

Abstract
Adenylosuccinate lyase (ADL) catalyzes the breakdown of 5-aminoimida-zole- (N-succinylocarboxamide) ribotide (SAICAR) to 5-aminoimidazole-4-carboxamide ribotide (AICAR) and fumarate, and of adenylosuccinate (ADS) to adenosine monophosphate (AMP) and fumarate in the de novo purine biosynthetic pathway. ADL belongs to the argininosuccinate lyase (ASL)/fumarase C superfamily of enzymes. Members of this family share several common features including: a mainly α-helical, homotetrameric structure; three regions of highly conserved amino acid residues; and a general acid-base catalytic mechanism with the overall β-elimination of fumarate as a product. The crystal structures of wild-type Escherichia coli ADL (ec-ADL), and mutant-substrate (H171A-ADS) and -product (H171N-AMP•FUM) complexes have been determined to 2.0, 1.85, and 2.0 Å resolution, respectively. The H171A-ADS and H171N-AMP•FUM structures provide the first detailed picture of the ADL active site, and have enabled the precise identification of substrate binding and putative catalytic residues. Contrary to previous suggestions, the ec-ADL structures implicate S295 and H171 in base and acid catalysis, respectively. Furthermore, structural alignments of ec-ADL with other superfamily members suggest for the first time a large conformational movement of the flexible C3 loop (residues 287–303) in ec-ADL upon substrate binding and catalysis, resulting in its closure over the active site. This loop movement has been observed in other superfamily enzymes, and has been proposed to be essential for catalysis. The ADL catalytic mechanism is re-examined in light of the results presented here.
Keywords: adenylosuccinate lyase, purine biosynthesis, β-elimination, argininosuccinate lyase/fumarase C superfamily
Introduction
Adenylosuccinate lyase (ADL)† is an enzyme with dual substrate specificity in the de novo purine biosynthetic pathway, where it catalyzes the breakdown of 5-aminoimidazole- (N-succinylocarboxamide) ribotide (SAICAR) to 5-aminoimidazole-4-carboxamide ribotide (AICAR) and fumarate, and of adenylosuccinate (ADS) to adenosine mono-phosphate (AMP) and fumarate. The de novo purine biosynthetic pathway is crucial for providing the purine nucleotides required for cellular replication. Along with adenylosuccinate synthetase and AMP deaminase, ADL also participates in the purine nucleotide cycle, which plays a role in regulating cellular metabolism by controlling the amounts of free AMP and the citric acid cycle intermediate, fumarate. Point mutations in the gene give rise to ADL deficiency in humans, which has been linked to adverse clinical effects including mental retardation with autistic features, muscle wasting, and/or epilepsy.1–3
ADL belongs to the argininosuccinate lyase (ASL)/fumarase C superfamily of enzymes, which consists of ASL/δ2-crystallin, class II fumarase, L-aspartase, and 3-carboxy-cis, cis-muconate lactonizing enzyme (CMLE). The structures of superfamily members has revealed that these enzymes are biologically active as homotetramers, with each monomer comprised of three distinct structural domains.4–9 Although the overall sequence identity between the superfamily members is only ~15–30%, the enzymes are characterized by three regions of highly conserved amino acid residues (denoted C1–C3, respectively). Conserved region C3 contains the signature sequence 294GSSxxPxKxN303 (Escherichia coli ADL numbering). While these regions are spatially remote from each other in the monomer, they are oriented in the tetramer such that three independent monomers contribute a different conserved region to each of the four active sites. Residues in these conserved regions have been implicated in catalysis.10–14
ADL catalyzes the cleavage of ADS through a general acid–base mechanism involving the β-elimination of fumarate (Figure 1(a)). The reaction is initiated by abstraction of the Cβ-proton from the substrate by the general base, resulting in the formation of a carbanion intermediate. Inhibition experiments with nitro-substrate analogs have confirmed the existence of such a species in the reaction mechanism.15 The intermediate is stabilized in the aci-carboxylate or resonance form, which carries two negative charges on its δ-carboxyl group.15 Subsequent proton donation by the catalytic acid to the N1 or N6 atom of the substrate results in cleavage of the Cα–N bond and product release (Figure 1(a)).16 The reaction proceeds via a uni-bi mechanism with fumarate leaving prior to AMP.17–19 Furthermore, the trans-stereochemistry of the reaction suggests that two separate groups participate in the proton abstraction and donation steps.20
Figure 1.

(a) Proposed reaction scheme for ADL. The ribose and phosphate groups of ADS are represented by RP. (b) The quaternary structure of E. coli ADL. Monomer A is shown in cartoon representation with domains 1–3 (residues 1–121, 122–381, and 382–456, respectively) indicated. The regions of highly conserved amino acid residues, which form the active sites of the protein, are colored black (C1, residues 117–130; C2, residues 168–178; C3, residues 294–307). Monomers A, B, C and D are labeled and are colored green, pink, yellow, and blue, respectively. PyMol was used for Figure preparation [http://pymol.sourceforge.net/].
Affinity labeling experiments on Bacillus subtilis ADL (bs-ADL) first implicated H91 and H171 (E. coli ADL numbering) as the acid and base catalysts in the ADL reaction mechanism, respectively,21,22 and subsequent mutagenesis and kinetic studies11,21 have confirmed the importance of these residues for enzymatic activity. The structure of apo Thermo-toga maritima ADL (tm-ADL)7 provided the first detailed structural description of the enzyme and allowed the substrate to be modeled into the active site region. This structure and a homology model of bs-ADL7,23–25 suggest that Q247, N303, and R335 bind the carboxylate groups of the succinyl moiety, while H118 and K301 interact with the sugar phosphate group. Site-directed mutagenesis of these residues in bs-ADL result in mutant proteins with severely compromised or no enzymatic activity.23–25 The current model of the bs-ADL enzyme–substrate complex24 also suggests that H91 is unlikely to act as the acid catalyst as its Nδ1 and Nε2 atoms are greater than 4Å away from the N1 or N6 atoms of ADS. Instead, H91 is poised to interact with one of the carboxylate groups of the substrate’s succinyl moiety.24
Although the studies described have provided us with substantial insight into the putative roles of various residues in ADL, the precise identification of substrate binding and catalytic residues has remained elusive. To enable us to map out the residues involved in ADS binding, and identify the putative catalytic groups, we have determined the three-dimensional structures of wild-type E. coli ADL (ec-ADL), and two mutant-substrate–product complexes, H171A–ADS and H171N–AMP•FUM, respectively. The H171A–ADS and H171N–AMP•FUM complexes represent the first structures of ADL with bound substrate or product. Comparison of these structures reveals that the flexible C3 loop (residues 287–303) undergoes a large conformational change upon substrate binding, resulting in its closure over the active site. The structures provide the first evidence that this event may be relevant to catalysis in ADL, and allow a detailed enzymatic mechanism to be presented.
Results
The H171 mutants and kinetic characterization
H171 has been implicated as the base catalyst in the ADL mechanism,7,22 and the presence of a “charge-relay” interaction between H171 and E308 suggested to facilitate the initial proton abstraction step.4,8,23,26 Similar roles have been proposed for the equivalent histidine/glutamate residues in ASL/δ2-crystallin8,27–29 and fumarase C.4,30 Mutation of H171 with a variety of substitutions (e.g. A, L, E, Q, R) in bs-ADL has highlighted the importance of this residue in catalysis, as residual activity was only detected in the H171R mutant, which was found to be 105-fold less active than the wild-type enzyme.11,23 These results suggested that mutation of H171 in ec-ADL would inactivate the protein and hence enable the structure of the enzyme–substrate complex to be determined. The ec-ADL H171 was therefore mutated to alanine, and the more conservative asparagine. The different mutations have the effect of assessing the importance of size, hydrogen bonding ability, and charge at this position.
