

Abstract
A substrate-induced stereocontrol strategy was used to gain access to the tetracyclic core of (-)-lemonomycin. An advanced intermediate was prepared from a known substituted tyrosinol through a 16-step sequence, which involved a Pictet-Spengler reaction, a [3+2] dipolar cycloaddition and an enamide hydrogenation.
Keywords: (-)-Lemonomycin, Tetrahydroisoquinoline antitumor antibiotics, Pictet-Spengler reaction, [3+2] dipolar cycloaddition
1. Introduction
Lemonomycin (1) is a member of the tetrahydroisoquinoline (THIQ) family of antitumor antibiotics.1 It was isolated from the fermentation broth of Streptomyces candidus (LL-AP191) in 1964,2 and its structure was reported by He and coworkers in 2000.3 This compound showed significant in vitro antimicrobial activities against both gram-negative and gram-positive bacteria, including antibiotic-resistant strains, as well as against the human colon tumor cell line HCT116.2,3 Structurally, the compound contains the tetracyclic core found in quinocarcin4 and tetrazomine,5 which includes a 3,8-diazabicyclo ring system and a rare bis-desoxy aminosugar portion, which has only been found in a few natural products.6-10 The structural complexity and biological activities of this substance have made lemonomycin an attractive target for the synthetic community. To date, there are two total syntheses by Stoltz11 and Fukuyama12 and synthetic studies by Magnus,13,14 Zhu,15,16,17 Mulzer18 and our laboratory.19
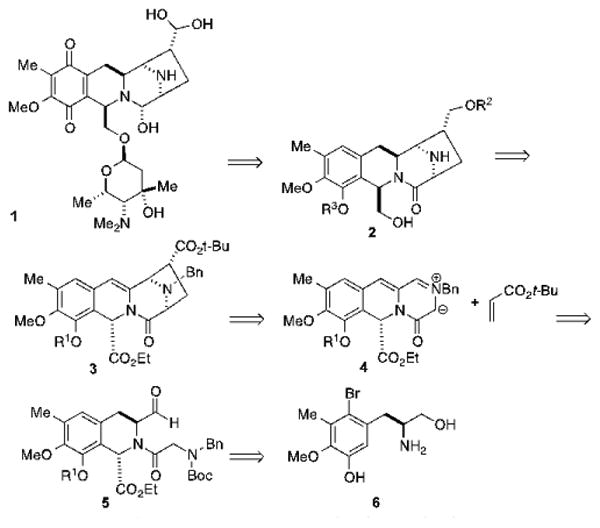
As shown in Scheme 1, we envisioned that the final steps in the synthesis of lemonomycin (1) would involve a late-stage glycosylation reaction, and the formation of the quinone, hemiaminal and aldehyde hydrate functional groups. Compound 2 could be accessed through the epimerization of the southern benzylic position and the reduction of the enamide double bond found in tetracycle 3. This key intermediate could be prepared from aldehyde 5 via azomethine ylide 4, using a [3+2] dipolar cycloaddition approach previously developed by our group.19 This key reaction was also used for the construction of the [3,8[-diazabicyclo ring system in our total syntheses of (-)-tetrazomine20 and (±)-quinocarcinamide.21 The tetra-hydroisoquinoline system of 5 could be formed through a Pictet-Spengler reaction involving a derivative of compound 6, which is a known compound.22
Scheme 1. Retrosynthetic analysis.
2. Results and discussion
Our synthetic sequence starts with substituted tyrosinol 6, which can be prepared from commercially available l-tyrosine methyl ester according to the procedure described by Liao.22 We initially attempted to perform the direct conversion of 6 into bissilyl ether 8 using two equivalents of TBS-Cl, but the yields were inconsistent and low (<30%).23,24 By increasing the relative amount of TBS-Cl to six equivalents, compound 6 was converted into the tris-silylated compound 7. Unexpectedly, the hydrolysis of the silylamine function required a prolonged vigorous stirring with aqueous NH4Cl at room temperature (∼ 2h) to form the bis-silyl ether 8 in 90% yield. The phenolic silyl ether was selectively cleaved with one equivalent of TBAF at 0 °C,25 to afford compound 9 in 98% yield (Scheme 2).
Scheme 2. Tetrahydroisoquinoline ring formation.
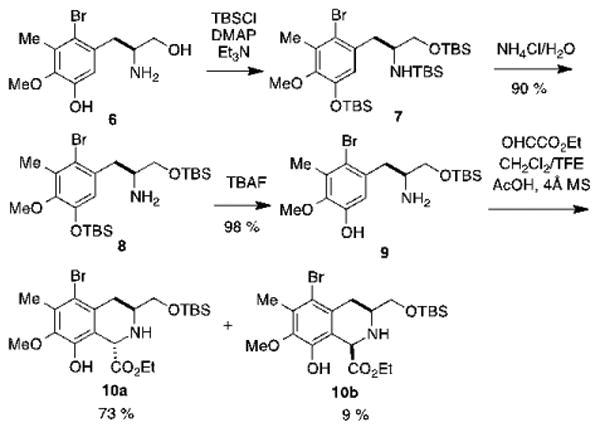
The next step entailed the formation of the trans-tetrahydroisoquinoline ring via a Pictet Spengler reaction between 9 and ethyl glyoxalate. Previously, our group reported a similar transformation, which was performed by stirring a solution of the starting materials in acetonitrile for 3.5 days at 50°C, which afforded the trans- product stereospecifically.26 A similar report by Zhu and coworkers involved the use of LiCl, hexafluorisopropanol and molecular sieves, and stirring the suspension in toluene at room temperature for 48 h. Since none of these mild conditions led to the formation of the desired tetrahydroisoquinoline ring system, we decided to adapt the reaction conditions that were originally described by Zhu27,28 to our substrate. The amount of acetic acid was reduced from 2.5 equivalents to 0.2 equivalents to prevent cleavage of the O-TBS ether due to the prolonged exposure to the acid. In the present system, treatment of a solution of compound 9 and ethyl glyoxylate with CF3CH2OH, AcOH (0.2 eq.) and 4 Å MS afforded an 8:1 mixture of 10a and 10b in 82% yield. These two diastereomers were separated via flash chromatography and 10a was subjected to selective acetylation,29 followed by hydrogenolysis of the C-Br bond22 to afford compound 12 (Scheme 3).
Scheme 3. Preparation of aldehyde 15.
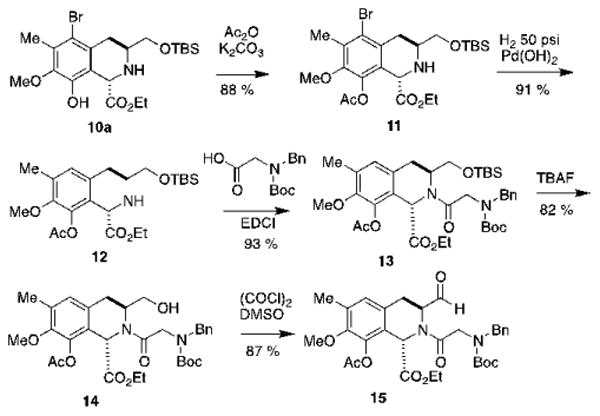
Following the conditions described in our previous report,19 we converted THIQ 12 into the [3+2] dipolar cycloaddition adducts 20a and 20b. Thus, THIQ 12 and N-Boc-N-Bn-Gly were coupled using EDCI, and the resulting amide was treated with TBAF to cleave the O-TBS ether, followed by a Swern oxidation30 to afford aldehyde 15 (Scheme 3).
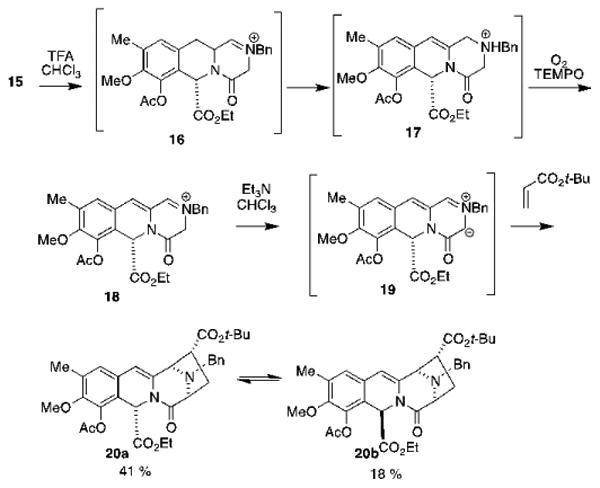
As illustrated in Scheme 4, aldehyde 15 was dissolved in CHCl3 and treated under aerobic conditions with TFA31,32,33 (50 eq.) and TEMPO (0.1 eq.), to generate iminuim ion 16, which tautomerizes to form ammonium ion 17. This intermediate is autoxidized in situ to afford conjugated iminium ion 18, which was concentrated to dryness and taken up in CHCl3. Addition of triethylamine induces the formation of azomethine ylide 19, which is trapped in situ by tert-butyl acrylate to give a 2.4:1 mixture of tetracycles 20a and 20b in a combined 59% yield.34
Scheme 4. Formation of cycloadducts 20a and 20b.
