

Significance
Repeatome, or the ensemble of all repeat sequences, with its enormous variability and internal epigenetic dynamics, emerges as a critical source of potentially adaptive changes and evolutionary novelties. This conclusion is exemplified here by the cosmopolitan Drosophila melanogaster from a sharp ecological contrast in North Israel. Flies derived from the opposing sides of this long-studied microsite exhibit a significant difference in the contents and distribution of mobile elements, as well as microsatellite allele frequencies, corresponding well with earlier reported phenotypic patterns of stress resistance and assortative mating in the system.
Keywords: adaptive evolution, genome sequencing, microsatellite, incipient speciation
Abstract
Repeat sequences, especially mobile elements, make up large portions of most eukaryotic genomes and provide enormous, albeit commonly underappreciated, evolutionary potential. We analyzed repeatomes of Drosophila melanogaster that have been diverging in response to a microclimate contrast in Evolution Canyon (Mount Carmel, Israel), a natural evolutionary laboratory with two abutting slopes at an average distance of only 200 m, which pose a constant ecological challenge to their local biotas. Flies inhabiting the colder and more humid north-facing slope carried about 6% more transposable elements than those from the hot and dry south-facing slope, in parallel to a suite of other genetic and phenotypic differences between the two populations. Nearly 50% of all mobile element insertions were slope unique, with many of them disrupting coding sequences of genes critical for cognition, olfaction, and thermotolerance, consistent with the observed patterns of thermotolerance differences and assortative mating.
One of the greatest surprises of comparative genomics has been the discovery that eukaryotic genomes are loaded with ubiquitous repeat sequences, such as transposable elements (TEs) and tandem array repeats (satellites). Once relegated to junk DNA, today many repeat elements emerge as potent functional genetic units, providing inspiration for new research initiatives (1). Although traditionally viewed as selfish DNA, TEs are increasingly being recognized as agents of adaptive change and a rich source of evolutionary novelties, including hybrid dysgenesis (2), insecticide resistance (3), mammalian placenta (4), and vertebrate adaptive immune system (4), as well as flower development and plant fitness (5). Domesticated TEs can be repurposed to function as transcription factors, whereas others play various roles in heterochromatin formation, genome stability, centromere binding, chromosome segregation, meiotic recombination, TE silencing, programmed genome rearrangement, V(D)J recombination, and translational regulation (4, 6). Even low-complexity sequence repeats, such as microsatellites, may act as novel regulatory elements (7), enhance alternative splicing (8), and provide new substrate for protein coding variation (9).
TEs have been implicated in adaptations to environmental changes either through somewhat elusive direct remobilization after stress (10) or more indirectly as a source of new and preexisting genetic variation (11). A striking example of adaptive TE insertion polymorphism has been found in Drosophila melanogaster from Evolution Canyon (lower Nahal Oren, Israel) (12, 13), in which slopes 100–400 m apart differ dramatically in aridity, solar radiation, and associated vegetation due to the higher insolation on the south-facing slope (SFS) relative to the north-facing slope (NFS).The promoter region of heat-shock-protein-70Ba (hsp70Ba), one of the five Hsp70 paralogs that encode a major inducible heat-shock protein in D. melanogaster, was polymorphic for a 1.2-kb P-element insertion, with the insert being 28 times more frequent in NFS- than SFS-inhabiting flies. The P-element disruption was associated with decreased Hsp70 expression and heat-shock survival, but increased reproductive success in temperate conditions (13). Although elevated levels of Hsp70 are beneficial during thermotolerance challenges, they tend to be deleterious for growth and development (14, 15).
In parallel with a common pattern of slope-specific adaptations observed across many other taxa inhabiting this remarkable ecological microgradient (16, 17), SFS-derived D. melanogaster outperform their conspecific neighbors from NFS not only in basal and inducible thermotolerance after diverse heat shocks (18–20), but also in resistance to desiccation and starvation (18, 21). In addition to stress resistance, the two populations differ in oviposition site preferences (18), male courtship song parameters (22), sexual and reproductive behavior (23) leading to partial assortative mating within slopes (24), and phenotypic plasticity for wing morphology (25)—all this despite the physical proximity and migration between slopes (26).
A recent genome analysis of the two populations (27) reveals a number of chromosomal regions of interslope divergence and low sequence polymorphism, suggestive of selective sweeps among genes enriched for functions related to stimulus responses and developmental and reproductive processes, remarkably consistent with our previous findings on the phenotypic patterns of stress responses, life history, and mating functions. Sequence divergence in genes underlying adaptive changes, coupled with assortative mating within populations, can be interpreted as signatures of incipient speciation (21). The picture of adaptive divergence in the natural system remains largely incomplete without looking into noncoding parts of the genomes, primarily those occupied by repeat elements. To fill the void, we have now used high-coverage (40×) genome sequencing and analyzed variation of TEs and microsatellites in the two D. melanogaster populations.
