. D and E: The ISO (100 nmol/L)-induced changes in the PKA FRET ratio and contractile shortening were recorded in adult rat cardiomyocytes expressing the GRK2 inhibitor βARKct after incubation with insulin (100 nmol/L for 30 min) as indicated. The maximal increases in PKA FRET ratio were plotted. *P < 0.05, **P < 0.01, and ***P < 0.001 by one-way ANOVA or Student t test relative to the ISO group or between group pairs as indicated; n indicates the number of cells tested. Scr., scramble; shRNA, small hairpin RNA.
The phosphorylation of the β2AR by GRK2 and PKA promotes receptor internalization and also switches the receptor coupling from Gs to Gi proteins in cardiomyocytes (28,37). In myocytes treated with insulin (100 nmol/L for 30 min), inhibition of Gi with pertussis toxin (PTX) or the specific Gi inhibitor Gi-CT (38) rescued the ISO-induced cAMP FRET response (Fig. 8A). Moreover, inhibition of Gi with PTX abolished the inhibitory effect of insulin on ISO-induced phosphorylation of PLB and contractile shortening in adult cardiomyocytes (Fig. 8B). Activation of IR has been reported to be linked to activation of phosphodiesterase 3 (PDE3) in oocytes and neurons (39,40). We therefore tested whether PDE3 plays any role in the inhibitory effect of insulin on βAR signaling in myocytes. Inhibition of PDE3 with cilostamide partially rescued the ISO-induced cAMP FRET response (Supplementary Fig. 6). These data suggest that insulin induces a β2AR-Gi coupling to inhibit cAMP production and myocyte contractility under adrenergic stimulation in part via activation of PDE3.
Figure 8.

Insulin induces Gi-dependent inhibition of βAR-induced cAMP activities and contractile shortening in cardiomyocytes. A: Neonatal cardiomyocytes expressing the cAMP biosensor ICUE3 together with Gi-ct, a Gi-specific inhibitor, were stimulated with ISO (100 nmol/L) after incubation with PTX (1 μg/mL for 3 h) and insulin (100 nmol/L for 30 min) as indicated. The maximal increases in cAMP FRET ratio were plotted. B: Adult rat cardiomyocytes were treated with ISO (100 nmol/L) after incubation with PTX (1 μg/mL for 3 h) and insulin (100 nmol/L for 30 min) as indicated, and the fractional shortening was recorded. *P < 0.05, **P < 0.01, and ***P < 0.001 by one-way ANOVA between groups; n indicates the number of cells tested. C: Working model of the IR-β2AR signaling network in cardiomyocytes for contractile responses. AC, adenylate cyclase; Gi, inhibitory G protein; Gs, stimulatory G protein.
Discussion
In both diabetes and heart failure, circulating insulin levels are chronically elevated, leading to persistent stimulation of IRs in these clinical conditions. Despite the resistance to Akt-mediated glucose metabolism in adipocytes and skeletal muscle cells, the heart retains its insulin sensitivity in terms of insulin’s ability to activate IR signaling cascades in type 2 diabetes (2,41). The increase in insulin signaling in myocardium promotes translocation of CD36, which exacerbates fatty acid uptake and lipotoxicity (42). Moreover, hyperactive insulin signaling also accelerates adverse LV remodeling in pressure overload hypertrophy (3). Here, we show that insulin can directly impair adrenergic signaling pathways for contractile function via an IR-β2AR signaling complex in animal hearts. This study offers a potential novel mechanism for cardiac dysfunction associated with hyperinsulinemia in diabetic cardiomyopathy and heart failure.
In animal hearts, this IR and β2AR signaling complex channels a direct and negative impact of insulin on β-adrenergic signaling pathways that stimulate cardiac contractile function. These data are consistent with previous reports showing that insulin inhibits β-adrenergic action in hearts after ischemia/reperfusion (23). Insulin stimulation promotes cross-talk with β2AR pathways via IRS-dependent and GRK2-mediated phosphorylation of the adrenergic receptor, which selectively activates a Gi-biased β2AR-signaling cascade to inhibit cAMP/PKA activities under β-adrenergic stimulation. Consequently, this IR-β2AR cross-talk leads to impaired β-adrenergic–induced contractile function in cardiomyocytes and perfused mouse hearts (Fig. 8C).
