

This paper describes an informatic analysis of RNA-seq data relevant to levels of mRNAs encoding ribosomal proteins. Major findings are that the vast majority of molar ratios of these mRNAs are within threefold of one another. In addition, some of these mRNAs, apparently transcribed from “pseudogenes,” are expressed in a tissue-specific manner.
Keywords: ribosome, ribosomal protein, RNA-seq, alternative splicing, pseudogene
Abstract
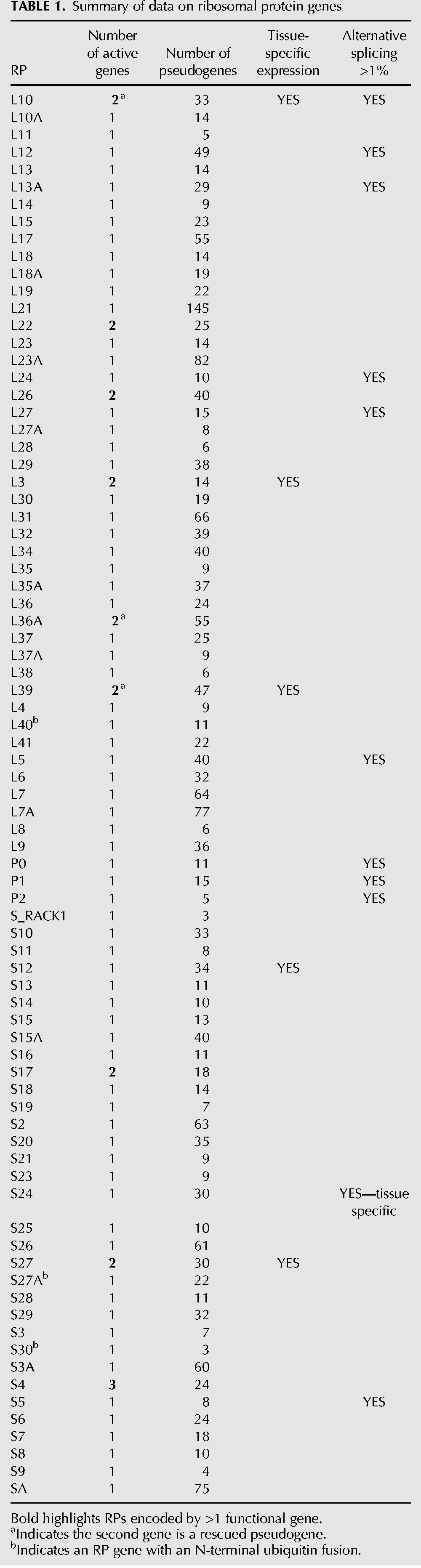
The torrent of RNA-seq data becoming available not only furnishes an overview of the entire transcriptome but also provides tools to focus on specific areas of interest. Our focus on the synthesis of ribosomes asked whether the abundance of mRNAs encoding ribosomal proteins (RPs) matched the equimolar need for the RPs in the assembly of ribosomes. We were at first surprised to find, in the mapping data of ENCODE and other sources, that there were nearly 100-fold differences in the level of the mRNAs encoding the different RPs. However, after correcting for the mapping ambiguities introduced by the presence of more than 2000 pseudogenes derived from RP mRNAs, we show that for 80%–90% of the RP genes, the molar ratio of mRNAs varies less than threefold, with little tissue specificity. Nevertheless, since the RPs are needed in equimolar amounts, there must be sluggish or regulated translation of the more abundant RP mRNAs and/or substantial turnover of unused RPs. In addition, seven of the RPs have subsidiary genes, three of which are pseudogenes that have been “rescued” by the introduction of promoters and/or upstream introns. Several of these are transcribed in a tissue-specific manner, e.g., RPL10L in testis and RPL3L in muscle, leading to potential variation in ribosome structure from one tissue to another. Of the 376 introns in the RP genes, a single one is alternatively spliced in a tissue-specific manner.
INTRODUCTION
As the central element in the production of proteins, the ribosome is key to the regulation of growth and development. Molecular structures from yeast (Ben-Shem et al. 2011) and humans (Anger et al. 2013) demonstrate that the ribosome is a fixed, unique arrangement of proteins and RNA molecules, mostly conserved over geological time scales. Yet recent work suggests that there may be more flexibility in both the structure and function of the ribosome than has been appreciated (for review, see Xue and Barna 2012).
The eukaryotic ribosome is composed of four RNA molecules and 80 ribosomal proteins (RPs), assembled through a complex series of steps requiring the participation of nearly 300 RNA and protein cofactors (for review, see Thomson et al. 2013; Woolford and Baserga 2013). Although much of the detailed analysis of this process has been carried out in S. cerevisiae, genome comparisons and recent experimental work suggest that much the same process occurs in mammalian cells, although with additional complexities (Tafforeau et al. 2013). The assembly process requires the nearly simultaneous presence of an equimolar amount of nearly all the 80 RPs. There are two types of exceptions: (1) In yeast, eight of the 79 RPs are not essential for reasonable growth (Steffen et al. 2012), and in mammals, a few RPs, such as L22 (O'Leary et al. 2013) and L40 (Lee et al. 2013) seem to be dispensable for ribosome assembly and for cell growth; none are known to be dispensable for the development of an intact animal; and (2) the stalk proteins, P1 and P2, are present in two copies per ribosome and exchange between a cytoplasmic pool and mature ribosomes (for review, see Gonzalo and Reboud 2003).
Ribosomes are abundant. An efficient cell would synthesize equimolar amounts of each of the RPs. Is that in fact the case? Indeed some evidence suggests that the cell produces far more of each of the RPs than needed, rapidly degrading any molecules not selected to form the complete ribosome (Lam et al. 2007). However, accumulating examples of pathological effects due to haploinsufficiency of genes encoding RPs (for review, see Raiser et al. 2014) question that notion. The discovery that Diamond-Blackfan anemia can be caused by haploinsufficiency of RPS19 (Draptchinskaia et al. 1999) or a number of other RP genes (Farrar et al. 2011), that 5q- myelodysplastic syndrome is caused by the loss of one copy of RPS14 (Ebert et al. 2008), and that haploinsufficiency for RPSa can lead to congenital asplenia (Bolze et al. 2013) suggests that adequate supplies of the RPs can be limiting.
Finally, increased focus on the role in disease of the ribosome and its synthesis has come from the realization that ribosome assembly is carefully monitored by the cell, when defective ribosome assembly due to imbalance in the production of RPs can lead to accumulation of p53, and in some cases to apoptosis (for review, see Vlatković et al. 2014).