The deacetylation of the 20a/20b mixture under standard methanolysis conditions provided a 5:1 mixture of 21a and 21b in 70% yield (Scheme 5). We suggest that 21b decomposes under the reaction conditions at a higher rate than 21a, which provides an explanation for both the moderate yield and the change in the diastereomeric ratio. The chemoselective reduction of the ethyl esters with one equivalent of LiAlH4 at -10 °C, afforded a 3:1 mixture of aldehydes 22a and 22b in 55% yield.35 We submit that the partial epimerization seen in this step is promoted by the slightly basic workup conditions. Treatment with BnBr and Na2CO3 formed the phenolic benzyl ethers and induced additional epimerization of the aldehyde's α carbon, to provide a 2.2:1 mixture of 23a and 23b36 which was then reacted with DBN in THF to invert the epimeric ratio.20,21
Scheme 5.
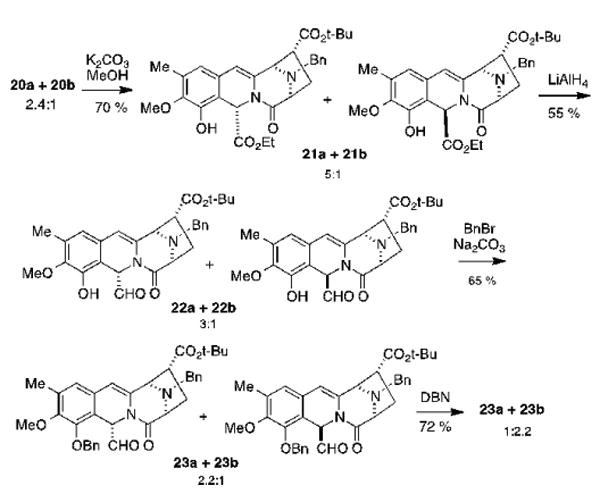
Synthesis of aldehydes 23a and 23b.
The 1:2.2 mixture of aldehydes 23a and 23b was then treated with sodium borohydride to afford a mixture of alcohols 24a and 24b, which were separated via flash chromatography to afford 24b in 65% yield (Scheme 6). The sequence used to transform the 20a/20b mixture into 24b not only provided the desired configuration in the benzylic position but also furnished an unhindered substrate for the N-debenzylation of the piperazinone amine. Thus, hydrogenolysis of 24b in glacial acetic acid (10% Pd/C, 1 atm.) effected the bis-debenzylation to afford 25 in 92% yield. Similarly, the removal of the N-benzyl group also provided an unhindered substrate for the hydrogenation of the enamide double bond from the Re face of C-3 (lemonomycin numbering). Gratifyingly, the hydrogenation of 25 with Raney® nickel at 100 psi20 provided compound 26 in 73% yield.
Scheme 6.
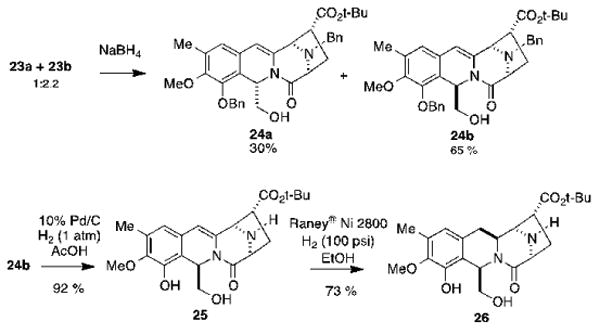
Synthesis of compound 26.
3. Conclusion
In summary, we have accomplished the construction of the tetracyclic core of (-)-lemonomycin. Compound 26 was prepared from known bromotyrosinol 6 in sixteen steps. Efforts to gain access to (-)-lemonomycin through this advanced intermediate are currently under investigation.
4. Experimental section
4.1. General Methods
Unless otherwise noted, all materials were obtained from commercial sources and used without purification. All reactions requiring anhydrous conditions were performed under a positive pressure of argon using flame-dried glassware. Organic solvents were degassed with argon and dried through a solvent purification system (Pure Process Technology). Flash chromatography was performed on silica gel grade 60 (230 × 400 mesh) from Sorbent Technologies. Thin layer chromatography was performed on glass plates coated with silica gel grade 60, from Merck. 1H NMR and 13C NMR spectra were recorded on Varian 300 or 400 MHz spectrometers as indicated. Proton spectra in CDCl3 were referenced to residual CHCl3 at 7.26 ppm. Carbon spectra in CDCl3 were referenced to 77.16 ppm. Proton spectra in DMSO-d6 were referenced to residual CD3SOCD2H at 2.50 ppm. Infrared spectra were recorded on a Bruker Tensor FT-IR spectrometer. High-resolution mass spectra were obtained using a TOF spectrometer using simultaneous electrospray (ESI) and atmospheric pressure chemical ionization (APCI). Optical rotations were recorded on a Rudolph Research Autopol polarimeter, at a wavelength of 589 nm.
4.2. (S)-1-(2-bromo-5-((tert-butyldimethylsilyl)oxy)-4-methoxy-3-methylphenyl)-3-((tert-butyldimethylsilyl)oxy)propan-2-amine (8)
To a stirred solution of compound 6 (1.55 g, 5.36 mmol, 1 eq.) (6) in CH2Cl2 (90 mL, 0.06 M), were added DMAP (327 mg, 2.68 mmol, 0.5 eq.), Et3N (4.48 mL, 32.2 mmol, 6.00 eq.) and TBS-Cl (4.86 g, 32.2 mmol, 6 eq.). The reaction was stirred under Ar for 3 h at RT, and then aq. sat. aq. NH4Cl (50 mL) was added and the mixture was stirred for 2h. The phases were separated, the aqueous layer was extracted with CH2Cl2 (2×50 mL) and the combined organic layers were rinsed with brine (50 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, hexane/EtOAc 5:1) to give the title compound 8 (2.50 g, 90%) as a colorless oil. 1H-NMR (400 MHz; CDCl3): δ 6.65 (s, 1H), 3.72 (s, 3H), 3.61 (1/2 ABX, J = 9.7, 4.1 Hz, 1H), 3.47 (1/2 ABX, J = 9.7, 6.5 Hz, 1H), 3.20-3.14 (m, 1H), 2.87 (1/2 ABX, J = 13.4, 5.4 Hz, 1H), 2.57 (1/2 ABX, J = 13.4, 8.0 Hz, 1H), 2.35 (s, 3H), 1.00 (s, 9H), 0.91 (s, 9H), 0.17 (s, 6H), 0.07 (s, 3H), 0.06 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 148.8, 147.6, 134.6, 133.1, 121.3, 119.4, 67.6, 60.2, 52.9, 41.3, 26.1, 25.8, 18.4, 18.4, 17.2, -4.4, -5.2; Rf (SiO2, 2:1 hexanes/EtOAc) 0.35; [α]D25 = + 0.9 ° (c=0.35, CHCl3); IR (film, CH2Cl2), νmax 2996, 2930, 2858, 2471, 839 cm-1; HRMS (MH+), found 520.2103. C23H45BrNO3Si2 requires 520.2101.