Rate determinations of the wild-type and ec-ADL H171 mutants were carried out at pH 7.0, and at pH 8.5, the pH at which the proteins were crystallized. The specific activities the wild-type protein were 4.65 and 16.47 μmol min−1 mg−1 at pH 7 and pH 8.5, respectively. The specific activities of the H171A and H171N mutants are on the order of ~1/500 of the wild-type enzyme at both pH values, suggesting an important role for H171 in the ec-ADL enzyme. The specific activities for the H171A and H171N proteins are 0.009 and 0.008 μmol min−1 mg−1 at pH 7, respectively, and 0.031 and 0.04 μmol min−1 mg−1 at pH 8.5, respectively. Interestingly, the specific activity of the ec-ADL proteins at pH 8.5 is 3.5–5 times higher than at pH 7. This result contrasts with the observation that the maximum velocity of bs-ADL is lower at pH 8.5 than at pH 7.0.
Kinetic characterization of the S295A mutant
Given our structural results described below and to probe the role of S295 in the catalytic mechanism, an S295A mutant was constructed and its specific activity measured to be 0.004 μmol min−1 mg−1 at pH 7.0, a reduction of more than 1000-fold compared to the wild-type enzyme. This result supports the proposal that S295 plays a critical catalytic role in the ADL reaction mechanism.
Structure determination
The structure of the wild-type selenomethionine (SeMet) protein was solved in space group I222 with one monomer in the asymmetric unit (ASU) and refined to an Rcrys and Rfree of 16.5 and 20.2%, respectively (Table 1). Application of the appropriate crystallographic symmetry operators to the contents of the ASU generates the biologically active tetramer. The final model consists of 445 residues per monomer, as residues 290–297 and 410–412 could not be modeled due to weak or absent electron density.
Table 1.
Data collection, phasing and refinement statistics
| WT-SeMet | H171A–ADS | H171N–AMP•FUM | |
|---|---|---|---|
| A. Data collection | |||
| Wavelength (λ) | 0.9794 | 1.5418 | 1.5418 |
| f′, f″ | −8.30, 3.55 | — | — |
| Space group | I222 | P21212 | P21212 |
| Unit cell dimensions (Å) | a=68.4, b=98.4, c=136.2 | a=98.4, b=143.2, c=69.5 | a=99.1 b=143.4, c=69.5 |
| Monomers per ASU | 1 | 2 | 2 |
| Resolution limits (Å) | 50.00–2.00 | 44.00–1.85 | 35–2.0 |
| Total data | 637,617 | 782,190 | 579,421 |
| Unique data | 60,017 | 84,199 | 67,697 |
| Mean redundancy | 10.6 (10.5)b | 9.29 (6.08)b | 8.56 (8.38)b |
| Completeness (%) | 100.0 (100.0)b | 99.7 (97.0)b | 99.9 (100.0)b |
| Average I/σI | 88.9 (58.8)b | 19.5 (5.6)b | 19.9 (7.4)b |
| % reflections for which I>3σI | 100.0 (100.0)b | 98.8 (95.2)b | 98.5 (97.8)b |
| Rsym (%)a | 4.8 (7.6)b | 6.8 (30.8)b | 6.7 (25.7)b |
| B. Phasing | |||
| Phasing power | 3.18 | — | — |
| FOM before solvent flattening | 0.512 | — | — |
| FOM after solvent flattening | 0.941 | — | — |
| C. Refinement | |||
| Resolution range (Å) | 50.00–2.00 | 44.00–1.85 | 35–2.0 |
| Rcryst (%)c | 16.5 | 18.3 | 17.5 |
| Rfree (%)d | 20.2 | 21.6 | 21.1 |
| No. reflections in refinement | 56,957 | 79,920 | 64,194 |
| No. reflections in test set | 2932 | 4220 | 3443 |
| No. non-hydrogen atoms | |||
| Protein/substrate/solvent | 3518/—/457 | 7188/62/610 | 7262/46/16/650 |
| Mean B-factor (Å2) | |||
| Per monomer (A/B) | 13.20 | 22.15/22.21 | 17.69/17.20 |
| Per domain (A/B) | |||
| Domain 1 | 15.41 | 22.63/22.64 | 18.44/18.28 |
| Domain 2 | 10.00 | 18.68/19.54 | 15.28/15.10 |
| Domain 3 | 20.96 | 34.63/30.44 | 24.68/22.71 |
| ADS substrate (A/B) | — | 21.06/28.22 | — |
| AMP, FUM (A/B) | — | 12.66, 17.22/13.95, 16.91 | |
| C3 loop (A/B) | — | 24.30/41.16 | 20.30/18.56 |
| Solvent | 25.40 | 29.21 | 25.12 |
| rms deviation from ideal | |||
| Bond lengths (Å)/angles (°) | 0.005/1.21 | 0.006/1.31 | 0.005/1.23 |
| Dihedral/improper angles (°) | 19.61/0.71 | 19.77/0.80 | 19.68/0.73 |
| Ramachandran Plote | |||
| Total favored (%) | 92.3 | 91.3 | 91.3 |
| Total allowed (%) | 7.2 | 8.1 | 8.2 |
Rsym = Σ|I−<I>|/ΣI, where I is the measured intensity for symmetry-related reflections and <I> is the mean intensity for the reflection.
Last resolution shell extends from 2.07 to 2.00 and from 1.92–1.85 Å, respectively.
Rcrys = Σ(|Fo|−|Fc|)/Σ(|Fo|).
Rfree = Σ(|Fos|−|Fcs|)/Σ(|Fos|) where s refers to a subset of data not used in the refinement, representing 5% of the total number of observations.
According to the Ramachandran plot in PROCHECK.31
The H171A and H171N structures have been refined to an Rcrys and Rfree of 18.3 and 21.6%, and 17.7 and 21.1%, respectively (Table 1). The mutant structures, solved in space group P21212, have two monomers in the ASU (monomers A and B in Figure 1(b)). Monomers C and D can be generated by applying the appropriate crystallographic symmetry to the contents of the H171A or H171N ASU (Figure 1(b)). The presence of the substrate or products in the H171A and H171N active sites, respectively, was unambiguously determined by the excellent quality of the difference electron density (Figure 2(a) and (c)). Monomers A and B in the final H171A–ADS model contain 451 and 453 residues as there was insufficient electron density present to model residues 295–297 and 409–413 in each monomer, respectively. All 456 residues were present in both monomers of the H171N–AMP•FUM complex. Structural alignments of monomers A and B of H171A–ADS or H171N–AMP•FUM yields Cα root-mean-square (rms) deviations of 0.39 Å and 0.16 Å, respectively, suggesting no large conformational differences exist between the two monomers in the ASU of each protein.
Figure 2.