Previous studies show that insulin induces phosphorylation of the β2AR at classic PKA phosphorylation sites for β2AR internalization and subsequent downregulation in human embryonic kidney (HEK)293 cells and adipocytes (25–27). Here, we identified additional phosphorylation of the β2AR at the classic GRK sites after insulin stimulation, which are also dependent on IRS expression (Fig. 6) (28). Notably, the GRK-mediated phosphorylation of β2AR in cardiomyocytes is different from those in HEK293 cells (36). In cardiomyocytes, GRK2 is necessary for agonist-induced phosphorylation of Ser355/356 (28), which we now show is promoted by activation of the IR. The phosphorylation at GRK sites is likely due to recruitment of GRK2 to the β2AR via increased association between GRK2 and IRS proteins after insulin stimulation (16). The signaling pathways involved in PKA phosphorylation of β2AR remain to be elucidated. A prior study reported that IR signaling intermediates downstream of phosphatidylinositol 3-kinase may play a role in inhibiting cAMP-mediated inotropic effects under adrenergic stimulation (24), which could be linked to phosphorylation at the PKA sites of β2AR.
After insulin stimulation, the phosphorylated β2AR dissociates from the complex and promotes receptor/Gi coupling. Thus, insulin signaling mimics a biased β2AR agonist that selectively activates a Gi-biased signaling pathway. This insulin-induced β2AR/Gi coupling is sufficient to attenuate adrenergic-induced cAMP activities in hearts. As a result, insulin blunts adrenergic-induced PKA phosphorylation of PLB, a critical protein involved in myocyte calcium cycling, and impairs β-adrenergic–induced contractility in both isolated myocytes and animal hearts. A prior study revealed that IGF-1 can promote β1AR internalization in HEK293 cells and inhibit β1AR signaling in canine adult cardiomyocytes (43). Conversely, overexpression of IGF-1 in the myocardium prevents streptozotocin-induced cardiac contractile dysfunction and restored β-adrenergic responsiveness in isolated myocytes (44). Therefore, it remains to be determined whether IGF-1 signaling modulates β1AR signaling in cardiomyocytes via mechanisms that are distinct from those of insulin.
Accumulating evidence also indicates that adrenergic signaling may modulate insulin signal transduction pathways that regulate glucose uptake in adipocytes and skeletal and cardiac muscle cells (8,16,18,20). Reduced β2AR expression in aged animals contributes to glucose intolerance, and overexpression of the β2AR can rescue this phenotype (45). Evidence also suggests that Akt could be a key mechanism underlying the impact of β-adrenergic stimulation on insulin-induced glucose uptake (46,47), although precise molecular signaling mechanisms remain to be elucidated. This newly identified IR-β2AR complex herein reported may provide a molecular basis to understand and reconcile recent studies showing that βAR activation impairs insulin-induced Akt activation, GLUT4 translocation, and glucose uptake in cardiomyocytes, adipocytes, and skeletal muscle cells (16,19,48,49).
The novel observations made in this report are of translational significance. Diabetes and insulin resistance are associated with altered cardiac structure and function independently of coronary artery disease and ischemia (50). Moreover, impaired inotropic reserve to dobutamine was observed in humans with type 2 diabetes in the absence of coronary artery disease (51). Given that type 2 diabetes in humans is associated with hyperinsulinemia and activation of myocardial insulin signaling (2), our findings provide a plausible mechanism for impaired myocardial inotropic reserve in individuals with type 2 diabetes. Diabetes also increases the risk of heart failure (52), and heart failure is an insulin-resistant state (2). If the present findings that hyperinsulinemia may inhibit β1AR signaling via Gi-biased β2AR signaling hold true in humans, then the possibility is raised that hyperinsulinemic subjects with type 2 diabetes and heart failure might have increased sensitivity to cardio-depressive effects of nonselective or β1 blockade, which currently represent the standard of care for managing patients with heart failure. Furthermore, the current study raises the interesting question of whether specific modulation of β2-adrenergic receptor signaling could influence the pathophysiology of diabetic cardiomyopathy or play a role in managing heart failure in diabetic or obese patients with hyperinsulinemia. More broadly, the IR-βAR signaling cross-talk herein described offers a new platform with which to explore the broad implications of myocardial insulin signaling in heart failure, which is an insulin-resistant and hyperinsulinemic state.