These considerations prompted us to exploit the massive amount of information now available through genomic initiatives to ask three specific questions about human ribosomes and their synthesis:
What genes are used to produce RPs? Are they the same in all tissues?
Is there equimolar representation of mRNAs encoding the 80 RPs?
To what degree does alternative splicing of RP gene transcripts lead to alternative RPs? Is there tissue specificity?
As will be evident below, the answers to these questions (partially summarized in Table 1) not only reveal limitations to conventional genomic analysis but also provide intriguing insights into potential ribosome heterogeneity.
TABLE 1.
Summary of data on ribosomal protein genes
RESULTS AND DISCUSSION
Proteome analysis of the ribosomal proteins
Can proteome analysis reveal whether the cell, indeed, has equimolar amounts of the 80 RPs? These are difficult experiments, but the Aebersold and Mann laboratories (Beck et al. 2011; Nagaraj et al. 2011) have used advanced mass spectrometry methods to measure the abundance of many thousands of individual proteins in two human cell lines. The results for RPs, extracted from their Supplemental Files, are shown in Supplemental Table S1. Two points are clear: RPs are abundant, and there remains a substantial scatter between the proteins and between the laboratories. Within one laboratory, however, the data are more consistent, allowing Geiger et al. (2012) to conclude that the ribosomal proteins are expressed at about the same levels in the 11 cell lines they studied. Nevertheless, these data are far from sufficient to confirm that cells have equimolar amounts of the 80 RPs.
Genes encoding ribosomal proteins and their derivatives
As in most mammals, the human genome carries a single copy of a gene encoding each of the 80 ribosomal proteins (Uechi et al. 2001), with the few exceptions to be described below. A valuable compilation of data on the ribosomal proteins and their genes in many organisms is available at http://ribosome.med.miyazaki-u.ac.jp (Nakao et al. 2004).
The exceptions include S4, which is encoded by one gene on the X chromosome, RPS4X, and two on the Y chromosome, RPS4Y1&2. Another is the dual copies of RPS17 due to a ∼300 kb tandemly duplicated region of chromosome 15. Neither S4 nor S17 has multiple genes in the mouse. In addition, there are several interesting variations on the canonical RP genes. RPL3L, RPL22L, RPL26L, and RPS27L are duplicated copies of the original gene, carrying introns in nearly identical positions. All but RPL26L have similar duplicates in the mouse. For convenience, we term these the “Like” genes, as indicated in their genetic names. RPL7L and RSL24D1 are related to RPL7 and RPL24, respectively, but are more likely to be involved in ribosome assembly and not present in the mature ribosome (Dunbar et al. 2000; Kappel et al. 2012; Babiano et al. 2013).
The human genome is not lacking for RP sequences, however. It carries more than 2000 pseudogenes derived from processed RP mRNAs (Zheng et al. 2007; Balasubramanian et al. 2009). Perhaps the abundant production of ribosomes in the developing oocyte provides the opportunity for reverse transcribed mRNAs to enter the germ line. Three of the pseudogenes have been rescued, in both man and mouse. These can be defined as pseudogenes because they have lost their normal introns and promoters. Yet they are active. RPL36aL is transcribed from a site some 1545 bp upstream, within <100 bp of the divergent initiation site of the MGAT2 gene. Since it is now clear that most promoters drive divergent transcription (Seila et al. 2009), we suggest that RPL36aL has been “fixed” in the “on” position, an example of a recent prediction (Wu and Sharp 2013). Excision of a 1422 nt intron from the 5′ UTR leaves the translation initiation site intact. RPL39L is similar except that there is no apparent gene to provide a transcription origin. Its two introns in the 5′ UTR total nearly 18,000 nt. Most intriguing is RPL10L, a processed pseudogene that has been activated without the help of either introns or external transcription start sites. Is it coincidence that each of the three activated pseudogenes is derived from a gene on the X chromosome?
Mapping RNA-seq output for RP genes
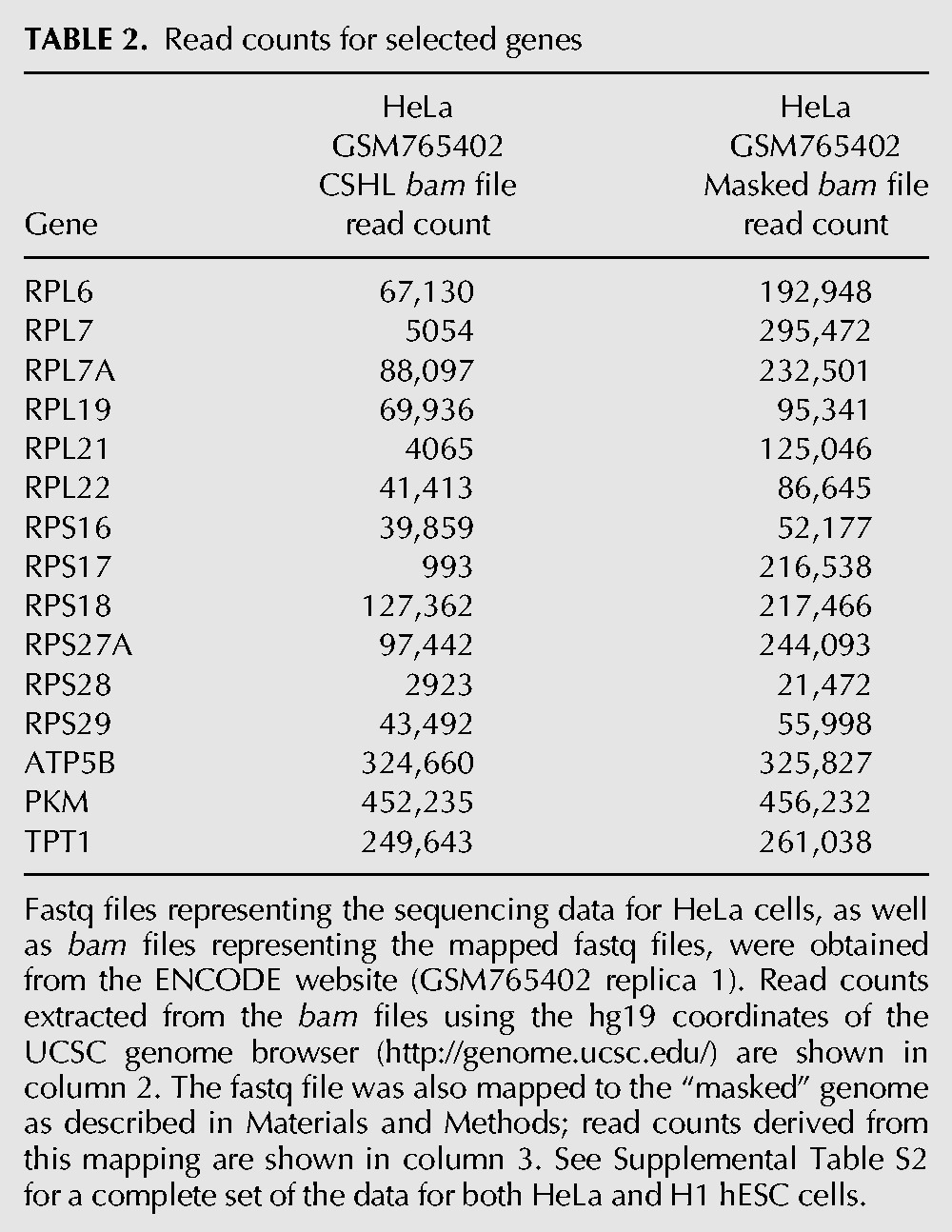
One aim of this study is to determine the level of mRNAs derived from each of the RP genes and to ask whether there is equimolar representation of the different mRNAs to yield the equimolar amount of RPs needed to construct ribosomes. However, examination of the data provided in a number of publications proved problematical due to enormous variability in the read counts for different RP genes from different sources. Many essential genes had few if any reads, whereas others had tens of thousands. An example is shown in Supplemental Table S2, from which a portion is excerpted for Table 2. Columns B and E of Supplemental Table S2 show read counts taken from the mapping (bam) file of RNA-seq analysis of HeLa and of H1hESC cell long PolyA+ RNA. It is evident that there is a >100-fold range in the read counts for different RP genes, a range that is not consistent between the two cell lines of Supplemental Table S2. Although it is possible that this wide range is a biological phenomenon, we suggest that it is due to mapping difficulties caused by the presence in the human genome of the numerous pseudogenes described above. Indeed, for some cases the original bam files attributed a larger fraction of the RP reads to pseudogenes than to the original gene.