Results and Discussion
In D. melanogaster, TEs make up 22% of the whole genome, with long terminal repeat (LTR) retrotransposons being the most abundant, followed by long interspersed nuclear element (LINE)-like non-LTR retrotransposons and terminal inverted repeat (TIR) DNA transposons (28). We identified a total of 14,190 and 15,025 TEs in SFS and NFS populations, respectively. NFS had consistently more TEs than SFS in all chromosomes and genomic regions, except TIRs and LTRs in coding sequences and TIRs and (marginally) non-LTRs in 3′-UTRs (Tables 1 and 2; Wilcoxon test, P = 0.047). Different from the results reported by Kofler et al. (29), but consistent with Bartolomé et al. (30), the X chromosome had the lowest concentration of TEs. 5′-UTRs contained overall less retroelement insertions than 3′-UTRs in both SFS and NFS, but the pattern was reversed for TIRs. Of the 5,222 known TE insertions present in the reference genome, 2,871 (55%) and 2,951 (57%) were identified in SFS and NFS, respectively. A total of 6,964 (48%) and 7,812 (51%) TE insertions were positioned uniquely in SFS and NFS, respectively (Tables 3 and 4), demonstrating the ubiquity of TE-induced polymorphism in the populations. Significant interslope differentiation was produced by retroelements (non-LTR: χ2 with Yates correction = 15.293, df = 1, P < 0.0001; LTR: 8.508, P = 0.0035) rather than DNA transposons (TIRs: 1.451, P = 0.228). Only 363 TEs in SFS and 518 TEs in NFS were slope unique and fixed (frequency > 95%). For more reliable sites after removing less than 10-read coverage sites and overlapping TE insertions, 9,877 and 10,926 TE insertions were identified in SFS and NFS, respectively. Of these filtered TE insertions, 2,174 (22%) and 2,452 (22%) were fixed (>95%) in SFS and NFS, respectively.
Table 1.
Chromosomal distribution of transposable elements in Evolution Canyon D. melanogaster
| Chr. | 5UTR | 3UTR | Intron | CDS | Promoter | Intergenic | Total |
| SFS | |||||||
| X | 26 | 51 | 778 | 78 | 20 | 1,054 | 2,007 |
| 2L | 36 | 48 | 1,162 | 94 | 25 | 1,485 | 2,850 |
| 2R | 49 | 72 | 1,308 | 112 | 28 | 1,418 | 2,987 |
| 3L | 34 | 51 | 1,301 | 108 | 30 | 1,449 | 2,973 |
| 3R | 44 | 80 | 1,262 | 120 | 30 | 1,261 | 2,797 |
| 4 | 4 | 13 | 361 | 22 | 1 | 175 | 576 |
| Total | 193 | 315 | 6,172 | 534 | 134 | 6,842 | 14,190 |
| NFS | |||||||
| X | 27 | 48 | 852 | 66 | 24 | 1,077 | 2,094 |
| 2L | 32 | 46 | 1,276 | 93 | 33 | 1,642 | 3,122 |
| 2R | 50 | 69 | 1,338 | 105 | 35 | 1,495 | 3,092 |
| 3L | 44 | 54 | 1,415 | 96 | 29 | 1,601 | 3,239 |
| 3R | 50 | 75 | 1,370 | 107 | 41 | 1,257 | 2,900 |
| 4 | 0 | 12 | 369 | 21 | 2 | 174 | 578 |
| Total | 203 | 304 | 6,620 | 488 | 164 | 7,246 | 15,025 |
Chr., chromosome; NFS, north-facing slope; SFS, south-facing slope.
Table 2.
Class I (LTR and non-LTR) and class II (TIRs) transposable elements and their genomic distribution in genomes of SFS- and NFS-derived D. melanogaster
| TE order | Region | SFS (n) | NFS (n) |
| Non-LTR | 5′-UTR | 22 | 25 |
| 3′-UTR | 55 | 54 | |
| Intron | 1,487 | 1,652 | |
| CDS | 67 | 86 | |
| Promoter | 22 | 32 | |
| Intergenic | 1,663 | 1,810 | |
| LTR | 5′-UTR | 47 | 49 |
| 3′-UTR | 151 | 153 | |
| Intron | 2,563 | 2,762 | |
| CDS | 281 | 252 | |
| Promoter | 45 | 56 | |
| Intergenic | 2,884 | 3,110 | |
| TIR | 5′-UTR | 124 | 129 |
| 3′-UTR | 109 | 97 | |
| Intron | 2,122 | 2,206 | |
| CDS | 186 | 150 | |
| Promoter | 67 | 76 | |
| Intergenic | 2,295 | 2,326 |
Table 3.
SFS- and NFS-unique transposable elements (LTRs, non-LTRs, and TIRs)
| Chr. | 5UTR | 3UTR | Intron | CDS | Promoter | Intergenic | Total |
| SFS | |||||||
| X | 20 | 28 | 476 | 29 | 8 | 536 | 1,097 |
| 2L | 23 | 30 | 566 | 51 | 16 | 705 | 1,391 |
| 2R | 35 | 32 | 581 | 54 | 12 | 519 | 1,233 |
| 3L | 24 | 34 | 649 | 56 | 20 | 671 | 1,454 |
| 3R | 22 | 46 | 810 | 77 | 19 | 734 | 1,708 |
| 4 | 0 | 4 | 49 | 6 | 1 | 21 | 81 |
| Total | 124 | 174 | 3,131 | 273 | 76 | 3,186 | 6,964 |
| NFS | |||||||
| X | 21 | 24 | 548 | 21 | 13 | 561 | 1,188 |
| 2L | 21 | 24 | 683 | 55 | 25 | 859 | 1,667 |
| 2R | 36 | 30 | 634 | 47 | 20 | 591 | 1,358 |
| 3L | 34 | 36 | 757 | 45 | 22 | 814 | 1,708 |
| 3R | 34 | 46 | 917 | 67 | 27 | 717 | 1,808 |
| 4 | 0 | 2 | 57 | 3 | 0 | 21 | 83 |
| Total | 146 | 162 | 3,596 | 238 | 107 | 3,563 | 7,812 |
Chr., chromosome.