4.3. (S)-5-(2-amino-3-((tert-butyldimethylsilyl)oxy)propyl)-4-bromo-2-methoxy-3-methylphenol (9)
To a stirred solution of compound 8 (1.59 g, 3.05 mmol, 1 eq.) in THF (100 mL, 0.03 M), under Ar, at 0 °C, was added a 1.0 solution M of TBAF in THF (3.05 mL, 3.05 mmol,, 1 eq.). The reaction was stirred for 25 minutes and quenched with sat. aq. NH4Cl (50 mL). The phases were allowed to warm to RT, the aqueous phase was extracted with EtOAc (2×50 mL) and the combined organic layers were rinsed with brine (50 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, CHCl3/MeOH 10:1) to give the title compound 9 (1.23 g, 98%) as a colorless oil. 1H-NMR (400 MHz; CDCl3): δ 6.77 (s, 1H), 3.73 (s, 3H), 3.71-3.66 (m, 1H), 3.56-3.50 (m, 1H), 3.27-3.24 (m, 1H), 2.92 (1/2 ABX, J = 13.5, 4.6 Hz, 1H), 2.62 (1/2 ABX, J = 13.5, 9.0 Hz, 1H), 2.34 (s, 3H), 0.92 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 148.4, 145.0, 134.6, 132.2, 117.2, 116.1, 67.1, 60.8, 52.9, 52.7, 40.5, 26.1, 18.4, 17.2, -5.2, -5.2. Rf (SiO2, CH2Cl2/MeOH 10:1) 0.4; [α]D25 = + 8.3 ° (c=0.41, CHCl3); IR (film, CH2Cl2), νmax 3263 (br), 2954, 2928, 2856, 1578, 1471, 1092 cm-1; HRMS (MH+), found 406.1233. C17H31BrNO3Si requires 406.1236
4.4. 1S,3S)-ethyl 5-bromo-3-(((tert-butyldimethylsilyl)oxy)-methyl)-8-hydroxy-7-methoxy-6-methyl-1,2,3,4-tetrahydro-isoquinoline-1-carboxylate (10a) and (1R,3S)-ethyl 5-bromo-3-(((tert-butyldimethylsilyl)oxy)methyl)-8-hydroxy-7-methoxy-6-methyl-1,2,3,4-tetrahydroisoquinoline-1-carboxylate (10b)
To a stirred solution of compound 9 (5.43 g, 13.4 mmol, 1.0 eq.) in CH2Cl2 (134 mL, 0.10 M), under Ar, were added, 4Å molecular sieves (2.72 g), CF3CH2OH (13.4 mL), AcOH (153 μL, 2.68 mmol, 0.20 eq.) and ethyl glyoxalate (50% solution in PhCH3, 2.93 mL, 14.8 mmol, 1.1 eq.). The reaction was stirred overnight, diluted with CH2Cl2 (50 mL), filtered through Celite® and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, hexanes/EtOAc 5:1) to give compound 10a (4.78g, 73%) as a white solid and compound 10b (610 mg, 9%) as a white solid. Compound 10 a:1H-NMR (400 MHz; CDCl3): δ 6.25 (br s, 1H), 4.89 (s, 1H), 4.26-4.18 (m, 2H), 3.82 (1/2 ABX, J = 9.8, 3.5 Hz, 1H), 3.76 (s, 3H), 3.54 (1/2 ABX, J = 9.8, 8.5 Hz, 1H), 3.13-3.07 (m, 1H), 2.71 (1/2 ABX, J = 16.9, 4.1 Hz, 1H), 2.35 (s, 3H), 2.27 (1/2 ABX, J = 16.9, 11.3 Hz, 1H), 1.29 (t, J = 7.1 Hz, 3H), 0.93 (s, 9H), 0.10 (s, 3H), 0.09 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 172.9, 145.6, 144.3, 130.8, 130.7, 120.0, 118.4, 66.9, 61.7, 61.2, 55.6, 51.6, 32.7, 26.0, 18.4, 17.0, 16.9, 14.4, 14.4, -5.1, -5.2, -5.2, -5.3; m.p. = 47 °C; Rf (SiO2, hexanes/EtOAc 4:1) 0.40; [α]D25 = -24.3 ° (c = 0.885, CHCl3); IR (film, CH2Cl2), νmax 3284 (br), 2955, 2931, 2857, 1739, 1462, 1178 cm-1; HRMS (MH+), found 490.1446. C21H35BrNO5Si requires 490.1447. Compound 10b: 1H-NMR (400 MHz; CDCl3): δ 5.86 (br s, 1H), 4.78 (s, 1H), 4.30-4.15 (m, 2H), 3.80 (1/2 ABX, J = 9.9, 4.1 Hz, 1H), 3.74 (s, 3H), 3.68 (1/2 ABX, J = 9.9, 6.6 Hz, 1H), 2.95 -2.89 (m, 1H), 2.77 (1/2 ABX, J = 16.6, 3.1 Hz, 1H), 2.44 (1/2 ABX, J = 16.6, 8.5 Hz, 2H), 2.36 (s, 3H), 1.27 (t, J = 7.1 Hz, 3H), 0.92 (s, 9H), 0.09 (s, 6H).; 13C-NMR (101 MHz, CDCl3): δ 172.8, 145.1, 143.9, 132.0, 130.4, 120.3, 118.4, 66.7, 61.5, 61.4, 58.4, 54.5, 33.0, 26.1, 26.0, 18.5, 17.0, 14.2, -5.1, -5.2; m.p. = 95 °C; Rf (SiO2, hexanes/EtOAc 4:1) 0.37; [α]D25 = -36.7 ° (c = 0.600, CHCl3); IR (film, CH2Cl2), νmax 3314 (br), 2955, 2931, 2858, 1738, 1463, 1257 cm-1; HRMS (MH+), found 490.1456. C21H35BrNO5Si requires 490.1447.
4.5. (1S,3S)-ethyl 8-acetoxy-5-bromo-3-(((tert-butyldimethyl-silyl)oxy)methyl)-7-methoxy-6-methyl-1,2,3,4-tetrahydro-isoquinoline-1-carboxylate (11)
To a stirred solution of compound 10a (840 mg, 1.72 mmol, 1.0 eq.) in acetone (34 mL, 0.05 M), under Ar, were added K2CO3 (1.20 g, 8.64 mmol, 5.0 eq.) and acetic anhydride (162 μL, 1.72 mmol, 1.0 eq.). The suspension was stirred overnight, the solvent was evaporated and the residue was partitioned between water (25 mL) and EtOAc (25 mL). The aqueous phase was extracted with EtOAc (2×25 mL) and the combined organic layers were rinsed with brine (50 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, hexanes/EtOAc 5:1) to give the title compound 11 (800 mg g, 88%) as a colorless oil. 1H-NMR (400 MHz; CDCl3): δ 4.66 (s, 1H), 4.18 (q, J = 7.1 Hz, 2H), 3.81 (1/2 ABX, J = 9.8, 3.5 Hz, 1H), 3.70 (s, 3H), 3.55 (1/2 ABX, J = 9.8, 7.6 Hz, 1H), 3.22-3.16 (m, 1H), 2.74 (1/2 ABX, J = 16.9, 4.1 Hz, 2H), 2.38 (s, 3H), 2.37-2.33 (m, 1H), 2.29 (s, 3H), 1.27 (t, J = 7.1 Hz, 3H), 0.921 (s, 9H), 0.10 (s, 3H), 0.07 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 171.8, 167.9, 148.6, 141.0, 132.5, 131.5, 125.9, 125.8, 66.7, 61.5, 61.1, 55.8, 51.0, 32.6, 26.0, 20.6, 18.4, 17.1, 14.4, -5.2, -5.3. Rf (SiO2, hexanes/EtOAc 4:1) 0.45; [α]D25 = -21.1 ° (c = 1.10, CHCl3); IR (film, CH2Cl2), νmax 2956, 2932, 2856, 1780, 1737, 1462, 1192 cm-1; HRMS (MH+), found 532.1561. C23H37BrNO6Si requires 530.1574
4.6. (1S,3S)-ethyl 8-acetoxy-3-(((tert-butyldimethylsilyl)oxy)-methyl)-7-methoxy-6-methyl-1,2,3,4-tetrahydroisoquinoline-1-carboxylate (12)
A solution of compound 11 (2.90 mg, 5.46 mmol) in MeOH (110 mL, 0.05 M), and Pearlman's catalyst (20% Pd(OH)2/C, 580 mg) were placed in a Fisher-Porter bottle, under Ar. The mixture was sparged with Ar for 5 minutes and the vessel was filled with hydrogen gas at 50 psi. The reaction was vigorously stirred overnight and then filtered through Celite® and the vessel was rinsed with MeOH (50 mL) and EtOAc (50 mL). The solution was concentrated under vacuum to dryness and partitioned between sat. aq. NaHCO3 (75 mL) and EtOAc (75 mL). The aqueous phase was extracted with EtOAc (2×75 mL) and the combined organic layers were rinsed with brine (50 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, hexanes/EtOAc 5:1, 4:1 and 3:1) to give the title compound 12 (2.25 mg, 91%) as a colorless oil. 1H-NMR (400 MHz; CDCl3): δ 6.85 (s, 1H), 4.65 (s, 1H), 4.17 (q, J = 7.1 Hz, 2H), 3.73 (1/2 ABX, J = 9.8, 3.7 Hz, 1H), 3.70 (s, 3H), 3.52 (1/2 ABX, J = 9.8, 7.2 Hz, 1H), 3.28-3.22 (m, 1H), 2.59 (1/2 ABX, J = 16.1, 4.2 Hz, 1H), 2.50 (1/2 ABX, J = 16.1, 10.7 Hz, 1H), 2.28 (s, 3H), 2.27 (s, 3H), 1.26 (t, J = 7.1 Hz, 3H), 0.90 (s, 9H), 0.07 (s, 3H), 0.06 (s, 2H). 13C-NMR (101 MHz, CDCl3): δ 172.2, 168.3, 148.2, 141.6, 131.5, 131.1, 129.2, 124.0, 66.7, 61.3, 60.6, 55.6, 50.7, 30.2, 26.0, 20.6, 18.4, 16.0, 14.4, -5.2, -5.3. Rf (SiO2, hexanes/EtOAc 4:1) 0.42; [α]D25 = -17 ° (c = 0.42, CHCl3); IR (film, CH2Cl2), νmax 2954, 2929, 2857, 1775, 1737, 1197 cm-1; HRMS (MH+), found 452.244. C23H38NO6Si requires 452.2468.