TABLE 2.
Read counts for selected genes
Several mapping programs have been devised to identify a region of the genome from which a specific sequencing read originated: TopHat (Trapnell et al. 2012), GSNAP (Wu and Nacu 2010), Bowtie2 (Langmead and Salzberg 2012), and STAR (Dobin et al. 2013), among others. However, the sequence of most of the pseudogenes is almost identical to that of the authentic mRNA. Thus, many reads derived from RP mRNAs will map to multiple locations in the genome, especially considering that most programs allow for two to three errors per 100 base calls. Although different programs deal with this problem in different ways, none has a failsafe way of linking a read to the authentic gene.
As an attempted solution to this problem, we mapped reads from selected data sets to a “masked” genome: the complete human genome (hg19), in which all the RP pseudogenes (derived from the list at http://pseudogenes.org/psidr/) as well as one of the duplicate copies of RPS17, were masked as N's (see Materials and Methods). Discrepancies between read counts, and identification of the sites to which “missing” reads had mapped, led to the identification of 46 additional pseudogenes, which have been added to the list (Supplemental Table S3). Since any individual read is limited to one “hit” in the genome, any read corresponding to an RP transcript will be credited to the original gene.
Read counts using our analysis compared to the read counts originally reported by The ENCODE Project Consortium (2011) are shown in Supplemental Table S2, with excerpts in Table 2. Mapping against the masked genome yields between 45% and 80% more reads for the RP genes, but the same number of reads for the control genes. The “lost” reads are not distributed uniformly. When mapped against the whole genome, genes such as RPL7 have lost practically all their reads; others have almost the same number. Since our interest is in the molar ratio of the mRNAs encoding RPs, we conclude that mapping with the masked genome provides more reliable estimates of the actual mRNA levels. We suggest that it is unwise to rely on the mapping data for ribosomal protein genes as provided in the bam files of many of the sequencing consortia.
Is it justified to mask the pseudogene sequences? Are any of the pseudogenes active? Since many of the pseudogenes derived from RP genes lie within the introns of expressed genes, they are being transcribed, but presumably not in a way that would provide functional mRNAs encoding RPs. As a general approach, we examined the RP pseudogenes for the chromatin signs of actual or potential transcription, H3K4me3 and H3K27Ac, using the ENCODE data (Djebali et al. 2012) displayed in the UCSC browser. Although each of the authentic RP genes has a robust signal for both histone marks, the pseudogenes for the most part have no signal or a very weak one. A typical example, comparing RPS19 with one of its pseudogenes, is shown in Figure 1. The only cases with substantial amounts of these chromatin marks over a pseudogene are when the pseudogene is within an intron near the transcription start site of another gene. On the other hand, for the cases described above in which a pseudogene has been “rescued,” such as RPL36aL1 and RPL39L, the chromatin marks are clearly evident. These chromatin marks are remarkably uniform from one cell type to another. In the case of RPL10L, the H3K4me3 and H3K27Ac chromatin marks are negative for cell lines but are robust in mouse testis, where the gene is transcribed.
FIGURE 1.
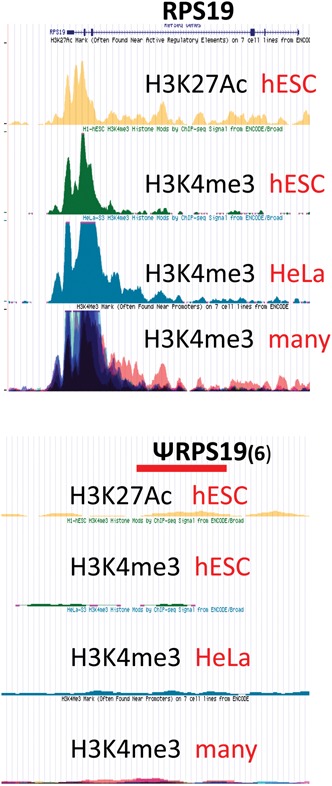
Chromatin signatures of authentic and pseudo RP genes. A snapshot from the UCSC browser showing the ChIP analyses of H3K27Ac and H3K4me3 from several cell lines at RPS19 and at the RPS19 pseudogene on chr6: 110,883,378–110,883,818. “Many” refers to a number of cell lines that have been overlaid.
In the end, any analysis of sequencing data requires some compromises. It is possible that some pseudogenes are marginally active, at least in some tissues. However, using the masked genome, their transcripts are likely to be counted as coming from the mother gene; such an attribution will still provide a measure of the coding capacity for a given RP. Only if the pseudogene has diverged sufficiently to encode a protein with different function will it be missed when using the masked genome.