Table 4.
Classes and chromosomal distribution of SFS- and NFS-unique transposable elements
| Chr. | LTR | Non-LTR | TIR | Total |
| SFS | ||||
| X | 610 | 224 | 263 | 1,097 |
| 2L | 809 | 320 | 262 | 1,391 |
| 2R | 670 | 306 | 257 | 1,233 |
| 3L | 814 | 340 | 300 | 1,454 |
| 3R | 988 | 419 | 301 | 1,708 |
| 4 | 18 | 10 | 53 | 81 |
| Total | 3,909 | 1,619 | 1,436 | 6,964 |
| NFS | ||||
| X | 676 | 265 | 247 | 1,188 |
| 2L | 913 | 436 | 318 | 1,667 |
| 2R | 739 | 344 | 275 | 1,358 |
| 3L | 948 | 435 | 325 | 1,708 |
| 3R | 1,041 | 468 | 299 | 1,808 |
| 4 | 20 | 11 | 52 | 83 |
| Total | 4,337 | 1,959 | 1,516 | 7,812 |
Chr., chromosome.
With a total of 84 differential insertion sites, I-elements were the most divergent TEs between SFS and NFS (Table S1). I-elements are ∼5.4-kb, non-LTR retroelements belonging to LINEs, having spread across natural populations of D. melanogaster in the early 20th century (31). I-elements are subject to female germ-line mobilization in the progeny of certain D. melanogaster intraspecific crosses, responsible for a maternal effect embryonic lethality known as the I-R type of hybrid dysgenesis (32). Quasimodo and roo LTRs were the next most slope-divergent TEs, followed by LINE-like jockey and Doc. The most slope-differential TIR was P-element, represented by 45 unique insertion sites. Interestingly, P-element insertions in the Hsp70Ba promoter reported earlier (12, 13) were absent in the current samples, a result of either demographic dynamics or selection against the insertion over the course of 13 y separating the fly collections in the field. However, there were 76 other unique TE insertions in SFS and 107 in NFS within the putative promoter regions, including a P-element in a gene from the small heat shock protein (HSP20) family, Hsp67Ba, in SFS flies. Although expression of Hsp67Ba was not directly investigated in D. melanogaster from Evolution Canyon, we have previously shown that small Hsps, such as Hsp40 and Hsp23, can contribute to thermotolerance in the system (33). There were at least 20 significant gene ontology (GO) term enrichments representing genes with slope-unique TE insertions in their putative promoters, with functional activities related to hydrolases and alternative splicing being most conspicuous (Tables S2 and S3).
An additional 426 genes were disrupted by 511 slope-specific TE insertions within coding sequences, presumably leading to the genes’ functional inactivation. Interestingly, there were 20 cognition-related and 17 sensory perception-related genes affected by the inserts, including 8 olfactory receptor and 8 gustatory receptor genes, all critical for detecting food and avoiding toxicants, as well as for courtship and mating. Cognition, sensory perception of chemical stimuli, and olfaction were among the most significantly overrepresented GO terms (Benjamini–Hochberg-adjusted P < 0.01) among genes with TE-disrupted coding sequences (Tables S4 and S5). This slope-divergent transposition of mobile elements may contribute to courtship changes and mating isolation. Indeed, we and others have observed various degrees of partial mating isolation between NFS- and SFS-derived D. melanogaster over many years of fly collections in Evolution Canyon (23, 24, 34) (see ref. 35 for an exception). Twenty-seven genes in SFS and 27 other genes in NFS were hit by multiple (2, 3) slope-unique TEs within coding DNA sequences (CDSs). For example, olfactory receptor gene Or92a in SFS was disrupted by two different TEs, roo and hobo, whereas CG11034 was affected by two independent F-element insertions and one jockey.
A question arises how transposition, which on average has deleterious effects, can result in a certain population-unique functional enrichment. First, it should be noted that transposition tends to be nonrandom with respect to insertion sites. For example, P-elements prefer a specific palindromic arrangement of hydrogen bonding sites over a 14-bp region centered on their insertion site (36). It is thus not entirely unexpected that certain gene families sharing common structural motifs and functionalities are preferentially attracting TEs. It has also been suggested that transcriptionally active housekeeping genes, such as Hsp70 paralogs, have a permanently accessible chromatin structure, making their promoters easier targets for mobile elements (37, 38). These intragenic TEs thereafter segregate in natural populations and provide a source of rampant genetic variation on which natural selection and demographic processes may act (11). Purifying selection against TEs or positive selection favoring particular TE insertions may have operated differently on NFS and SFS, dependent on ecological conditions of the contrasting slopes. Alternatively to differential selection regimes, it is possible that demographic processes may have starkly different dynamics on the opposite slopes of Evolution Canyon. Such environmental events as droughts and forest fires, like that one in December of 2010 that ravaged the Carmel mountains, including Evolution Canyon habitats (39), presumably drive local Drosophila populations to very low numbers, followed by rapid repopulations and expansions. Even without major environmental disasters, D. melanogaster natural populations have been known to be subject to boom and bust cycles (40), boosting short-term Ne and enabling short-term evolution act primarily on preexisting intermediate-frequency genetic variants that are swept the remainder of the way to fixation in a process known as soft sweep (41).