4.7. (1S,3S)-ethyl 8-acetoxy-2-(2-(benzyl(tert-butoxycarbonyl)-amino)acetyl)-3-(((tert-butyldimethylsilyl)oxy)methyl)-7-methoxy-6-methyl-1,2,3,4-tetrahydroisoquinoline-1-carboxylate (13)
A solution of compound 12 (2.20 g, 4.87 mmol, 1.0 eq.), N-Bn-N-Boc-glycine (2.58 g, 9.74 mmol, 2.0 eq.) and EDCI (1.40 g, 7.31 mmol, 1.5 eq.) in CH2Cl2 (2.5 mL, 2 M), under Ar, was stirred for 2.5 days. The reaction was diluted with EtOAc (200 mL), and the solution was extracted with water (100 mL), sat. aq. NaHCO3 (2 × 100 mL) and brine (100 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, hexanes/EtOAc 4:1, 3:1 and 2:1) to give the title compound 13 (3.15 g, 93%) as a colorless oil. 1H-NMR (300 MHz; DMSO-d6, 393 K, mixture of rotamers): δ 7.36-7.24 (m, 5H), 6.94 (s, 1H), 6.88 (s, 1H, minor rotamer), 5.48 (s, 1H), 4.49 (1/2 AB, J = 15.6 Hz, 1H), 4.39 (1/2 AB, J = 15.6.0 Hz, 1H), 4.33-4.27 (m, 2H), 4.13-3.87 (m, 3H), 3.69 (s, 3H), 3.66 (s, 1H, minor rotamer), 3.34-3.10 (br m, 2H), 3.07-2.91 (br m, 2H), 2.33 (s, 3H), 2.24 (s, 3H), 2.23 (s, 3H, minor rotamer), 2.23 (s, 1H, minor rotamer), 1.41 (s, 9H), 1.21 (t, J = 7.0 Hz, 3H, minor rotamer), 1.12 (t, J = 7.1 Hz, 3H), 0.92 (d, J = 0.6 Hz, 2H), 0.79 (s, 9H), 0.08 (s, 3H, minor rotamer), 0.04 (s, 3H, minor rotamer), 0.03 (m, 3H, minor rotamer), -0.11 (s, 3H), -0.14 (s, 3H). 13C-NMR (101 MHz, CDCl3, mixture of rotamers): δ 170.1, 169.7, 168.1, 168.0, 156.1, 149.6, 129.2, 129.1, 128.7, 128.4, 127.8, 127.5, 127.5, 127.4, 121.6, 80.5, 80.4, 71.6, 61.9, 61.3, 60.7, 53.6, 53.6, 53.5, 53.0, 53.0, 52.9, 52.9, 50.9, 47.7, 29.5, 28.5, 28.4, 26.0, 26.0, 25.9, 20.9, 18.3, 16.1, 16.0, 14.0, 13.9, -5.3, -5.4, -5.4, -5.7. Rf (SiO2, hexanes/EtOAc 3:1) 0.30; [α]D25 = 26.8 ° (c = 0.995, CHCl3); IR (film, CH2Cl2), νmax 2956, 2931, 2857, 1781, 1743, 1703, 1668, 1199 cm-1; HRMS (MH+), found 699.3666. C37H55N2O9Si requires 699.3677.
4.8. (1S,3S)-ethyl 2-(2-(benzyl(tert-butoxycarbonyl)amino)acetyl)-8-hydroxy-3-(hydroxymethyl)-7-methoxy-6-methyl-1,2,3,4-tetrahydroisoquinoline-1-carboxylate (14)
To a solution of compound 13 (765 mg, 1.09 mmol, 1.0 eq.) in THF (10 mL, 0.11 M), under Ar, were added MeOH (625 μL) and TBAF (1.0 M solution in THF, 2.18 mL, 2.0 eq.). The reaction was stirred overnight and quenched with sat. aq. NH4Cl (50 mL) and then diluted with EtOAc (100 mL). The phases were separated, the aqueous phase was extracted with EtOAc (2 × 25 mL) and the combined organic layers were rinsed with brine (50 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude material was dissolved in the minimal amount of CH2Cl2 and purified by flash chromatography (silica gel, hexanes/EtOAc 2:1, then 1:1) to give the title compound 14 (525 mg, 82%) as a white amorphous solid. 1H-NMR (300 MHz; DMSO-d6, 393 K): δ 7.36-7.26 (m, 5H), 6.96 (s, 1H), 5.50 (s, 1H), 4.49-4.40 (br m, 2H), 4.27-4.17 (br m, 2H), 4.07-3.89 (m, 3H), 3.70 (s, 3H), 3.19-3.03 (br m, 2H), 2.94-2.81 (m, 2H, overlapped with H2O signal), 2.34 (s, 3H), 2.25 (s, 3H), 1.41 (s, 9H), 1.13 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3, mixture of rotamers): δ 170.2, 170.1, 168.0, 168.0, 156.4, 154.8, 152.8, 149.7, 145.0, 141.7, 141.5, 138.1, 138.1, 138.0, 132.9, 132.8, 130.2, 130.0, 128.7, 128.5, 128.4, 128.3, 128.1, 127.8, 127.8, 127.7, 127.5, 127.5, 127.1, 124.2, 121.2, 80.9, 80.4, 65.1, 63.8, 62.0, 60.6, 53.7, 53.5, 52.7, 52.0, 51.0, 47.9, 30.6, 30.0, 29.5, 28.5, 28.4, 20.9, 16.2, 13.8; m.p. 80 °C; Rf (SiO2, hexanes/EtOAc 1:1) 0.35; [α]D25 = 78 ° (c = 0.44, CHCl3); IR (film, CH2Cl2), νmax 3455 (br), 2977, 2935, 1780, 1742, 1698, 1663, 1200 cm-1; HRMS (MH+), found 585.2816. C31H41N2O9 requires 585.2812.
4.9. (1S,3S)-ethyl 2-(2-(benzyl(tert-butoxycarbonyl)amino)-acetyl)-3-formyl-8-hydroxy-7-methoxy-6-methyl-1,2,3,4-tetrahydroisoquinoline-1-carboxylate (15)
A solution of oxalyl chloride (825 μL, 9.75 mmol, 3.0 eq.) in CH2Cl2 (22.5 mL), under Ar, was cooled to -78 C, and DMSO (921 μL, 13.0 mmol, 4.0 eq.) was added dropwise. The resulting mixture was stirred an additional 30 min at -78 °C. A solution of compound 14 (1.90 mg, 3.25 mmol, 1.0 eq.) in CH2Cl2 (10 mL) at RT was then added slowly by cannula, and the mixture continued to stir at -78 °C for 30 min. Triethylamine (4.50 mL, 32.5 mmol, 10 eq.) was then added dropwise, and the solution was stirred for 15 min at -78 °C and an additional 30 min at 0 °C. The reaction was quenched with sat. aq. NH4Cl (50 mL) and allowed to warm to RT. The layers were separated, the aqueous phase was extracted with CH2Cl2 (3×50 mL) and the combined organic layers were rinsed with brine (50 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, hexanes/EtOAc 2:1, then 1:1) to give the title compound 15 (1.65 g, 87%) as a colorless oil, which solidifies upon standing to afford a colorless amorphous solid. 1H-NMR (300 MHz; DMSO-d6, 373 K, mixture of rotamers): δ 9.54 (s, 1H, minor rotamer), 9.28 (s, 1H), 7.35-7.23 (m, 5H), 7.08 (s, 1H, minor rotamer), 7.01 (s, 1H), 6.94 (s, 1H, minor rotamer), 6.91 (s, 1H, minor rotamer), 5.70 (s, 1H), 5.10-4.88 (m, 1H), 4.52-4.28 (m, 3H), 4.26-4.14 (m, 1H), 4.13-3.98 (m, 2H), 3.97-3.86 (m, 1H), 3.69 (s, 3H, minor rotamer), 3.68 (s, 3H, minor rotamer), 3.67 (s, 3H), 3.38-3.27 (m, 1H), 2.34 (s, 3H), 2.32 (s, 3H, minor rotamer), 2.25 (s, 3H, minor rotamer), 2.24 (s, 3H, minor rotamer), 2.22 (s, 3H), 1.41 (s, 9H, minor rotamer), 1.40 (s, 9H), 1.36 (s, 9H, minor rotamer), 1.34 (s, 9H, minor rotamer), 1.12 (t, J = 7.2 Hz, 3H). 13C-NMR (101 MHz, CDCl3, mixture of rotamers): δ 201.2, 199.6, 199.2, 169.9, 167.8, 155.9, 155.8, 150.2, 149.7, 141.5, 141.0, 137.6, 137.5, 137.5, 133.6, 133.3, 128.6, 128.6, 128.5, 128.5, 128.4, 128.1, 128.0, 127.9, 122.6, 122.1, 81.0, 80.8, 62.7, 62.2, 60.9, 60.6, 60.6, 60.2, 53.7, 53.5, 53.3, 50.9, 47.8, 47.6, 47.1, 29.6, 28.5, 28.5, 28.4, 28.2, 20.9, 16.2, 13.8; m.p. 78 °C; Rf (SiO2, hexanes/EtOAc 1:1) 0.40; [α]D25 = 35 ° (c = 0.23, CHCl3); IR (film, CH2Cl2), νmax 2978, 2937, 1780, 1742, 1699, 1673, 1200 cm-1; HRMS (MH+), found 583.2654. C31H39N2O9 requires 583.2656.
