

Significance
Leukocyte migration to sites of inflammation involves “braking” and attachment of blood-borne leukocytes onto target endothelium (vessel surfaces), a process mediated principally by the endothelial molecule E-selectin binding to leukocyte E-selectin ligands. We report here that the cytokine granulocyte-colony stimulating factor (G-CSF) induces expression of a novel E-selectin ligand on human leukocytes, a glycovariant of the enzyme myeloperoxidase (MPO), which we call “MPO–E-selectin ligand” (MPO–EL). MPO is normally found in lysosomes, but G-CSF triggers both MPO-EL production and its presentation on the leukocyte surface. MPO–EL is catalytically active. The production of toxic oxidizing agents by MPO–EL in apposition to E-selectin–bearing endothelium kills the cell. This previously unrecognized function of G-CSF and MPO raises additional insights into the biology of vascular injury during inflammatory reactions.
Keywords: glycosylation, leukemia, hematopoietic stem cell transplantation
Abstract
The host defense response critically depends on the production of leukocytes by the marrow and the controlled delivery of these cells to relevant sites of inflammation/infection. The cytokine granulocyte-colony stimulating factor (G-CSF) is commonly used therapeutically to augment neutrophil recovery following chemo/radiation therapy for malignancy, thereby decreasing infection risk. Although best known as a potent inducer of myelopoiesis, we previously reported that G-CSF also promotes the delivery of leukocytes to sites of inflammation by stimulating expression of potent E-selectin ligands, including an uncharacterized ∼65-kDa glycoprotein. To identify this ligand, we performed integrated biochemical analysis and mass spectrometry studies of G-CSF–treated primary human myeloid cells. Our studies show that this novel E-selectin ligand is a glycoform of the heavy chain component of the enzyme myeloperoxidase (MPO), a well-known lysosomal peroxidase. This specialized MPO glycovariant, referred to as “MPO–E-selectin ligand” (MPO–EL), is expressed on circulating G-CSF–mobilized leukocytes and is naturally expressed on blood myeloid cells in patients with febrile leukocytosis. In vitro biochemical studies show that G-CSF programs MPO–EL expression on human blood leukocytes and marrow myeloid cells via induction of N-linked sialofucosylations on MPO, with concomitant cell surface display of the molecule. MPO–EL is catalytically active and mediates angiotoxicity on human endothelial cells that express E-selectin. These findings thus define a G-CSF effect on MPO chemical biology that endows unsuspected functional versatility upon this enzyme, unveiling new perspectives on the biology of G-CSF and MPO, and on the role of E-selectin receptor/ligand interactions in leukocyte migration and vascular pathology.
Granulocyte-colony stimulating factor (G-CSF) serves a critical role in native immunity by directing elaboration of myeloid cells and their cytocidal arsenal (1), and the characteristic leukocytosis (the “leukemoid reaction”) of inflammatory reactions is fueled by physiologic up-regulation of G-CSF (2). Binding of G-CSF to its receptor on early hematopoietic progenitors and on intermediate-stage myeloid progenitors activates proliferation pathways, resulting in programmed expansion and release (“mobilization”) of progenitors and mature myeloid cells into the peripheral circulation (2). This cytokine is in routine clinical use, administered to stimulate myelopoiesis after chemo- or radiotherapy, to treat cyclic neutropenia, and to mobilize progenitor cells for hematopoietic stem cell transplantation (HSCT) (3).
Leukocyte production stimulated by G-CSF is not sufficient to mediate host defense, as it is essential to deliver the leukocyte cytocidal arsenal at pertinent sites of inflammation. Leukocyte recruitment begins with tethering and rolling of blood-borne cells on the endothelial lining under hemodynamic shear conditions, followed by activation of integrins and firm adhesion to the vessel wall, culminating in transendothelial migration (4). The initial shear-resistant adherence of leukocytes to the endothelial surface is principally mediated by selectin receptor/ligand interactions (4). The selectin family consists of “leukocyte-specific” L-selectin and “vascular selectins” P- and E-selectin, each of which binds to specialized carbohydrate determinants containing an α2,3-linked sialic acid substitution on galactose, and an α1,3-linked fucose modification on N-acetylglucosamine, prototypically displayed as sialyl Lewis x (sLex), a tetrasaccharide recognized by monoclonal antibody, clone HECA-452 (4). Various glycoproteins that display sLex have been described as E-selectin ligands in mouse and human leukocytes. The principal E-selectin ligand on human and mouse myeloid cells is the molecule known as cutaneous lymphocyte antigen (CLA), a heavily sialofucosylated glycoform of P-selectin glycoprotein ligand-1 (PSGL-1) (5, 6). A molecule known as E-selectin ligand-1 (ESL-1) serves as an E-selectin ligand on mouse neutrophils (7), but it is not expressed on human neutrophils nor on human hematopoietic stem and progenitor cells (HSPCs) (8). Biochemical assays have indicated that the leukocyte β2 integrins LFA-1 (lymphocyte function-associated antigen 1 or CD11a/CD18) and MAC-1 (macrophage-1 antigen or CD11b/CD18) can serve as E-selectin ligands on human leukocytes (9). CD43 has also been implicated to be an E-selectin ligand on mouse neutrophils and activated T cells (10), and we have reported that CD43 on human T cells can bind E-selectin, but not P-selectin (11). On human hematopoietic cells, two glycoproteins function as major counter receptors for E-selectin: the CLA molecule (5, 6) and a glycoform of CD44 known as hematopoietic cell E-/L-selectin ligand (HCELL) (6, 12). Studies from our laboratory have shown that following clinical G-CSF administration to mobilize hematopoietic progenitors for HSCT, circulating myeloid cells exhibit increased adherence to TNF-stimulated vascular endothelium compared with native leukocytes (NLs). Biochemical analysis showed that this augmented endothelial adherence of G-CSF–mobilized human leukocytes (MLs) was due to increased E-selectin ligand activity, in part mediated by expression of a novel E-selectin ligand, a membrane glycoprotein with a molecular weight of ∼65 kDa (12).