To investigate the potential relationship between molecular adaptations and TEs, we tested whether TEs are found in the range of nonneutral SNP regions of Tajima’s D < −2.0 and significant interslope differentiation, suggestive of disruptive positive selection, as we reported elsewhere (27). Except for chromosome X (and 2L in SFS), the density of TEs was increased in the regions relative to mean TE density per chromosome, even as much as three times on 2R. The highest number of TEs (18 in NFS and 16 in SFS) was found in the window 2R (230,000–240,000), all inserted in intergenic sequences. The ∼60-kb region (9,069,408–9,127,928 bp) of interest on 3R that showed a high excess of SNPs assigned by a hidden Markov model analysis to high-differentiating state contained four TEs in SFS (hobo, roo, and two Quasimodos) and only two TEs in NFS (roo and Quasimodo), all within intergenic DNA. The highest interslope differentiation was found in a nonneutral region of the X chromosome (positions 10,520,000–10,530,000): six TEs in SFS and only one in NFS (Fig. 1), affecting an exon of CG9806 (SFS), an intron of X11Lbeta (SFS and NFS), and the intergenic sequence between the two genes (SFS).
Fig. 1.
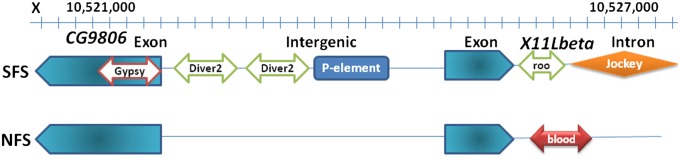
An X chromosome region characterized by high sequence divergence, coupled with low Tajima’s D values, as well as high TE differentiation.
Similar to other studies (29, 30), the highest concentration of TEs in both NFS and SFS was on chromosome 4, and intergenic sequences accumulated more TEs than other genomic regions. In D. melanogaster, chromosome 4 is a small (5–6 Mb) genetic element that normally does not recombine (42), free to accumulate TEs and other repetitive elements. Although the TE density tends to be locally higher in regions with low recombination rates, the relationship between the recombination rate and transposable site occupancy frequency exhibits a less consistent pattern, seemingly dependent on TE type (43). Consistent with Rizon et al. (43), we observed that the abundance of transposons (TIRs) but not retroelements (both LTR and non-LTR), significantly negatively correlated with recombination rate along chromosome arms. However, this effect again depended on the TE type. TIRs were significantly correlated with recombination in all chromosomes in both NFS and SFS populations (Spearman r, −0.82 to −0.44; P < 0.05), whereas LTR and non-LTR retroelements showed a significant correlation only along the X chromosome in SFS (−0.44 to −0.42, P < 0.05). The significant negative correlation observed between transposons and recombination rate suggests that selection acts against these TEs and that TIR remobilization is less efficient to compensate for recombination and selection effects relative to retroelements. An opposite pattern was observed in Caenorhabditis elegans (44), wherein TIR (but not LTR or non-LTR retrotransposon) density was positively correlated with recombination rate.
Traditionally, microsatellite alleles have been used as neutral markers in population genetic research (45), despite the fact that many microsatellites, especially those within coding and regulatory sequences, may produce distinct morphological and behavioral phenotypes with adaptive significance (9, 46, 47). Although intronic microsatellites were most abundant (39,127; 46%) in the sequenced genomes, we identified as many as 8,562 (10%) and 692 (0.8%) microsatellite loci in exons and putative promoter regions, respectively (Fig. 2). There was a large asymmetry in the number of microsatellites in 5′- (14,141) compared with 3′-UTR introns (500), a result of a 22 times higher number of introns in the former, as well as the average intron size difference between the two regions. Previous surveys of microsatellite divergence between NFS- and SFS-derived D. melanogaster have provided reports of high (12) and low (48) genetic differentiation (Fst), an inconsistency probably due to demographic fluctuations in the populations, as well as a very limited choice of marker loci in the pregenomics past. Here we used a total of 65,396 microsatellite loci to estimate Fst values in comparison with Fst estimates from all genomic SNPs (27). The average Fst estimate across all microsatellites was 0.012, one order of magnitude higher than that reported by others (48) but consistently lower than our estimates of Fst from SNPs for the same genomes (Fig. 3). The exclusion of exonic microsatellites slightly increases the mean Fst estimate (0.014). There was a significant positive correlation between microsatellite- and SNP-derived Fst values across 1-Mb intervals on all chromosomal arms (Spearman rank correlation, r = 0.447–0.521; P < 0.05), except for 3R (r = 0.129, P = 0.513; Fig. 3). A monomer (Tn) locus within a 5′-UTR intron of CG42686 (3R chromosome), with one NFS-exclusive allele and two SFS-exclusive alleles, was the most divergent microsatellite, followed by 11 other loci that remained significantly different between slopes after Benjamini–Hochberg correction of individual P values, all in noncoding sequences (Table S6).