4.10. (5S,8S,10R,11S)-10-tert-butyl 5-ethyl 4-acetoxy-13-benzyl-3-methoxy-2-methyl-7-oxo-5,7,8,9,10,11-hexahydro-8,11-epiminoazepino[1,2-b[isoquinoline-5,10-dicarboxylate (20a) and (5R,8S,10R,11S)-10-tert-butyl 5-ethyl 4-acetoxy-13-benzyl-3-methoxy-2-methyl-7-oxo-5,7,8,9,10,11-hexahydro-8,11-epiminoazepino[1,2-b[isoquinoline-5,10-dicarboxylate (20b)
To solution of compound 15 (1.65 g, 2.83 mmol, 1.0 eq.) in CHCl3 (28 mL, 0.1 M), under air, were added TEMPO (44 mg, 0.28 mmol, 0.10 eq.), and trifluoroacetic acid (10.8 mL, 142 mmol, 50 eq.) and the flask was loosely capped with a Teflon® stopper. The solution was stirred for 4h, the solvent was evaporated to dryness under vacuum and the residue was taken up in CHCl3. The solution was cooled to 0 °C and then tert-butyl acrylate (8.20 mL, 56.6 mmol, 20 eq.) and triethylamine (3.95 mL, 28.3 mmol, 10 eq.) were added. The reaction was allowed to warm to RT and stirred overnight. The solution was diluted with EtOAc (200 mL), rinsed with sat. aq. NH4Cl (50 mL) and brine (50 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, hexanes/EtOAc 4:1, 3:1) to afford a 2.4:1 mixture of the title compounds 20a and 20b (985 mg, 59%) as a yellow oil, which was used in the next step without further purification. 1H-NMR (400 MHz; CDCl3): δ 7.41-7.22 (m, 5H), 6.74 (s, 1H, minor diastereomer), 6.73 (s, 1H), 6.36 (s, 1H, minor diastereomer), 6.27 (s, 1H), 5.51 (s, 1H, minor diastereomer), 5.50 (s, 1H), 4.28-3.96 (m, 6H), 3.89-3.71 (m, 2H), 3.75 (s, 3H, minor diastereomer), 3.72 (s, 3H), 2.80-2.67 (m, 2H), 2.45 (dd, J = 13.0, 9.8 Hz, 1H, minor diastereomer), 2.40 (s, 3H), 2.39 (s, 3H, minor diastereomer), 2.28 (s, 3H, minor diastereomer), 2.26 (s, 3H), 2.13 (dd, J = 13.3, 9.5 Hz, 1H), 1.46 (s, 9H, minor diastereomer), 1.42 (s, 9H), 1.24 (t, J = 7.1 Hz, 3H), 1.20 (t, J = 7.2 Hz, 3H, minor diastereomer); 13C-NMR (101 MHz, CDCl3): δ 172.4, 171.7, 168.7, 167.9, 149.9, 141.6, 133.1, 129.0, 128.6, 128.4, 128.4, 127.4, 127.3, 126.6, 125.0, 124.8, 117.3, 116.7, 104.6, 103.1, 81.4, 81.3, 65.1, 64.1, 63.1, 62.6, 62.4, 62.3, 60.7, 60.6, 52.7, 51.8, 51.3, 50.7, 50.0, 48.0, 34.3, 31.9, 31.7, 28.2, 22.8, 21.0, 16.1, 14.2, 14.0; Rf (SiO2, hexanes/EtOAc 3:1) 0.5; [α]D25 = -65.0 ° (c = 0.320, CH2Cl2); IR (film, CH2Cl2), νmax 2980, 2936, 1781, 1741, 1693, 1651 cm-1; HRMS (MH+), found 591.2712. C33H39N2O8 requires 591.2706.
4.11. (5S,8S,10R,11S)-10-tert-butyl 5-ethyl 13-benzyl-4-hydroxy-3-methoxy-2-methyl-7-oxo-5,7,8,9,10,11-hexahydro-8,11-epiminoazepino[1,2-b[isoquinoline-5,10-dicarboxylate (21a) and (5R,8S,10R,11S)-10-tert-butyl 5-ethyl 13-benzyl-4-hydroxy-3-methoxy-2-methyl-7-oxo-5,7,8,9,10,11-hexahydro-8,11-epiminoazepino[1,2-b[isoquinoline-5,10-dicarboxylate (21b)
To a stirred solution of a 2.6:1 mixture of compounds 20a and 20b (410 mg, 0.695 mmol, 1.0 eq.) in THF/MeOH 1:1 (14 mL, 0.05 M), under Ar, was added K2CO3 (192 mg, 1.39 mmol, 2.0 eq.). The suspension was stirred for 2.5 h, the solvent was evaporated and the residue was partitioned between phosphate buffer (0.1 M, pH = 7.5, 50 mL) and EtOAc (33 mL). The aqueous phase was extracted with EtOAc (2×33 mL) and the combined organic layers were rinsed with brine (50 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, hexanes/EtOAc 4:1) to afford a 5:1 mixture of the title compounds 21a and 21b (255 mg, 67%) as a pale yellow oil, which was used in the next step without further purification. 1H-NMR (400 MHz; CDCl3): δ 7.38-7.24 (m, 5H), 6.76 (s, 1H), 6.63 (s, 1H, minor diastereomer), 6.49 (s, 1H, minor diastereomer), 6.43 (s, 1H, minor diastereomer), 6.42 (s, 1H), 6.39 (s, 5H), 5.46 (s, 1H, minor diastereomer), 5.45 (s, 1H. minor diastereomer), 4.24 (q, J = 7.1 Hz, 2H), 4.19-3.9 (m, 5H), 4.07 (s, 1H), 3.86 (d, J = 7.5 Hz, 1H), 3.84 (s, 1H, minor diastereomer), 3.82 (s, 3H), 3.31 (dd, J = 9.8, 6.0 Hz, 1H, minor diastereomer), 2.79 (dd, J = 9.5, 4.6 Hz, 1H), 2.74-2.66 (m, 1H), 2.46 (dd, J = 13.0, 9.9 Hz, 1H, minor diastereomer), 2.26 (s, 3H, minor diastereomer), 2.24 (s, 3H), 2.13 (dd, J = 13.4, 9.6 Hz, 6H), 1.46 (s, 9H, minor diastereomer), 1.42 (s, 9H), 1.27 (t, J = 7.1 Hz, 3H), 1.24 (t, J = 7.2 Hz, 3H, minor diastereomer); 13C-NMR (101 MHz, CDCl3): δ 172.4, 171.9, 170.7, 170.6, 170.4, 168.9, 146.9, 146.8, 146.2, 138.5, 138.0, 136.1, 134.2, 131.9, 131.7, 128.5, 127.4, 127.3, 126.6, 126.0, 119.1, 119.1, 110.8, 103.4, 81.3, 64.3, 62.7, 62.6, 60.8, 60.8, 52.2, 52.2, 52.1, 51.5, 50.8, 48.2, 32.1, 28.2, 28.1, 22.8, 15.9, 14.3; Rf (SiO2, hexanes/EtOAc 2:1) 0.45; [α]D25 = -73.6 ° (c = 0.282, CH2Cl2); IR (film, CH2Cl2), νmax 3374 (br), 2980, 2938, 1736, 1689, 1647, 1154 cm-1; HRMS (MH+), found 549.2606. C31H37N2O7 requires 549.2601.
4.12. (5S,8S,10R,11S)-tert-butyl 13-benzyl-5-formyl-4-hydroxy-3-methoxy-2-methyl-7-oxo-5,7,8,9,10,11-hexahydro-8,11-epimino-azepino[1,2-b[isoquinoline-10-carboxylate (22a) and (5R,8S,10R,11S)-tert-butyl 13-benzyl-5-formyl-4-hydroxy-3-methoxy-2-methyl-7-oxo-5,7,8,9,10,11-hexahydro-8,11-epiminoazepino[1,2-b[isoquinoline-10-carboxylate (22b)
A solution of LiAlH4 in THF (1.0 M, 447 μL, 0.447 mmol, 1.0 eq.) was added dropwise to a solution of a 5:1 mixture of compounds 21a and 21b (245 mg, 0.447 mmmol, 1.0 eq.) in THF (9 mL, 0.05 M), under Ar, at -10 °C. The solution was stirred for 10 minutes at this temperature, quenched with EtOAc (12 mL) and sat. aq. Rochelle's salt (12 mL) and allowed to warm to RT. The flask was covered with aluminum foil and stirred overnight under a stream of Ar. The solution was diluted with phosphate buffer (0.1 M, pH = 7.5, 50 mL), the phases were separated and aqueous phase was extracted with EtOAc (3×33 mL) and the combined organic layers were rinsed with brine (25 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, hexanes/EtOAc 4:1) to afford a 3:1 mixture of the title compounds 22a and 22b (124 mg, 55%) as a pale yellow oil, which was used in the next step without further purification. 1H-NMR (400 MHz; CDCl3): δ 9.48 (s, 1H), 9.38 (s, 1H, minor diastereomer), 7.41-7.23 (m, 5H), 6.58 (s, 1H), 6.57 (s, 1H, minor diastereomer), 6.43 (s, 1H, minor diastereomer), 6.41 (s, 1H), 6.18 (s, 1H, minor diastereomer), 6.17 (s, 1H, minor diastereomer), 4.25 (1/2 AB, J = 13.5 Hz, 1H), 4.18 (1/2 AB, J = 13.5 Hz, 1H), 4.07 (s, 1H), 4.00 (s, 1H, minor diastereomer), 3.83 (s, 3H, minor diastereomer), 3.81 (s, 3H, minor diastereomer), 3.40 (dd, J = 9.7, 6.0 Hz, 1H, minor diastereomer), 2.81 (dd, J = 9.5, 4.7 Hz, 1H), 2.73-2.67 (m, 1H), 2.59 (dd, J = 13.0, 9.8 Hz, 1H, minor diastereomer), 2.28 (s, 3H, minor diastereomer), 2.26 (s, 3H), 2.15 (dd, J = 13.4, 9.6 Hz, 1H), 1.48 (s, 9H, minor diastereomer), 1.46 (s, 9H); 13C-NMR (101 MHz, CDCl3): δ 192.1, 191.3, 172.5, 171.9, 170.1, 145.7, 144.6, 138.6, 138.0, 136.6, 135.3, 131.5, 128.8, 128.4, 127.5, 127.2, 119.4, 119.2, 107.3, 104.0, 102.7, 102.6, 81.4, 64.1, 64.0, 62.9, 62.8, 61.2, 61.1, 58.6, 58.5, 51.6, 50.8, 48.8, 34.7, 32.4, 31.7, 29.8, 28.1, 22.8, 16.0, 14.3; Rf (SiO2, hexanes/EtOAc 2:1) 0.42; [α]D25 = -64.8 ° (c = 0.250, CH2Cl2); IR (film, CH2Cl2), νmax 3331 (br), 2977, 2935, 1733, 1679, 1642, 1154 cm-1; HRMS (MH+), found 505.2345. C29H33N2O6 requires 505.2339.