To identify this previously uncharacterized E-selectin ligand, we used lectin affinity chromatography, combined with mass spectrometry peptide fingerprinting and Western blot analysis. Our studies show that this G-CSF–induced glycoprotein is a unique glycoform of myeloperoxidase (MPO), designated here as “MPO–E-selectin ligand” (MPO–EL), which contains E-selectin binding determinants on the ∼65-kDa heavy chain. Although MPO is usually expressed within the lysosome (azurophilic granules), our data show that G-CSF induces cell membrane expression of MPO concomitant with the elaboration of a glycovariant of the heavy chain, carrying specialized N-linked sialofucosylated determinants. MPO–EL is catalytically active and coincubation of MPO–EL-bearing myeloid cells with E-selectin–bearing endothelial cells engenders angiotoxicity. Our findings thus highlight the capacity of posttranslational carbohydrate modifications to shape protein function, revealing previously unrecognized versatility of MPO biology tuned by G-CSF that could incite vascular injury in vivo.
Results
Physiologic Expression of the Novel E-Selectin Ligand on Circulating Myeloid Cells.
Our prior studies showed that pharmacologic doses of G-CSF induce expression of the uncharacterized ∼65 kDa ligand on circulating human myeloid cells. To determine whether expression of this novel E-selectin ligand on circulating human myeloid cells is a general effect of G-CSF stimulation (i.e., not solely consequent to pharmacologic G-CSF use), we analyzed E-selectin ligands displayed on circulating leukocytes of patients presenting to hospital with fever and leukocytosis (leukocyte counts >10 × 106/mL; febrile leukocytosis, FL). To this end, Western blot analysis using E-selectin–Ig chimera (E–Ig) as probe was performed on lysates from human peripheral blood leukocytes mobilized with G-CSF (MLs), from blood leukocytes of healthy subjects (NLs) and from FL. In each group, over 14 separate samples were analyzed, with consistent results among all samples. The loaded material was normalized among samples depending on the cell number (Fig.1A) or protein quantity (Fig. S1). As shown by E–Ig reactivity on Western blot (Fig. 1A), both ML and FL display an increased expression of E-selectin ligands in comparison with NLs, with both cell populations prominently displaying the ∼65-kDa structure. Thus, G-CSF, whether physiologically generated or clinically administered, programs human myeloid cell expression of this E-selectin ligand.
Fig. 1.
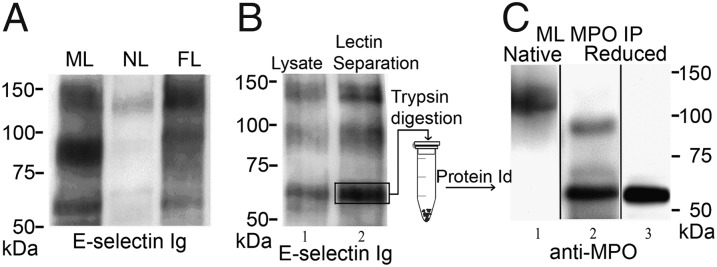
Expression of the ∼65-kDa E-selectin ligand is characteristic of MLs and circulating myeloid cells during leukemoid reactions (FL), and comprises the heavy chain of MPO. (A) Lysates of mobilized leukocytes (MLs), native leukocytes (NLs), and circulating myeloid cells of patients with febrile leukocytosis (FL), normalized for input cell number, were resolved by SDS/PAGE and stained in Western blot with E-selectin Ig chimera (E–Ig). A representative blot of multiple patient samples (n > 14 for each group) is shown. Expression of the ∼65-kDa glycoprotein is characteristic of G-CSF–primed cells (MLs and FLs), but not of NLs. (B) Lysates of MLs (lane 1) and WGA lectin chromatography-purified glycoproteins (lane 2) resolved on reducing 7.5% SDS/PAGE gel and immunoblotted with E-selectin–Ig chimera. E-selectin ligands are present at ∼140 kDa (PSGL-1), ∼100 kDa (HCELL), and at ∼65 kDa. (C) Representative results of Western blots of MPO immunoprecipitates (IPs) from MLs resolved under nonreducing (lane 1) or reducing conditions (lanes 2 and 3) and stained with anti-MPO mAb. In nonreduced gels, the mature homodimer of ∼140 kDa is evident (lane 1). Under reducing conditions, Western blot with anti-MPO mAb 2C7 (lane 2) reveals bands at ∼90 kDa (precursor) and ∼65 kDa (heavy chain) or only the ∼65-kDa band when stained with anti-MPO mAb 3D3 (lane 3).
Identification of the ∼65-kDa E-Selectin Ligand Expressed on Myeloid Cells.
To enrich for sialylated glycoproteins, lysates of MLs were passed over wheat germ agglutinin (WGA) lectin columns. The bound glycoprotein fraction was eluted, resolved by SDS/PAGE, and E-selectin ligands were detected by staining with E–Ig. As shown in parallel gel lanes normalized for input protein content, the WGA chromatography step preserved and concentrated E-selectin ligands present in ML lysates (Fig. 1B, lanes 1 and 2). A series of gel bands were then excised from the 60- to 70-kDa region, trypsin digested, and subjected to peptide mass fingerprinting. Mass spectrometry coupled with bioinformatics analysis identified the ∼65-kDa protein as the heavy chain of MPO, a well-recognized lysosomal enzyme whose synthesis is known to be induced by G-CSF (1, 13, 14). To further characterize the ∼65-kDa molecule, MPO was immunoprecipitated (IP) from ML lysates and resolved by SDS/PAGE under nonreducing or reducing conditions. Under nonreducing conditions, Western blotting with anti-MPO mAb (clone 2C7) revealed the mature molecule, a heme-containing glycoprotein of ∼140 kDa (Fig. 1C, lane 1), which consists of two catalytically active monomers of ∼70–90 kDa, each composed of a ∼55- to 65-kDa heavy chain and a ∼10- to 15-kDa light chain (15, 16). Under reducing conditions, the same MPO antibody revealed the monomer at ∼90 kDa and the heavy chain at ∼65 kDa (Fig. 1C, lane 2), whereas another anti-MPO mAb (clone 3D3) stained only the heavy chain (Fig. 1C, lane 3). Thus, the ∼65-kDa E-selectin ligand is the heavy chain component of a glycoform of mature MPO.
