

Abstract
Background
Cycas simplicipinna (T. Smitinand) K. Hill. (Cycadaceae) is an endangered species in China. There were seven populations and 118 individuals that we could collect were genotyped in this study. Here, we assessed the genetic diversity, genetic structure and demographic history of this species.
Results
Analyses of data of DNA sequences (two maternally inherited intergenic spacers of chloroplast, cpDNA and one biparentally inherited internal transcribed spacer region ITS4-ITS5, nrDNA) and sixteen microsatellite loci (SSR) were conducted in the species. Of the 118 samples, 86 individuals from the seven populations were used for DNA sequencing and 115 individuals from six populations were used for the microsatellite study. We found high genetic diversity at the species level, low genetic diversity within each of the seven populations and high genetic differentiation among the populations. There was a clear genetic structure within populations of C. simplicipinna. A demographic history inferred from DNA sequencing data indicates that C. simplicipinna experienced a recent population contraction without retreating to a common refugium during the last glacial period. The results derived from SSR data also showed that C. simplicipinna underwent past effective population contraction, likely during the Pleistocene.
Conclusions
Some genetic features of C. simplicipinna such as having high genetic differentiation among the populations, a clear genetic structure and a recent population contraction could provide guidelines for protecting this endangered species from extinction. Furthermore, the genetic features with population dynamics of the species in our study would help provide insights and guidelines for protecting other endangered species effectively.
Keywords: Cycas simplicipinna, Pleistocene, Genetic differentiation, Population contraction, In situ, Ex situ conservation
Background
Historical processes leave imprints on the genetic structure of existing populations, especially those of long-lived and sessile organisms. The present genetic structure of many species has therefore been used to estimate the relationship between historical vicariance and geological change [1], dispersal history [2] and episodes of expansion and contraction associated with global climate change [3]. Climate can influence genetic variation by controlling the demography of a species [4]. The influence of Quaternary climate change on present patterns of genetic variation of some species has been studied [5,6]. Gugger [7] verified that late Quaternary glacial cycles played an important role in shaping the genetic structure and diversity of the present population of Quercus lobata Nee. The results showed that Quercus lobata maintained a stable distribution with local migration from the last interglacial period (~120 ka) through the Last Glacial Maximum (~21 ka, LGM) to the present. This contrasts with large-scale range shifts in Quercus alba L [7]. More recent climatic oscillations have had profound effects on the dynamics of population expansion and contraction, causing populations to contract into glacial refugia, become extinct and possibly to adapt locally [8,9]. Cycads are an ancient plant form, and their current genetic structure and population dynamic history are not fully understood. Therefore, they are valuable for contemporary researchers to study what they experienced in history and how they respond to historic climate change.
Cycads are the most primitive living seed plants. Fossil evidence shows that cycads originated approximately 275–300 million years ago [10,11]. Molecular evidence also shows that cycads originated much earlier than flowering plants [12,13], which originated approximately 125 million years ago [14,15]. Although cycads are generally long-lived [16,17], they presently comprise a relatively small group with two families (Cycadaceae, Zamiaceae) and ten genera [18]. They are currently considered to be the most threatened groups of organisms on the planet [19]. Cycads are distributed in Africa, Asia, Australia and South and Central America; 62% of the known cycad species are threatened with extinction [19]. There is only one cycad genus, Cycas, in China, and it is considered to be the oldest cycads genus [20]. All cycads have been given ‘First Grade’ conservation status in China [21].
Cycas simplicipinna (T. Smitinand) K. Hill was formally described in 1995. It is distinguishable by having the morphological characteristics of a shrub, an unremarkable trunk, and lanceolate cataphylls and is distributed in the Yunnan Province of China, Laos, Northern Thailand, and Vietnam. The species is dioecious and allogamous. Their seeds are mainly distributed by weight and usually distribute around the mother plant. So the phenomenon of severe inbreeding is common in the species, resulting in the expected high genetic differentiation and structure by using maternally inherited DNA. Despite being a national key protected plant, the genetic diversity and genetic structure of C. simplicipinna has not been studied in detail. The reasons for its endangerment are unclear. This study was undertaken to provide better understanding of the species’ genetic diversity and genetic structure and the reasons for its endangerment. Field surveys showed that there are two populations with fewer than 20 individuals. It is urgent to develop effective protection measures that are based on a comprehensive study of its genetic diversity and population structure.
The organelle DNA of cycads is maternally inherited and is dispersed only in seeds [22]. Their nuclear DNA (nDNA) is biparentally inherited and is dispersed by both seeds and pollen. Microsatellite markers (SSRs) are known to be codominant and to have more genetic variation than other molecular markers. In this study, we used cpDNA (psbA-trnH and trnL-trnF), nrDNA (ITS4-ITS5) and SSR markers. The main aim of the study was to evaluate the genetic diversity, genetic structure and demographic history of C. simplicipinna and to provide basic guidelines for its conservation.
Methods
Study species
A total of 118 individual samples were collected from seven populations of C. simplicipinna (four populations were sampled in Yunnan Province, China and three populations were sampled in Laos). Of the 118 samples, 86 individuals from the seven populations were used for chloroplast and nuclear DNA sequencing. The population known as BOL was eliminated from SSR analysis because there were only 3 individuals. A total of 115 individuals from six populations were used for the microsatellite study. Information on each sampling location and the number of individuals from each population that were used in DNA sequences and SSR analyses are presented in Table 1 and Figure 1, respectively.
Table 1.
Details of sample locations, sample sizes (n), haplotype diversity ( H d) and nucleotide diversity ( P i) surveyed for cpDNA and nrDNA of C. simplicipinna
| Population code | Population location | Latitude (N°) | Longitude (E°) | Altitude (m) | Individuals for DNA sequences/SSR (n) |
cpDNA |
nrDNA |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Haplotypes (No.) | H d | P i × 10 3 | Haplotypes (No.) | H d | P i × 10 3 | ||||||
| BOL |
Bolikhamxay, Laos |
18.456 |
103.802 |
140 |
3/0 |
Hap A (3) |
0 |
0 |
Hap 1 (3) |
0 |
0 |
| LUA |
LuangPrabang, Laos |
19.897 |
102.161 |
300 |
20/25 |
Hap F (20) |
0 |
0 |
Hap 5 (20) |
0 |
0 |
| LU |
LuangPrabang, Laos |
19.831 |
102.144 |
280 |
20/23 |
Hap E (20) |
0 |
0 |
Hap 5 (20) |
0 |
0 |
| MM |
Mengman town, Yunnan province |
21.128 |
101.315 |
640 |
20/20 |
Hap B (20) |
0 |
0 |
Hap 2 (20) |
0 |
0 |
| NZD |
Nuozhadu Hydropower Station, Yunnan province |
22.690 |
100.419 |
780 |
12/12 |
Hap C(5) Hap D(7) |
0.530 |
0.37 |
Hap 3 (12) Hap 4 (9) |
0.514 |
0.95 |
| ML |
Menglun town, Yunnan province |
21.932 |
101.253 |
550 |
11/11 |
Hap G (11) |
0 |
0 |
Hap 2 (20) |
0 |
0 |
| NBH |
Nature reserve of Nabanhe, Yunnan province |
22.166 |
100.663 |
694 |
10/24 |
Hap H (10) |
0 |
0 |
Hap 3 (10) |
0 |
0 |
| Total | 7 | 96/115 | 0.864 | 2.59 | 0.723 | 8.00 | |||||
Figure 1.
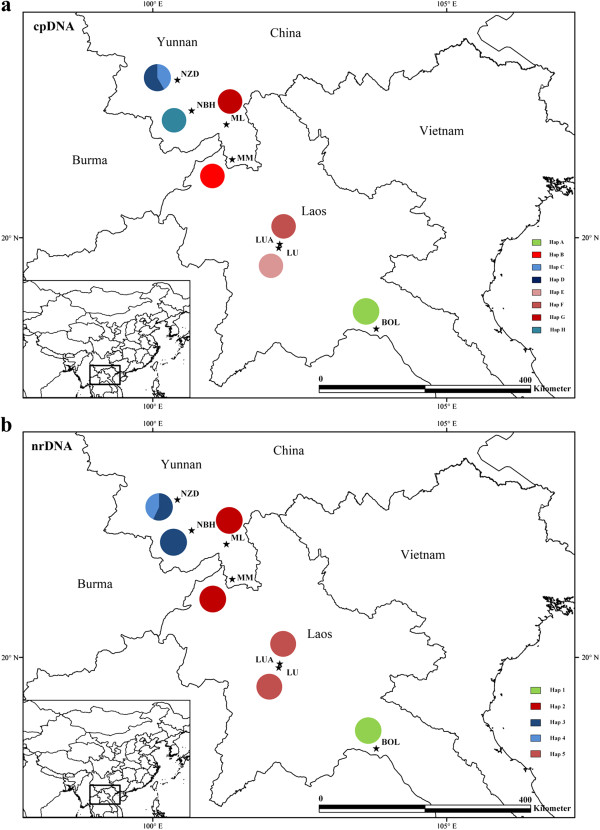
Distribution of cpDNA (a) and nrDNA (b) haplotypes detected among seven populations of C. simplicipinna. Full names of the abbreviations for the populations are shown in Table 1.