Stereo view of the superimposed H171A (pink) and H171N (green) active sites, showing the interactions involving the AMP (a) and fumarate groups (c). The σA weighted Fo–Fc omit maps for the substrate (orange) and products (blue) in the H171A and H171N proteins, respectively, are shown contoured at 3σ. Water molecules are shown as spheres. The corresponding schematic representations for the AMP and fumarate groups are shown in (b) and (d), respectively. Hydrogen bonds are represented as red broken lines with the distances indicated in angstroms (Å). Distances for the H171N protein are in bold, and the distances for active site 2 of each protein are given in parentheses. The letter following the residue number denotes the monomer to which each residue belongs, with those for active site 2 of the proteins shown in parentheses. In (b), residues involved in coordinating water molecules are colored blue. Poor electron density did not allow residues S295 and S296 in active site 2 of the H171A protein to be modeled. PyMol was used for Figure preparation.
Greater than 90% of all residues in the three structures reside in the most favored regions of the Ramachandran plot as determined by the program PROCHECK31 (Table 1). W333 and R335 are found in generously allowed and disallowed regions, respectively, in the wild-type SeMet and mutant proteins. W333 and R335 are located in a helix-turn comprised of residues 331–337. The electron density for the main-chain and side-chain atoms of W333 and R335, as well as those of the surrounding residues is well defined, thus enabling both residues to be modeled with a high degree of confidence in all structures.
Overall fold and comparison to previously determined ADL structures
The overall fold of the ec-ADL protein is reminiscent of other ASL/fumarase C superfamily members with three distinct domains (Figure 1(b)).4–9 However, while the general structure of the ec-ADL protein is similar to those of the previously determined tm- and Pyrobaculum aerophilum (pa-) ADL enzymes,7,32 significant conformational differences are observed between the proteins. A structural alignment of wild-type ec-ADL with monomers A of tm- or pa-ADL yields rms deviations of 2.9 Å and 3.0 Å for all equivalent Cα positions, respectively. When the individual domains (1–3) of ec-ADL are aligned with the corresponding domains from tm- and pa-ADL independently of the rest of the protein, Cα rms deviations of between 2.1 Å –3.1 Å are still observed. This suggests significant conformational differences are present even on the level of the individual domains between the different ADL enzymes.
Mutant–substrate and product complexes of E. coli ADL
Given the significant reduction in residual ADL activity in the H171N and H171A mutants we co-crystallized these proteins with ADS in the hopes of being able to capture the structure of a protein–substrate complex. While the difference electron density in the H171A active site does unambiguously verify the presence of the ADS substrate (Figure 2(a) and (c)), a clear break was seen in the difference electron density for the H171N mutant. This corresponds to the cleavage of the Cα–N6 bond in the substrate (Figure 2(a) and (c)). As a result, the products AMP and fumarate were modeled into the electron density in both active sites present in the ASU of the H171N protein. In both H171A and H171N structures, active site 1 is formed by monomers A, B and symmetry related monomer D, while active site 2 is composed of monomers A, B, and symmetry related monomer C. The H171A and H171N tetramers each contain four active sites, two of which are generated by applying the appropriate crystallographic symmetry to the contents of the ASU. Given that the substrate or products are bound in very similar conformations in both active sites of the H171A or H171N proteins, respectively, the following discussion will refer to the interactions observed in active site 1 of each protein.
A structural alignment of the H171A–ADS and H171N–AMP•FUM complexes reveals that the substrate and products are bound in very similar conformations in the proteins. The phosphate and ribose moieties of ADS and AMP are found to superimpose very well, with only a small deviation observed in the conformation of the adenine rings (Figure 2(a) and (c)). In addition, the conformation of the H171A–ADS and H171N–AMP•FUM active sites are highly similar. Only the side-chain of L337 is observed to deviate significantly (Figure 2(a)), and this is likely due to the different substitutions at position 171 in the proteins. In both structures, the AMP group makes hydrogen bonds to residues R15, Y16, N309 from monomer A and N90, D92, Q247, R335, S340, R344 from monomer B, in addition to six ordered water molecules which are in turn anchored in the active site by residues E86, T122, E124, N127, L337, and T341 from monomer B. In the H171N structure, the side-chain of N171 also makes hydrogen bonds to the N6 and N7 atoms of the substrate (Figure 2(a) and (b)). The fumarate group in both structures interacts with S295, S296, K301, N303 from monomer A, H91, T122, S123, Q247 from monomer B, and T170 from monomer D. A number of the residues involved in binding the fumarate group are highly or strictly conserved across the ASL/fumarase C superfamily. T122 and S123 of conserved region C1 are 100 and ≥60% conserved, respectively, while S295 and S296 of conserved region C3 are invariant across all superfamily members. Residues 294–297 of monomer B could not be modeled in the H171A protein and as a result, the interactions involving S295 and S296 and the substrate are not observed for the second active site in this structure.
Conformational differences
A structural alignment of the apo SeMet protein, H171A–ADS and H171N–AMP•FUM complexes reveal several regions of conformational variability. The most notable difference involves an intrinsically flexible loop encompassing residues 287–303 (C3 loop), which comprises conserved region C3 of the proteins. In the apo SeMet protein, electron density was not observed for residues 290–297 of this loop. Difficulty in modeling this region of the protein has been reported for various members of the ASL/fumarase C superfamily.7,9,32 However, in the presence of bound substrate or product, the C3 loop becomes ordered closing over the active site. With the exception of monomer B of the H171A protein for which residues 294–297 could not be modeled, the B-factors for the C3 loop of each H171A and H171N monomer are comparable to the average values for the rest of the protein (Table 1). Interestingly, the conformation of the C3 loop in the H171A–ADS and H171N–AMP•FUM structures resembles the conformation observed previously in the sulfate bound dδc1 protein33 (Figure 3).
Figure 3.
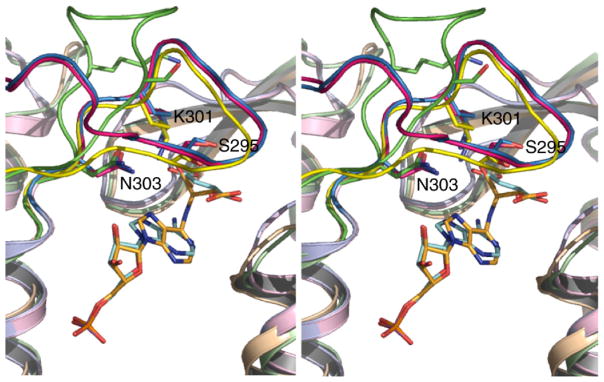
Stereo view of the superimposed H171A–ADS (pink), H171N–AMP•FUM (blue) and (yellow) active sites, showing the conformation of the C3 loop in the proteins. Since the C3 loop in the SeMet protein could not be modeled due to the absence of electron density, the open conformation of the loop observed in the dδc2-S281A mutant12 is also shown for comparison (green). The side-chains of selected C3 loop residues are shown and numbered according to the E. coli ADL sequence. PyMol was used for Figure preparation.