Supplementary Material
Article Information
Acknowledgments. The authors thank Dr. Donald Bers (University of California, Davis) for reagents.
Funding. This study was supported by National Institutes of Health grants RO1 HL082846 to Y.K.X. and DK092065 to E.D.A., who is an established investigator of the American Heart Association (AHA); AHA established investigator grant 12EIA8410007 to Y.K.X. and AHA 0730347N to X.C.; National Natural Science Foundation of China grant 81102438 to Q.F.; and a postdoctoral fellowship from the German Research Foundation to C.R.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. Q.F. wrote and edited the manuscript and researched data. B.X., Y.Liu, D.P., J.L., Y.Li, Y.Zha., Y.Zhu, T.R., Q.S., and X.C. researched data. C.R. and R.B.C. provided essential reagents and materials. E.D.A. and Y.K.X. wrote and edited the manuscript and researched data. Y.K.X. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-1763/-/DC1.
References
- 1.DeFronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009;58:773–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cook SA, Varela-Carver A, Mongillo M, et al. Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. Eur Heart J 2010;31:100–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shimizu I, Minamino T, Toko H, et al. Excessive cardiac insulin signaling exacerbates systolic dysfunction induced by pressure overload in rodents. J Clin Invest 2010;120:1506–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chokshi A, Drosatos K, Cheema FH, et al. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation 2012;125:2844–2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation 2007;115:3213–3223 [DOI] [PubMed] [Google Scholar]
- 6.Abel ED, O’Shea KM, Ramasamy R. Insulin resistance: metabolic mechanisms and consequences in the heart. Arterioscler Thromb Vasc Biol 2012;32:2068–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mancia G, Bousquet P, Elghozi JL, et al. The sympathetic nervous system and the metabolic syndrome. J Hypertens 2007;25:909–920 [DOI] [PubMed] [Google Scholar]
- 8.Morisco C, Lembo G, Trimarco B. Insulin resistance and cardiovascular risk: New insights from molecular and cellular biology. Trends Cardiovasc Med 2006;16:183–188 [DOI] [PubMed] [Google Scholar]
- 9.Masuo K, Rakugi H, Ogihara T, Esler MD, Lambert GW. Cardiovascular and renal complications of type 2 diabetes in obesity: role of sympathetic nerve activity and insulin resistance. Curr Diabetes Rev 2010;6:58–67 [DOI] [PubMed] [Google Scholar]
- 10.Xiang Y, Kobilka BK. Myocyte adrenoceptor signaling pathways. Science 2003;300:1530–1532 [DOI] [PubMed] [Google Scholar]
- 11.Xiao RP, Zhu W, Zheng M, et al. Subtype-specific alpha1- and beta-adrenoceptor signaling in the heart. Trends Pharmacol Sci 2006;27:330–337 [DOI] [PubMed] [Google Scholar]
- 12.Perrino C, Rockman HA. Reversal of cardiac remodeling by modulation of adrenergic receptors: a new frontier in heart failure. Curr Opin Cardiol 2007;22:443–449 [DOI] [PubMed] [Google Scholar]
- 13.Brittsan AG, Kranias EG. Phospholamban and cardiac contractile function. J Mol Cell Cardiol 2000;32:2131–2139 [DOI] [PubMed] [Google Scholar]
- 14.O’Neill BT, Abel ED. Akt1 in the cardiovascular system: friend or foe? J Clin Invest 2005;115:2059–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nevzorova J, Evans BA, Bengtsson T, Summers RJ. Multiple signalling pathways involved in beta2-adrenoceptor-mediated glucose uptake in rat skeletal muscle cells. Br J Pharmacol 2006;147:446–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciccarelli M, Chuprun JK, Rengo G, et al. G protein-coupled receptor kinase 2 activity impairs cardiac glucose uptake and promotes insulin resistance after myocardial ischemia. Circulation 2011;123:1953–1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song X, Zheng X, Malbon CC, Wang H. Galpha i2 enhances in vivo activation of and insulin signaling to GLUT4. J Biol Chem 2001;276:34651–34658 [DOI] [PubMed] [Google Scholar]