G-CSF Up-Regulates Expression of Catalytically Active MPO on the Surface of Mobilized Human, but Not Mouse, Myeloid Cells.
To assess cell surface expression of MPO, granulocyte, monocyte, lymphocyte, and CD34+ cell fractions of human NLs and MLs were analyzed by flow cytometry. Native granulocytes (NGs) and native monocytes (NMs) express minimal surface MPO, whereas mobilized granulocytes (MGs) and mobilized monocytes (MMs) display uniformly high levels of membrane MPO (Fig. 2 A and B); lymphocytes and CD34+ cells do not express cell surface MPO. To determine whether in vivo G-CSF administration stimulates MPO surface expression in mouse myeloid cells, C57BL/6 or BALB/c mice were treated for 5 d with recombinant human G-CSF or with saline (control) and blood was collected for colony-forming unit (cfu) assays and for flow cytometry analysis. In mice treated with G-CSF but not in those treated with saline, cfu assays showed that hematopoietic progenitors were mobilized, and there was increase in circulating Sca-1+/c-kit+/lin− cells (Fig. S2A). However, flow cytometry analysis using anti-MPO mAb indicated that, in contrast to results in humans, G-CSF–mobilized mouse leukocytes do not express cell surface MPO (Fig. S2B).
Fig. 2.

G-CSF up-regulates expression of catalytically active MPO on the surface of mobilized myeloid cells. (A) Representative MPO flow cytometry histograms are shown of native granulocytes (NGs) and native mononuclear cells (NMs) in comparison with mobilized granulocytes (MGs) and mobilized mononuclear cells (MMs). (B) Cumulative results of flow cytometry analysis for MPO cell surface expression typical for mononuclear cells and granulocytes. Values represent mean ± SD of percent positive cells from multiple donors (n = 15), *P < 0.001. (C) Representative Western blot of MPO immunoprecipitates (mAb 1A1) from lysates of surface biotinylated (+) or nonbiotinylated (−) MLs and NLs resolved on reducing SDS/PAGE gel. Biotin-labeled (membrane expressed) MPO was revealed with streptavidin-HRP. (D) MPO activity on the surface of NL and ML was evaluated by spectrophotometric detection of o-phenylenediamine dihydrochloride (OPD), a chromogenic peroxidase product (n = 5).
Extensive cell washes under high salt conditions (1.5 M NaCl) did not alter ML surface MPO levels, nor did digestion of glycosylphosphatidylinositol (GPI) anchors with phosphatidylinositol-specific phospholipase C, indicating that MPO is a membrane-expressed protein. To confirm MPO surface expression in human myeloid cells, a complementary approach was undertaken whereby cell membrane proteins from freshly collected NLs and MLs were labeled with biotin. MPO immunoprecipitates from lysates of surface biotinylated cells were then resolved by electrophoresis and blotted with streptavidin-HRP. Western blots revealed biotinylated MPO prominently in MLs, whereas NLs showed only trace amounts on the cell surface (Fig. 2C). To determine whether surface MPO is enzymatically active, ML membrane proteins labeled with biotin were captured with streptavidin-conjugated beads and incubated with peroxidase substrate. Spectrophotometric analysis revealed a linear correlation between MPO activity and input cell number (Fig. 2D). Collectively, these results indicate that in vivo G-CSF treatment markedly up-regulates expression of catalytically active MPO on the surface of mobilized human myeloid cells.
G-CSF Induces Expression of Cell Surface MPO That Binds E-Selectin.
To examine the effects of in vivo G-CSF administration on MPO membrane expression and E-selectin binding activity, lysates of equivalent numbers of leukocytes from bone marrow aspirates (BMs), NLs and MLs were immunoprecipitated with anti-MPO mAb 1A1 and blotted with anti-MPO mAb 3D3, which recognizes the heavy chain (Fig. 3A) or with E–Ig (Fig. 3B). As shown in Fig. 3 A and B, although there was little variation in MPO staining among the samples, MPO from MLs displayed markedly more E–Ig reactivity than that of BMs or NLs. This E–Ig-reactive MPO glycoform was recognized by HECA-452 in Western blots of immunoprecipitated MPO from different samples of MLs (Fig. S3). Alternatively, surface biotinylated proteins were captured with strepavidin-coupled agarose beads and blotted with mAb HECA-452 or with anti-MPO mAb 3D3 (Fig. S4). Two previously characterized E-selectin ligands, CLA at ∼130 kDa and HCELL at ∼90 kDa, were stained with HECA-452 in MLs and NLs, but HECA-452 staining at a ∼65-kDa band was only observed in ML lysates (Fig. S4A); this band was recognized by mAb 3D3 (Fig. S4B). None of these molecules were detected in streptavidin pull-downs from lysates of cells that were not biotinylated (control). Thus, G-CSF induces membrane expression of MPO containing a heavy chain glycovariant that binds E-selectin (MPO–EL).
Fig. 3.