Domain 3 also appears to be conformationally flexible, as judged by the higher than average B-factors (Table 1) and absence of electron density for a number of residues. With the available structures, it is difficult to differentiate whether the structural differences observed in domain 3 are the result of substrate or product binding, or the consequence of the mobility of this region. Domain 3 has been hypothesized to undergo a rigid body movement in ASL/δ2-crystallin upon substrate binding and catalysis, with the effect of partially sequestering the substrate from solvent.33
Discussion
Role of H171 in acid catalysis
The H171A–ADS and H171N–AMP•FUM structures represent the first enzyme–substrate and product complexes of ADL, and the first structures of any superfamily member with bound substrate or product with the C3 loop “closed” over the active site. This loop conformation has only been observed in the sulfate and phosphate bound forms of dδc1 and E. coli ASL (ec-ASL), respectively.33,34 A closed C3 loop is thought to be the catalytically relevant conformation, as the highly conserved residues in this region are positioned to interact with the substrate. In the “open” conformation these residues are up to 8 Å away from the active site region.
Although H91 has been suggested to be the catalytic acid in bs-ADL,21 its amide nitrogen atoms are more than 5 Å away from the N1 or N6 atom of the substrate in the H171A–ADS complex. Instead, the Nε2 atom of H91 is 2.7 Å from the Oδ2 atom of the fumarate moiety, suggesting a role in substrate binding rather than catalysis. Previously available data have implicated H171 as the base catalyst in the ADL enzymatic mechanism.11,22 However, examination of the H171A–ADS and H171N–AMP•FUM complexes reveals that the Cβ of A171 and the Oδ1 of N171 are ~8 Å and 6 Å away from the Cβ-atom of the substrate, respectively, too far from the substrate for proton abstraction. Furthermore in the apo-wild-type structure, the charge-relay interaction between H171 and E308 observed in other superfamily members4,8,26 is not conserved, as H171 is rotated away from the active site region (Figure 4). These results are both surprising and unexpected, and suggest that the role of H171 is one other than in base catalysis.
Figure 4.
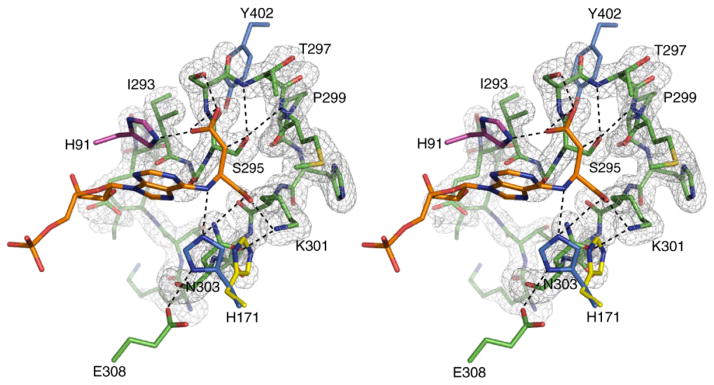
Stereo view of the σA weighted Fo–Fc omit map of residues 287–303 of the H171A–ADS protein, contoured at 3.0σ. Additional residues within 5 Å of S295 are shown. Also illustrated is the conformation of H171 as observed in the SeMet wild-type protein (yellow), and when modeled to reflect the conformation of N171 in the H171N–AMP•FUM complex (blue). Residues contributed by monomers A, B, and D are colored green, pink, and blue, respectively. Hydrogen bonds are represented by broken lines. PyMol was used for Figure preparation.
When the apo-wild-type, H171A–ADS and H171N–AMP•FUM structures are superimposed and the position of H171 examined, steric clashes are found between H171 and residues K301 and N303 of the C3 loop of the H171A and H171N structures (Figure 4). This suggests that the conformation of H171 on closure of the C3 loop is different from that found in the apo-wild-type protein. Interestingly, the steric clashes can be eliminated by simple rotation about χ1 of H171 and L337. When these residues are rotated to reflect the conformations of N171 and L337 in the H171N protein, the Nε2 atom of H171 is 2.7 Å away from the N6 atom of the substrate, while its Nδ1 atom is within 3.0 Å of the Oε2 atom of E308 (Figure 4). Together, our structural data suggest that upon ADS binding and closure of the C3 loop, the side-chains of H171 and L337 undergo large rotations, bringing H171 within hydrogen bonding distance of E308 and the N6 atom of the substrate. Cleavage of the Cα–N6 bond may occur through protonation of the N6 atom by the imidazolium form of H171, which is facilitated through the interaction of its Nδ1 and Nε2 atoms with E308 and the Oγ1 atom of ADS (Figure 4). The presence of AMP and fumarate rather than substrate in the H171N active site and the ability to measure a reaction rate for this mutant enzyme imply that the substrate–product conformations observed in both mutant structures are catalytically relevant, but that H171 is not absolutely required for cleavage of the Cα–N6 bond in ADS. Hanson & Havir16 suggest that bond cleavage could be facilitated via protonation of the N1 ring nitrogen (Figure 1(a)). Examination of the H171A and H171N active sites suggest that a water molecule, W59, is ideally positioned 3.0 Å away from the N1 ring nitrogen of ADS or AMP, respectively, and could therefore play the role of the acid catalyst. The presence of AMP and fumarate in the H171N mutant suggests that the interactions involving N171, or H171 in the native ec-ADL enzyme are important for productive bond cleavage. As efficient catalysis requires the specific microenvironment created by the interactions involving all active site residues, and not only those directly involved in catalysis, the absence of these interactions in the H171A protein may explain the presence of uncleaved substrate in the H171A active site.
Role of S295 in base catalysis
Given the position of A/H171 in the H171A–ADS complex and our proposal that H171 plays the role of the acid catalyst, the identity of the catalytic base remains to be resolved. Our mutant complexes are the first structures of any superfamily member with substrate or products bound and the C3 loop in the closed conformation. The presence of the AMP and fumarate products in the H171N structure also provides convincing evidence that the H171A–ADS structure represents a catalytically relevant enzyme–substrate conformation. The excellent quality of the electron density for not only the AMP but also the fumarate moiety of ADS has enabled us to unambiguously identify, for the first time, interactions between the C3 loop and the substrate. In the H171A–ADS and H171N–product complexes the Oγ of S295 is the closest atom to the Cβ of the substrate (3.0 Å –3.3Å) (Figure 2(c) and (d)). S295 is strictly conserved across all ASL/fumarase C superfamily members and is present on the flexible C3 loop. Mutation of S295 and the equivalent S281 in dδc226 demonstrate the importance of this residue for enzymatic activity, but its exact role in catalysis has until now been unclear. S281 has been proposed to be the acid catalyst in ASL/δ2-crystallin,33 while in ec-ASL (S277)34 and Bacillus sp. YM55-1 aspartase (S318)10 it has been suggested to be involved in base catalysis. However, it is important to note that the role of S295 (or its equivalent) was previously inferred from modeling studies.