4.13. (5S,8S,10R,11S)-tert-butyl 13-benzyl-4-(benzyloxy)-5-formyl-3-methoxy-2-methyl-7-oxo-5,7,8,9,10,11-hexahydro-8,11-epiminoazepino[1,2-b[isoquinoline-10-carboxylate (23a) and (5R,8S,10R,11S)-tert-butyl 13-benzyl-4-(benzyloxy)-5-formyl-3-methoxy-2-methyl-7-oxo-5,7,8,9,10,11-hexahydro-8,11-epiminoazepino[1,2-b[isoquinoline-10-carboxylate (5R,8S,10R,11S)-tert-butyl 13-benzyl-4-(benzyloxy)-5-formyl-3-methoxy-2-methyl-7-oxo-5,7,8,9,10,11-hexahydro-8,11-epiminoazepino[1,2-b[isoquinoline-10-carboxylate (23b)
To a stirred solution of a 3:1 mixture of compounds 22a and 22b (115 mg, 0.228 mmol, 1.0 eq.) and benzyl bromide (108 μL, 0.912 mmol, 4.0 eq.) in DMF (7.6 mL, 0.03 M), under Ar, were added tetrabutylammonium iodide (9.0 mg, 0.023 mmol, 0.10 eq.) and finely ground anhydrous Na2CO3 (241 mg, 2.28 mmol, 10 eq.). The mixture was vigorously stirred for 2h and diluted with water (25 mL) and phosphate buffer (0.1 M, pH = 7.5, 25 mL). The aqueous phase was extracted with EtOAc (3×33 mL) and the combined organic layers were rinsed with brine (25 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, hexanes/EtOAc 6:1, 4:1) to afford a 2.2:1 mixture of compounds 23a and 23b (88 mg, 65%) as a pale yellow oil, which was used in the next step without further purification. 1H-NMR (400 MHz; CDCl3): δ 9.32 (s, 1H), 9.19 (s, 1H minor diastereomer), 7.48-7.22 (m, 10H), 6.63 (s, 1H, minor diastereomer), 6.62 (s, 1H), 6.38 (s, 1H, minor diastereomer), 6.36 (s, 1H), 5.37 (s, 1H), 5.36 (s, minor diastereomer), 5.28 (1/2 AB, J = 11.1 Hz, 1H, minor diastereomer), 5.26 (1/2 AB, J = 11.1 Hz, 2H), 5.20 (1/2 AB, J = 11.1 Hz, 1H, minor diastereomer), 5.14 (1/2 AB, J = 11.1 Hz, 1H), 4.20 (1/2 AB, J = 13.5 Hz, 1H), 4.14 (1/2 AB, J = 13.5 Hz, 1H), 4.05 (s, 1H), 3.97 (s, 1H, minor diastereomer), 3.86 (s, 3H, minor diastereomer), 3.84 (s, 3H), 3.80 (1/2 AB, J = 13.5 Hz, 1H) 3.79 (d, J = 7.4 Hz, 1H), 3.75 (d, J = 6.9 Hz, minor diastereomer), 3.68 (1/2 AB, J = 13.5 Hz, 1H, minor diastereomer), 3.37 (dd, J = 9.7, 6.1 Hz, 1H, minor diastereomer), 2.78 (dd, J = 9.6, 4.7 Hz, 1H), 2.71-2.64 (m, 2H), 2.53 (1/2 ABX, J = 13.1, 9.9 Hz, 1H, minor diastereomer), 2.28 (s, 3H, minor diastereomer), 2.26 (s, 3H), 2.10 (dd, J = 13.3, 9.7 Hz, 1H), 1.45 (s, 9H minor diastereomer), 1.44 (s, 9H); 13C-NMR (101 MHz, CDCl3): δ 192.5, 191.7, 171.9, 169.8, 168.8, 150.4, 150.3, 148.3, 148.1, 138.6, 138.1, 136.9, 136.9, 136.6, 135.3, 133.7, 128.9, 128.8, 128.8, 128.6, 128.6, 128.5, 128.5, 128.4, 128.3, 127.4, 127.2, 126.9, 123.1, 122.8, 115.0, 114.6, 103.9, 102.5, 81.4, 81.2, 75.0, 75.0, 65.0, 63.9, 63.1, 62.8, 60.5, 58.8, 57.3, 52.8, 51.6, 48.8, 34.7, 32.2, 28.2, 16.0, 16.0; Rf (SiO2, hexanes/EtOAc 4:1) 0.45; [α]D25 = -64 ° (c = 0.32, CH2Cl2); IR (film, CH2Cl2), νmax 3030, 2976, 2934, 1733, 1688, 1646, 1154 cm-1; HRMS (MH+), found 595.2801. C36H39N2O6 requires 595.2808.
To a stirred solution of a 2.2:1 mixture of compounds 23a and 23b (88 mg, 0.15 mmol, 1.0 eq.) in THF (2 mL, 0.08 M), under Ar, was added DBN (19 μL, 0.15 mmol, 1.0 eq.). The mixture was stirred for 30 minutes and then diluted with phosphate buffer (0.1 M, pH = 7.5, 50 mL) and water (50 mL). The aqueous phase was extracted with EtOAc (3×33 mL) and the combined organic layers were rinsed with brine (25 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude material was dissolved in the minimal amount of EtOAc purified by flash chromatography (silica gel, hexanes/EtOAc 4:1) to afford a 1:2.2 mixture of compounds 23a and 23b (64 mg, 72%) as a pale yellow oil, which was used in the next step without further purification. 1H-NMR (400 MHz; CDCl3): δ 9.32 (s, 1H, minor diastereomer), 9.19 (s, 1H), 7.48-7.24 (m, 10H), 6.63 (s, 1H), 6.62 (s, 1H, minor diastereomer), 6.38 (s, 1H), 6.36 (s, 1H, minor diastereomer), 5.37 (s, 1H, minor diastereomer), 5.36 (s, 1H), 5.28 (1/2 AB, J = 11.1 Hz, 1H), 5.26 (1/2 AB, J = 11.1 Hz, 1H, minor diastereomer), 5.19 (1/2 AB, J = 11.1 Hz, 1H), 5.13 (1/2 AB, J = 11.2 Hz, 2H, minor diastereomer), 4.20 (1/2 AB, J = 13.4 Hz, 1H, minor diastereomer), 4.14 (1/2 AB, J = 13.5 Hz, 1H minor diastereomer), 4.05 (s, 1H, minor diastereomer), 3.97 (s, 1H), 3.86 (s, 3H, minor diastereomer), 3.84 (s, 3H, minor diastereomer), 3.81 (1/2 AB, J = 13.6 Hz, 1H), 3.79 (d, J = 6.2 Hz, 4H), 3.75 (d, J = 6.6 Hz, 3H), 3.68 (1/2 AB, J = 13.4 Hz, 3H), 3.37 (dd, J = 9.8, 6.0 Hz, 1H), 2.78 (dd, J = 9.5, 4.7 Hz, 1H, minor diastereomer), 2.71-2.65 (m, 2H), 2.53 (1/2 ABX, J = 13.0, 9.9 Hz, 3H), 2.28 (s, 3H), 2.26 (s, 3H, minor diastereomer), 2.10 (dd, J = 13.4, 9.5 Hz, 1H, minor diastereomer), 1.45 (s, 9H), 1.44 (s, 9H, minor diastereomer); 13C-NMR (101 MHz, CDCl3): δ 192.5, 191.7, 172.5, 171.9, 168.8, 150.4, 148.1, 138.0, 136.9, 135.3, 133.7, 128.9, 128.8, 128.8, 128.6, 128.5, 128.4, 128.3, 127.4, 127.2, 126.9, 123.1, 122.8, 115.0, 114.6, 103.9, 102.5, 81.4, 81.2, 75.0, 75.0, 65.0, 63.9, 63.1, 62.7, 60.4, 58.8, 57.3, 52.7, 51.6, 50.9, 48.8, 34.7, 32.2, 28.2, 28.1, 16.0, 16.0; Rf (SiO2, hexanes/EtOAc ); [α]D25 = +27 ° (c = 0.22, CHCl3); IR (film, CH2Cl2), νmax 3029, 2969, 2935, 1732, 1688, 1647, 1154 cm-1; HRMS (MH+), 595.2789. C36H39N2O6 requires 595.2808.