Once the number of reads has been determined, the conventional measure of the relative number of mRNAs derived from different genes is the RPKM (Reads Per Kilobase of mRNA per Million mapped reads). An additional compromise is necessary for the appropriate value of K. The RefSeq mRNAs for a number of RP genes have long 3′ UTR sequences, increasing the length used for K. However, in some cases, we find few if any reads within the extended 3′ UTR. Therefore, based on such analysis of a number of data sets, we have corrected the K values for a few RP mRNAs (Supplemental Table S4). A recent analysis of polyadenylation sites largely confirms these corrections and provides interesting examples of tissue-specific differences in 3′ termination sites for some RP transcripts (Lianoglou et al. 2013).
Finally, to facilitate comparison of the data on RP transcripts over many data sets, we have normalized most of the data. The normalized value for RPx is RPKMx*80/ΣRPKM for any given data set. In this way, if all the 80 RPs were represented by equal numbers of mRNAs, then each would have a value of 1.0. The extent to which the actual value differs from 1 is a measure of the deficiency or excess mRNA for that RP.
Stoichiometry of mRNAs encoding RPs
As an example of the protocol, the analysis of data sets of three tissues from the Illumina Human Body Map (GSE30611), heart, liver, and testis, is shown in Supplemental Table S5, with a more complete analysis in Supplemental Table S6. A number of points are clear.
The read counts are substantial, consistent with ribosomal protein genes being among the most active in the cell. In this case, they make up from 1.2% to 3.3% of the total reads, depending on tissue. Consistently, brain and heart have less representation of the RP genes, with more active tissues and cell lines usually approaching 5% of the total (Supplemental Tables S2, S6).
From the normalized RPKM columns, it is clear that mRNAs encoding most of the RPs are within a fairly small spread, between ∼0.5 and ∼1.5. Thus, there is about a threefold variation in the relative numbers of the mRNAs encoding the different RPs, but with a few outliers. In any individual data set, however, the range can be much larger, perhaps five- to sixfold. Overall, there is substantial consistency between the Body Map data and other comparable tissue data (Supplemental Table S6, indicated in red; Brawand et al. 2011) (GSM752691). Comparison of these data sets with a number of others, mostly from a variety of tissue culture cells, shows substantial uniformity. Certain proteins such as L41and S27 have two- to threefold more mRNAs than the average (see far right columns of Supplemental Table S6, where the RP genes are sorted according to overall abundance of their mRNAs). Surprisingly, S17, in spite of its gene being present at twice the copy number because of a duplicated chromosome fragment, has nearly the average level of mRNA. Other proteins, such as L36, S26, S5, and S30, have less than half the average number of mRNAs. Interestingly, S19 mRNA is consistently low in both of these data sets but not in some others. Nevertheless, this is a provocative finding in light of the effects of haploinsufficiency of RPS19 in Diamond-Blackfan anemia (Draptchinskaia et al. 1999). Finally, P1 and P2 are represented by above average mRNA as might be expected for genes encoding proteins found in more than one copy per ribosome.
These results are at significant odds with the report that there can be 100-fold differences in the levels of the various RP mRNAs in several tissues of the developing mouse embryo (Kondrashov et al. 2011). Whether this represents differences in experimental protocol or intriguing aspects of tissue differentiation remains to be seen.
Unfortunately, differences in RNA and/or library preparation between different laboratories can lead to inconsistent results. Thus, for a number of genes, one data set consistently records higher read counts than the other, e.g., RPL15, RPL22, RPL34, RPL9, and RPS3a are higher in the Body Map than in the Brawand et al. (2011) data, whereas RPL13, RPL28, and RPL36 are lower in the Body Map data (Supplemental Table S6). We observed a similar situation in comparing data on the same cell line generated by different laboratories. An additional cause of potential variation may arise from the observation that RP mRNA abundance is subject to circadian rhythm (Jouffe et al. 2013), rarely recorded for RNA-seq data sets.
The scatter in the data from different sources is sufficiently large that it is generally not possible to identify tissue-specific differences in the abundance of mRNAs derived from individual RP genes. One clear exception is the low level of mRNA from RPL3 in both skeletal and heart muscle, which is somewhat offset by the increased transcription of RPL3L in those tissues (Supplemental Tables S3, S4). Perhaps this is another example of autoregulation as has been reported for the RPL22/RPL22L pair (O'Leary et al. 2013).
The three tissues shown in Supplemental Table S5 are all from males. The one functioning RP gene on the Y chromosome, RPS4Y1, generally contributes ∼20%–25% of the total mRNA encoding S4. RPS4Y2 appears silent, although other analyses suggest it is active in testis and prostate (Lopes et al. 2010). Comparing the data across the tissues represented in the Body Map (Supplemental Table S6), the expression of RPS4X is consistently greater in female than in male tissues. Surprisingly, a data set from Xist negative cells (Vallot et al. 2013) showed no increase in the expression either of RPS4X, or of RPL36A and RPL39, both of which are on the X chromosome.
As mentioned above, there are seven “Like” genes, from second copies of RP genes or from rescued pseudogenes derived from RP genes. Contributions of these “Like” genes are summarized in Supplemental Table S7. Although this table represents only the Body Map data set, similar values and tissue specificities were observed in a variety of human as well as mouse data sets. Although the “rescued” pseudogene RPL36AL contributes ∼40% or more to the supply of L36a in most tissues, other “Like” genes make only a low, relatively tissue-nonspecific contribution to the supplies of that protein. Nevertheless, RPL22L has some key function in hematopoietic stem cell development (Zhang et al. 2013), and its expression is substantially increased in RPL22−/− mice (O'Leary et al. 2013). There are several cases of tissue specificity: RPL39L is highly expressed in testis, somewhat in brain. RPL10L is expressed almost exclusively in testis, as has been observed biochemically (Sugihara et al. 2010). RPL3L is highly expressed in heart and skeletal muscle, but barely at all elsewhere. This partially makes up for a substantial reduction in transcripts of RPL3 in those tissues. Because of the overall similarity of the “Like” proteins with their parents, we assume that they can take their place in the ribosome, as has been shown directly for L10L, L22L, and L39L (Sugihara et al. 2010; Stadanlick et al. 2011). Thus, one of the most intriguing results from the tissue comparisons in Supplemental Tables S6 and S7 is that there are clearly distinct tissue differences in ribosomes, e.g., L3L has likely replaced L3 in ∼25% of the ribosomes of muscle tissue. Since about a quarter of the amino acids differ between the two proteins, the effects on the structure of the ribosome could be significant. Testis, which seems to be the most active in expressing the “Like” proteins, could have a variety of different ribosomes. Aside from the case of L38, whose absence appears to affect the translation of a specific class of mRNAs (Kondrashov et al. 2011), we know basically nothing about the influence of RPs on the translation of specific cellular mRNAs.