G-CSF induces cell surface expression and E-selectin ligand activity of MPO. (A) MPO immunoprecipitates (IPs) from lysates of bone marrow myeloid cells (BMs), NLs, and MLs stained in Western blot with anti-MPO mAb clone 3D3. (B) MPO IPs resolved on reducing SDS/PAGE and stained with E-selectin Ig chimera. (C) Flow cytometry analysis of NLs, BMs, and MLs cultured for 48 h without G-CSF (−) or with G-CSF (+). Values represent mean ± SD of percent MPO-positive cells from multiple donors (n > 30), *P < 0.001. (D) Western blots of MPO IPs from BMs, NLs, and MLs treated (+) or not treated (−) with G-CSF and stained with anti-MPO mAb 3D3. (E) MPO IPs from BMs, NLs, and MLs treated (+) or not treated (−) with G-CSF were probed with E-selectin–Ig chimera in Western blots.
To assess whether induction of myeloid cell surface MPO expression by G-CSF in vivo is a primary effect of this cytokine, we examined the effect of G-CSF in vitro on MPO surface expression in BM, NL, and ML populations (Fig. S5). The various cell types were then incubated for 48 h with human G-CSF at 10 ng/mL, a dose reflecting serum concentrations of G-CSF following in vivo administration (12, 17). Changes in membrane expression of MPO were measured by cell surface staining with anti-MPO mAb. G-CSF treatment of NL and BM cells dramatically increased expression of surface MPO (Fig. 3C), but had no significant effect on the already high levels in ML (Fig. 3C). From these cells (equivalent cell number), MPO was immunoprecipitated, then resolved on SDS/PAGE, and blotted with anti-MPO mAb 3D3 (Fig. 3D) or blotted with E–Ig (Fig. 3E). As shown in Fig. 3E, G-CSF markedly stimulated E–Ig reactivity of the MPO heavy chain in both BMs and NLs, without significant increase in MPO quantity (Fig. 3D). Notably, in vitro exposure to G-CSF did not further increase E–Ig reactivity of MPO in MLs (Fig. 3E). Altogether, these results show that G-CSF induces MPO–EL expression on human myeloid cells by inducing E-selectin binding determinants exclusively on the MPO heavy chain.
To determine whether MPO–EL is expressed on pathologic myeloid cells, we performed Western blot analysis of acute myeloid leukemia (AML) myeloblasts of various subtypes: one M1, one M2, and two M3 (i.e., acute promyelocytic leukemia, APL). Equal amounts of total protein from cell lysates were stained in Western blots with E–Ig and with anti-MPO mAb 3D3, and, as loading control, Grb2 expression level was monitored with anti-Grb2 mAb (Fig. S6). We observed high MPO signal intensity in Western blot in all AML samples, relatively more intense in M3 specimens. As shown by staining with E-selectin–Ig, all AML samples tested express the MPO–EL heavy chain at ∼65 kDa (Fig. S6A).
N-Glycan Processing Is Required for G-CSF–Induced Sialofucosylations and MPO–EL Membrane Expression.
To assess the molecular basis of G-CSF effects on MPO structural biology, we used metabolic inhibitors of glycosylation. Glycoproteins bearing high mannose N-glycans, such as those typically found on lysosomal MPO, require glycosidase remodeling to display sialofucosylated determinants on lactosamine scaffolds. This remodeling normally takes place in the ER and Golgi complex and is a prerequisite for cell surface expression of E-selectin ligands. To assess whether such glycan processing is needed for cell membrane expression of MPO, we used deoxymannojirimycin (DMJ), an α-mannosidase inhibitor that hinders remodeling of high mannose N-glycans into complex carbohydrates. To this end, BM cells were first treated with sialidase to eliminate existing E-selectin binding determinants or treated with buffer alone. Thereafter, cells were cultured with or without G-CSF in the presence or absence of DMJ to analyze de novo expression of HECA-452 determinants and of membrane MPO. After 48 h, in the absence of G-CSF and DMJ, sialidase-treated BM cells recovered expression of HECA-452–reactive glycans to a level approximating that of buffer-treated cells (Fig. 4A). Importantly, cells treated with sialidase followed by incubation with G-CSF showed increased expression of HECA-452 determinants compared with cells not treated with G-CSF (Fig. 4B). However, addition of DMJ markedly diminished the G-CSF–induced expression of HECA-452–reactive glycans (Fig. 4 B and C). These findings suggest that G-CSF stimulates de novo expression of HECA-452–reactive moieties predominantly on N-glycans. The residual HECA-452 determinants displayed in the presence of DMJ likely reflect the native contribution(s) of sialofucosylated O-linked glycans found on both glycoproteins and glycolipids (18, 19).
Fig. 4.

N-glycan processing is required for G-CSF–induced surface HECA-452 reactivity and MPO–EL expression. Flow cytometry analysis of BM cells treated with sialidase and cultured with G-CSF or G-CSF and DMJ: (A–C) HECA-452 reactivity and (D–F) MPO expression. (A and C) BMs in buffer alone (buffer treated) or treated with sialidase and cultured for 48 h (48-h recovery after sialidase). (A) Sialidase efficiency was confirmed before cell culturing (sialidase treated). (B and E) BM cells treated with sialidase and cultured for 48 h without G-CSF (48-h recovery after sialidase) or with G-CSF (48 h after sialidase + G-CSF). (B and E) BM cells treated with sialidase and cultured for 48 h with G-CSF and DMJ (48 h after sialidase + G-CSF + DMJ). (C) Cumulative results for expression of HECA-452 determinants on cells treated with sialidase and then cultured ± G-CSF and ± DMJ. Cells were isolated from different donors (n = 5), *P < 0.001. (F) Cumulative results for MPO surface expression on cells treated with sialidase and then cultured ± G-CSF and ± DMJ. Cells were isolated from multiple donors (n = 5), *P < 0.001.