Molecular procedures
Young and healthy leaves were collected and dried immediately in silica gel for DNA extraction. Genomic DNA was extracted from dried leaves using the modified CTAB method [23]. After preliminary screening of 21–28 samples (representing approximately 3–4 individuals from each population) with universal chloroplast and nuclear primers, we chose two cpDNA intergenic spacers, psbA-trnH [24] and trnL-trnF [25], and one nrDNA internal transcribed spacer, ITS4-ITS5 [26], for complete analysis. The three pairs of fragments were amplified for the most polymorphic sites of the 86 individuals. PCR amplification was carried out in 40 μL reactions. For cpDNA, the PCR reactions contained 20 ng DNA, 2.0 μL MgCl2 (25 mM), 2.0 μL dNTPs (10 mM), 4.0 μL 10 × PCR buffer, 0.6 μL of each primer, 0.6 μL Taq DNA polymerase (5 U/μL) (Takara, Shiga, Japan) and 26 μL double-distilled water. For nrDNA, the PCR reactions contained 40 ng DNA, 2.4 μL MgCl2 (25 mM), 2.0 μL dNTPs (10 mM), 2.0 DMSO, 4.0 μL 10 × PCR buffer, 0.7 μL of each primer, 0.7 μL Taq DNA polymerase (5 U/μL) (Takara, Shiga, Japan) and 24.6 μL double-distilled water. PCR amplifications were performed in a thermocycler under the following conditions: an initial 5 min denaturation at 80°C, followed by 29 cycles of 1 min at 95°C, 1 min annealing at 50°C, and a 1.5 min extension at 65°C, and a final extension for 5 min at 65°C for cpDNA intergenic spacers. For nrDNA sequences we used an initial 4 min denaturation at 94°C, which was followed by 29 cycles of 45 s at 94°C, 1 min annealing at 50°C, and a 1.5 min extension at 72°C, and a final extension for 9 min at 72°C. All PCR products were sequenced in both directions with the same primers for the amplification reactions, using an ABI 3770 automated sequencer at Shanghai Sangon Biological Engineering Technology & Services Company Ltd. For nrDNA, we cloned individuals which had one or more heterozygous sites in the first sequencing round. Six to ten clones were randomly selected and sequenced until the heterozygous site split into two alleles.
Microsatellite markers were selected from recently developed nuclear microsatellites in Cycas[27-33]. PCR amplification was carried out in a 20 μL reaction, containing 10 ng DNA, 1.5 μL MgCl2 (25 mM), 1 μL dNTPs (10 mM), 1.5 μL 10 × PCR buffer, 0.6 μL of each primer, 0.16 μL Taq DNA polymerase (5 U/μL) (Takara, Shiga, Japan) and 12.14 μL double-distilled water. PCR amplifications were performed in a thermocycler under the following conditions: an initial 4 min denaturation at 94°C, which was followed by 29 cycles of 40 s each at 94°C, 25 s annealing at 48–60°C, and a 30 s extension at 72°C, and a final extension for 10 min at 72°C. PCR products were checked with 8% non-denaturing polyacrylamide gel electrophoresis. Then, we made preliminary screening microsatellite loci for C. simplicipinna. The selected microsatellite loci were stained with a fluorescent dye at the 5' end, their PCR products were separated and visualized using an ABI 3770 automated sequencer, and their profiles were read with the GeneMapper software. An individual was declared null (nonamplifying) at a locus and was treated as missing data after two or more amplification failures. Finally, we chose polymorphic microsatellite loci for C. simplicipinna after calculating polymorphism indices.
Data analysis
Data analysis of DNA sequences
Sequences were edited and assembled using SeqMan. Multiple alignments of the DNA sequences were performed manually with Clustal X, version 1.83 [34], with subsequent adjustment in Bioedit, version 7.0.4.1 [35]. Two cpDNA regions were combined. A congruency test for the two combined cpDNA regions showed a significant rate of homogeneity (P > 0.5) by PAUP* 4.0b10 [36], suggesting a high degree of homogeneity between the two cpDNA regions. The combined cpDNA sequences were therefore used in the following analysis.
Haplotypes were calculated from aligned DNA sequences by DnaSP, version 5.0 [37]. Within- and among-population genetic diversity were estimated by calculating Nei’s nucleotide diversity (Pi) and haplotype diversity (Hd) indices using DnaSP, version 5.0 [37]. We calculated within-population gene diversity (HS), gene diversity in total populations (HT = HS + DST, DST, gene diversity between populations [38]), and two measures of population differentiation, GST and NST, according to the methods described by Pons & Petit [39] using the Permut, 1.0 (http://www.pierroton.inra.fr/genetics/labo/Software/Permut). We used the program Arlequin, version 3.11 [40] to conduct an analysis of molecular variance (AMOVA) [41] and to estimate the genetic variation that was assigned within and among populations.
Phylogenetic relationships among cpDNA and nrDNA haplotypes of C. simplicipinna were inferred using maximum parsimony (MP) in PAUP* 4.0b10 [36] and Bayesian methods implemented in MrBayes, version 3.1.2 [42]. Cycas diannanensis was used as the outgroup. We used Mega, version 5 [43], to construct a neighbor-joining (NJ) tree that was based on the neighbor-joining method without using an outgroup. The degree of relatedness among cpDNA and among nrDNA haplotypes was also estimated using Network, version 4.2.0.1 [44]. In network analysis, indels were treated as single mutational events.
A well-documented evolutionary rate is needed to estimate coalescent time between lineages within populations. We used the evolutionary rates that had previously been estimated for seed plants to be 1.01 × 10-9 and 5.1-7.1 × 10-9[45] mutation per site per year for synonymous sites for cpDNA and nDNA, respectively. We used BEAST, version 1.6.1 [46], to estimate the time of divergence by using the HKY model and a strict molecular clock. We also used the BEAST program to create a Bayesian skyline plot with seven steps to infer the historical demography of C. simplicipinna. Posterior estimates of the mutation rate and time of divergence were obtained by Markov Chain Monte Carlo (MCMC) analysis. The analysis was run for 107 iterations with a burn-in of 106 under the HKY model and a strict clock. Genealogies and model parameters were sampled every 1,000 iterations. Convergence of parameters and mixing of chains were followed by visual inspection of parameter trend lines and checking of effective sampling size (ESS) values in three pre-runs. The ESS parameter was found to exceed 200, which suggests acceptable mixing and sufficient sampling. Adequate sampling and convergence to the stationary distribution were checked using TRACER, version 1.5 [47]. We used a pairwise mismatch distribution to test for population expansion in DnaSP, version 5.0 [37], to further investigate the demography of the species. The sum-of-squared deviations (SSD) between the observed and expected mismatch distributions were computed, and P-values were calculated as the proportion of simulations producing a larger SSD than the observed SSD. Arlequin, version 3.11 [40], was also used to calculate the raggedness index and its significance to quantify the smoothness of the observed mismatch distribution. The signatures of demographic change were examined by neutrality tests, Fu’s FS[48] to detect departures from population equilibrium. They were calculated using DnaSP, version 5.0 [37].
Data analysis of SSR markers
Dataset editing and formatting was performed in GenAlEx, version 6.3 [49]. We tested for evidence of preliminarily selection of our selected loci because our microsatellites had been derived from recently developed nuclear microsatellites of Cycas. We also used the Fst-outlier approach to test for signs of positive and balancing selection on those loci [50,51] by LOSITAN [52]. The outlier loci were identified by the expected distribution of Wright’s inbreeding coefficient Fst compared with HE[53]. As recommended by Antao [52], we ran LOSITAN to identify the loci under neutral selection by using the infinite allele model and 10,000 simulations. Twenty microsatellites were first selected after detecting the levels of genetic diversity in the sample of 115 individuals of C. simplicipinna in the six populations. The results of positive and balancing selection on the twenty microsatellites detected balancing selection on locus A16 and positive selection on four other loci (A3, A9, A13, and A14). However, locus A13 did not reach the significant level of an Fst-outlier (Figure 2). Therefore, four loci (A3, A9, A14, and A16) with significant levels as Fst-outliers were removed from further analysis. Finally, we selected sixteen microsatellites with high polymorphism, stability, and conformity with neutral selection for our research (Additional file 1: Table S1).
Figure 2.
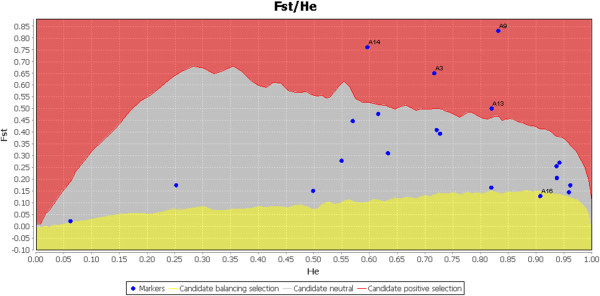
Test for selection on SSR loci. Red area represent positive selection, gray area represent neutral selection, and yellow area represent balancing selection. Four loci (A3, A9, A13, A14) subject to positive selection and one locus (A16) subject to balancing selection.
The number of alleles (NA), private alleles (AP), effective number of alleles (NE), expected heterozygosity (HE = 1-∑Pi2, Pi, population allele frequencies), observed heterozygosity (HO = No. of Hets/N), information index (I), and fixation index (F = 1-(HO/HE)) were calculated using GenAlEx, version 6.3 [49], and POPGENE, version 1.32 [54], with mutual correction. Allelic richness (AR) was estimated with FSTAT, version 2.9.3 [55], and percentage of polymorphic loci (PPB) was calculated with GenAlEx, version 6.3 [49]. Differentiation between pairs of populations was computed using FST and tested with GenAlEx, version 6.3 [49]. Isolation by distance (IBD) was tested on SSR data by computing Mantel tests in GenAlEx, version 6.3 [49] using a correlation of FST/(1-FST) with geographic distance for all pairs of populations. FST/(1-FST) was caculated with Genepop, version 4.1.4 [56]. Gene flow between pairs of populations was estimated using Wright’s principles Nm = (1-FST)/4FST[57]. Hardy-Weinberg equilibrium (HWE) was tested for each locus and each population using Genepop, version 4.1.4 [56].
The genetic structures of sampled populations and individuals were estimated by unweighted pair group mean analysis (UPGMA) using TEPGA, version 1.3 [58], with 5,000 of permutations. An individual-based principal coordinate analysis (PCO) was visualized by the program MVSP, version 3.12 [59], using genetic distances among SSR phenotypes. We also conducted a Bayesian analysis of population structure on the SSR data using STRUCTURE, version 2.2 [60]. Ten independent runs were performed for each set, with values of K ranging from 1 to 6, a burn-in of 1 × 105 iterations and 1 × 105 subsequent MCMC steps. The combination of an admixture and a correlated-allele frequencies model was used for the analysis. The second-order rate of change of the log probability of the data with respect to the number of clusters (ΔK) was used as an additional estimator of the most likely number of genetic clusters [61]. The best-fit number of grouping was evaluated using ΔK by STRUCTURE HARVESTER, version 0.6.8 [62]. Finally, we identified geographical locations where major genetic barriers among populations might occur with a barrier boundary analysis, using BARRIER, version 2.2 [63], based on genetic distance matrices.