Fig. 2.

Genomic distribution of microsatellites in Evolution Canyon D. melanogaster.
Fig. 3.

NFS-SFS interpopulation differentiation (Fst) in SNPs and microsatellites across the major chromosome arms.
Overall, our results show substantial interslope divergence in repeat sequences, in parallel with differentiation of coding sequences (27). Although we find no evidence that microsatellites contribute to local adaptations in Evolution Canyon, mobile elements emerge as potential surrogates of adaptive divergence along the sharp microclimate gradient. A similar conclusion has been reached in a study of wild barley (Hordeum spontaneum) from Evolution Canyon (49, 50), showing that more BARE-1 retrotransposon copies and proportionally fewer solo LTRs were found in the upper, drier sites of the canyon, particularly at the top of the SFS, than at lower sites. Interestingly, the pattern seems to be roughly opposite to D. melanogaster whose NFS genomes carry a heavier TE load.
Materials and Methods
Sampling and DNA Extractions.
A total of 16 NFS and 16 SFS D. melanogaster isofemale lines were obtained from females collected in Evolution Canyon (Mount Carmel, Israel) in October 2010. Approximately 1 µg of DNA from each line was pooled to make population representations. Illumina paired-end libraries were constructed and sequenced with HiSeq, 100-cycle, at ∼40× coverage per population, as described elsewhere (27).
Mapping Reads and Data Processing.
Paired-end reads were filtered for minimum average base quality score of 20 and a minimum length of 50 bp (see ref. 27 for details). Trimmed reads were mapped to the D. melanogaster reference genome using Burrows–Wheeler Aligner (BWA) (0.5.9-r16) (51). Paired-end data were converted to binary alignment map format using SAMtools. For each detected SNP, differentiation between populations was calculated using both Fst and the Fisher exact test. Footprints of natural selection were detected using Tajima's D calculated over nonoverlapping 10-kb windows along chromosome arms.
Detection of Interslope Selective Differentiated Genomic Regions and GO Term Enrichment.
The hidden Markov model (HMM) analysis in R was used to discriminate between the distributions of three hidden states corresponding to high, moderate, and low interslope differentiation and to assign each SNP to a corresponding state, using interslope Fst values (see ref. 27 for details). To search for high differentiation regions along chromosome arms, we used 10-kb nonoverlapping windows. For each window, the level of differentiation was represented by two parameters, LI = nL/nT (low island) and HI = nH/nT (high island), where nT is the total number of SNPs within each window, and nH and nL are the numbers of SNPs assigned in the HMM step to high- and low-differentiation states, respectively. Following this step, a permutation test was conducted to detect genomic regions with significant enrichment of HI scores. Significantly differentiated genomic regions between slopes were then inspected for the type and strength of selection using the Tajima's D scores for each window as obtained with Popoolation (52). Trimmed files of each population were converted to a pileup format separately and used to calculate population measures over a nonoverlapping window of size 10 kb in PoPoolation. A score of D < −2 is indicative of a recent selective sweep (fixation of novel mutation) followed by a slow recovery of variation and hence an excess of rare alleles. On the other hand, D > 2 is indicative of small allele frequency differences due to balancing selection. Thus, combining both differentiation and selection scores can be used for detection of genomic regions corresponding to interslope differentiation caused by alternative selection (small D and significant HI).
To study the biological significance of genes under diversifying selection, an enrichment analysis of GO terms was conducted with DAVID 6.7 (53).
Identification of TEs.
To identify TEs, we used a PoPoolationTE software package that allows finding TE insertions present in a reference genome, as well as novel TE insertions (29). As a reference, the D. melanogaster genome v5.31, TE sequences v5.31, and a GFF file (v5.31) from FlyBase were used. We used the same classification of TE insertions proposed by Kofler et al. (29), with three major orders: TIR elements and LTR and non-LTR retrotransposons, further grouped into 115 families and 5,222 insertions. However, unlike Kofler et al. (29), we added P-element (FlyBase) to the list of analyzed TEs. To measure differentiation between SFS and NFS, which is expressed by the fixation index (Fst), we used PoPoolation2 software (54).
Creating a Set of Microsatellites with Uniquely Mappable Flanking Sequences.
A set of microsatellite loci with unique flanking sequences was identified in the dmel reference (BDGP R5/dm3) genome using methods that were used before to create a unique microsatellite set for the human genome (55, 56). Tandem Repeat Finder (57) was run on the dmel reference genome to identify a starting set of 875,647 microsatellites using the following parameters: 2, 5, 5, 80, 10, 14, 6. A Perl script reduced this set to only 1–6 mer microsatellites, which were at least 12 nucleotides in length with a minimum 90% identity score. This set contained 209,591 microsatellite loci. Using the long interspersed nuclear element locations for dmel from the UCSC Genome Browser (58), we removed microsatellites from the set that were within one nucleotide of any of these large repetitive regions. This set was further reduced to only contain those microsatellites with unique flanking sequences. The resulting set contained 72,603 microsatellites. Locations of RefSeq genes in the dmel reference (Flybase and Drosophila Heterochromatin Genome Project, Lawrence Berkeley National Laboratory) were downloaded from the UCSC Genome Browser (58). A Perl script was written to identify the position of each microsatellite with respect to the RefSeq genes. The Perl script used the first nucleotide of each microsatellite to determine its position. The first RefSeq gene that contained this position in the list of RefSeq genes was used. We ignored any genes that followed in the list that might also contain this point. Upstream was defined as the region spanning 1,000 nucleotides before the transcription start nucleotide.