To ask whether the differences in mRNA level encoding the different RPs are reflected, or compensated, in translation, we have analyzed several sets of data from ribosome profiling experiments (Hsieh et al. 2012; Loayza-Puch et al. 2013; Stumpf et al. 2013). In general, the relative amount of mRNA encoding each RP that is protected by ribosomes is similar to that in the total transcriptome. This is interesting because RP mRNAs are the original members of the “tracks of pyrimidine” (TOP) mRNA class whose translation is regulated under the influence of growth conditions (Meyuhas and Dreazen 2009), yet there is no evidence for the specific suppression of translation of those mRNAs that appear to be in excess. Regulation of translation can occur through upstream ORFs (Hinnebusch and Lorsch 2012). Only two of the RP gene transcripts, those encoding L3 and L29, have an AUG upstream of the authentic translation initiation site; neither of these has an unusually high level of mRNA. Although it is clear that many factors influence the rate of translation, such as specific initiation factors (Mahoney et al. 2009) or the secondary structure near the initiation codon (Gu et al. 2010), as yet there is insufficient data to establish whether there is control of the translation of individual RPs at this level.
One mRNA about which we can speculate is that encoding L41, which consistently has substantially more mRNA than any of the other RP genes and also has by far the highest value in ribosome profiling experiments. L41 is a unique protein, with only 25 amino acids, of which 17 are arginine or lysine, highly conserved throughout the eukaryotes, and occupying an intimate location at the interface between the 60S and 40S subunits (Jenner et al. 2012). With such a short ORF, the mRNA encoding L41 is translated by only one ribosome at a time (Yu and Warner 2001). Furthermore, stretches of basic amino acids are translated inefficiently, perhaps because of their difficulty in traversing the ribosome “tunnel” (Charneski and Hurst 2013). For these two reasons, it is likely that the efficiency of translation of L41 mRNA is lower and the need for L41 mRNA is greater than for the other RPs. Presumably specific features of many RPs or their mRNAs contribute to influencing their rate of translation.
In summary, our analysis of these data sets suggest that RP mRNAs are highly abundant, each representing >0.03% of total mRNA number. Although there are not large differences in the numbers of mRNAs for the different RPs, the reproducible differences that are apparent imply either that there are substantial variations in the efficiency of translation of different mRNAs and/or that there are differences in the amounts of proteins actually produced. Although there is substantial evidence of nonribosomal functions for a limited number of RPs (for review, see Warner and McIntosh 2009), it seems likely that such functions would require only a small fraction of the abundant output of those RPs. Thus, such functions could be markedly affected by small fluctuations in the amounts or the translation of RP mRNAs.
Splicing of ribosomal protein gene transcripts
It has been suggested that >95% of human genes undergo alternative splicing (Pan et al. 2008; Wang et al. 2008). On the other hand, ribosomal proteins are conserved across highly diverged species, a necessity for their positioning in the compact, complex structure of the ribosome. We approach alternative splicing of RP gene transcripts with two questions: To what degree does alternative splicing affect the nature and the structure of the ribosome? Are there situations in which alternative splicing of transcripts of an RP gene yields a protein with an RNA binding motif fused to peptide element that provides a nonribosomal function?
Most mapping programs provide a “junctions” file in which are specified the locations and number of splicing reads that span two exons. Thorough examination of the mapping data from both cell lines and tissues reveals that among all the RP transcripts, there is only a single case of tissue-specific alternative splicing. As shown in Figure 2A, the transcripts of RPS24 are spliced in three alternative ways, including or excluding microexons of 19 or 22 nt, and yielding predicted proteins that differ in their C-terminal amino acids (Fig. 2B). Muscle and heart tissue largely splice AA′, whereas liver and kidney are evenly divided between B and C. The tissue specificity is reproducible not only between data sets but also between species (Fig. 2C). Remarkably, it is possible to observe the shift in splicing pattern as mouse ESC cells differentiate into myoblasts (Fig. 2D; Trapnell et al. 2010). There is also a significant difference between HeLa and H1 hESC cells (see Supplemental Table S8). The S24 protein products due to alternative splicing differ rather little, and the C terminus of S24 is not highly conserved (Fig. 2E). Unfortunately, the recent cryo-EM structure of human ribosomes (Anger et al. 2013) does not include the C terminus of S24, and the structure of the yeast ribosome (Jenner et al. 2012) is uninformative because of the differences in sequence (Fig. 2E). Is the alternative splicing of RPS24 transcripts functional or is it simply a by-product of the splicing factors characteristic of skeletal muscle (Llorian and Smith 2011)?
FIGURE 2.
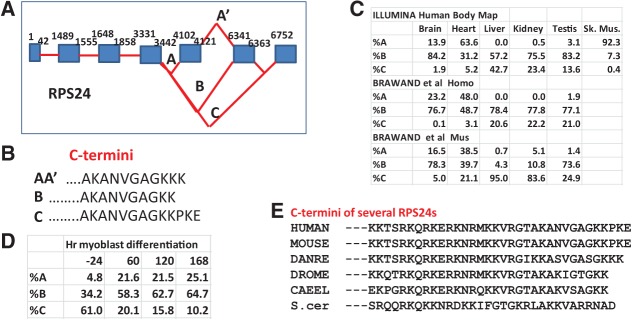
Alternative splicing of RPS24 transcripts: (A) A cartoon representing the alternative splices observed for transcripts of RPS24 (not to scale). (B) The predicted C termini of S24 resulting from the three alternative splicing variations. (C) The percentage of splicing in each of the three variations as a function of tissue and organism. (D) The percentage of splicing in each of the three variations as myoblasts differentiate (data from Trapnell et al. 2010). (E) The C termini of S24 in a number of organisms.
Of the 376 introns in the RP genes, we have identified only 14 that exhibit alternative splicing at a >1% level (Supplemental Table S8). If the cutoff is lowered to ∼0.2%, a number of additional introns demonstrate alternative (erroneous?) splicing. Although, as described above, mRNA quantitation often exhibits substantial scatter between data sets generated by different laboratories, there is far better agreement on the presence and the level of alternative splicing events. With the exception of RPS24, there does not appear to be significant tissue specificity to the alternative splicing, but in most cases the data is insufficiently deep to lead to confident conclusions. There may, however, be some underlying regulation, since a longitudinal study of one individual over a long time span (Chen et al. 2012) shows substantial variation in some splicing variants (Fig. 3). Initiation of transcription at different sites is often followed by different splices that lead to the same exon2, which contains the initiating ATG. Thus, for RPL17 there are reproducibly six 5′ splice sites that all splice to the same 3′ site, upstream of the initiator ATG. We have not considered these as alternative splices.