Given the proximity of S295 to the Cβ-atom of ADS in our mutant complexes, in addition to its strict conservation across the superfamily members, our results suggest that S295 is involved in base catalysis in the ADL mechanism. However, this proposal raises a number of questions given the lack of precedence for serine as a base catalyst, and the absence of any obvious candidates in the active site that could serve to prime S295 for proton abstraction (Figure 4). The pKa value of S295 may be perturbed by the interaction of its Oγ atom with the backbone NH groups of T297 and M298.35,36 While the precise details surrounding the activation of S295 remain unclear and the involvement of serine in base catalysis uncommon given its intrinsically high pKa value, serine has been reported to play this role in other enzymes.37 In line with our proposal is the increase in specific activity observed at pH 8.5 for ec-ADL, which would certainly be expected if S295 acts as the base catalyst. Although it is unlikely that the reaction mechanisms for orthologous bacterial enzymes are different, it is interesting to note that the Vmax value for bs-ADL, contrary to what we observe for ec-ADL, is lower at pH 8.5 than at pH 7.0.25
Stabilization of the aci-carboxylate intermediate
Another residue in the C3 loop of ec-ADL of particular interest is K301. This residue is strictly conserved across the superfamily and has been suggested to play a role in stabilizing the aci-carboxylate intermediate in several of the superfamily members.12,13,30 This proposal is supported by mutagenesis experiments in E. coli L-aspartase13 and dδc2 (M. T., unpublished work), and by the dδc1-sulfate complex, which demonstrated that closure of the C3 loop over the active site enabled the Nζ atom of the equivalent lysine residue to interact with the sulfate ion.33 However, in the H171A–ADS complex K301 is not in a position to interact with the δ-carboxylate group of the substrate, on which the negative charges accumulate in the aci-carboxylate intermediate. Instead the Nζ atom of K301 is 2.9Å away from the Oγ2 atom of the succinyl moiety, suggesting a role in coordinating the γ-carboxylate group of ADS (Figure 2(c) and (d)). The network of interactions between K301, T170, and N303, all of which are highly conserved across the ASL/fumarase C superfamily, is likely a major determinant in binding this end of the substrate.
No other positively charged residues are observed in close proximity to the δ-carboxylate group of ADS. However, the Oδ1 and Oδ2 atoms do participate in extensive hydrogen bonding interactions with T122, S123, S296, and H91 (Figure 2(c) and (d)), which may contribute partially to stabilizing the aci-carboxylate intermediate. When the residues equivalent to T122 and S123 are replaced by alanine in bs-ADL, the catalytic activity of the enzyme is adversely affected.38 S123 is also found at the N-terminal end of one of the core helices in domain 2, suggesting the partial positive charge resulting from the dipole moment of the helix may further stabilize the negative charges on the δ-carboxylate group of the intermediate.
Catalytic mechanism
The H171A–ADS and H171N–AMP•FUM complexes reveal a complex network of interactions responsible for substrate binding. Our current results suggest that upon ADS binding, the C3 loop undergoes a large conformational movement, which results in its closure over the active site. This event is coupled to rearrangements of active site residues, including H171 and L337. The resulting interactions serve to restrict the conformational freedom of the substrate, in particular the fumarate moiety, which ensures its precise orientation for catalysis. In the closed conformation of the C3 loop, S295 is properly positioned to abstract the Cβ-proton from the substrate, which results in the formation of the carbanion intermediate. The hydrogen-bonding network involving H91, T122, S123, and S296 stabilizes the negative charges on the aci-carboxylate intermediate. Given that the leaving group is not strongly basic, bond cleavage is facilitated by protonation at either the N6 or N1 atoms of the substrate, by H171 or a water molecule, respectively. The inductive withdrawal of electrons may further assist in the elimination process.16 The interactions between H/N171 and the adenine ring of the substrate appear to be important for efficient catalysis. The sequential release of AMP and fumarate from the active site requires the C3 loop to return to the open conformation although the exact driving force for loop opening other than its intrinsic flexibility is not immediately apparent.
The H171A–ADS and H171N–AMP•FUM complexes represent the first time the closed conformation of the C3 loop is observed in a superfamily member with bound substrate or product. These results provide the first structural evidence that this large loop movement may occur in all ASL/fumarase C superfamily members upon substrate binding and catalysis. While our ec-ADL structures have provided significant new insights into the ADL enzymatic mechanism, these structures have not been able to confirm unambiguously the presence of a common enzymatic mechanism across the ASL/fumarase C superfamily. The strict conservation of S295 in all superfamily members makes this residue an attractive candidate for the base catalyst, but H171 is replaced by a glutamine residue in all species of aspartase except B. subtilis and by tryptophan in CMLE. A more comprehensive analysis of the ASL/fumarase C superfamily as a whole is required to determine if a common catalytic mechanism exists for all the enzymes.
Materials and Methods
Expression vector
The purB gene encoding the ec-ADL protein in the pUC118 vector was obtained from Dr Howard Zalkin.39 The gene was subcloned into the pET-28a expression vector (Novagen) at the NcoI and XhoI sites upstream of a six-histidine tag. DNA sequencing (ACGT Corporation, Toronto) revealed a number of spurious mutations in the purB gene, which were subsequently corrected with the QuikChange Site-Directed Mutagenesis kit (Stratagene), with the exception of a conservative I154L mutation. This mutation is mapped to one of the core helices in domain 2 away from the active site region, and is therefore not expected to have any effect on the enzymatic activity of the protein. For the purpose of this manuscript, the pET-28a vector containing the purB gene with the I154L mutation will be referred to as the wild-type construct.
Preparation of wild-type selenomethionine protein
The pET-28a-vector containing the purB gene was transformed into B834 (DE3) E. coli cells. A 20 ml overnight culture from a single colony was used to inoculate 1 l of 2YT containing 100 μg/ml kanamycin. The cells were grown at 37 °C until an A600 of ~0.4 was reached before they were spun down (8000 rpm for 25 min at 4 °C) and transferred to 1 l of M9 medium containing 100 μg/mL kanamycin. Cells were grown for 1 hour at 30 °C prior to supplementing the M9 medium with 0.4% (v/v) glucose, 2 mM Mg2SO4, 25 μg/ml of Fe2SO4·7H2O, 1 μg/ml of vitamin mixture (riboflavin, niacinamide, pyridoxine monohydrochloride, thiamine), 40 μg/ml of amino acid mixture (except Met), 40 μg/ml of seleno-L-methionine, and 1 mM IPTG. Cells were harvested (8000 rpm for 25 min at 4 °C) approximately 3.5 h post-induction.
The pellet was resuspended in buffer A (50 mM Na3PO4 (pH 8.0), 300 mM NaCl, 0.5 mM TCEP) and sonicated four times for 60 s in the presence of 1 mM PMSF with 60 s of cooling on ice between each round. The cell lysate was centrifuged (17,000 rpm for 25 min at 4 °C) and the supernatant applied onto a Ni-NTA agarose resin (Qiagen) column charged with 50 mM Ni2SO4 and pre-equilibrated with buffer A. The column was washed extensively with 150 ml of buffer A followed by 200 ml of buffer B (50 mM Na3PO4 (pH 6.0), 300 mM NaCl, 10% (v/v) glycerol, 0.5 mM TCEP). The protein was eluted from the column with 15 ml of buffer B containing 150 mM imidazole. After concentrating to ~1.0 ml with an Amicon Ultra® centrifugal filter device (Millipore), the protein was applied to a MonoQ FPLC column (Pharmacia) pre-equilibrated with 50 mM Na3PO4 (pH 8.0), and 0.5 mM TCEP. A linear NaCl gradient (0–1 M) was used to elute the protein. The peak fractions, corresponding to ~300 mM NaCl, were pooled and concentrated to 500 μl and buffer exchanged into buffer C (20 mM Na3PO4 (pH 7.0), 20 mM NaCl, 10% (v/v) glycerol, 0.5 mM TCEP). Approximately 5 mg of >95% pure SeMet protein was obtained from 1 l of cell culture. The incorporation of eight selenium atoms into the wild-type protein was verified by mass spectrometry (Advanced Protein Technology Centre, The Hospital for Sick Children, Toronto).