Microsatellite Calling.
All raw Illumina reads were mapped to the dmel reference with BWA (0.5.9-r16) (51). Using our microsatellite calling software that includes local realignment and read filtering (55, 56), we were able to call 67,565 and 66,276 microsatellite loci in the NFS and SFS samples, respectively. We required at least three reads to call each microsatellite allele. Because the samples contained multiple genomes, we did not limit each microsatellite locus to a maximum of two alleles.
Supplementary Material
Acknowledgments
The project was supported by United States–Israel Binational Science Foundation Grant 2011438 (to P.M., A.B.K., and E.R.) and the Ancell Teicher Research Foundation for Genetics and Molecular Evolution (E.N.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1410372111/-/DCSupplemental.
References
- 1.Wang T, et al. Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc Natl Acad Sci USA. 2007;104(47):18613–18618. doi: 10.1073/pnas.0703637104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashburner M, Golic KG, Hawley RS. Drosophila: A Laboratory Handbook. 2nd Ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2005. p. xxviii. [Google Scholar]
- 3.Aminetzach YT, Macpherson JM, Petrov DA. Pesticide resistance via transposition-mediated adaptive gene truncation in Drosophila. Science. 2005;309(5735):764–767. doi: 10.1126/science.1112699. [DOI] [PubMed] [Google Scholar]
- 4.Sinzelle L, Izsvák Z, Ivics Z. Molecular domestication of transposable elements: From detrimental parasites to useful host genes. Cell Mol Life Sci. 2009;66(6):1073–1093. doi: 10.1007/s00018-009-8376-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joly-Lopez Z, Forczek E, Hoen DR, Juretic N, Bureau TE. A gene family derived from transposable elements during early angiosperm evolution has reproductive fitness benefits in Arabidopsis thaliana. PLoS Genet. 2012;8(9):e1002931. doi: 10.1371/journal.pgen.1002931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feschotte C. Transposable elements and the evolution of regulatory networks. Nat Rev Genet. 2008;9(5):397–405. doi: 10.1038/nrg2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iglesias AR, Kindlund E, Tammi M, Wadelius C. Some microsatellites may act as novel polymorphic cis-regulatory elements through transcription factor binding. Gene. 2004;341:149–165. doi: 10.1016/j.gene.2004.06.035. [DOI] [PubMed] [Google Scholar]
- 8.Sirand-Pugnet P, Durosay P, Brody E, Marie J. An intronic (A/U)GGG repeat enhances the splicing of an alternative intron of the chicken beta-tropomyosin pre-mRNA. Nucleic Acids Res. 1995;23(17):3501–3507. doi: 10.1093/nar/23.17.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li YC, Korol AB, Fahima T, Nevo E. Microsatellites within genes: Structure, function, and evolution. Mol Biol Evol. 2004;21(6):991–1007. doi: 10.1093/molbev/msh073. [DOI] [PubMed] [Google Scholar]
- 10.Capy P, Gasperi G, Biémont C, Bazin C. Stress and transposable elements: Co-evolution or useful parasites? Heredity (Edinb) 2000;85(Pt 2):101–106. doi: 10.1046/j.1365-2540.2000.00751.x. [DOI] [PubMed] [Google Scholar]
- 11.González J, Karasov TL, Messer PW, Petrov DA. Genome-wide patterns of adaptation to temperate environments associated with transposable elements in Drosophila. PLoS Genet. 2010;6(4):e1000905. doi: 10.1371/journal.pgen.1000905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Michalak P, et al. Genetic evidence for adaptation-driven incipient speciation of Drosophila melanogaster along a microclimatic contrast in “Evolution Canyon,” Israel. Proc Natl Acad Sci USA. 2001;98(23):13195–13200. doi: 10.1073/pnas.231478298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lerman DN, Michalak P, Helin AB, Bettencourt BR, Feder ME. Modification of heat-shock gene expression in Drosophila melanogaster populations via transposable elements. Mol Biol Evol. 2003;20(1):135–144. doi: 10.1093/molbev/msg015. [DOI] [PubMed] [Google Scholar]
- 14.Krebs RA, Feder ME. Deleterious consequences of Hsp70 overexpression in Drosophila melanogaster larvae. Cell Stress Chaperones. 1997;2(1):60–71. doi: 10.1379/1466-1268(1997)002<0060:dcohoi>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zatsepina OG, et al. A Drosophila melanogaster strain from sub-equatorial Africa has exceptional thermotolerance but decreased Hsp70 expression. J Exp Biol. 2001;204(Pt 11):1869–1881. doi: 10.1242/jeb.204.11.1869. [DOI] [PubMed] [Google Scholar]
- 16.Nevo E. Asian, African and European biota meet at Evolution-Canyon Israel: Local tests of global biodiversity and genetic diversity patterns. Proc Roy Soc Lond B Bio. 1995;262(1364):149–155. [Google Scholar]