FIGURE 3.
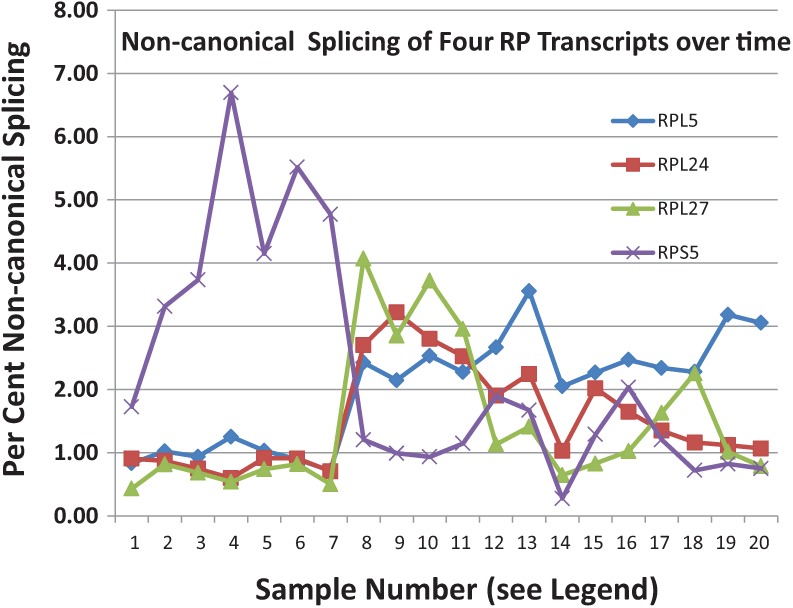
Alternative/aberrant splicing of transcripts of several RP genes: Junction files were obtained from mapping of the fastq files obtained in the analyses of the leukocytes of a single individual over many months (Chen et al. 2012). The proportion of the noncanonical splice events are indicated as a percentage. The time points are erratic (see Chen et al. 2012).
The alternative splices usually involve exon skipping, although some involve aberrant 5′ or 3′ sites. Supplemental Table S8 shows there are six cases—RPL10, RPL12, RPL13a, RPLP0, RPLP1 and RPS12—in which the predicted mRNA would encode a protein made up of a portion of a RP fused to a substantial number of downstream amino acids. Although these alternatively spliced transcripts represent only 1%–2% of the level of the RP mRNAs, the fused proteins should have significant abundance since RP mRNAs themselves are so abundant. Thus, transcripts of RPS12 are spliced, at about a 1%–3% level with no apparent tissue specificity, to form an mRNA encoding a protein with 41AA of the RP fused to 33 additional AA. This is conserved in mouse tissues. Is this RNA binding region used to carry the C-terminal sequences to a target?
However, we have been unable to identify any of the novel peptides predicted in Supplemental Table S8, either in the gpmdb proteome database (http://gpmdb.thegpm.org), or among the more than 150,000 human peptides analyzed in Geiger et al. (2012). If synthesized, they must be rapidly degraded. What is unknowable at present is whether some, or all, of these aberrant splice events are truly “alternative splicing” with a biological function or “erroneous splicing” due to noise in the splicing process.
Since many of the aberrant splicing events lead to termination codons within the open reading frame, the degree of aberrant splicing may be somewhat masked by nonsense-mediated decay (NMD). Indeed, a regulated alternative splice of intron 3 of the transcripts of RPL3 leads to rapid degradation by NMD (Cuccurese et al. 2005). Nevertheless, while data sets from mouse cells in which NMD has been abolished by deletion of either SMG1 (McIlwain et al. 2010) or UPF2 (Weischenfeldt et al. 2012) clearly show the accumulation of the alternative splice junction in the RPL3 transcript, they fail to show any substantial change in the amounts of noncanonical splice junctions of RP genes described above.
Conclusions
The analyses described above lead to several conclusions regarding the products of the genes encoding human ribosomal proteins.
There is a roughly fivefold difference between the most and least abundant mRNAs. This difference is reasonably consistent over many data sets but can be exceeded in any individual data set. Since each ribosome contains a single copy of each RP, this variation implies that there is unequal translation of the different mRNAs and/or that there is significant degradation of unassembled RPs. Note that either overexpression or underexpression of individual RPs can lead to accumulation of p53 and apoptosis, and in some cases to tumorigenesis (for review, see Raiser et al. 2014). It is interesting that one of several breast tumor lines, BT474, has a 10-fold amplification of a segment of chromosome 17 that includes RPL17 and RPL23, accompanied by a 10-fold excess of the mRNA for each (Sun et al. 2011). It would be interesting to know if such an excess has any effect on the cell.
There seems to be little tissue specificity for the major RP genes, except that muscle has reduced transcripts from RPL3, perhaps in compensation for the presence of transcripts from RPL3L. On the other hand, there is substantial tissue specificity for some of the “Like” RP genes (Supplemental Table S7). Thus, there clearly are different populations of ribosomes in different tissues. Whether this leads to differences in the translation of specific mRNAs is not known.
The presence of so many pseudogenes derived from RP genes is intriguing. Although we have shown that very few of them contribute to the formation of ribosomes, there are arguments that they may to serve to modulate the activity of their parental genes (Muro et al. 2011; Li et al. 2013).
With S24 the single product of tissue-specific alternative splicing, it seems clear that transcripts of RP genes are far less subject to alternative splicing than those of the average gene. It remains to be seen whether the several instances of different splices at the 1%–3% level and the many more at the <0.5% level represent splicing errors or encode functional proteins. Indeed, because RP mRNAs are among the most abundant of the cell, alternate splicing of even a very small proportion, yielding the fusion of an RNA binding domain to some functional domain, could lead to enough product to carry out important functions.
MATERIALS AND METHODS
Fastq files were downloaded from the Gene Expression Omnibus (GEO). Because several of the RP mRNAs are quite short, we only analyzed data from sources where the insert size was <300 bp.
Mapping of reads was carried out using STAR (Dobin et al. 2013), with the parameters: outFilterMismatchNmax :1 mismatch per 25 bases read, and outFilterMultimapNmax :1 hit per read. The output from STAR not only provides read count but also junction files that identify the endpoints of splicing events.