Preparation of H171 and S295 mutant proteins
The QuikChange Site-Directed Mutagenesis method (Stratagene) was used to construct the H171A, H171N and S295A mutants. The wild-type gene in the pET-28a vector served as the starting template and DNA sequencing (ACGT Corporation, Toronto) confirmed the presence of the desired mutations.
The pET-28a vectors encoding the mutant genes were transformed into BL21C+ E. coli cells. In each case, a 10 ml overnight culture from a single colony was used to inoculate 1 l of LB media containing 100 μg/ml kanamycin. The cells were grown at 37 °C to an A600 of ~0.4 before lowering the temperature to 25 °C and inducing expression with 0.5 mM IPTG. The cells were grown overnight prior to harvesting. The purification protocol for the mutant proteins was similar to that of the SeMet protein with the following modifications. TCEP was not added to any of the buffers. Furthermore to improve protein purity, 5 mM and 20 mM imidazole was added to buffers A and B, respectively, and the proteins were eluted off the Ni-NTA column using a three-step imidazole gradient (50 mM, 75 mM, and 100 mM). The protein containing fractions corresponding to 75 mM or 100 mM imidazole, depending on the protein preparation were concentrated to ~1.5 ml and dialyzed against 1 l of buffer C. Additional purification steps were not necessary given the high purity of the proteins. Approximately 15 mg of ~99% pure protein was obtained per 1 l of cell culture.
Crystallization
Crystals of the wild-type SeMet protein, H171A–substrate, and H171N–product complexes were grown using the hanging drop vapor diffusion method at room temperature by mixing 3 μl of protein (7 mg/ml in buffer C) with 1 μl of reservoir buffer (85 mM Tris–HCl (pH 8.5), 170 mM NaCH3COOH•3H2O, 25.5% (w/v) PEG 4000, 15–23% glycerol). The H171A and H171N proteins (7 mg/ml in buffer C) were incubated with 25 mM adenylosuccinic acid (disodium salt; Sigma Aldrich) for 30 min at room temperature prior to setting up the crystallization plates. After one to two weeks, chunky three-dimensional crystals (~0.2 mm×0.2 mm×0.3 mm) were observed for the SeMet protein, while rod-shaped crystals (~0.3 mm×0.6 mm×0.1 mm) were seen for the mutant complexes.
Structure determination and refinement of the wild-type selenomethionine protein
Single anomalous diffraction (SAD) data were collected on a single SeMet crystal to 2.0 Å resolution at beamline X8C of the National Synchrotron Light Source (NSLS, Brookhaven, NY). Additional cryo-protectant was not required prior to flash freezing the crystal (−170 °C). The data were processed using HKL200040 treating the Bijvoet-related reflections as individual reflections (Table 1). The program BnP v0.9741 was used to determine the anomalous substructure and the positions of the selenium atoms used to calculate initial phases with CNS.42 The phases were refined by density modification using the solvent flipping method (Table 1). The excellent quality of the initial electron density maps enabled most of the main-chain and side-chain atoms to be manually built into the experimental maps using the program Xfit.43
All rounds of refinement were performed using the 2.0 Å SeMet data in CNS42 with a maximum likelihood target function,44,45 a flat bulk solvent correction,46 and no low resolution or σ cutoff applied to the data. Five percent of the reflections were randomly selected to compute an Rfree for cross-validation of the model. The refinement protocol consisted of simulated annealing and grouped or individual B-factor refinement, calculation of σA-weighted 2Fo–Fc and Fo–Fc maps, followed by manual rebuilding of the model in Xfit. After the third round of refinement, peaks greater than 3.0σ on the σA-weighted Fo–Fc map and with proper hydrogen bonding coordination were picked automatically as water molecules. Water molecules were modeled manually into additional peaks with correct hydrogen bonding geometry and greater than 1σ and 2.5σ on the σA-weighted 2Fo–Fc and Fo–Fc maps, respectively. Eighteen residues were truncated to alanine due to weak or absent electron density (D18, D62, R196, E223, T288, I289, M298, K400, Y402, E403, K406, E407, T409, R413, V414, K420, E430, E431).
Structure determination and refinement of the H171A and H171N mutants
Diffraction data for the H171A and H171N–complexes were collected on our in house X-ray diffraction facilities at the Hospital for Sick Children, Toronto. The crystals were transferred directly to the continuous cold stream (−170 °C). After processing the data with the program d*TREK v9.447 (Table 1), the DREAR program package48 was used to apply Baysian statistics to the reduced data to improve the weak data and to eliminate or correct negative intensities. The structure of the H171A mutant was solved using molecular replacement and the coordinates of the wild-type SeMet protein as the initial search model. The cross-rotation and translation searches using all data between 15 Å –5 Å, resulted in a correlation coefficient and an initial Rcrys of 0.66 and 39%, respectively. Given that the H171A and H171N mutants crystallize in the same space group (P21212), difference Fourier methods were used to determine the structure of the H171N–complex.
The refinement protocols for the mutant proteins were as described for the apo SeMet protein, except that in each case, an initial round of rigid body refinement was performed. During the later rounds of refinement, strong positive density in the σA-weighted Fo–Fc maps allowed ADS, and AMP and fumarate (FUM), to be modeled into the active sites of the H171A (H171A–ADS) and H171N (H171N–AMP•FUM) proteins, respectively. Due to absent or weak electron density for the side-chains, residues E223, E399, K400, Y402, E403, K406, E407, L408, V414, E417, K420, Q421, E430, E431, K437 in monomer A and I289, R413, E430, E431 in monomer B of the H171A mutant, and R196, E223, E399, E407, R413, K420, E430, E431 in monomer A and K54, R413, E417, K420, E431 in monomer B of the H171N protein were truncated to alanine.
Structural and sequence alignments
Amino acid sequences were retrieved from the SWISS-PROT database, and CLUSTAL W49 was used for the multiple sequence alignment. Structural alignments were performed with PROFIT (version 6.0) written by G. D. Smith. Structurally equivalent residues in five of the core helices in domain 2 of the proteins were chosen and subjected to an iterative least-squares fitting procedure. The SSM Superpose option in the program Coot50 was used for additional structural alignments.
Kinetic characterization
The rate of enzyme catalyzed conversion of adenylo-succinate to AMP and fumarate was determined by monitoring the decrease in absorbance at 282 nm over time.51 Rate determinations at pH 7.0 and pH 8.5 were performed in 50 mM Hepes and 54 mM Taps, respectively, and with 60 and 120 μM adenylosuccinate present, respectively. The reactions at pH 7.0 were initiated by adding 4 μg of wild-type enzyme or 150 μg –250 μg of the mutant proteins. At pH 8.5, 0.75 μg of wild-type enzyme or 200 μg –270 μg of the H171N or H171A mutant proteins was added to start the reaction. Specific activities were expressed as the amount of adenylosuccinate, in μmol, converted to AMP and fumarate per minute per mg of enzyme present. A circulating water bath was used to maintain the temperature at 25 °C for all assays.
Protein Data Bank accession numbers
The coordinates and structure factors have been deposited in the RCSB Protein Data Bank, as entries 2PTS, 2PTR, and 2PTQ for the wild-type SeMet, H171A–ADS, and H171N–AMP•FUM complexes, respectively.