- 17.Nevo E. “Evolution Canyon,” a potential microscale monitor of global warming across life. Proc Natl Acad Sci USA. 2012;109(8):2960–2965. doi: 10.1073/pnas.1120633109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nevo E, Rashkovetsky E, Pavlicek T, Korol A. A complex adaptive syndrome in Drosophila caused by microclimatic contrasts. Heredity (Edinb) 1998;80(Pt 1):9–16. doi: 10.1046/j.1365-2540.1998.00274.x. [DOI] [PubMed] [Google Scholar]
- 19.Lupu A, Pechkovskaya A, Rashkovetsky E, Nevo E, Korol A. DNA repair efficiency and thermotolerance in Drosophila melanogaster from “Evolution Canyon”. Mutagenesis. 2004;19(5):383–390. doi: 10.1093/mutage/geh045. [DOI] [PubMed] [Google Scholar]
- 20.Rashkovetsky E, et al. Adaptive differentiation of thermotolerance in Drosophila along a microclimatic gradient. Heredity (Edinb) 2006;96(5):353–359. doi: 10.1038/sj.hdy.6800784. [DOI] [PubMed] [Google Scholar]
- 21.Korol A, Rashkovetsky E, Iliadi K, Nevo E. Drosophila flies in “Evolution Canyon” as a model for incipient sympatric speciation. Proc Natl Acad Sci USA. 2006;103(48):18184–18189. doi: 10.1073/pnas.0608777103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iliadi KG, et al. Peculiarities of the courtship song in the Drosophila melanogaster populations adapted to gradient of microecological conditions. J Evol Biochem Physiol. 2009;45(5):579–589. [PubMed] [Google Scholar]
- 23.Iliadi K, et al. Sexual and reproductive behaviour of Drosophila melanogaster from a microclimatically interslope differentiated population of “Evolution Canyon” (Mount Carmel, Israel) Proc Biol Sci. 2001;268(1483):2365–2374. doi: 10.1098/rspb.2001.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korol A, et al. Nonrandom mating in Drosophila melanogaster laboratory populations derived from closely adjacent ecologically contrasting slopes at “Evolution Canyon”. Proc Natl Acad Sci USA. 2000;97(23):12637–12642. doi: 10.1073/pnas.220041397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Debat V, et al. Multidimensional analysis of Drosophila wing variation in Evolution Canyon. J Genet. 2008;87(4):407–419. doi: 10.1007/s12041-008-0063-x. [DOI] [PubMed] [Google Scholar]
- 26.Pavlicek T, Frenkel Z, Korol AB, Beiles A, Nevo E. Drosophila at the “Evolution Canyon” microsite, Mt. Carmel, Israel: Selection overrules migration. Isr J Ecol Evol. 2008;54(2):165–180. [Google Scholar]
- 27.Hübner S, et al. Genome differentiation of Drosophila melanogaster from a microclimate contrast in Evolution Canyon, Israel. Proc Natl Acad Sci USA. 2013;110(52):21059–21064. doi: 10.1073/pnas.1321533111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clark AG, et al. Drosophila 12 Genomes Consortium Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450(7167):203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- 29.Kofler R, Betancourt AJ, Schlötterer C. Sequencing of pooled DNA samples (Pool-Seq) uncovers complex dynamics of transposable element insertions in Drosophila melanogaster. PLoS Genet. 2012;8(1):e1002487. doi: 10.1371/journal.pgen.1002487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartolomé C, Maside X, Charlesworth B. On the abundance and distribution of transposable elements in the genome of Drosophila melanogaster. Mol Biol Evol. 2002;19(6):926–937. doi: 10.1093/oxfordjournals.molbev.a004150. [DOI] [PubMed] [Google Scholar]
- 31.Bucheton A, et al. I elements and the Drosophila genome. Genetica. 1992;86(1-3):175–190. doi: 10.1007/BF00133719. [DOI] [PubMed] [Google Scholar]
- 32.Orsi GA, Joyce EF, Couble P, McKim KS, Loppin B. Drosophila I-R hybrid dysgenesis is associated with catastrophic meiosis and abnormal zygote formation. J Cell Sci. 2010;123(Pt 20):3515–3524. doi: 10.1242/jcs.073890. [DOI] [PubMed] [Google Scholar]
- 33.Carmel J, Rashkovetsky E, Nevo E, Korol A. Differential expression of small heat shock protein genes Hsp23 and Hsp40, and heat shock gene Hsr-omega in fruit flies (Drosophila melanogaster) along a microclimatic gradient. J Hered. 2011;102(5):593–603. doi: 10.1093/jhered/esr027. [DOI] [PubMed] [Google Scholar]
- 34.Singh SR, Rashkovetsky E, Iliadi K, Nevo E, Korol A. Assortative mating in Drosophila adapted to a microsite ecological gradient. Behav Genet. 2005;35(6):753–764. doi: 10.1007/s10519-005-6119-2. [DOI] [PubMed] [Google Scholar]
- 35.Panhuis TM, Swanson WJ, Nunney L. Population genetics of accessory gland proteins and sexual behavior in Drosophila melanogaster populations from Evolution Canyon. Evolution. 2003;57(12):2785–2791. doi: 10.1111/j.0014-3820.2003.tb01520.x. [DOI] [PubMed] [Google Scholar]