- 18.Luan B, Zhao J, Wu H, et al. Deficiency of a beta-arrestin-2 signal complex contributes to insulin resistance. Nature 2009;457:1146–1149 [DOI] [PubMed] [Google Scholar]
- 19.Cipolletta E, Campanile A, Santulli G, et al. The G protein coupled receptor kinase 2 plays an essential role in beta-adrenergic receptor-induced insulin resistance. Cardiovasc Res 2009;84:407–415 [DOI] [PubMed] [Google Scholar]
- 20.Usui I, Imamura T, Satoh H, et al. GRK2 is an endogenous protein inhibitor of the insulin signaling pathway for glucose transport stimulation. EMBO J 2004;23:2821–2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rane S, He M, Sayed D, Yan L, Vatner D, Abdellatif M. An antagonism between the AKT and beta-adrenergic signaling pathways mediated through their reciprocal effects on miR-199a-5p. Cell Signal 2010;22:1054–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferrara N, Abete P, Corbi G, et al. Insulin-induced changes in beta-adrenergic response: an experimental study in the isolated rat papillary muscle. Am J Hypertens 2005;18:348–353 [DOI] [PubMed] [Google Scholar]
- 23.Yu QJ, R Si, N Zhou, et al. Insulin inhibits beta-adrenergic action in ischemic/reperfused heart: a novel mechanism of insulin in cardioprotection. Apoptosis 2008;13:305–317 [DOI] [PubMed]
- 24.Leblais V, Jo SH, Chakir K, et al. Phosphatidylinositol 3-kinase offsets cAMP-mediated positive inotropic effect via inhibiting Ca2+ influx in cardiomyocytes. Circ Res 2004;95:1183–1190 [DOI] [PubMed] [Google Scholar]
- 25.Doronin S, Wang Hy HY, Malbon CC. Insulin stimulates phosphorylation of the beta 2-adrenergic receptor by the insulin receptor, creating a potent feedback inhibitor of its tyrosine kinase. J Biol Chem 2002;277:10698–10703 [DOI] [PubMed] [Google Scholar]
- 26.Hadcock JR, Port JD, Gelman MS, Malbon CC. Cross-talk between tyrosine kinase and G-protein-linked receptors. Phosphorylation of beta 2-adrenergic receptors in response to insulin. J Biol Chem 1992;267:26017–26022 [PubMed] [Google Scholar]
- 27.Shumay E, Song X, Wang HY, Malbon CC. pp60Src mediates insulin-stimulated sequestration of the beta(2)-adrenergic receptor: insulin stimulates pp60Src phosphorylation and activation. Mol Biol Cell 2002;13:3943–3954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu R, Ramani B, Soto D, De Arcangelis V, Xiang Y. Agonist dose-dependent phosphorylation by protein kinase A and G protein-coupled receptor kinase regulates beta2 adrenoceptor coupling to G(i) proteins in cardiomyocytes. J Biol Chem 2009;284:32279–32287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.MacDonnell SM, García-Rivas G, Scherman JA, et al. Adrenergic regulation of cardiac contractility does not involve phosphorylation of the cardiac ryanodine receptor at serine 2808. Circ Res 2008;102:e65–e72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belke DD, Betuing S, Tuttle MJ, et al. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J Clin Invest 2002;109:629–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazumder PK, O’Neill BT, Roberts MW, et al. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes 2004;53:2366–2374 [DOI] [PubMed] [Google Scholar]
- 32.Soto D, De Arcangelis V, Zhang J, Xiang Y. Dynamic protein kinase a activities induced by beta-adrenoceptors dictate signaling propagation for substrate phosphorylation and myocyte contraction. Circ Res 2009;104:770–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu S, Li Y, Kim S, et al. Phosphodiesterases coordinate cAMP propagation induced by two stimulatory G protein-coupled receptors in hearts. Proc Natl Acad Sci U S A 2012;109:6578–6583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou YY, Wang SQ, Zhu WZ, et al. Culture and adenoviral infection of adult mouse cardiac myocytes: methods for cellular genetic physiology. Am J Physiol Heart Circ Physiol 2000;279:H429–H436 [DOI] [PubMed] [Google Scholar]