Treatment of cells with sialidase did not induce MPO expression (Fig. 4D) nor did it alter the G-CSF–induced expression of membrane MPO (Fig. 4E). However, G-CSF–induced MPO expression was significantly attenuated in the presence of DMJ, to levels equivalent to that of cells not treated with G-CSF (Fig. 4 E and F). Collectively, these data show that surface expression of MPO–EL is dependent on G-CSF–induced remodeling of MPO N-glycans into complex sialofucosylated structures.
MPO–EL Is Catalytically Active and Mediates Myeloid Cell Angiotoxicity.
To evaluate the effect of G-CSF on cell surface MPO enzymatic activity, NLs were cultured in the presence or absence of G-CSF for 48 h. Cells were then surface biotinylated, lysed, and membrane proteins were captured with streptavidin-agarose beads. The membrane proteins were incubated with peroxidase substrate and enzymatic activity was measured by spectrophotometric analysis of colorimetric product. This analysis showed that G-CSF stimulation increases surface presentation of MPO, which engenders enhanced peroxidase activity on cells treated with G-CSF in vitro (Fig. 5A). However, this induction was inhibited by the addition of DMJ, demonstrating that N-glycan remodeling is required for the G-CSF–induced surface expression of catalytically active MPO–EL (Fig. 5A).
Fig. 5.

Inhibition of E-selectin receptor/ligand interactions and of MPO activity suppresses G-CSF–induced myeloid cell angiotoxicity. (A) Spectrophotometric detection of membrane MPO activity from NLs cultured in the presence or absence of G-CSF and DMJ. Values represent the relative change in absorbance with respect to substrate alone (n = 5), *P < 0.05. (B) Endothelial cell death of TNF-stimulated HUVEC cultures was evaluated in the absence of leukocytes (no L) or after coculture with MLs, NLs, or with G-CSF–treated NLs without or with DMJ. DMJ treatment reduces G-CSF–induced myeloid cell angiotoxicity to levels observed with NL coincubation. Values represent mean ± SD of percent endothelial cell death (n = 5), *P < 0.05. (C) Percentage endothelial cell death was evaluated in TNF-stimulated HUVEC monolayers in the presence (+) or absence (−) of G-CSF for endothelial cells alone (no L) or in coculture with myeloid cells (NLs, BMs, or MLs). Myeloid cell angiotoxicity was evaluated in the presence (+) or absence (–) of an E-selectin blocking antibody or MPO inhibitor (4-ABAH). Values represent mean ± SD of percent endothelial cell death from multiple donors (n > 20), *P < 0.05.
When released into the extracellular milieu, the cytotoxic oxidants produced by MPO damage surrounding cells (20). To evaluate whether the presence of membrane-expressed MPO–EL could directly trigger cytotoxicity on juxtaposed E-selectin–expressing endothelial cells, we incubated primary human umbilical vein endothelial cells (HUVECs) with TNF-α for 6 h to mimic inflammation conditions. Stimulated HUVECs were then cultured in absence of leukocytes (no L), or with addition of MLs or NLs, and endothelial cell viability was monitored by trypan blue exclusion. As shown in Fig. 5B, a basal level of endothelial cell death was observed after 48 h in the HUVEC cultures that were stimulated with TNF-α, and coincubation with NLs increased cell death above this baseline. However, angiotoxicity was significantly enhanced by incubation with MLs or with G-CSF–treated NLs (Fig. 5B). Importantly, coincubation with DMJ significantly reduced G-CSF–induced angiotoxicity (Fig. 5B).
Inhibition of E-Selectin Receptor/Ligand Interactions or MPO Activity Prevents G-CSF–Induced Myeloid Cell Angiotoxicity.
To determine the mediators of G-CSF–induced angiotoxicity, TNF-α–stimulated HUVECs were incubated with NLs, BMs, or MLs in the presence or absence of G-CSF. In all cultures, endothelial cell death was markedly increased by coincubation with MLs or G-CSF–treated NLs or BMs, compared with untreated NL or BM cells (Fig. 5C). There was no significant increase in angiotoxicity with G-CSF–treated MLs compared with untreated MLs. Coincubation with an E-selectin function-blocking antibody reduced endothelial cell death in ML cultures and in NL or BM cultures treated with G-CSF (Fig. 5C). Importantly, incubation with the inhibitor 4-aminobenzoic acid hydrazide (4-ABAH), a suicidal substrate for MPO (21), most effectively decreased endothelial cell death in all cultures (Fig. 5C).
Discussion
Under physiologic conditions, MPO synthesis occurs at the promyelocyte stage and stops as myeloid cells reach maturity. MPO biosynthesis follows a complex succession of processing steps that direct accumulation of this glycoprotein within intracellular azurophilic granules (22). Although other studies have shown that G-CSF triggers increased expression of MPO (13), the results here present first evidence of a specific effect of G-CSF on MPO structural biology, highlighting the role of posttranslational glycosylations in conferring both cell surface expression and function as an adhesion molecule. We previously reported that G-CSF stimulates expression of glycosyltransferases within myeloid cells that direct synthesis of sialofucosylated glycans on various proteins (12). Our present dataset shows that a major target for these glycosyltransferases is the heavy chain of the enzyme MPO. Studies using the metabolic inhibitor DMJ show that N-glycan processing of the MPO heavy chain within the Golgi is required for both G-CSF–induced decoration of MPO with sialofucosylated glycans and MPO membrane expression. Thus, G-CSF programs E-selectin binding activity and also promotes release of this host defense enzyme from constraint of lysosomal storage to presentation at the cell surface. Heretofore, a leukocyte surface molecule mediating both leukocyte trafficking and cytotoxicity has not been identified.