We calculated the effective population sizes of each population to establish the degree of endangerment of the species. We used the program LDNe at three levels of the lowest allele frequency (=0.01, 0.02, 0.05) at a 95% confidence interval [64]. We tested the bottleneck statistic at the population level to explore the demographic history of populations by using different models and testing methods implemented in BOTTLENECK, version 1.2.02 [65]. The computation was performed under a stepwise mutation model (SMM) and a two-phased model (TPM). We did not use the standardized differences test in this study because the test was usually used at the condition of having at least twenty polymorphic loci. Two other methods (Sign tests and Wilcoxon tests) were applied to the two models. We also used a mode shift model [66] to test for bottlenecks in each population. These methods implemented in BOTTLENECK have low power unless the decline is greater than 90% [66]. They are most powerful when bottlenecks are severe and recent [67]. In addition, a genetic bottleneck was further investigated with the Garza-Williamsion index (also called M-ratio [68], the ratio of number of alleles to range in allele size). When seven or more loci are analyzed, the Garza-Williamsion index is lower than the critical Mc value of 0.68, a value obtained by simulations based on the empirical data in bottlenecked populations, suggesting a reduction in population size [40,68]. The Garza-Williamsion index is more powerful to detect genetic bottlenecks if the bottleneck lasted several generations or if the population made a rapid demographic recovery [67]. The index was analyzed by Arlequin, version 3.11 [40].
Results
DNA sequences
The combined length of cpDNA (psbA-trnH and trnL-trnF) varied from 1,408 to 1,438 bp and aligned with a 1,452 bp consensus length that contained 14 polymorphic sites and 16 indels (Additional file 2: Table S2). A total of eight chloroplast haplotypes was identified, and each population was fixed for one particular haplotype, except for population NZD, in which two unique haplotypes was detected (Table 1). The aligned nrDNA (ITS4-ITS5) matrix ranged from 1,079 to 1,087 bp with a consensus length of 1,100 bp that contained 32 polymorphic sites and 11 indels (Additional file 3: Table S3). A total of five nuclear haplotypes was derived. Population BOL had one unique haplotype (Hap 1), MM and ML shared haplotype 2, LUA and LU shared haplotype 5, and NZD had two haplotypes (one was unique and another shared with NBH) (Table 1).
Genetic diversity indices of total nucleotide (Pi) and haplotype (Hd) diversity in all populations were, respectively, 0.00259 and 0.864 as inferred from cpDNA and 0.008 and 0.723 as infered from nrDNA (Table 1). Only population NZD showed substantial genetic diversity. Total genetic diversity (HT = 1.000, 0.878 from cpDNA and nrDNA, respectively) was higher than the average intrapopulation diversity (HS = 0.076, 0.073 from cpDNA and nrDNA, respectively), resulting in high levels of genetic differentiation (GST = 0.924, 0.916; NST = 0.985, 0.992, from cpDNA and nrDNA, respectively Table 2). U tests showed that NST was not significantly greater than GST (P > 0.05) (Table 2), which suggests that there is no correspondence between haplotype similarities and their geographic distribution in C. simplicipinna.
Table 2.
Genetic diversity, differentiation parameters for the combined cpDNA sequences and nrDNA (ITS4-ITS5) sequences in all populations of C. simplicipinna
| Markers | H s | H T | G ST | N ST |
|---|---|---|---|---|
| cpDNA |
0.076 |
1.000 |
0.924 |
0.985 |
| (0.0758) |
(0.0198) |
(0.0758) |
(0.0166) |
|
| ITS4-ITS5 | 0.073 |
0.878 |
0.916 |
0.992 |
| (0.0735) | (0.0491) | (0.0828) | (0.0085) |
The AMOVA revealed that 98.67% of the genetic variation was partitioned among populations and 1.33% was within populations at the cpDNA level. At the nrDNA level, 97.95% of the genetic variation was partitioned among populations and 2.05% was within populations (Table 3). These results indicate that C. simplicipinna has high levels of genetic variation among populations and so high population structure.
Table 3.
Analysis of molecular variance (AMOVA) based on cpDNA and nrDNA haplotype frequencies for populations of C. simplicipinna
| Markes | Source of variation | d.f. | Sum of squares | Variance components | Percentage of variation (%) |
|---|---|---|---|---|---|
| cpDNA |
Among populations |
6 |
194.292 |
2.43825 |
98.67 |
| |
Within populations |
89 |
2.917 |
0.03277 |
1.33 |
| ITS4-ITS5 |
Among populations |
6 |
437.219 |
5.00785 |
97.95 |
| Within populations | 98 | 10.286 | 0.10496 | 2.05 |
A phylogeny of cpDNA and nrDNA haplotypes was constructed by both maximum parsimony (MP) and Bayesian methods, using C. diannanensis as an outgroup. Both analyses produced phylogenetic trees with consistent topologies (Figure 3). Eight cpDNA haplotypes appeared as a comb-like structure because they lacked enough information sites (Figure 3, a). Five nrDNA haplotypes were clustered into three clades, showing that Hap 2 is more closely related to Hap 5, and Hap 3 is more closely related to Hap 4 (Figure 3, b). The neighbor-joining trees (NJ) supported the congruent phylogenetic relationship of the cpDNA and nrDNA haplotypes (Figure 4). The haplotype network analysis of cpDNA and nrDNA also yielded the same topological relationships (Figure 5). Most haplotypes were distributed in the outside nodes of the reticulate evolutionary diagram, and many missing haplotypes, specifically between Hap 1 and Hap 2, were evident in the reticulate evolutionary diagram of the nrDNA haplotypes (Figure 5, b).
Figure 3.
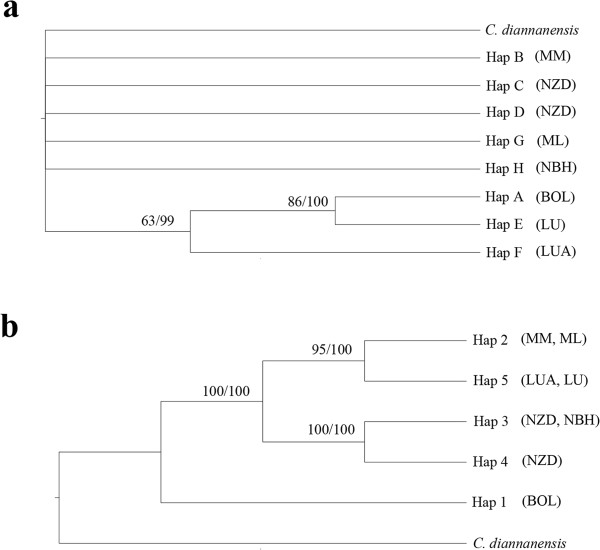
Strict consensus tree obtained by analysis of eight cpDNA haplotypes (a) and five nrDNA haplotypes (b) of C. simplicipinna, with C. diannanensis used as the outgroup. The numbers on branches indicate bootstrap values from the Maximum Parsimony principle (left) and the Bayesian analysis (right). The symbols BOL-NBH in the bracket represent population codes.
Figure 4.
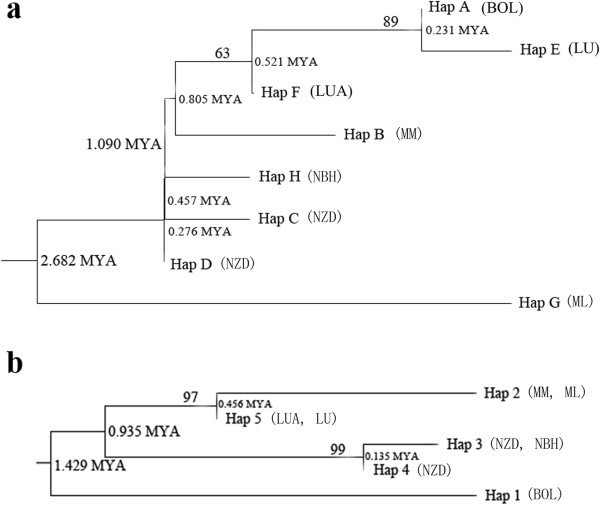
Neighbor-joining trees were built by using genetic distance based on eight cpDNA (a) and five nrDNA (b) haplotypes of C. simplicipinna. Bootstrap values were shown on branches and divergency times were shown on the nodes. MYA represent million years ago. The symbols BOL-NBH in the bracket represent population codes.
Figure 5.
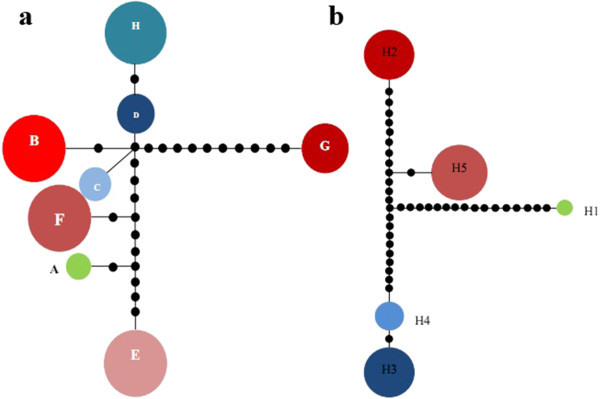
Network of haplotypes of C. simplicipinna based on cpDNA (a) and nrDNA (b). The size of the circles corresponds to the frequency of each haplotype, the small black circles represents one mutational step.
We derived the estimated time of divergence of C. simplicipinna with the Bayesian method, using BEAST, version 1.6.1 [46]. The estimated time of divergence ranged from 0.276 MYA to 2.682 MYA according to the cpDNA data and 0.135 MYA to 1.429 MYA according to the nrDNA data (Figure 4). The cpDNA haplotype G (Hap G) was the earliest to diverge. Its time of divergence was estimated to have been 2.682 MYA. The time of divergence of the clade comprising Hap A, E, F, and B and the clade comprising Hap H, C, and D was 1.090 MYA (Figure 4, a). The phylogenetic tree of nrDNA shows that Hap 1 was the earliest haplotype to diverge. Its time of divergence was 1.429 MYA. The time of divergence between the clade comprising Hap 2 and 5 and the clade comprising Hap 3 and 4 was 0.935 MYA (Figure 4, b). These results imply that the C. simplicipinna haplotypes were diverged during the Pleistocene (2.6 Ma to 11 ka).