The presence of many pseudogenes derived from RP mRNAs complicates the task of mapping RNA-seq reads to authentic RP genes. To avoid this problem, we constructed two artificial genomes: an RP genome containing only the RP genes and some controls and a masked genome for which the maskFastaFromBed utility from BEDtools was used to mask, with Ns, all the pseudogenes listed in the database http://pseudogenes.org/psidr/. On comparing the read counts obtained from mapping against the masked and the RP genomes, it was clear that some reads were lost using the masked genome. To resolve this issue, we used the FilterSamReads utility from PICARD to identify locations in the masked genome that mapped to RP genes in the RP genome. In this way, we identified an additional 46 processed pseudogenes (Supplemental Table S3), which were then masked to generate the final masked genome against which the fastq files were mapped. We also selected a set of control genes on the basis of abundant, relatively non-tissue-specific transcription. Pseudogenes derived from the set of control genes were also masked. Masked FASTA files for human (hg19) and mouse (mm9) genomes are available upon request.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We are grateful to the many researchers who generated the data analyzed in this paper, especially G. Schroth for early access to the Illumina Body Map data, to A. Dobin for advice on STAR, to D. Zheng for discussions on pseudogenes, and to J. Woolford, U. Maitra, and C. Query for critical reading of the manuscript. This work was partially supported by the Einstein HPC Core and by NIH Grant GM25532 to J.R.W.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.043653.113.
REFERENCES
- Anger AM, Armache JP, Berninghausen O, Habeck M, Subklewe M, Wilson DN, Beckmann R 2013. Structures of the human and Drosophila 80S ribosome. Nature 497: 80–85 [DOI] [PubMed] [Google Scholar]
- Babiano R, Badis G, Saveanu C, Namane A, Doyen A, Díaz-Quintana A, Jacquier A, Fromont-Racine M, de la Cruz J 2013. Yeast ribosomal protein L7 and its homologue Rlp7 are simultaneously present at distinct sites on pre-60S ribosomal particles. Nucleic Acids Res 41: 9461–9470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian S, Zheng D, Liu YJ, Fang G, Frankish A, Carriero N, Robilotto R, Cayting P, Gerstein M 2009. Comparative analysis of processed ribosomal protein pseudogenes in four mammalian genomes. Genome Biol 10: R2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, Herzog F, Rinner O, Ellenberg J, Aebersold R 2011. The quantitative proteome of a human cell line. Mol Syst Biol 7: 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M 2011. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science 334: 1524–1529 [DOI] [PubMed] [Google Scholar]
- Bolze A, Mahlaoui N, Byun M, Turner B, Trede N, Ellis SR, Abhyankar A, Itan Y, Patin E, Brebner S, et al. 2013. Ribosomal protein SA haploinsufficiency in humans with isolated congenital asplenia. Science 340: 976–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawand D, Soumillon M, Necsulea A, Julien P, Csárdi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M, et al. 2011. The evolution of gene expression levels in mammalian organs. Nature 478: 343–348 [DOI] [PubMed] [Google Scholar]
- Charneski CA, Hurst LD 2013. Positively charged residues are the major determinants of ribosomal velocity. PLoS Biol 11: e1001508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Mias GI, Li-Pook-Than J, Jiang L, Lam HY, Chen R, Miriami E, Karczewski KJ, Hariharan M, Dewey FE, et al. 2012. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 148: 1293–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuccurese M, Russo G, Russo A, Pietropaolo C 2005. Alternative splicing and nonsense-mediated mRNA decay regulate mammalian ribosomal gene expression. Nucleic Acids Res 33: 5965–5977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draptchinskaia N, Gustavsson P, Andersson B, Pettersson M, Willig TN, Dianzani I, Ball S, Tchernia G, Klar J, Matsson H, et al. 1999. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet 21: 169–175 [DOI] [PubMed] [Google Scholar]
- Dunbar DA, Dragon F, Lee SJ, Baserga SJ 2000. A nucleolar protein related to ribosomal protein L7 is required for an early step in large ribosomal subunit biogenesis. Proc Natl Acad Sci 97: 13027–13032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, Raza A, Root DE, Attar E, Ellis SR, et al. 2008. Identification of RPS14 as a 5q− syndrome gene by RNA interference screen. Nature 451: 335–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2011. A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 9: e1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar JE, Vlachos A, Atsidaftos E, Carlson-Donohoe H, Markello TC, Arceci RJ, Ellis SR, Lipton JM, Bodine DM 2011. Ribosomal protein gene deletions in Diamond-Blackfan anemia. Blood 118: 6943–6951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger T, Wehner A, Schaab C, Cox J, Mann M 2012. Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol Cell Proteomics 11: M111.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo P, Reboud JP 2003. The puzzling lateral flexible stalk of the ribosome. Biol Cell 95: 179–193 [DOI] [PubMed] [Google Scholar]
- Gu W, Zhou T, Wilke CO 2010. A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput Biol 6: e1000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG, Lorsch JR 2012. The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb Perspect Biol 4: a011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, Shi EY, Stumpf CR, Christensen C, Bonham MJ, et al. 2012. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485: 55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner L, Melnikov S, Garreau de Loubresse N, Ben-Shem A, Iskakova M, Urzhumtsev A, Meskauskas A, Dinman J, Yusupova G, Yusupov M 2012. Crystal structure of the 80S yeast ribosome. Curr Opin Struct Biol 22: 759–767 [DOI] [PubMed] [Google Scholar]
- Jouffe C, Cretenet G, Symul L, Martin E, Atger F, Naef F, Gachon F 2013. The circadian clock coordinates ribosome biogenesis. PLoS Biol 11: e1001455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappel L, Loibl M, Zisser G, Klein I, Fruhmann G, Gruber C, Unterweger S, Rechberger G, Pertschy B, Bergler H 2012. Rlp24 activates the AAA-ATPase Drg1 to initiate cytoplasmic pre-60S maturation. J Cell Biol 199: 771–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov N, Pusic A, Stumpf CR, Shimizu K, Hsieh AC, Xue S, Ishijima J, Shiroishi T, Barna M 2011. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 145: 383–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YW, Lamond AI, Mann M, Andersen JS 2007. Analysis of nucleolar protein dynamics reveals the nuclear degradation of ribosomal proteins. Curr Biol 17: 749–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS, Burdeinick-Kerr R, Whelan SP 2013. A ribosome-specialized translation initiation pathway is required for cap-dependent translation of vesicular stomatitis virus mRNAs. Proc Natl Acad Sci 110: 324–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Yang W, Wang XJ 2013. Pseudogenes: pseudo or real functional elements? J Genet Genomics 40: 171–177 [DOI] [PubMed] [Google Scholar]