- 36.Liao GC, Rehm EJ, Rubin GM. Insertion site preferences of the P transposable element in Drosophila melanogaster. Proc Natl Acad Sci USA. 2000;97(7):3347–3351. doi: 10.1073/pnas.050017397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walser JC, Chen B, Feder ME. Heat-shock promoters: Targets for evolution by P transposable elements in Drosophila. PLoS Genet. 2006;2(10):e165. doi: 10.1371/journal.pgen.0020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haney RA, Feder ME. Contrasting patterns of transposable element insertions in Drosophila heat-shock promoters. PLoS ONE. 2009;4(12):e8486. doi: 10.1371/journal.pone.0008486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen Y, Lansky E, Traber M, Nevo E. Increases in both acute and chronic temperature potentiate tocotrienol concentrations in wild barley at ‘Evolution Canyon’. Chem Biodivers. 2013;10(9):1696–1705. doi: 10.1002/cbdv.201300133. [DOI] [PubMed] [Google Scholar]
- 40.Harry M, et al. Fine-scale biodiversity of Drosophilidae in “Evolution Canyon” at the Lower Nahal Oren microsite, Israel. Biologia. 1999;54(6):685–705. [Google Scholar]
- 41.Burke MK, et al. Genome-wide analysis of a long-term evolution experiment with Drosophila. Nature. 2010;467(7315):587–590. doi: 10.1038/nature09352. [DOI] [PubMed] [Google Scholar]
- 42.Hochman B. The fourth chromosome of Drosophila melanogaster. In: Ashburner M, Novitski E, editors. The Genetics and Biology of Drosophila. Vol 1b. New York: Academic Press; 1976. pp. 903–928. [Google Scholar]
- 43.Rizzon C, Marais G, Gouy M, Biémont C. Recombination rate and the distribution of transposable elements in the Drosophila melanogaster genome. Genome Res. 2002;12(3):400–407. doi: 10.1101/gr.210802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duret L, Marais G, Biémont C. Transposons but not retrotransposons are located preferentially in regions of high recombination rate in Caenorhabditis elegans. Genetics. 2000;156(4):1661–1669. doi: 10.1093/genetics/156.4.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tautz D, Schlötterer Simple sequences. Curr Opin Genet Dev. 1994;4(6):832–837. doi: 10.1016/0959-437x(94)90067-1. [DOI] [PubMed] [Google Scholar]
- 46.Fondon JW, 3rd, Garner HR. Molecular origins of rapid and continuous morphological evolution. Proc Natl Acad Sci USA. 2004;101(52):18058–18063. doi: 10.1073/pnas.0408118101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fondon JW, 3rd, Hammock EA, Hannan AJ, King DG. Simple sequence repeats: Genetic modulators of brain function and behavior. Trends Neurosci. 2008;31(7):328–334. doi: 10.1016/j.tins.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 48.Schlötterer C, Agis M. Microsatellite analysis of Drosophila melanogaster populations along a microclimatic contrast at lower Nahel Oren canyon, Mount Carmel, Israel. Mol Biol Evol. 2002;19(4):563–568. doi: 10.1093/oxfordjournals.molbev.a004112. [DOI] [PubMed] [Google Scholar]
- 49.Kalendar R, Tanskanen J, Immonen S, Nevo E, Schulman AH. Genome evolution of wild barley (Hordeum spontaneum) by BARE-1 retrotransposon dynamics in response to sharp microclimatic divergence. Proc Natl Acad Sci USA. 2000;97(12):6603–6607. doi: 10.1073/pnas.110587497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bedada G, Westerbergh A, Nevo E, Korol A, Schmid KJ. DNA sequence variation of wild barley Hordeum spontaneum (L.) across environmental gradients in Israel. Heredity (Edinb) 2014;112(6):646–655. doi: 10.1038/hdy.2014.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kofler R, et al. PoPoolation: A toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS ONE. 2011;6(1):e15925. doi: 10.1371/journal.pone.0015925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 54.Kofler R, Pandey RV, Schlötterer C. PoPoolation2: Identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq) Bioinformatics. 2011;27(24):3435–3436. doi: 10.1093/bioinformatics/btr589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McIver LJ, Fondon JW, 3rd, Skinner MA, Garner HR. Evaluation of microsatellite variation in the 1000 Genomes Project pilot studies is indicative of the quality and utility of the raw data and alignments. Genomics. 2011;97(4):193–199. doi: 10.1016/j.ygeno.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McIver LJ, McCormick JF, Martin A, Fondon JW, 3rd, Garner HR. Population-scale analysis of human microsatellites reveals novel sources of exonic variation. Gene. 2013;516(2):328–334. doi: 10.1016/j.gene.2012.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benson G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karolchik D, et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014;42(Database issue):D764–D770. doi: 10.1093/nar/gkt1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.