Acknowledgments
We thank Dr Yura Lobsanov and Dr Jeffrey E. Lee for assistance with data collection at X8C, and Shao Yang Ku for use of the ChaChe program. This work is supported by grants from the Canadian Institutes for Health Research (CIHR) and Natural Sciences Engineering Council of Canada (NSERC) (to P.L. H.). M.T. is supported by a CIHR Canada Graduate Scholarship (CGS) Doctoral award, and by the Ontario Student Opportunity Trust Fund, Hospital for Sick Children Foundation Student Scholarship Program. P.L.H. is the recipient of a Canada Research Chair. Beam line X8C at the National Synchrotron Light Source is supported by the Department of Energy, USA, and by major facilities access grants from the Natural Sciences and Engineering Research Council (NSERC) of Canada and CIHR.
Abbreviations
- ADL
adenylosuccinate lyase
- SAICAR
5-aminoimidazole-(N-succinylocarboxamide) ribotide
- AICAR
5-aminoimidazole-4-carboxamide ribotide
- ADS
adenylosuccinate
- AMP
adenosine mono-phosphate
- CMLE
3-carboxy-cis, cis-muconate lactonizing enzyme
- CX
conserved amino acid region X, where X corresponds to 1, 2 or 3
- bs-ADL
Bacillus subtilis adenylosuccinate lyase
- tm-ADL
Thermotoga maritima adenylosuccinate lyase
- pa-ADL
Pyrobaculum aerophilum adenylosuccinate lyase
- ec-ADL
Escherichia coli adenylosuccinate lyase
- dδc1
duck δ1-crystallin
- SeMet
selenomethionine
- ASU
asymmetric unit
- ec-ASL
E. coli argininosuccinate lyase
- rms
root-mean-square
- AS
argininosuccinate substrate
Footnotes
For clarity, the amino acid numbering used throughout for B. subtilis ADL is that of the E. coli ADL protein.
References
- 1.Verginelli D, Luckow B, Crifo C, Salerno C, Gross M. Identification of new mutations in the adenylosuccinate lyase gene associated with impaired enzyme activity in lymphocytes and red blood cells. Biochim Biophys Acta. 1998;1406:81–84. doi: 10.1016/s0925-4439(97)00086-0. [DOI] [PubMed] [Google Scholar]
- 2.Maaswinkel-Mooij PD, Laan LA, Onkenhout W, Brouwer OF, Jaeken J, Poorthuis BJ. Adenylosuccinase deficiency presenting with epilepsy in early infancy. J Inherit Metab Dis. 1997;20:606–607. doi: 10.1023/a:1005323512982. [DOI] [PubMed] [Google Scholar]
- 3.Stone RL, Aimi J, Barshop BA, Jaeken J, Van den Berghe G, Zalkin H, Dixon JE. A mutation in adenylosuccinate lyase associated with mental retardation and autistic features. Nature Genet. 1992;1:59–63. doi: 10.1038/ng0492-59. [DOI] [PubMed] [Google Scholar]
- 4.Weaver TM, Levitt DG, Donnelly MI, Stevens PP, Banaszak LJ. The multisubunit active site of fumarase C from Escherichia coli. Nature Struct Biol. 1995;2:654–662. doi: 10.1038/nsb0895-654. [DOI] [PubMed] [Google Scholar]
- 5.Shi W, Dunbar J, Jayasekera MM, Viola RE, Farber GK. The structure of L-aspartate ammonia-lyase from Escherichia coli. Biochemistry. 1997;36:9136–9144. doi: 10.1021/bi9704515. [DOI] [PubMed] [Google Scholar]
- 6.Simpson A, Bateman O, Driessen H, Lindley P, Moss D, Mylvaganam S, et al. The structure of avian eye lens delta-crystallin reveals a new fold for a superfamily of oligomeric enzymes. Nature Struct Biol. 1994;1:724–734. doi: 10.1038/nsb1094-724. [DOI] [PubMed] [Google Scholar]
- 7.Toth EA, Yeates TO. The structure of adenylosuccinate lyase, an enzyme with dual activity in the de novo purine biosynthetic pathway. Structure Fold Des. 2000;8:163–174. doi: 10.1016/s0969-2126(00)00092-7. [DOI] [PubMed] [Google Scholar]
- 8.Turner MA, Simpson A, McInnes RR, Howell PL. Human argininosuccinate lyase: a structural basis for intragenic complementation. Proc Natl Acad Sci USA. 1997;94:9063–9068. doi: 10.1073/pnas.94.17.9063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang J, Wang Y, Woolridge EM, Arora V, Petsko GA, Kozarich JW, Ringe D. Crystal structure of 3-carboxy-cis,cis-muconate lactonizing enzyme from Pseudomonas putida, a fumarase class II type cycloisomerase: enzyme evolution in parallel pathways. Biochemistry. 2004;43:10424–10434. doi: 10.1021/bi036205c. [DOI] [PubMed] [Google Scholar]
- 10.Fujii T, Sakai H, Kawata Y, Hata Y. Crystal structure of thermostable aspartase from Bacillus sp YM55-1: structure-based exploration of functional sites in the aspartase family. J Mol Biol. 2003;328:635–654. doi: 10.1016/s0022-2836(03)00310-3. [DOI] [PubMed] [Google Scholar]
- 11.Lee TT, Worby C, Bao ZQ, Dixon JE, Colman RF. His68 and His141 are critical contributors to the intersubunit catalytic site of adenylosuccinate lyase of Bacillus subtilis. Biochemistry. 1999;38:22–32. doi: 10.1021/bi982299s. [DOI] [PubMed] [Google Scholar]
- 12.Sampaleanu LM, Yu B, Howell PL. Mutational analysis of duck delta 2 crystallin and the structure of an inactive mutant with bound substrate provide insight into the enzymatic mechanism of argininosuccinate lyase. J Biol Chem. 2002;277:4166–4175. doi: 10.1074/jbc.M107465200. [DOI] [PubMed] [Google Scholar]
- 13.Saribas AS, Schindler JF, Viola RE. Mutagenic investigation of conserved functional amino acids in Escherichia coli L-aspartase. J Biol Chem. 1994;269:6313–6319. [PubMed] [Google Scholar]
- 14.Weaver T, Lees M, Banaszak L. Mutations of fumarase that distinguish between the active site and a nearby dicarboxylic acid binding site. Protein Sci. 1997;6:834–842. doi: 10.1002/pro.5560060410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porter DJ, Rudie NG, Bright HJ. Nitro analogs of substrates for adenylosuccinate synthetase and adenylosuccinate lyase. Arch Biochem Biophys. 1983;225:157–163. doi: 10.1016/0003-9861(83)90019-x. [DOI] [PubMed] [Google Scholar]
- 16.Hanson KR, Havir EA. The enzymic elimination of ammonia. In: Boyer PD, editor. The Enzymes. 3. Vol. 7. Academic Press; New York: 1972. pp. 75–166. [Google Scholar]
- 17.Bridger WA, Cohen LH. The kinetics of adenylosuccinate lyase. J Biol Chem. 1968;243:644–650. [PubMed] [Google Scholar]
- 18.Casey PJ, Lowenstein JM. Purification of adenylosuccinate lyase from rat skeletal muscle by a novel affinity column. Stabilization of the enzyme, and effects of anions and fluoro analogues of the substrate. Biochem J. 1987;246:263–269. doi: 10.1042/bj2460263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ratner S. Argininosuccinases and adenylo-succinases. In: Boyer PD, editor. The Enzymes. 3. Vol. 7. Academic Press; New York: 1972. pp. 167–197. [Google Scholar]