Population dynamic analysis using cpDNA and nrDNA data showed that the population demography of C. simplicipinna was stable until approximately 50,000 years ago, at which time a contraction event occurred (Figure 6). The results of the mismatch analysis for all C. simplicipinna populations displayed a multimodal distribution pattern (Figure 7) with significant SSD and raggedness values (Table 4), which indicates that C. simplicipinna has not undergone a recent population expansion. This conclusion is also supported by the results of the Neutrality Test, Fu’s F S , which yielded positive values (Table 4). Based on a Bayesian simulation, the skyline plot showed recent declines in population size of all populations of C. simplicipinna during Quaternary glaciations and no subsequent expansion (Figure 6).
Figure 6.
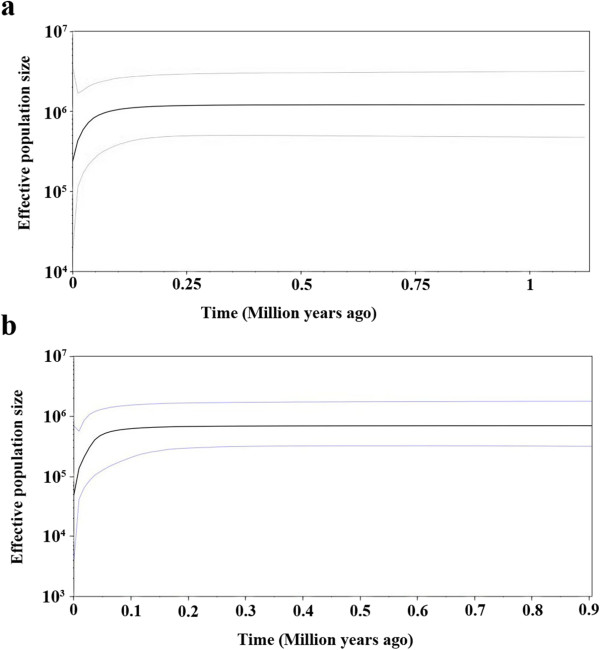
Bayesian skyline plot based on cpDNA (a) and nrDNA (b) for the effective population size fluctuation throughout time. Black line: median estimation; area between gray lines: 95% confidence interval.
Figure 7.
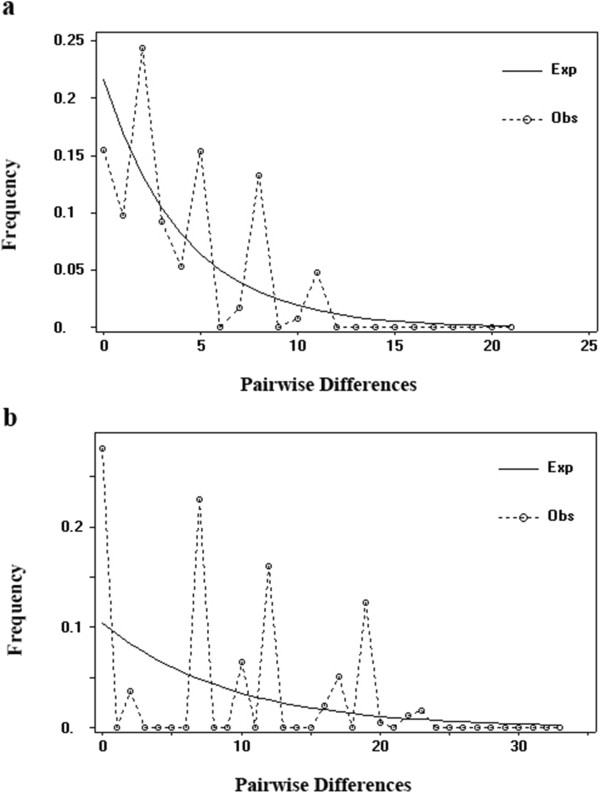
Mismatch distribution of cpDNA (a) and nrDNA (b) haplotypes based on pairwise sequence difference against the frequency of occurrence for C. simplicipinna.
Table 4.
Parameters of neutrality tests and mismatch analysis based on cpDNA and nrDNA of C. simplicipinna
| Markers | Fu and Li’ F * | Fu’ F s | SSD | Raggedness |
|---|---|---|---|---|
| cpDNA |
1.570 |
0.362 |
0.029* |
0.112** |
| ITS4-ITS5 | 2.022* | 0.439 | 0.028* | 0.109** |
Note: * is P < 0.05, significant difference; ** is P < 0.01, the most significant difference.
SSR data
A total of 169 alleles were identified at the sixteen loci. Diversity estimates varied in different populations (Table 5). Allelic richness was lowest in population MM (AR, 2.628) and highest in population LUA (AR, 5.014). The number of alleles (NA) ranged from 2.875 to 6.063, the number of private alleles (AP) ranged from 1 to 14, the effective number of alleles (NE) ranged from 1.925 to 3.521, the information index (I) ranged from 0.635 to 1.268, observed heterozygosity (HO) ranged from 0.306 to 0.473, and expected heterozygosity (HE) ranged from 0.353 to 0.603. These indices all showed a similar trend, with the lowest values in MM and the highest values in LUA. Fixation indices (F) were positive for all six populations, with a mean value F = 0.170, which suggests a high level of inbreeding within each population. The percentage of polymorphic loci (PPB) was high, ranging from 75% to 100%. Population MM had the lowest genetic diversity, and LUA had the highest. The genetic differentiation coefficient FST varied from 0.036 to 0.467, with a mean value 0.261. No significant effect of isolation by distance (IBD) was detected (Figure 8), as the correlation between genetic and geographic distances was non-significant (P > 0.05), which was supported by the result of Mantel test. Estimates of gene flow between each pair of the six populations are showed in Table 6. Population LUA had the most gene flow with the other populations, and MM had the least. Excesses of homozygotes caused five populations and nine loci to deviate from Hardy-Weinberg equilibrium (Table 5, Additional file 4: Table S4).
Table 5.
Genetic diversity and effective population size of six populations of C. simplicpinna based on sixteen SSR loci
| Population | N T | N P | A R | N A | N E | I | H O | H E | F | HWE ( P ) | PPB (%) | Ne |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LUA |
97 |
14 |
5.014 |
6.063 |
3.521 |
1.268 |
0.473 |
0.603 |
0.197 |
0.000*** |
100 |
164.9 |
| LU |
91 |
6 |
4.835 |
5.688 |
3.402 |
1.220 |
0.459 |
0.589 |
0.228 |
0.000*** |
100 |
33.9 |
| MM |
46 |
1 |
2.628 |
2.875 |
1.925 |
0.635 |
0.306 |
0.353 |
0.156 |
0.000*** |
81.25 |
56.6 |
| ML |
53 |
6 |
3.312 |
3.313 |
2.310 |
0.773 |
0.330 |
0.418 |
0.190 |
0.002** |
75.00 |
24.8 |
| NZD |
64 |
6 |
3.889 |
4.000 |
2.385 |
0.885 |
0.441 |
0.457 |
0.093 |
0.477ns |
93.75 |
43.7 |
| NBH |
63 |
8 |
3.417 |
3.938 |
2.292 |
0.846 |
0.387 |
0.442 |
0.154 |
0.000*** |
93.75 |
106.1 |
| Mean | 69 | 6.83 | 3.849 | 4.313 | 2.639 | 0.938 | 0.394 | 0.447 | 0.170 | 90.63 | 71.7 |
Note: NT, number of total alleles; NP, number of private alleles; AR, allelic richness; NA, number of alleles; NE, effective number of alleles; I, information index; HO, observed heterozygosity; HE, expected heterozygosity; F, fixation index; HWE, Hardy-Weinberg equilibrium; PPB, percentage of polymorphic loci; Ne, effective population size.-, Monomorphic; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Figure 8.
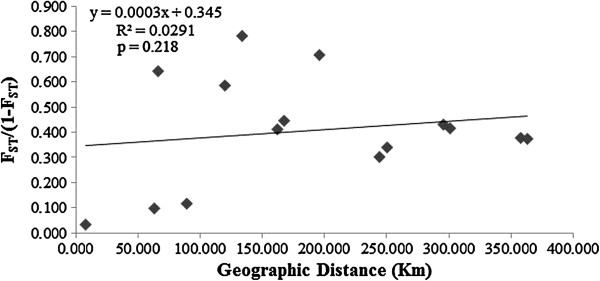
Plot of geographical distance against genetic distance for six populations of C. simplicipinna .
Table 6.
Estimates of gene flow between each pair of the six populations of C. simplicipinna
| Population | LUA | LU | MM | ML | NZD | NBH |
|---|---|---|---|---|---|---|
| LUA |
0.000 |
|
|
|
|
|
| LU |
6.555 |
0.000 |
|
|
|
|
| MM |
0.602 |
0.558 |
0.000 |
|
|
|
| ML |
0.816 |
0.731 |
2.073 |
0.000 |
|
|
| NZD |
0.653 |
0.666 |
0.353 |
0.425 |
0.000 |
|
| NBH |
0.577 |
0.595 |
0.319 |
0.387 |
2.47 |
0.000 |
| Mean | 1.534 | 1.518 | 0.651 | 0.739 | 0.761 | 0.725 |
The STUCTURE analysis, using the ΔK method, showed that the optimal K value was K = 3 (Figure 9), which showed that the six populations were clustered into three groups. Populations LUA and LU were grouped into one cluster (Cluster I), MM and ML were grouped into another cluster (Cluster II), and NZD and NBH were grouped into a third cluster (Cluster III). The result of K = 6 was also present here to detect whether or not has further subdivision in the species. From the Figure 9 we can see that there is only further subdivision at K = 6 between the population LUA and LU. In contrast with K = 6, it is clear that K value was K = 3 is a better solution, because the existence of three groups was also supported by the PCO analysis (Figure 10). Two-dimensional PCO separated all individuals into three clusters along the two axes. The dendrogram (Additional file 5: Figure S1) obtained with the UPGMA clustering method showed that the six populations were separated into three clades with high bootstrap values (100). It is the same as STRUCTURE (K = 3) and PCO analysis. In the UPGMA clustering dendrogram, populations LUA, LU, MM, and ML were clustered into one large clade with a bootstrap value of 78.7. The BARRIER analysis showed that there was only one major genetic boundary (Barrier I), with a 52.7% mean bootstrap value, separating the six populations into two clusters (Figure 11).