Engagement of vascular E-selectin with corresponding ligands on the surface of circulating leukocytes guarantees that the host defense arsenal is delivered to the correct inflammatory “address.” On their way to membrane presentation, the canonical E-selectin ligands HCELL and CLA are decorated by Golgi enzymes with sialofucosylated glycans, which render E-selectin adhesive properties. Under G-CSF influence, MPO is similarly processed by the Golgi enzymes and gains E-selectin binding activity but, unlike the canonical E-selectin ligands, MPO–EL is a catalytically active enzyme that generates powerful reactive oxygen species. The finding here that MPO is presented on the leukocyte surface as a glycoform that can localize this cytocidal agent in direct contact with endothelial cells suggests the possibility that E-selectin–mediated MPO–EL binding and consequent production of oxidizing agents could serve to affix toxic metabolites to the endothelium, thereby heightening vascular injury within inflamed tissue(s). Moreover, MPO catalyzes oxidation of vascular nitric oxide (NO), resulting in consumption of NO (23, 24) and formation of highly reactive nitrite species that participate in deleterious protein tyrosine nitration (25). Endogenous NO is a critical anti-inflammatory and antiatherogenic factor that inhibits endothelial activation (26), supports vasodilatation, and prevents leukocyte adhesion (27) by suppressing expression of E-selectin. Thus, through its enzymatic role in reducing NO bioavailability, MPO sustains expression of E-selectin on endothelial cells that further supports binding of leukocytes bearing E-selectin ligands such as MPO–EL. This loop would serve to compound vascular injury. Our in vitro studies of myeloid cells coincubated with HUVECs indicate that G-CSF–treated myeloid cells are angiotoxic to E-selectin–expressing endothelial cells; this effect is ameliorated by interruption of E-selectin receptor/ligand interactions and by inhibition of MPO activity. Although further studies are necessary to directly assess the role(s) of MPO–EL in vasculopathy, these findings offer novel perspectives on the association between MPO expression and vascular injury: in vivo, MPO–EL could serve as a key effector of sickle cell crises (28), myocardial infarction and coronary artery disease (29, 30), atherosclerosis (31, 32), and stroke (33), conditions which have all been linked to MPO. Indeed, G-CSF administration can trigger angina and myocardial infarction (34, 35). Importantly, life-threatening sickle cell crisis is induced by G-CSF administration in humans (36) and, notably, anti–E-selectin agents can ameliorate this process (37), suggesting a role for MPO–EL in this vasculopathy.
Our data showing increased MPO membrane expression in leukocytes isolated from G-CSF–mobilized human blood and from patients with febrile leukocytosis (leukomoid reactions), but not in leukocytes from G-CSF–mobilized mouse blood, highlight key species disparities that can limit parallels drawn regarding effects of G-CSF on vascular integrity in mouse and in man. It is known that MPO levels in mouse leukocytes are ∼10% that of humans (38) and it is also well recognized that humans and mice differ critically in the types of glycoproteins that serve as E-selectin ligands (8, 39). Collectively, these differences may account for the fact that, in contrast to the well-recognized life-threatening complications associated with G-CSF administration in humans with sickle cell disease (36), mice bearing human hemoglobin S (“sickle cell” mice) tolerate G-CSF administration without any adverse effects (40).
Apart from providing new insights regarding the molecular basis of complications associated with pharmacologic administration of G-CSF, the finding that MPO–EL expression is characteristic of circulating myeloid cells in leukemoid reactions offers novel perspectives on the role(s) of MPO in vascular injury associated with various sepsis and other systemic inflammatory reactions. Notably, G-CSF administration induces flares in patients with vasculitis (41, 42) and can induce cutaneous leukocytoclastic vasculitis in otherwise healthy donors for hematopoietic stem cell transplantation (43), raising the possibility that MPO–EL may be etiologic in such exacerbations. In fact, leukocyte surface MPO expression is known to be associated with development of systemic vasculitides such as Wegener’s granulomatosis and Churg–Strauss syndrome (44–46), and although our understanding of MPO surface presentation is incomplete, it is well known that membrane MPO serves as an antigenic target for antineutrophil cytoplasmic autoantibodies (ANCAs).
The identification of MPO–EL serves as a prime example of the capacity of posttranslational glycan modifications to engender distinct biology on target molecules. The data presented here provide new insights on the biology of G-CSF and also unveil previously unrecognized biologic pleiotropism of MPO and of E-selectin ligands. Specifically, G-CSF induces both structural and topological changes in MPO expression, placing this well-recognized “lysosomal” effector of cytotoxicity onto the leukocyte surface in a form that is primed to engage endothelial beds expressing E-selectin. Among leukocyte adhesion molecules, MPO–EL stands out in its capacity to serve the dual function of mediating both leukocyte trafficking and elaboration of cytotoxic products. These findings focus new attention on the multipurpose role(s) of E-selectin ligands in vascular pathology and provide novel mechanistic insights on how interruption of E-selectin receptor/ligand interactions may serve to prevent vascular complications associated with G-CSF administration and, more broadly, sickle cell crises, ischemic vascular events, sepsis-related vascular compromise, and systemic vasculitic syndromes.
Materials and Methods
Cells.
All human cells were obtained and used in accordance with the procedures approved by the Human Experimentation and Ethics Committees of Partners Cancer Care Institutions (Massachusetts General Hospital, Brigham and Women’s Hospital, and Dana-Farber Cancer Institute). To ensure consistency in sample preparation and obtain data most relevant to native human biology, cells from blood and marrow samples were uniformly processed for relevant analyses within 2 h of collection.
Additional Methods.
Please refer to SI Materials and Methods for details.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants P01 HL107146 (National Heart, Lung, and Blood Institute Program of Excellence in Glycosciences), R21 DK075012, and R01 HL073714, and by the Team Jobie Leukemia Research Fund.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1320833111/-/DCSupplemental.