4.14. (5S,8S,10R,11S)-tert-butyl 13-benzyl-4-(benzyloxy)-5-(hydroxymethyl)-3-methoxy-2-methyl-7-oxo-5,7,8,9,10,11-hexahydro-8,11-epiminoazepino[1,2-b[isoquinoline-10-carboxylate (24a) and (5R,8S,10R,11S)-tert-butyl 13-benzyl-4-(benzyloxy)-5-(hydroxymethyl)-3-methoxy-2-methyl-7-oxo-5,7,8,9,10,11-hexahydro-8,11-epiminoazepino[1,2-b[isoquinoline-10-carboxylate (24b)
To a stirred solution of a mixture of compounds 23a and 23b (60 mg, 0.10 mmol) in EtOH (5 mL, 0.20 M), at 0 °C, under Ar, was added NaBH4 (30 mg, 0.80 mmol 8.0 eq.). The reaction was stirred at RT for 2 hours, quenched with 1N HCl (2.4 mL, 2.40 mmol, 24 eq.) and diluted with phosphate buffer (0.1 M, pH = 7.5, 50 mL). The aqueous phase was extracted with EtOAc (3×25 mL) and the combined organic layers were rinsed with brine (25 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, hexanes/EtOAc 4:1) to afford compounds 23a (18 mg, 30%) as a colorless oil and compound 23b (38 mg, 65%) as a colorless oil. Compound 23a: 1H-NMR (400 MHz; CDCl3): δ 7.50-7.22 (m, 10H), 6.62 (s, 1H), 6.14 (t, J = 6.1 Hz, 1H), 5.48 (s, 1H), 5.18 (1/2 AB, J = 11.1 Hz, 1H), 5.09 (1/2 AB, J = 11.1 Hz, 1H), 4.10 (s, 1H), 3.97 (1/2 AB, J = 13.3 Hz, 1H), 3.87 (1/2 AB, J = 13.3 Hz, 1H), 3.79 (d, J = 7.7 Hz, 1H), 3.73 (s, 1H), 2.69 (ddt, J = 27.0, 9.0, 4.6 Hz, 2H), 2.25 (s, 3H), 2.06 (dd, J = 13.3, 9.5 Hz, 1H), 1.90 (br t, 6.0 Hz, 1H), 1.44 (s, 9H); 13C-NMR (101 MHz, CDCl3): δ 171.9, 171.7, 150.5, 148.2, 138.3, 137.2, 135.6, 132.6, 128.7, 128.5, 128.4, 128.4, 127.6, 127.3, 122.1, 120.1, 103.7, 81.3, 75.1, 65.4, 64.2, 62.7, 60.4, 51.7, 51.3, 50.7, 31.6, 28.1, 28.1, 15.9, 14.3; Rf (SiO2, hexanes/EtOAc 4:1) 0.12; [α]D25 = -60.0 ° (c = 0.895, CHCl3); IR (film, CH2Cl2), νmax 3447 (br), 3063, 3030, 2934, 2870, 1730, 1676, 1636, 1154 cm-1; HRMS (MH+), found 597.2971. C36H41N2O6 requires 597.2965. Compound 24b: 1H-NMR (400 MHz; CDCl3): δ 6.62 (s, 1H), 6.09 (dd, J = 8.4, 4.4 Hz, 1H), 5.45 (s, 1H), 5.17 (1/2 AB, J = 11.1 Hz, 1H), 5.14 (1/2 AB, J = 11.1 Hz, 1H), 3.95 (s, 1H), 3.83 (s, 3H), 3.78 (d, J = 13.5 Hz, 1H), 3.73 (d, J = 6.6 Hz, 1H), 3.63 (d, J = 13.4 Hz, 1H), 3.63-3.50 (m, 1H), 3.15 (dd, J = 9.8, 6.1 Hz, 1H), 2.63 (dt, J = 12.8, 6.5 Hz, 1H), 2.45 (dd, J = 13.0, 9.8 Hz, 1H), 1.77-1.74 (br m, 1H), 1.45 (s, 9H); 13C-NMR (101 MHz, CDCl3): δ 172.3, 170.4, 150.6, 147.9, 138.1, 137.2, 134.0, 132.5, 128.7, 128.7, 128.6, 128.6, 128.5, 128.5, 128.4, 128.4, 127.3, 126.5, 122.8, 122.7, 120.6, 105.3, 105.3, 81.3, 75.0, 65.5, 65.5, 63.1, 63.0, 60.4, 52.7, 49.4, 49.4, 48.4, 34.8, 28.2, 16.0, 16.0; Rf (SiO2, hexanes/EtOAc 4:1) 0.10; [α]D25 = +64 ° (c = 0.31, CHCl3); IR (film, CH2Cl2), νmax 3444 (br), 3062, 3029, 2970, 2927, 1729, 1682, 1639, 1154 cm-1; HRMS (MH+), found 597.2974. C36H41N2O6 requires 597.2965.
4.15. (5R,8S,10R,11S)-tert-butyl 4-hydroxy-5-(hydroxymethyl)-3-methoxy-2-methyl-7-oxo-5,7,8,9,10,11-hexahydro-8,11-epimino-azepino[1,2-b[isoquinoline-10-carboxylate (25)
A solution of compound 24b (7.0 mg, 0.012 mmol) in glacial acetic acid (1 mL) and 10% Pd/C (7 mg) were placed in round bottom flask and sparged with Ar for 5 minutes. The vessel was evacuated and filled with hydrogen three times. The reaction was vigorously stirred overnight under hydrogen (1 atm). The suspension was diluted with CH2Cl2 (25 mL) and then filtered through Celite® and the flask was rinsed with CH2Cl2 (3×5 mL). The solution was extracted with sat. aq. NaHCO3 (3×15 mL). The combined aqueous layers were diluted with phosphate buffer (0.1 M, pH = 7.5, 25 mL) and extracted with CH2Cl2 (3×15 mL). The combined organic layers were rinsed with brine (50 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, CHCl3/MeOH 97:3) to afford compound 25 (4.6 mg, 92%) as a colorless oil. 1H-NMR (400 MHz; CDCl3): δ 6.40 (s, 1H), 6.05 (dd, J = 7.9, 4.1 Hz, 1H), 5.53 (s, 1H), 4.30 (s, 1H), 4.09 (d, J = 6.7 Hz, 1H), 3.78-3.74 (m, 2H), 3.76 (s, 3H), 3.65-3.60 (m, 1H), 3.17 (dd, J = 9.3, 6.2 Hz, 1H), 2.61 (dd, J = 13.1, 9.4 Hz, 1H), 2.32 (dt, J = 13.2, 6.6 Hz, 1H) 2.24 (s, 3H), 1.47 (s, 9H); 13C-NMR (101 MHz, CDCl3): δ 173.4, 171.1, 145.2, 144.7, 144.7, 136.9, 136.8, 130.3, 127.1, 119.1, 112.9, 112.9, 102.7, 81.6, 65.1, 62.4, 61.8, 61.0, 49.5, 48.1, 37.0, 29.8, 29.8, 28.2, 15.9; Rf (SiO2, CHCl3/MeOH 95:5) 0.17; [α]D25 = +4.3 ° (c = 0.23, CHCl3); IR (film, CH2Cl2), νmax 3262 (br), 2969, 2925, 2854, 1719, 1683, 1646, 1154 cm-1; HRMS (MH+), found 417.2033. C22H29N2O6 requires 417.2026.