Figure 9.
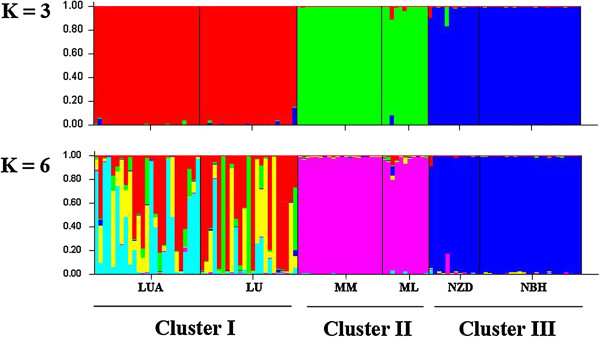
Estimated genetic clustering (K = 3 and 6) obtained with the STRUCTURE program for six populations of C. simplicipinna based on SSR data. Black lines separate different populations.
Figure 10.
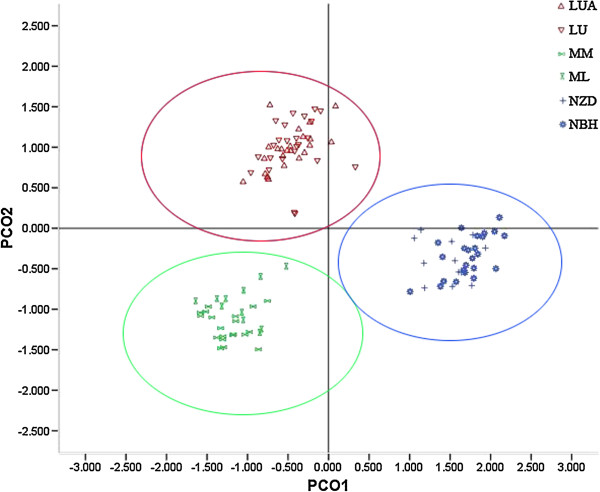
Principal coordinate analysis (PCO) of SSR phenotypes from six populations and 115 individuals of C. simplicipinna. The symbols LUA-NBH on the figure represent population codes. Colour coding corresponds to the STRUCTURE analysis.
Figure 11.
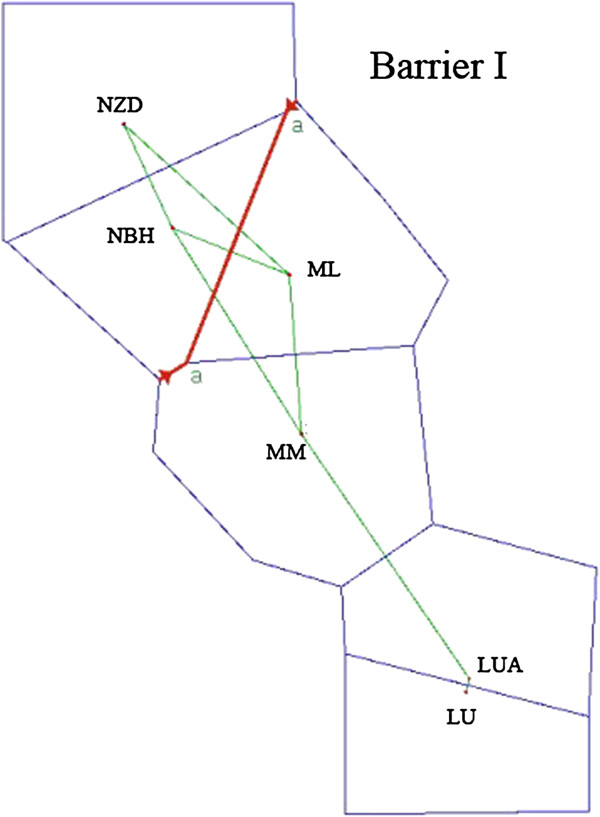
The boundaries detected using the BARRIER program based on matrices of Nei’s (1983) unbiased genetic distance.
Estimates of effective population sizes with the lowest allele frequency (=0.02) as shown by the LDNe analysis are listed in Table 5. The effective population size of LUA and NBH was more than 100 and was less than 50 in three other populations. The BOTTLENECK analysis was used to calculate mutation-drift equilibrium as estimated with different models and different methods (Table 7). This analysis indicates that C. simplicipinna did not experience a bottleneck. When TPM was used, only MM had a significant excess of heterozygosity as estimated with the two methods (P < 0.05), suggesting that MM deviated from mutation-drift equilibrium. When SMM was used, only ML showed a significant excess of heterozygosity (Wilcoxon text testing). Mode shift models showed that all populations had normal L-shaped distributions, which suggests that C. simplicipinna has not experienced a recent severe bottleneck. While all the Garza-Williamson indices (Table 7) of the six populations are lower than the critical Mc value of 0.68, which indicate that there was a past reduction of effective population size in the species. Populations of C. simplicipinna underwent a demographic bottleneck in history.
Table 7.
Bottleneck analysis for six populations of C. simplicipinna under different models and different methods
| Population |
T.P.M |
S.M.M |
Mode shift |
Garza-Williamson |
||
|---|---|---|---|---|---|---|
| Sign test | Wilcoxon test | Sign test | Wilcoxon test | index | ||
| LUA |
0.156 |
0.058 |
0.499 |
0.430 |
L |
0.466 |
| LU |
0.160 |
0.037* |
0.497 |
0.490 |
L |
0.462 |
| MM |
0.040* |
0.008** |
0.432 |
0.116 |
L |
0.379 |
| ML |
0.294 |
0.012* |
0.468 |
0.047* |
L |
0.349 |
| NZD |
0.556 |
0.470 |
0.310 |
0.719 |
L |
0.450 |
| NBH | 0.481 | 0.232 | 0.333 | 0.702 | L | 0.417 |
Note: P is test for heterozigosity excess, *:P < 0.05, significant difference; **:P < 0.01, most significant difference.
Discussion
Genetic variation and genetic structure
The genetic variation of a species is a product of its long-term evolution and represents its evolutionary potential for survival and development [69,70]. Cycads, as ancient gymnosperms with millions of years of evolutionary history, a long life cycle, and overlapping generations, would be expected to have genomes that are responsive to different selective pressures. High levels of genetic variation would be expected to have accumulated during a long evolutionary history. As expected, we found that C. simplicipinna has high genetic diversity (Table 1, 2 and 5) at a species level compared with other species of Cycas by using similar markers e.g., an average value of HT = 0.564 and Pi = 0.00132 were reported for two markers of type cpDNA in C. debaoensis[5], and an average value of HO = 0.349 and HE = 0.545 and the maximum value of Ap = 2.1, NA = 5.8 were reported for 14 markers of type EST-microsatellites in C. micronesica[53]. Cycas simplicipinna also has higher genetic diversity than many conifers. Many individual conifer species show lower genetic diversity, e.g., an average value of HT = 0.234 and Hs = 0.190 were reported for two markers of type cpDNA in Pinus tabulaeformis[71], an average value of π = 0.000573 and π = 0.006131 were reported for two markers of type cpDNA and one marker of type nDNA in Tsuga dumosa, respectively [72], and an average value of HT = 0.77, Hs = 0.66, NR = 3.98, HE = 0.62 were reported for seven markers of type nuclear microsatellites in Taxus baccata[73]. The mean genetic diversity value of 170 plant species that was estimated from cpDNA-based studies was HT = 0.67 [74]. However, at a population level, C. simplicipinna shows low genetic diversity; only population NZD has a relatively high genetic diversity.
The genetic diversity of C. simplicipinna among all populations (HT = 1.000, 0.878 from cpDNA and nrDNA, respectively Table 2) is also higher than the average intra-population diversity (HS = 0.076, 0.073 from cpDNA and nrDNA, respectively Table 2), which indicates that there are high levels of genetic differentiation among populations (GST = 0.924, 0.916, NST = 0.985, 0.992 from cpDNA and nrDNA, respectively Table 2). U tests showed that NST was not significantly greater than GST, suggesting that there is no distinct phylogeographical structure in C. simplicipinna. The FST value of C. simplicipinna (nSSR: FST = 0.261, GST = 0.246, Table 5) was higher than the mean value of outcrossing species (FST = 0.22) that was inferred from SSR [75]. Wright [76] had proposed that an FST value greater than 0.25 (C. simplicipinna: FST = 0.26 > 0.25) would indicate that there was significant genetic differentiation among populations. Additionally, according to the results of deviation from Hardy-Weinberg equilibrium test (Table 5, Additional file 4: Table S4), only population NZD was in Hardy-Weinberg equilibrium. The remaining five populations deviated significantly from Hardy-Weinberg equilibrium, and the fixation indices (F) were greater than zero. We therefore conclude that there is a notable deficit in heterozygosity and severe inbreeding in C. simplicipinna populations, resulting in a high among-population genetic differentiation as a whole.