References
- 1.Berliner N, et al. Granulocyte colony-stimulating factor induction of normal human bone marrow progenitors results in neutrophil-specific gene expression. Blood. 1995;85(3):799–803. [PubMed] [Google Scholar]
- 2.Panopoulos AD, Watowich SS. Granulocyte colony-stimulating factor: Molecular mechanisms of action during steady state and ‘emergency’ hematopoiesis. Cytokine. 2008;42(3):277–288. doi: 10.1016/j.cyto.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elfenbein GJ, Sackstein R. Primed marrow for autologous and allogeneic transplantation: A review comparing primed marrow to mobilized blood and steady-state marrow. Exp Hematol. 2004;32(4):327–339. doi: 10.1016/j.exphem.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 4.Sackstein R. The lymphocyte homing receptors: Gatekeepers of the multistep paradigm. Curr Opin Hematol. 2005;12(6):444–450. doi: 10.1097/01.moh.0000177827.78280.79. [DOI] [PubMed] [Google Scholar]
- 5.Fuhlbrigge RC, Kieffer JD, Armerding D, Kupper TS. Cutaneous lymphocyte antigen is a specialized form of PSGL-1 expressed on skin-homing T cells. Nature. 1997;389(6654):978–981. doi: 10.1038/40166. [DOI] [PubMed] [Google Scholar]
- 6.Sackstein R. The bone marrow is akin to skin: HCELL and the biology of hematopoietic stem cell homing. J Invest Dermatol. 2004;122(5):1061–1069. doi: 10.1111/j.0022-202X.2004.09301.x. [DOI] [PubMed] [Google Scholar]
- 7.Hidalgo A, Peired AJ, Wild MK, Vestweber D, Frenette PS. Complete identification of E-selectin ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity. 2007;26(4):477–489. doi: 10.1016/j.immuni.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merzaban JS, et al. Analysis of glycoprotein E-selectin ligands on human and mouse marrow cells enriched for hematopoietic stem/progenitor cells. Blood. 2011;118(7):1774–1783. doi: 10.1182/blood-2010-11-320705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kotovuori P, et al. The vascular E-selectin binds to the leukocyte integrins CD11/CD18. Glycobiology. 1993;3(2):131–136. doi: 10.1093/glycob/3.2.131. [DOI] [PubMed] [Google Scholar]
- 10.Alcaide P, et al. The 130-kDa glycoform of CD43 functions as an E-selectin ligand for activated Th1 cells in vitro and in delayed-type hypersensitivity reactions in vivo. J Invest Dermatol. 2007;127(8):1964–1972. doi: 10.1038/sj.jid.5700805. [DOI] [PubMed] [Google Scholar]
- 11.Fuhlbrigge RC, King SL, Sackstein R, Kupper TS. CD43 is a ligand for E-selectin on CLA+ human T cells. Blood. 2006;107(4):1421–1426. doi: 10.1182/blood-2005-05-2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dagia NM, et al. G-CSF induces E-selectin ligand expression on human myeloid cells. Nat Med. 2006;12(10):1185–1190. doi: 10.1038/nm1470. [DOI] [PubMed] [Google Scholar]
- 13.Allen RC, Stevens PR, Price TH, Chatta GS, Dale DC. In vivo effects of recombinant human granulocyte colony-stimulating factor on neutrophil oxidative functions in normal human volunteers. J Infect Dis. 1997;175(5):1184–1192. doi: 10.1086/595866. [DOI] [PubMed] [Google Scholar]
- 14.Sato N, Kashima K, Tanaka Y, Shimizu H, Mori M. Effect of granulocyte-colony stimulating factor on generation of oxygen-derived free radicals and myeloperoxidase activity in neutrophils from poorly controlled NIDDM patients. Diabetes. 1997;46(1):133–137. doi: 10.2337/diab.46.1.133. [DOI] [PubMed] [Google Scholar]
- 15.Strömberg K, Persson AM, Olsson I. The processing and intracellular transport of myeloperoxidase. Modulation by lysosomotropic agents and monensin. Eur J Cell Biol. 1986;39(2):424–431. [PubMed] [Google Scholar]
- 16.Koeffler HP, Ranyard J, Pertcheck M. Myeloperoxidase: Its structure and expression during myeloid differentiation. Blood. 1985;65(2):484–491. [PubMed] [Google Scholar]
- 17.Faulkner LB, et al. G-CSF serum pharmacokinetics during peripheral blood progenitor cell mobilization: Neutrophil count-adjusted dosage might potentially improve mobilization and be more cost-effective. Bone Marrow Transplant. 1998;21(11):1091–1095. doi: 10.1038/sj.bmt.1701241. [DOI] [PubMed] [Google Scholar]
- 18.Handa K, Stroud MR, Hakomori S. Sialosyl-fucosyl Poly-LacNAc without the sialosyl-Lex epitope as the physiological myeloid cell ligand in E-selectin-dependent adhesion: Studies under static and dynamic flow conditions. Biochemistry. 1997;36(41):12412–12420. doi: 10.1021/bi971181t. [DOI] [PubMed] [Google Scholar]
- 19.Nimrichter L, et al. E-selectin receptors on human leukocytes. Blood. 2008;112(9):3744–3752. doi: 10.1182/blood-2008-04-149641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang C, et al. Endothelial dysfunction is induced by proinflammatory oxidant hypochlorous acid. Am J Physiol Heart Circ Physiol. 2001;281(4):H1469–H1475. doi: 10.1152/ajpheart.2001.281.4.H1469. [DOI] [PubMed] [Google Scholar]
- 21.Kettle AJ, Gedye CA, Winterbourn CC. Mechanism of inactivation of myeloperoxidase by 4-aminobenzoic acid hydrazide. Biochem J. 1997;321(Pt 2):503–508. doi: 10.1042/bj3210503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansson M, Olsson I, Nauseef WM. Biosynthesis, processing, and sorting of human myeloperoxidase. Arch Biochem Biophys. 2006;445(2):214–224. doi: 10.1016/j.abb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 23.Eiserich JP, et al. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science. 2002;296(5577):2391–2394. doi: 10.1126/science.1106830. [DOI] [PubMed] [Google Scholar]