4.16. (5R,8S,10R,11S,11aS)-tert-butyl 4-hydroxy-5-(hydroxymethyl)-3-methoxy-2-methyl-7-oxo-5,7,8,9,10,11,11a, 12- octahydro-8,11-epiminoazepino[1,2-b[isoquinoline-10-carboxylate (26)
To a solution of compound 25 (4.6 mg, 0.011 mmol) in EtOH (1 mL) in a 5 mL vial, was added a slurry of Raney® nickel 2800 (500 μL of commercially available water slurry, washed with EtOH (3×1 mL) and suspended in EtOH (1 mL)). The vial was placed in a Fisher-Porter bottle, under Ar, the suspension was sparged with Ar for 5 minutes and the vessel was filled with hydrogen gas at 100 psi. The reaction was vigorously stirred overnight diluted with EtOAc (10 mL) and sat. aq. Rochelle's salt (10 mL), and stirred vigorously for 2 h. The biphasic suspension was filtered through Celite®, the phases separated and the aqueous phase extracted with EtOAc (3×10 mL). The combined organic phases were rinsed with brine (25 mL), dried (Na2SO4), filtered, and concentrated under vacuum. The crude material was purified by flash chromatography (silica gel, CHCl3/MeOH 97:3) to afford compound 26 (3.4 mg, 74%) as a colorless oil. 1H-NMR (400 MHz; CDCl3): δ 6.51 (s, 1H), 5.59 (dd, J = 5.6, 3.4 Hz, 1H), 3.96 (d, J = 6.1 Hz, 1H), 3.88 (dd, J = 10.9, 3.2 Hz, 1H), 3.78 (s, 3H), 3.77-3.76 (m, 1H), 3.67 (dt, J = 12.4, 2.6 Hz, 1H), 3.61 (dd, J = 11.1, 5.8 Hz, 1H), 3.16 (dd, J = 9.0, 6.4 Hz, 1H), 2.84 (t, J = 13.5 Hz, 1H), 2.54 (dd, J = 14.7, 2.2 Hz, 1H), 2.50 (dd, J = 13.2, 9.0 Hz, 1H), 2.27 (s, 3H), 2.18 (dt, J = 13.2, 6.6 Hz, 1H), 1.53-1.45 (m, 9H); 13C-NMR (101 MHz, CDCl3): δ 174.4, 172.4, 145.7, 132.0, 129.7, 121.2, 120.2, 118.0, 81.5, 67.8, 63.0, 62.2, 61.0, 60.8, 52.6, 42.8, 38.8, 32.1, 29.9, 28.2, 15.9; Rf (SiO2, CHCl3/MeOH 95:5) 0.20; [α]D25 = -36 ° (c = 0.080, CHCl3); IR (film, CH2Cl2), νmax 3286 (br), 2958, 2925, 2855, 1729, 1652, 1456 cm-1; HRMS (MH+), found 419.2174. C22H31N2O6 requires 419.2182.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from the National Institutes of Health (Grant RO1CA085419) and Bristol Myers Squibb Co (doctoral fellowship to A.J.).
Footnotes
Supplementary Material:1H and 13C NMR spectra of all compounds.
References and notes
- 1.Scott JD, Williams RM. Chem Rev. 2002;102:1669–1730. doi: 10.1021/cr010212u. [DOI] [PubMed] [Google Scholar]
- 2.Whaley HA, P EL, Dann M, Shay AJ, Porter JN. Antimicrob Agents Chemother. 8:83–86. 164. [PubMed] [Google Scholar]
- 3.He HY, Shen B, Carter GT. Tetrahedron Lett. 2000;41:2067–2071. [Google Scholar]
- 4.Takahashi K, Tomita F. J Antibiot. 1983;36:468–470. doi: 10.7164/antibiotics.36.468. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki K, Sato T, Morioka M, Nagai K, Abe K, Yamaguchi H, Saito T, Ohmi Y, Susaki K. J Antibiot. 1991;44:479–485. doi: 10.7164/antibiotics.44.479. [DOI] [PubMed] [Google Scholar]
- 6.Hegde VR, Patel MG, Das PR, Pramanik B, Puar MS. J Antibiot. 1997;50:126–134. doi: 10.7164/antibiotics.50.126. [DOI] [PubMed] [Google Scholar]
- 7.Li WY, Leet JE, Ax HA, Gustavson DR, Brown DM, Turner L, Brown K, Clark J, Yang H, Fung-Tomc J, Lam KS. J Antibiot. 2003;56:226–231. doi: 10.7164/antibiotics.56.226. [DOI] [PubMed] [Google Scholar]
- 8.Northcote PT, Siegel M, Borders DB, Lee MD. J Antibiot. 1994;47:901–908. doi: 10.7164/antibiotics.47.901. [DOI] [PubMed] [Google Scholar]
- 9.Sasaki T, Otani T, Matsumoto H, Unemi N, Hamada M, Takeuchi T, Hori M. J Antibiot. 1998;51:715–721. doi: 10.7164/antibiotics.51.715. [DOI] [PubMed] [Google Scholar]
- 10.Zhang CW, Herath K, Jayasuriya H, Ondeyka JG, Zink DL, Occi J, Birdsall G, Venugopal J, Ushio M, Burgess B, Masurekar P, Barrett JF, Singh SB. J Nat Prod. 2009;72:841–847. doi: 10.1021/np800783b. [DOI] [PubMed] [Google Scholar]
- 11.Ashley ER, Cruz EG, Stoltz BM. J Am Chem Soc. 2003;125:15000–15001. doi: 10.1021/ja039223q. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida A, Akaiwa M, Asakawa T, Hamashima Y, Yokoshima S, Fukuyama T, Kan T. Chemistry – A European Journal. 2012 doi: 10.1002/chem.201202073. n/a-n/a. [DOI] [PubMed] [Google Scholar]
- 13.Magnus P, Matthews KS. J Am Chem Soc. 2005;127:12476–12477. doi: 10.1021/ja0535817. [DOI] [PubMed] [Google Scholar]
- 14.Magnus P, Matthews KS. Tetrahedron. 2012;68:6343–6360. [Google Scholar]
- 15.Couturier C, Schlama T, Zhu JP. Synlett. 2006:1691–1694. [Google Scholar]
- 16.Wu YC, Bernadat G, Masson G, Couturier C, Schlama T, Zhu JP. J Org Chem. 2009;74:2046–2052. doi: 10.1021/jo8027449. [DOI] [PubMed] [Google Scholar]
- 17.Bernadat G, George N, Couturier C, Masson G, Schlama T, Zhu JP. Synlett. 2011:576–578. [Google Scholar]
- 18.Siengalewicz P, Brecker L, Mulzer J. Synlett. 2008:2443–2446. [Google Scholar]
- 19.Vincent G, Chen YY, Lane JW, Williams RM. Heterocycles. 2007;72:385–398. [Google Scholar]
- 20.Scott JD, Williams RM. Angew Chem Int Edit. 2001;40:1463–+. doi: 10.1002/1521-3773(20010417)40:8<1463::AID-ANIE1463>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 21.Flanagan ME, Williams RM. J Org Chem. 1995;60:6791–6797. [Google Scholar]
- 22.Liao XW, Liu W, Dong WF, Guan BH, Chen SZ, Liu ZZ. Tetrahedron. 2009;65:5709–5715. [Google Scholar]
- 23.The lack of reactivity of the primary hydroxyl of 7 is consistent with the regioselectivity observed in the reaction between TBS-Cl and diols bearing a β-aminoalcohol motif (ref. 24). We concur with the explanation provided by the authors, which stated that the nucleophilicity of the primary hydroxyl is reduced by internal hydrogen bonding to the neighboring amino group
- 24.Sales M, Charette AB. Org Lett. 2005;7:5773–5776. doi: 10.1021/ol052436v. [DOI] [PubMed] [Google Scholar]
- 25.Frie JL, Jeffrey CS, Sorensen EJ. Org Lett. 2009;11:5394–5397. doi: 10.1021/ol902168g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lane JW, Chen YY, Williams RM. J Am Chem Soc. 2005;127:12684–12690. doi: 10.1021/ja0535918. [DOI] [PubMed] [Google Scholar]
- 27.Chen JC, Chen XC, Bois-Choussy M, Zhu JP. J Am Chem Soc. 2006;128:87–89. doi: 10.1021/ja0571794. [DOI] [PubMed] [Google Scholar]
- 28.Zhu's conditions were also used by Liao (ref. 22) to convert compound into a trans-THIQ system, using 2-benzyloxyacetaldehyde
- 29.Fukuyama T, Nunes JJ. J Am Chem Soc. 1988;110:5196–5198. [Google Scholar]
- 30.Mancuso AJ, Huang SL, Swern D. J Org Chem. 1978;43:2480–2482. [Google Scholar]
- 31.Vishnetskaya MV, Yakimova IY, Sidorenkova IA. Russ J Phys Chem. 2006;80:176–180. [Google Scholar]
- 32.Vishnetskaya MV, Yakimova IY, Sidorenkova IA. Russ J Phys Chem. 2006;80:173–175. [Google Scholar]
- 33.Vishnetskaya MV, Ivanova MS, Solkan VN, Zhidomirov GM, Mel'nikov MY. Russ J Phys Chem A. 2012;86:889–891. [Google Scholar]
- 34.As illustrated in Scheme 4, we propose that the dipolarophile adds from the Re face of the iminium ion carbon to form 20a, which epimerizes under the reaction conditions to form 20b
- 35.We propose that the observed chemoselectivity can be explained by the initial formation of a phenoxyaluminum hydride species, which upon delivery of one hydride to the ester, forms a stable 7-membered ring alcoxy(phenoxy)aluminum hydride species
- 36.Compounds 23a and 23b are unstable to silica gel. Consequently, we did not attempt their separation for the purpose of recycling of 23a
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.