The genetic structure of C. simplicipinna based on SSR markers showed that the six study populations were divided into three clades (I, II and III). BARRIER analysis (Figure 11) showed that only one barrier (BS > 50%) exists among the six populations, suggesting that clade I and clade II are genetically more closely related to each other than either is to clade III. Genetic structures of C. simplicipinna derived from organelle and nuclear markers are different. It displays high differentiation at cpDNA markers and high distinct structure at biparental markers. This is caused by different features of the markers, such as mutation rates and the biased migration between organelle (pollen migration) and nuclear markers (pollen migration and seed migration, seed migration is very little). Comparisons of genetic with paleoecological data of temperate woody species are known to reveal unique genetic lineages and⁄ or endemic haplotypes in separate refuge populations [77,78]. Although C. simplicipinna is a tropical or subtropical woody species, the species similarly possesses unique genetic lineages and endemic cpDNA haplotypes in its separate refuge populations. Duminil [79] had proposed that the level of genetic structure in temperate trees and the potential to reflect historical population isolation are determined in part by life history. Specifically, species with large geographical ranges and wide-ranging seed dispersal display low differentiation at maternally inherited cpDNA markers, and long-lived outcrossing species display low structure at biparental markers. However, C. simplicipinna displays the opposite results. This outcome may be due to its having a limited geographic distribution and severe population inbreeding or to its being a tropical or subtropical woody species, which differs from temperate trees.
The correlation between genetic and geographic distances was non-significant (Figure 8), which indicates that there is no significant effect of isolation by distance (IBD). The species lacks of clear geographic structure. It does not automatically mean that the genetic information has no value in directing management. Yang and Meerow [80] estimated that gene flow distance among local populations in Cycas was 2–7 km. However, the distances between extant populations of C. simplicipinna are all greater than 7 km (the geographic distance between LUA and LU was the smallest of all population pairs (7.52 km)). Because effective gene flow higher than 1 is often regarded as high enough to prevent population differentiation due to genetic drift, while gene flow less than 1 may be a major reason caused the genetic differentiation among populations[81], many gene exchanges between populations less than 1 reflect a low level of gene flow (Table 6). Cycas simplicipinna is dioecious and pollenated by insect (Curculionoidea, weevils) [82]. Unlike the birds, weevils can’t spread pollen over a long distance. Its seeds are too large to disperse naturally over such a long distance. Most seeds disperse near the mother plant, which increases inbreeding. Inter-population could not exchange gene flow easily. We therefore conclude that the strong genetic differentiation and structure observed in C. simplicipinna is due mainly to its limited gene flow and severe inbreeding. Our analysis therefore suggests that C. simplicipinna has high genetic diversity at the species level, low genetic diversity within populations, high genetic differentiation among populations and a clear genetic structure. This conclusion is also supported by the AMOVA analysis (Table 3), which shows that almost all of the genetic variation exists among populations (DNA sequences). Our results support the conclusion that low genetic variation within populations is biologically typical for Cycas, unlike other gymnosperms [83].
Phylogeny and demographic history
Phylogenetic trees constructed on the basis of DNA sequences with different criteria in four different software systems all suggested a consistent systematic relationship of haplotypes (Figure 3 and 4). The comb-like structure of cpDNA haplotypes is likely to be the result of insufficient information site due to insufficient evolutionary time [1,84]. The network analysis (Figure 5) showed that most of the haplotypes were distributed in the outside nodes of the reticulate evolutionary diagrams and there were many missing haplotypes among them. The reason for the observed haplotypes distribution pattern is that diversity within populations is extremely low and differentiation among populations is high. With the exception of the population NZD, the populations have no haplotype and nucleotide polymorphism. This is because that missing haplotypes in the network are due to the species’ long evolutionary history during which climate variations, geological activities, and human activities formed the several scattered populations that currently exist.
Understanding a species’ demographic history aids in understanding its ancient evolutionary environment. Quaternary glaciers are known to have profoundly affected the distribution of plant species [3]. Previous studies have shown that different plant species had different responses to glacial and interglacial influences. Most plant taxa are believed to have shifted the latitude or elevation of their ranges in response to glaciation [8]. Some plant species, e.g., Taxus wallichiana[85], C. revoluta and C. taitungensis[6], experienced population expansion during the most recent glacial period, and others, e.g., C. debaoensis[5], showed population contraction. Although there is growing evidence for population demographic stability or expansion throughout the Last Glacial Maximum (LGM) in a range of different organisms [86-91], C. simplicipinna appears to have exhibited population demographic contraction during the LGM and no later expansion. We infer this from the Bayesian skyline plot (Figure 6), a divergence of the observed mismatch distribution from the expected distribution (Figure 7). The significant positive values of SSD and raggedness and non-significant positive Fu’s Fs (Table 4) also imply C. simplicipinna did not undergo a population expansion, so it maybe undergo a population contraction or stay a population dynamic equilibrium. Bottleneck analysis (Table 7) showed that populations of C. simplicipinna have not experienced recent bottleneck event but a reduction in population size, which was in accord with Bayesian skyline plot (Figure 6). The divergence times of C. simplicipinna haplotypes fall generally in the Pleistocene. We therefore conclude that C. simplicipinna was widely and continuously distributed before the glacials and contracted into several isolated surviving populations during the glacials. Refugium populations typically have relatively high genetic diversity and unique haplotypes [77,92]. In our study, C. simplicipinna had high genetic diversity at the species level. Each of its population had a unique haplotype for the cpDNA data but low level genetic diversity. The existing C. simplicipinna populations are distributed mainly in the tropics or subtropics. Because they are adapted to warm temperatures, temperature is the main factor affecting their growth. The average annual temperature in Chinese subtropical and tropical areas during the last glacial maximum was 4-6°C lower than it is today, which caused change and migration in vegetation [93]. This could have had a strong influence on the distribution of C. simplicipinna. The low Ice Age temperatures were not suitable for C. simplicipinna, which led to decline or even local extinction of populations. Some cpDNA and nuclear haplotypes were lost in the process, leading us to deduce that the present areas of distribution are its Ice Age glacial refugia. Cycas simplicipinna migrated to the scatter refugia that form its haplotypes current distribution pattern. They did not migrate to a common refugium during the Quaternary glacial period but instead survived in their original locales.
We conclude that the reduction in effective population size and limited gene flow were the main factors promoting genetic differentiation among populations of C. simplicipinna, which in turn led to the current population structure and distribution pattern.
Conservation implications
One goal for the conservation of threatened plants is to maintain the genetic diversity of native plant species [94]. Ex situ collections are also important because they provide a setting for breeding for introduction, which is an important way to increase the genetic diversity of native plant populations. We found that three (NBH, BOL, ML) of our seven study populations were distributed in protection zones in which there has been no overexploitation and habitat destruction. However, the other four populations (NZD, LU, LUA, and MM) occur outside of protected areas and should be protected. Our study, which revealed a clear population structure of C. simplicipinna that show low genetic variation within populations and high genetic differentiation among populations, has significant implications for conservation of this species. The population structure and demographic history of C. simplicipinna imply that conservation efforts cannot focus on only one part of the species’ range. We suggest that habitat protection be strengthened immediately by establishing protection zones or plots in the distribution areas of C. simplicipinna to improve the conservation awareness of local farmers and to prohibit deforestation in Cycas distribution areas. Protection of populations with the highest genetic diversity, such as populations NZD and LUA, should be of highest priority. Priority should also be given to populations with unique haplotypes. Based on the analysis of the two cpDNA sequences in our study, six of the seven sampled populations of C. simplicipinna each had one unique cpDNA haplotype. Therefore, every C. simplicipinna population should be given maximum protection to prevent losing any haplotypes. The reduction of effective population size in the Ice Age has led to a small effective population size for the species as a whole, which jeopardizes the species’ current genetic diversity. Different populations and/or individuals should be moved from current areas of rich genetic diversity to secure remote areas for ex situ conservation. These measures taken together could protect and enrich the genetic diversity of C. simplicipinna.
Conclusions
This study shows that this cycas species underwent a past population contraction during Pleistocene with high genetic differentiation among populations and a clear genetic structure. In addition, unique haplotype was detected in all populations in this study. These populations need to be protected for sustaining high genetic diversity in C. simplicipinna. Furthermore, the reconstruction of population demographic history in C. simplicipinna provides insights and guidelines for protecting C. simplicipinna and other endangered cycas species effectively.
Availability of supporting data
The data set of the DNA sequencing data in our study are deposited in GenBank under accession numbers KM065478-KM065496.
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
XG participated in the design of the study as well as collected study materials. XF carried out the molecular genetic studies, participated in the data analysis and drafted the manuscript. YW participated in the study’s design and coordination. All authors read and approved the final manuscript.
Supplementary Material
Information of sixteen SSR makers used to study the population genetics of C. simplicipinna.
Variable sites from the two combined cpDNA in C. simplicipinna.
Variable sites from the nrDNA in C. simplicipinna.
P-value of Hardy-Weinberg equilibrium test for six populations of C. simplicipinna.
An unweighted pair-group method with arithmetic averages (UPGMA) phenogram of six populations of C. simplicipinna based on SSR markers. Numbers on branches indicated bootstrap values from 5000 replicates.
{kind=link}
Contributor Information
Xiuyan Feng, Email: fengxiuyan@mail.kib.ac.cn.
Yuehua Wang, Email: wangyh58212@126.com.
Xun Gong, Email: gongxun@mail.kib.ac.cn.
Acknowledgements
The authors thank Yuezhi Pan, Wei Zhou and Longqian Xiao for their assistance with field sampling and thank Wei Zhou for his help in data analysis. This research was supported by the United Fund of the NSFC and the Yunnan Natural Science Foundation (U1136602 to X.G.).