- 24.Eiserich JP, et al. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 1998;391(6665):393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- 25.Lau D, Baldus S. Myeloperoxidase and its contributory role in inflammatory vascular disease. Pharmacol Ther. 2006;111(1):16–26. doi: 10.1016/j.pharmthera.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 26.De Caterina R, et al. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995;96(1):60–68. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kubes P, Suzuki M, Granger DN. Nitric oxide: An endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA. 1991;88(11):4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saleh AW, Hillen HF, Duits AJ. Levels of endothelial, neutrophil and platelet-specific factors in sickle cell anemia patients during hydroxyurea therapy. Acta Haematol. 1999;102(1):31–37. doi: 10.1159/000040964. [DOI] [PubMed] [Google Scholar]
- 29.Baldus S, et al. CAPTURE Investigators Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation. 2003;108(12):1440–1445. doi: 10.1161/01.CIR.0000090690.67322.51. [DOI] [PubMed] [Google Scholar]
- 30.Zhang R, et al. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA. 2001;286(17):2136–2142. doi: 10.1001/jama.286.17.2136. [DOI] [PubMed] [Google Scholar]
- 31.Hazell LJ, et al. Presence of hypochlorite-modified proteins in human atherosclerotic lesions. J Clin Invest. 1996;97(6):1535–1544. doi: 10.1172/JCI118576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malle E, et al. Immunohistochemical evidence for the myeloperoxidase/H2O2/halide system in human atherosclerotic lesions: Colocalization of myeloperoxidase and hypochlorite-modified proteins. Eur J Biochem. 2000;267(14):4495–4503. doi: 10.1046/j.1432-1327.2000.01498.x. [DOI] [PubMed] [Google Scholar]
- 33.Breckwoldt MO, et al. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc Natl Acad Sci USA. 2008;105(47):18584–18589. doi: 10.1073/pnas.0803945105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukumoto Y, et al. Angina pectoris occurring during granulocyte colony-stimulating factor-combined preparatory regimen for autologous peripheral blood stem cell transplantation in a patient with acute myelogenous leukaemia. Br J Haematol. 1997;97(3):666–668. doi: 10.1046/j.1365-2141.1997.842724.x. [DOI] [PubMed] [Google Scholar]
- 35.Hill JM, et al. Outcomes and risks of granulocyte colony-stimulating factor in patients with coronary artery disease. J Am Coll Cardiol. 2005;46(9):1643–1648. doi: 10.1016/j.jacc.2005.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fitzhugh CD, Hsieh MM, Bolan CD, Saenz C, Tisdale JF. Granulocyte colony-stimulating factor (G-CSF) administration in individuals with sickle cell disease: Time for a moratorium? Cytotherapy. 2009;11(4):464–471. doi: 10.1080/14653240902849788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang J, et al. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood. 2010;116(10):1779–1786. doi: 10.1182/blood-2009-12-260513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noguchi N, et al. Role of myeloperoxidase in the neutrophil-induced oxidation of low density lipoprotein as studied by myeloperoxidase-knockout mouse. J Biochem. 2000;127(6):971–976. doi: 10.1093/oxfordjournals.jbchem.a022713. [DOI] [PubMed] [Google Scholar]
- 39.Sackstein R. Engineering cellular trafficking via glycosyltransferase-programmed stereosubstitution. Ann N Y Acad Sci. 2012;1253(1):193–200. doi: 10.1111/j.1749-6632.2011.06421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ieremia J, Blau CA. Limitations of a mouse model of sickle cell anemia. Blood Cells Mol Dis. 2002;28(2):146–151. doi: 10.1006/bcmd.2002.0503. [DOI] [PubMed] [Google Scholar]
- 41.Iking-Konert C, et al. Granulocyte colony-stimulating factor induces disease flare in patients with antineutrophil cytoplasmic antibody-associated vasculitis. J Rheumatol. 2004;31(8):1655–1658. [PubMed] [Google Scholar]
- 42.Vasiliu IM, Petri MA, Baer AN. Therapy with granulocyte colony-stimulating factor in systemic lupus erythematosus may be associated with severe flares. J Rheumatol. 2006;33(9):1878–1880. [PubMed] [Google Scholar]
- 43.Glass LF, Fotopoulos T, Messina JL. A generalized cutaneous reaction induced by granulocyte colony-stimulating factor. J Am Acad Dermatol. 1996;34(3):455–459. doi: 10.1016/s0190-9622(96)90439-9. [DOI] [PubMed] [Google Scholar]
- 44.Falk RJ, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med. 1988;318(25):1651–1657. doi: 10.1056/NEJM198806233182504. [DOI] [PubMed] [Google Scholar]
- 45.Tervaert JW, et al. Detection of autoantibodies against myeloid lysosomal enzymes: A useful adjunct to classification of patients with biopsy-proven necrotizing arteritis. Am J Med. 1991;91(1):59–66. doi: 10.1016/0002-9343(91)90074-8. [DOI] [PubMed] [Google Scholar]
- 46.Bansal PJ, Tobin MC. Neonatal microscopic polyangiitis secondary to transfer of maternal myeloperoxidase-antineutrophil cytoplasmic antibody resulting in neonatal pulmonary hemorrhage and renal involvement. Ann Allergy Asthma Immunol. 2004;93(4):398–401. doi: 10.1016/S1081-1206(10)61400-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.