- Lianoglou S, Garg V, Yang JL, Leslie CS, Mayr C 2013. Ubiquitously transcribed genes use alternative polyadenylation to achieve tissue-specific expression. Genes Dev 27: 2380–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorian M, Smith CW 2011. Decoding muscle alternative splicing. Curr Opin Genet Dev 21: 380–387 [DOI] [PubMed] [Google Scholar]
- Loayza-Puch F, Drost J, Rooijers K, Lopes R, Elkon R, Agami R 2013. p53 induces transcriptional and translational programs to suppress cell proliferation and growth. Genome Biol 14: R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes AM, Miguel RN, Sargent CA, Ellis PJ, Amorim A, Affara NA 2010. The human RPS4 paralogue on Yq11.223 encodes a structurally conserved ribosomal protein and is preferentially expressed during spermatogenesis. BMC Mol Biol 11: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney SJ, Dempsey JM, Blenis J 2009. Cell signaling in protein synthesis ribosome biogenesis and translation initiation and elongation. Prog Mol Biol Transl Sci 90: 53–107 [DOI] [PubMed] [Google Scholar]
- McIlwain DR, Pan Q, Reilly PT, Elia AJ, McCracken S, Wakeham AC, Itie-Youten A, Blencowe BJ, Mak TW 2010. Smg1 is required for embryogenesis and regulates diverse genes via alternative splicing coupled to nonsense-mediated mRNA decay. Proc Natl Acad Sci 107: 12186–12191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyuhas O, Dreazen A 2009. Ribosomal protein S6 kinase from TOP mRNAs to cell size. Prog Mol Biol Transl Sci 90: 109–153 [DOI] [PubMed] [Google Scholar]
- Muro EM, Mah N, Andrade-Navarro MA 2011. Functional evidence of post-transcriptional regulation by pseudogenes. Biochimie 93: 1916–1921 [DOI] [PubMed] [Google Scholar]
- Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Paabo S, Mann M 2011. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol 7: 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao A, Yoshihama M, Kenmochi N 2004. RPG: the Ribosomal Protein Gene database. Nucleic Acids Res 32: D168–D170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary MN, Schreiber KH, Zhang Y, Duc AC, Rao S, Hale JS, Academia EC, Shah SR, Morton JF, Holstein CA, et al. 2013. The ribosomal protein Rpl22 controls ribosome composition by directly repressing expression of its own paralog, Rpl22l1. PLoS Genet 9: e1003708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40: 1413–1415 [DOI] [PubMed] [Google Scholar]
- Raiser DM, Narla A, Ebert BL 2014. The emerging importance of ribosomal dysfunction in the pathogenesis of hematologic disorders. Leuk Lymphoma 55: 491–500 [DOI] [PubMed] [Google Scholar]
- Seila AC, Core LJ, Lis JT, Sharp PA 2009. Divergent transcription: a new feature of active promoters. Cell Cycle 8: 2557–2564 [DOI] [PubMed] [Google Scholar]
- Stadanlick JE, Zhang Z, Lee SY, Hemann M, Biery M, Carleton MO, Zambetti GP, Anderson SJ, Oravecz T, Wiest DL 2011. Developmental arrest of T cells in Rpl22-deficient mice is dependent upon multiple p53 effectors. J Immunol 187: 664–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen KK, McCormick MA, Pham KM, MacKay VL, Delaney JR, Murakami CJ, Kaeberlein M, Kennedy BK 2012. Ribosome deficiency protects against ER stress in Saccharomyces cerevisiae. Genetics 191: 107–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf CR, Moreno MV, Olshen AB, Taylor BS, Ruggero D 2013. The translational landscape of the mammalian cell cycle. Mol Cell 52: 574–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugihara Y, Honda H, Iida T, Morinaga T, Hino S, Okajima T, Matsuda T, Nadano D 2010. Proteomic analysis of rodent ribosomes revealed heterogeneity including ribosomal proteins L10-like, L22-like 1, and L39-like. J Proteome Res 9: 1351–1366 [DOI] [PubMed] [Google Scholar]
- Sun Z, Asmann YW, Kalari KR, Bot B, Eckel-Passow JE, Baker TR, Carr JM, Khrebtukova I, Luo S, Zhang L, et al. 2011. Integrated analysis of gene expression, CpG island methylation, and gene copy number in breast cancer cells by deep sequencing. PLoS One 6: e17490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafforeau L, Zorbas C, Langhendries JL, Mullineux ST, Stamatopoulou V, Mullier R, Wacheul L, Lafontaine DL 2013. The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of pre-rRNA processing factors. Mol Cell 51: 539–551 [DOI] [PubMed] [Google Scholar]
- Thomson E, Ferreira-Cerca S, Hurt E 2013. Eukaryotic ribosome biogenesis at a glance. J Cell Sci 126: 4815–4821 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uechi T, Tanaka T, Kenmochi N 2001. A complete map of the human ribosomal protein genes: assignment of 80 genes to the cytogenetic map and implications for human disorders. Genomics 72: 223–230 [DOI] [PubMed] [Google Scholar]
- Vallot C, Huret C, Lesecque Y, Resch A, Oudrhiri N, Bennaceur-Griscelli A, Duret L, Rougeulle C 2013. XACT, a long noncoding transcript coating the active X chromosome in human pluripotent cells. Nat Genet 45: 239–241 [DOI] [PubMed] [Google Scholar]
- Vlatković N, Boyd MT, Rubbi CP 2014. Nucleolar control of p53: a cellular Achilles’ heel and a target for cancer therapy. Cell Mol Life Sci 71: 771–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner JR, McIntosh KB 2009. How common are extra-ribosomal functions of ribosomal proteins? Mol Cell 34: 3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt J, Waage J, Tian G, Zhao J, Damgaard I, Jakobsen JS, Kristiansen K, Krogh A, Wang J, Porse BT 2012. Mammalian tissues defective in nonsense-mediated mRNA decay display highly aberrant splicing patterns. Genome Biol 13: R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolford JL Jr, Baserga SJ 2013. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 195: 643–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Nacu S 2010. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26: 873–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Sharp PA 2013. Divergent transcription: a driving force for new gene origination? Cell 155: 990–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue S, Barna M 2012. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat Rev Mol Cell Biol 13: 355–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Warner JR 2001. Expression of a micro-protein. J Biol Chem 276: 33821–33825 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Duc AC, Rao S, Sun XL, Bilbee AN, Rhodes M, Li Q, Kappes DJ, Rhodes J, Wiest DL 2013. Control of hematopoietic stem cell emergence by antagonistic functions of ribosomal protein paralogs. Dev Cell 24: 411–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, Frankish A, Baertsch R, Kapranov P, Reymond A, Choo SW, Lu Y, Denoeud F, Antonarakis SE, Snyder M, et al. 2007. Pseudogenes in the ENCODE regions: consensus annotation, analysis of transcription, and evolution. Genome Res 17: 839–851 [DOI] [PMC free article] [PubMed] [Google Scholar]