References
- Avise JC. Phylogeography: the history and formation of species. Cambridge: Harvard University Press; 2000. [Google Scholar]
- Petit RJ, Pineau E, Demesure B, Bacilieri R, Ducousso A, Kremer A. Chloroplast DNA footprints of postglacial recolonization by oaks. Proc Natl Acad Sci. 1997;94:9996–10001. doi: 10.1073/pnas.94.18.9996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt G. The genetic legacy of the Quaternary ice ages. Nature. 2000;405:907–913. doi: 10.1038/35016000. [DOI] [PubMed] [Google Scholar]
- James PM, Coltman DW, Murray BW, Hamelin RC, Sperling FA. Spatial genetic structure of a symbiotic beetle-fungal system: toward multi-taxa integrated landscape genetics. PLoS One. 2011;6:e25359. doi: 10.1371/journal.pone.0025359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan QQ, Wang JF, Gong X, Peng H. Patterns of chloroplast DNA variation in Cycas debaoensis (Cycadaceae): conservation implications. Conservat Genet. 2011;12:959–970. doi: 10.1007/s10592-011-0198-9. [DOI] [Google Scholar]
- Chiang YC, Hung KH, Moore SJ, Ge XJ, Huang S, Hsu TW, Schaal BA, Chiang TY. Paraphyly of organelle DNAs in Cycas Sect. Asiorientales due to ancient ancestral polymorphisms. BMC Evol Biol. 2009;9:161. doi: 10.1186/1471-2148-9-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gugger PF, Ikegami M, Sork VL. Influence of late Quaternary climate change on present patterns of genetic variation in valley oak, Quercus lobata Née. Mol Ecol. 2013;22:3598–3612. doi: 10.1111/mec.12317. [DOI] [PubMed] [Google Scholar]
- Davis MB, Shaw RG. Range shifts and adaptive responses to Quaternary climate change. Science. 2001;292:673–679. doi: 10.1126/science.292.5517.673. [DOI] [PubMed] [Google Scholar]
- Hewitt G. Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond B Biol Sci. 2004;359:183–195. doi: 10.1098/rstb.2003.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhifeng G, Thomas BA. A review of fossil cycad megasporophylls, with new evidence of Crossozamia pomel and its associated leaves from the lower permian of Taiyuan, China. Rev Palaeobot Palynol. 1989;60:205–223. doi: 10.1016/0034-6667(89)90044-4. [DOI] [Google Scholar]
- Axsmith BJ, Serbet R, Krings M, Taylor TN, Taylor EL, Mamay SH. The enigmatic paleozoic plants spermopteris and phasmatocycas reconsidered. Am J Bot. 2003;90:1585–1595. doi: 10.3732/ajb.90.11.1585. [DOI] [PubMed] [Google Scholar]
- Samigullin TK, Martin WF, Troitsky AV, Antonov AS. Molecular data from the chloroplast rpoC1 gene suggest a deep and distinct dichotomy of contemporary spermatophytes into two monophyla: gymnosperms (including Gnetales) and angiosperms. J Mol Evol. 1999;49:310–315. doi: 10.1007/PL00006553. [DOI] [PubMed] [Google Scholar]
- Soltis DE, Soltis PS, Zanis MJ. Phylogeny of seed plants based on evidence from eight genes. Am J Bot. 2002;89:1670–1681. doi: 10.3732/ajb.89.10.1670. [DOI] [PubMed] [Google Scholar]
- Friis EM, Pedersen KR, Crane PR. Early angiosperm diversification: the diversity of pollen associated with angiosperm reproductive structures in Early Cretaceous floras from Portugal. Ann Mo Bot Gard. 1999;86:259–296. doi: 10.2307/2666179. [DOI] [Google Scholar]
- Sun G, Ji Q, Dilcher DL, Zheng S, Nixon KC, Wang X. Archaefructaceae, a new basal angiosperm family. Science. 2002;296:899–904. doi: 10.1126/science.1069439. [DOI] [PubMed] [Google Scholar]
- Vovides AP. Spatial distribution, survival, and fecundity of Dioon edule (Zamiaceae) in a tropical deciduous forest in Veracruz, Mexico, with notes on its habitat. Am J Bot. 1990;77:1532–1543. doi: 10.2307/2444486. [DOI] [Google Scholar]
- Raimondo DC, Donaldson JS. Responses of cycads with different life histories to the impact of plant collecting: simulation models to determine important life history stages and population recovery times. Biol Conserv. 2003;111:345–358. doi: 10.1016/S0006-3207(02)00303-8. [DOI] [Google Scholar]
- Christenhusz M, Reveal J, Farjon A, Gardner MF, Mill RR, Chase MW. A new classification and linear sequence of extant gymnosperms. Phytotaxa. 2011;19:55–70. [Google Scholar]
- Hoffmann M, Hilton-Taylor C, Angulo A, Böhm M, Brooks TM, Butchart SH, Carpenter KE, Chanson J, Collen B, Cox NA. The impact of conservation on the status of the world’s vertebrates. Science. 2010;330:1503–1509. doi: 10.1126/science.1194442. [DOI] [PubMed] [Google Scholar]
- Hill KD, Stevenson DW, Osborne R. The world list of cycads. Bot Rev. 2004;70:274–298. doi: 10.1663/0006-8101(2004)070[0274:TWLOC]2.0.CO;2. [DOI] [Google Scholar]
- XIAO LQ, GE XJ, Gong X, Hao G, ZHENG SX. ISSR variation in the endemic and endangered plant Cycas guizhouensis (Cycadaceae) Ann Bot. 2004;94:133–138. doi: 10.1093/aob/mch119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Hsieh HT, Fang K, Chiang YC. Patterns of genetic variation and demography of Cycas taitungensis in Taiwan. Bot Rev. 2004;70:86–92. doi: 10.1663/0006-8101(2004)070[0086:POGVAD]2.0.CO;2. [DOI] [Google Scholar]
- Doyle J. In: Molecular Techniques in Taxonomy. Hewitt GM, editor. Berlin: Springer; 1991. DNA protocols for plants-CTAB total DNA isolation; pp. 283–293. [Google Scholar]
- Chiang T, Peng C. In: Proceeding of the symposium on Conservation of Endemic Species. Yeng SD, editor. Taipei, Taiwan: Research Institute of Taiwan Endemic Species; 1998. Phylogeography of the endemic plants in Taiwan; pp. 148–155. [Google Scholar]
- Taberlet P, Gielly L, Pautou G, Bouvet J. Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol. 1991;17:1105–1109. doi: 10.1007/BF00037152. [DOI] [PubMed] [Google Scholar]
- White TJ, Bruns T, Lee S, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR. 1990;18:315–322. [Google Scholar]
- Ju LP, Kuo CC, Chao YS, Cheng YP, Gong X, Chiang YC. Microsatellite primers in the native perennial cycad Cycas taitungensis (Cycadaceae) Am J Bot. 2011;98:e84–e86. doi: 10.3732/ajb.1000504. [DOI] [PubMed] [Google Scholar]
- Yang Y, Li Y, LI LF GEXJ, Gong X. Isolation and characterization of microsatellite markers for Cycas debaoensis YC Zhong et CJ Chen (Cycadaceae) Mol Ecol Resour. 2008;8:913–915. doi: 10.1111/j.1755-0998.2008.02114.x. [DOI] [PubMed] [Google Scholar]
- Zhang F, Su T, Yang Y, Zhai Y, Ji Y, Chen S. Development of seven novel EST–SSR markers from Cycas panzhihuaensis (Cycadaceae) Am J Bot. 2010;97:e159–e161. doi: 10.3732/ajb.1000377. [DOI] [PubMed] [Google Scholar]
- Zhang M, Wang ZF, Jian SG, Ye WH, Cao HL, Zhu P, Li L. Isolation and characterization of microsatellite markers for Cycas hainanensis CJ Chen (Cycadaceae) Conservat Genet. 2009;10:1175–1176. doi: 10.1007/s10592-008-9737-4. [DOI] [Google Scholar]
- Wang ZF, Ye WH, Cao HL, Li ZC, Peng SL. Identification and characterization of EST-SSRs and cpSSRs in endangered Cycas hainanensis. Conservat Genet. 2008;9:1079–1081. doi: 10.1007/s10592-007-9461-5. [DOI] [Google Scholar]
- Li L, Wang ZF, Jian SG, Zhu P, Zhang M, Ye H, Ren H. Isolation and characterization of microsatellite loci in endangered Cycas changjiangensis (Cycadaceae) Conservat Genet. 2009;10:793–795. doi: 10.1007/s10592-008-9664-4. [DOI] [Google Scholar]
- Cibrián-Jaramillo A, Marler TE, DeSalle R, Brenner ED. Development of EST-microsatellites from the cycad Cycas rumphii, and their use in the recently endangered Cycas micronesica. Conservat Genet. 2008;9:1051–1054. doi: 10.1007/s10592-007-9447-3. [DOI] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT Nucleic acids symposium series 199995–98.10780396
- Swofford DL. PAUP*: Phylogenetic Analysis Using Parsimony (and Other Methods), Version 4.0b10. Massachusetts: Sinauer Associates, Inc, Sunderland; 2002. [Google Scholar]
- Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Nei M. Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci. 1973;70:3321–3323. doi: 10.1073/pnas.70.12.3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons OPR. Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics. 1996;144:1237–1245. doi: 10.1093/genetics/144.3.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online. 2005;1:47. [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster M, Forster P, Watson J. NETWORK (version 4.2. 0.1): a software for population genetics data analysis. [ http://www.fluxusengineering.com/sharenet.htm].
- Graur D, Li W-H. Fundamentals of molecular evolution. MA: Sinauer Associates Sunderland; 2000. [Google Scholar]
- Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Drummond A. Tracer, MCMC Trace Analysis Tool. Oxford, UK: University of Oxford; 2004. [Google Scholar]
- Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–925. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peakall R, Smouse PE. GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes. 2006;6:288–295. doi: 10.1111/j.1471-8286.2005.01155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont MA, Nichols RA. Evaluating loci for use in the genetic analysis of population structure. Proc Roy Soc Lond B Biol Sci. 1996;263:1619–1626. doi: 10.1098/rspb.1996.0237. [DOI] [Google Scholar]
- Beaumont MA. Adaptation and speciation: what can Fst tell us? Trends Ecol Evol. 2005;20:435–440. doi: 10.1016/j.tree.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Antao T, Lopes A, Lopes R, Beja-Pereira A, Luikart G. LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinform. 2008;9:323. doi: 10.1186/1471-2105-9-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CIBRIÁN‒JARAMILLO A, Daly A, Brenner E, Desalle R, Marler T. When North and South don’t mix: genetic connectivity of a recently endangered oceanic cycad, Cycas micronesica, in Guam using EST‒microsatellites. Mol Ecol. 2010;19:2364–2379. doi: 10.1111/j.1365-294X.2010.04638.x. [DOI] [PubMed] [Google Scholar]
- Yeh FC, Yang R, Boyle TB, Ye Z, Mao JX. POPGENE, the user-friendly shareware for population genetic analysis. Mol Biol Biotechnol Centre. 1997. University of Alberta, Alberta.