- 35.Riehle C, Wende AR, Sena S, et al. Insulin receptor substrate signaling suppresses neonatal autophagy in the heart. J Clin Invest 2013;123:5319–5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nobles KN, Xiao K, Ahn S, et al. Distinct phosphorylation sites on the β(2)-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci Signal 2011;4:ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiang Y, Kobilka B. The PDZ-binding motif of the beta2-adrenoceptor is essential for physiologic signaling and trafficking in cardiac myocytes. Proc Natl Acad Sci U S A 2003;100:10776–10781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeGeorge BR, Jr, Gao E, Boucher M, et al. Targeted inhibition of cardiomyocyte Gi signaling enhances susceptibility to apoptotic cell death in response to ischemic stress. Circulation 2008;117:1378–1387 [DOI] [PubMed] [Google Scholar]
- 39.Han SJ, Vaccari S, Nedachi T, et al. Protein kinase B/Akt phosphorylation of PDE3A and its role in mammalian oocyte maturation. EMBO J 2006;25:5716–5725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao AZ, Huan JN, Gupta S, Pal R, Sahu A. A phosphatidylinositol 3-kinase phosphodiesterase 3B-cyclic AMP pathway in hypothalamic action of leptin on feeding. Nat Neurosci 2002;5:727–728 [DOI] [PubMed] [Google Scholar]
- 41.Wright JJ, Kim J, Buchanan J, et al. Mechanisms for increased myocardial fatty acid utilization following short-term high-fat feeding. Cardiovasc Res 2009;82:351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glatz JF, Luiken JJ, Bonen A. Membrane fatty acid transporters as regulators of lipid metabolism: implications for metabolic disease. Physiol Rev 2010;90:367–417 [DOI] [PubMed] [Google Scholar]
- 43.Gavi S, Yin D, Shumay E, Wang HY, Malbon CC. Insulin-like growth factor-I provokes functional antagonism and internalization of beta1-adrenergic receptors. Endocrinology 2007;148:2653–2662 [DOI] [PubMed] [Google Scholar]
- 44.Norby FL, Aberle NS, 2nd, Kajstura J, Anversa P, Ren J. Transgenic overexpression of insulin-like growth factor I prevents streptozotocin-induced cardiac contractile dysfunction and beta-adrenergic response in ventricular myocytes. J Endocrinol 2004;180:175–182 [DOI] [PubMed] [Google Scholar]
- 45.Santulli G, Lombardi A, Sorriento D, et al. Age-related impairment in insulin release: the essential role of β(2)-adrenergic receptor. Diabetes 2012;61:692–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morisco C, Condorelli G, Trimarco V, et al. Akt mediates the cross-talk between beta-adrenergic and insulin receptors in neonatal cardiomyocytes. Circ Res 2005;96:180–188 [DOI] [PubMed] [Google Scholar]
- 47.Stuenaes JT, Bolling A, Ingvaldsen A, et al. Beta-adrenoceptor stimulation potentiates insulin-stimulated PKB phosphorylation in rat cardiomyocytes via cAMP and PKA. Br J Pharmacol 2010;160:116–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pessin JE, Gitomer W, Oka Y, Oppenheimer CL, Czech MP. beta-Adrenergic regulation of insulin and epidermal growth factor receptors in rat adipocytes. J Biol Chem 1983;258:7386–7394 [PubMed] [Google Scholar]
- 49.Zhang J, Hupfeld CJ, Taylor SS, Olefsky JM, Tsien RY. Insulin disrupts beta-adrenergic signalling to protein kinase A in adipocytes. Nature 2005;437:569–573 [DOI] [PubMed] [Google Scholar]
- 50.Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014;57:660–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galderisi M, de Simone G, Innelli P, et al. Impaired inotropic response in type 2 diabetes mellitus: a strain rate imaging study. Am J Hypertens 2007;20:548–555 [DOI] [PubMed] [Google Scholar]
- 52.Nichols GA, Hillier TA, Erbey JR, Brown JB. Congestive heart failure in type 2 diabetes: prevalence, incidence, and risk factors. Diabetes Care 2001;24:1614–1619 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.