- 20.Miller RW, Buchanan JM. Biosynthesis of the purines. 28 Mechanism of action of adenylosuccinase. J Biol Chem. 1962;237:491–496. [PubMed] [Google Scholar]
- 21.Lee TT, Worby C, Bao ZQ, Dixon JE, Colman RF. Implication of His68 in the substrate site of Bacillus subtilis adenylosuccinate lyase by mutagenesis and affinity labeling with 2- [(4-bromo-2,3-dioxobutyl)thio]adenosine 5′-monophosphate. Biochemistry. 1998;37:8481–8489. doi: 10.1021/bi9805339. [DOI] [PubMed] [Google Scholar]
- 22.Lee TT, Worby C, Dixon JE, Colman RF. Identification of His141 in the active site of Bacillus subtiliadenylosuccinate lyase by affinity labeling with 6-(4-bromo-2,3-dioxobutyl)thioadenosine 5′-monophosphate. J Biol Chem. 1997;272:458–465. [PubMed] [Google Scholar]
- 23.Brosius JL, Colman RF. Three subunits contribute amino acids to the active site of tetrameric adenylosuccinate lyase: Lys268 and Glu275 are required. Biochemistry. 2002;41:2217–2226. doi: 10.1021/bi011998t. [DOI] [PubMed] [Google Scholar]
- 24.Segall ML, Colman RF. Gln212, Asn270, and Arg301 are critical for catalysis by adenylosuccinate lyase from Bacillus subtilis. Biochemistry. 2004;43:7391–7402. doi: 10.1021/bi0494774. [DOI] [PubMed] [Google Scholar]
- 25.Brosius JL, Colman RF. A key role in catalysis for His89 of adenylosuccinate lyase of Bacillus subtilis. Biochemistry. 2000;39:13336–13343. doi: 10.1021/bi001488j. [DOI] [PubMed] [Google Scholar]
- 26.Chakraborty AR, Davidson A, Howell PL. Mutational analysis of amino acid residues involved in argininosuccinate lyase activity in duck delta II crystallin. Biochemistry. 1999;38:2435–2443. doi: 10.1021/bi982150g. [DOI] [PubMed] [Google Scholar]
- 27.Abu-Abed M, Turner MA, Vallee F, Simpson A, Slingsby C, Howell PL. Structural comparison of the enzymatically active and inactive forms of delta crystallin and the role of histidine 91. Biochemistry. 1997;36:14012–14022. doi: 10.1021/bi971407s. [DOI] [PubMed] [Google Scholar]
- 28.Patejunas G, Barbosa P, Lacombe M, O’Brien WE. Exploring the role of histidines in the catalytic activity of duck delta-crystallins using site-directed mutagenesis. Exp Eye Res. 1995;61:151–154. doi: 10.1016/s0014-4835(05)80034-x. [DOI] [PubMed] [Google Scholar]
- 29.Vallee F, Turner MA, Lindley PL, Howell PL. Crystal structure of an inactive duck delta II crystallin mutant with bound argininosuccinate. Biochemistry. 1999;38:2425–2434. doi: 10.1021/bi982149h. [DOI] [PubMed] [Google Scholar]
- 30.Weaver T, Banaszak L. Crystallographic studies of the catalytic and a second site in fumarase C from Escherichia coli. Biochemistry. 1996;35:13955–13965. doi: 10.1021/bi9614702. [DOI] [PubMed] [Google Scholar]
- 31.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 32.Toth EA, Worby C, Dixon JE, Goedken ER, Marqusee S, Yeates TO. The crystal structure of adenylosuccinate lyase from Pyrobaculum aerophilum reveals an intracellular protein with three disulfide bonds. J Mol Biol. 2000;301:433–450. doi: 10.1006/jmbi.2000.3970. [DOI] [PubMed] [Google Scholar]
- 33.Sampaleanu LM, Vallee F, Slingsby C, Howell PL. Structural studies of duck delta 1 and delta 2 crystallin suggest conformational changes occur during catalysis. Biochemistry. 2001;40:2732–2742. doi: 10.1021/bi002272k. [DOI] [PubMed] [Google Scholar]
- 34.Bhaumik P, Koski MK, Bergmann U, Wierenga RK. Structure determination and refinement at 2.44 Å resolution of argininosuccinate lyase from Escherichia coli. Acta Crystallog sect D. 2004;60:1964–1970. doi: 10.1107/S0907444904021912. [DOI] [PubMed] [Google Scholar]
- 35.Li H, Robertson AD, Jensen JH. The determinants of carboxyl pKa values in turkey ovomucoid third domain. Proteins: Struct Funct Genet. 2004;55:689–704. doi: 10.1002/prot.20032. [DOI] [PubMed] [Google Scholar]
- 36.Naor MM, Jensen JH. Determinants of cysteine pKa values in creatine kinase and alpha1-antitrypsin. Proteins: Struct Funct Genet. 2004;57:799–803. doi: 10.1002/prot.20261. [DOI] [PubMed] [Google Scholar]
- 37.Zheng L, Kennedy MC, Beinert H, Zalkin H. Mutational analysis of active site residues in pig heart aconitase. J Biol Chem. 1992;267:7895–7903. [PubMed] [Google Scholar]
- 38.Segall ML, Cashman MA, Colman RF. Important roles of hydroxylic amino acid residues in the function of Bacillus subtilis adenylosuccinate lyase. Protein Sci. 2007;16:441–448. doi: 10.1110/ps.062650007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He B, Smith JM, Zalkin H. Escherichia coli purB gene: cloning, nucleotide sequence, and regulation by purR. J Bacteriol. 1992;174:130–136. doi: 10.1128/jb.174.1.130-136.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW, Sweet RM, editors. Methods in Enzymology. 276A. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 41.Weeks CM, Blessing RH, Miller R, Mungee R, Potter SA, Rappleye J, et al. Towards automated protein structure determination: BnP, the SnB-PHASES interface. Zeitschrift Fur Kristallographie. 2002;217:686–693. [Google Scholar]
- 42.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallog sect D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 43.McRee DE. XtalView/Xfit–A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 44.Adams PD, Pannu NS, Read RJ, Brunger AT. Cross-validated maximum likelihood enhances crystallographic simulated annealing refinement. Proc Natl Acad Sci USA. 1997;94:5018–5023. doi: 10.1073/pnas.94.10.5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pannu NS, Murshudov GN, Dodson EJ, Read RJ. Incorporation of prior phase information strengthens maximum-likelihood structure refinement. Acta Crystallog sect D. 1998;54:1285–1294. doi: 10.1107/s0907444998004119. [DOI] [PubMed] [Google Scholar]
- 46.Wang BC. Resolution of phase ambiguity in macromolecular crystallography. Methods Enzymol. 1985;115:90–112. doi: 10.1016/0076-6879(85)15009-3. [DOI] [PubMed] [Google Scholar]
- 47.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallog sect D. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 48.Blessing RH. DREADD - data reduction and error analysis for single-crystal diffractometer data. J Appl Crystallogr. 1989;22:396–397. [Google Scholar]
- 49.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallog sect D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 51.Tornheim K, Lowenstein JM. The purine nucleotide cycle. The production of ammonia from aspartate by extracts of rat skeletal muscle. J Biol Chem. 1972;247:162–169. [PubMed] [Google Scholar]