- Goudet J. FSTAT (version 1.2): a computer program to calculate F-statistics. J Hered. 1995;86:485–486. [Google Scholar]
- Rousset F. genepop’007: a complete re‒implementation of the genepop software for Windows and Linux. Mol Ecol Resour. 2008;8:103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- Wright S. Evolution in Mendelian populations. Genetics. 1931;16:97. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MP. Tools for population genetic analyses (TFPGA) 1.3: A Windows program for the analysis of allozyme and molecular population genetic data. Comput Softw. 1997;4:157. [Google Scholar]
- Kovach W. MVSP-A multivariate statistical Package for Windows, ver. 3.1. Kovach Comput Serv. 1999. Pentraeth, Wales.
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Earl DA. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservat Genet Resour. 2012;4:359–361. doi: 10.1007/s12686-011-9548-7. [DOI] [Google Scholar]
- Manni F, Guerard E, Heyer E. Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier's algorithm. Hum Biol. 2004;76:173–190. doi: 10.1353/hub.2004.0034. [DOI] [PubMed] [Google Scholar]
- Waples RS, Do C. LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour. 2008;8:753–756. doi: 10.1111/j.1755-0998.2007.02061.x. [DOI] [PubMed] [Google Scholar]
- Piry S, Luikart G, Cornuet J. BOTTLENECK: a computer program for detecting recent reductions in the effective size using allele frequency data. J Hered. 1999;90:502–503. doi: 10.1093/jhered/90.4.502. [DOI] [Google Scholar]
- Cornuet JM, Luikart G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics. 1996;144:2001–2014. doi: 10.1093/genetics/144.4.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson-Natesan EG. Comparison of methods for detecting bottlenecks from microsatellite loci. Conservat Genet. 2005;6:551–562. [Google Scholar]
- Garza J, Williamson E. Detection of reduction in population size using data from microsatellite loci. Mol Ecol. 2001;10:305–318. doi: 10.1046/j.1365-294x.2001.01190.x. [DOI] [PubMed] [Google Scholar]
- Gitzendanner MA, Soltis PS. Patterns of genetic variation in rare and widespread plant congeners. Am J Bot. 2000;87:783–792. doi: 10.2307/2656886. [DOI] [PubMed] [Google Scholar]
- Soltis PS, Soltis DE, Tucker TL, Lang FA. Allozyme variability is absent in the narrow endemic Bensoniella oregona (Saxifragaceae) Conserv Biol. 1992;6:131–134. doi: 10.1046/j.1523-1739.1992.610131.x. [DOI] [Google Scholar]
- Chen K, ABBOTT RJ, MILNE RI, TIAN XM, Liu J. Phylogeography of Pinus tabulaeformis Carr. (Pinaceae), a dominant species of coniferous forest in northern China. Mol Ecol. 2008;17:4276–4288. doi: 10.1111/j.1365-294X.2008.03911.x. [DOI] [PubMed] [Google Scholar]
- Cun YZ, Wang XQ. Plant recolonization in the Himalaya from the southeastern Qinghai-Tibetan Plateau: Geographical isolation contributed to high population differentiation. Mol Phylogenet Evol. 2010;56:972–982. doi: 10.1016/j.ympev.2010.05.007. [DOI] [PubMed] [Google Scholar]
- González-Martínez SC, Dubreuil M, Riba M, Vendramin G, Sebastiani F, Mayol M. Spatial genetic structure of Taxus baccata L. in the western Mediterranean Basin: Past and present limits to gene movement over a broad geographic scale. Mol Phylogenet Evol. 2010;55:805–815. doi: 10.1016/j.ympev.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Petit RJ, Duminil J, Fineschi S, Hampe A, Salvini D, Vendramin GG. Invited review: comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol Ecol. 2005;14:689–701. doi: 10.1111/j.1365-294X.2004.02410.x. [DOI] [PubMed] [Google Scholar]
- Nybom H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol. 2004;13:1143–1155. doi: 10.1111/j.1365-294X.2004.02141.x. [DOI] [PubMed] [Google Scholar]
- Wright S. Evolution and the Genetics of Populations: A Treatise in Four Volumes. Vol. 4, Variability Within and Among Natural Populations. Chicago: University of Chicago Press; 1978. [Google Scholar]
- Petit RJ, Aguinagalde I, De Beaulieu J-L, Bittkau C, Brewer S, Cheddadi R, Ennos R, Fineschi S, Grivet D, Lascoux M. Glacial refugia: hotspots but not melting pots of genetic diversity. Science. 2003;300:1563–1565. doi: 10.1126/science.1083264. [DOI] [PubMed] [Google Scholar]
- Heuertz M, Carnevale S, Fineschi S, Sebastiani F, Hausman J, Paule L, Vendramin G. Chloroplast DNA phylogeography of European ashes, Fraxinus sp. (Oleaceae): roles of hybridization and life history traits. Mol Ecol. 2006;15:2131–2140. doi: 10.1111/j.1365-294X.2006.02897.x. [DOI] [PubMed] [Google Scholar]
- Duminil J, Fineschi S, Hampe A, Jordano P, Salvini D, Vendramin GG, Petit RJ. Can population genetic structure be predicted from life‒history traits? Am Nat. 2007;169:662–672. doi: 10.1086/513490. [DOI] [PubMed] [Google Scholar]
- Yang SL, Meerow AW. The Cycas pectinata (Cycadaceae) complex: genetic structure and gene flow. Int J Plant Sci. 1996;157:468–483. doi: 10.1086/297364. [DOI] [Google Scholar]
- Slatkin M. Gene flow in natural populations. Annu Rev Ecol Systemat. 1985;16:393–430. doi: 10.1146/annurev.ecolsys.16.1.393. [DOI] [Google Scholar]
- Chen JR. In: Proceedings of the Fifth Chinese Academic Conference on Cycads: October 2007; Shenzhen. Li N, Tang MY, editor. 2007. Insect pollination of Cycas simplicipinna [abstract] p. 34. [Google Scholar]
- Walters TW, Decker-Walters DS. Patterns of allozyme diversity in the West Indies cycad Zamia pumila (Zamiaceae) Am J Bot. 1991;78:436–445. doi: 10.2307/2444966. [DOI] [Google Scholar]
- Neigeli JE, Avise JC. Phylogenetic relationships of mitochondrial DNA under various demographic models of speciation. New York: Academic Press; 1986. [Google Scholar]
- Liu J, Möller M, Provan J, Gao LM, Poudel RC, Li DZ. Geological and ecological factors drive cryptic speciation of yews in a biodiversity hotspot. New Phytol. 2013;199:1093–1108. doi: 10.1111/nph.12336. [DOI] [PubMed] [Google Scholar]
- King MG, Horning ME, Roalson EH. Range persistence during the last glacial maximum: Carex macrocephala was not restricted to glacial refugia. Mol Ecol. 2009;18:4256–4269. doi: 10.1111/j.1365-294X.2009.04280.x. [DOI] [PubMed] [Google Scholar]
- Marko PB, Hoffman JM, Emme SA, McGovern TM, Keever CC, Nicole Cox L. The ‘Expansion–Contraction’model of Pleistocene biogeography: rocky shores suffer a sea change? Mol Ecol. 2010;19:146–169. doi: 10.1111/j.1365-294X.2009.04417.x. [DOI] [PubMed] [Google Scholar]
- Bisconti R, Canestrelli D, Colangelo P, Nascetti G. Multiple lines of evidence for demographic and range expansion of a temperate species (Hyla sarda) during the last glaciation. Mol Ecol. 2011;20:5313–5327. doi: 10.1111/j.1365-294X.2011.05363.x. [DOI] [PubMed] [Google Scholar]
- Cunha RL, Lopes EP, Reis DM, Castilho R. Genetic structure of Brachidontes puniceus populations in Cape Verde archipelago shows signature of expansion during the last glacial maximum. J Molluscan Stud. 2011;77:175–181. doi: 10.1093/mollus/eyr001. [DOI] [Google Scholar]
- Pinheiro F, De Barros F, Palma‒Silva C, Fay MF, Lexer C, Cozzolino S. Phylogeography and genetic differentiation along the distributional range of the orchid Epidendrum fulgens: a Neotropical coastal species not restricted to glacial refugia. J Biogeogr. 2011;38:1923–1935. doi: 10.1111/j.1365-2699.2011.02539.x. [DOI] [Google Scholar]
- Batalha-Filho H, Cabanne GS, Miyaki CY. Phylogeography of an Atlantic forest passerine reveals demographic stability through the last glacial maximum. Mol Phylogenet Evol. 2012;65:892–902. doi: 10.1016/j.ympev.2012.08.010. [DOI] [PubMed] [Google Scholar]
- CHENG YP, HWANG SY, LIN TP. Potential refugia in Taiwan revealed by the phylogeographical study of Castanopsis carlesii Hayata (Fagaceae) Mol Ecol. 2005;14:2075–2085. doi: 10.1111/j.1365-294X.2005.02567.x. [DOI] [PubMed] [Google Scholar]
- Harrison S, Yu G, Takahara H, Prentice I. Palaeovegetation (Communications arising): diversity of temperate plants in east Asia. Nature. 2001;413:129–130. doi: 10.1038/35093166. [DOI] [PubMed] [Google Scholar]
- Montalvo AM, Williams SL, Rice KJ, Buchmann SL, Cory C, Handel SN, Nabhan GP, Primack R, Robichaux RH. Restoration biology: a population biology perspective. Restor Ecol. 1997;5:277–290. doi: 10.1046/j.1526-100X.1997.00542.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Information of sixteen SSR makers used to study the population genetics of C. simplicipinna.
Variable sites from the two combined cpDNA in C. simplicipinna.
Variable sites from the nrDNA in C. simplicipinna.
P-value of Hardy-Weinberg equilibrium test for six populations of C. simplicipinna.
An unweighted pair-group method with arithmetic averages (UPGMA) phenogram of six populations of C. simplicipinna based on SSR markers. Numbers on branches indicated bootstrap values from 5000 replicates.