

Abstract
Extracellular matrix (ECM) remodeling is a critical aspect of cardiac remodeling following myocardial infarction. Tissue inhibitors of metalloproteinases (TIMPs) are physiological inhibitors of matrix metalloproteinases (MMPs) that degrade the ECM proteins. TIMP3 is highly expressed in the heart, and is markedly downregulated in patients with ischemic cardiomyopathy. We therefore examined the time- and region-dependent role of TIMP3 in the cardiac response to myocardial infarction (MI). TIMP3−/− and wild-type (WT) mice were subjected to MI by ligation of the left anterior descending artery. TIMP3−/−-MI mice exhibited a significantly compromised rate of survival compared with WT-MI mice, primarily due to increased left ventricular (LV) rupture, greater infarct expansion, exacerbated LV dilation, and greater systolic and diastolic dysfunction. Second harmonic generation imaging of unfixed and unstained hearts revealed greater collagen disarray and reduced density in the TIMP3−/− infarct myocardium compared with the WT group. Gelatinolytic and collagenolytic activities increased in TIMP3−/− compared with WT hearts at 1 day post-MI but not at 3 days or 1 wk post-MI. Neutrophil infiltration and inflammatory MMPs were significantly increased in the infarct and peri-infarct regions of TIMP3−/−-MI hearts. Treatment of TIMP3−/− mice with a broad-spectrum MMP inhibitor (PD-166793) for 2 days before and 2 days after MI markedly improved post-MI infarct expansion, LV rupture incident, LV dilation, and systolic dysfunction in these mice up to 1 wk post-MI. Our data demonstrate that the initial rise in proteolytic activities early post-MI is a triggering factor for subsequent LV adverse remodeling, LV rupture, and dilated cardiomyopathy. Hence, timing of treatments to improve cardiac response to MI may be critical in producing favorable outcome.
Keywords: tissue inhibitor of metalloptroteinase-3, inflammation, extracellular matrix
coronary artery disease is the most common cause of heart failure and remains a major cause of morbidity and mortality worldwide (20). Myocardial infarction (MI) leads to a region- and time-dependent adverse myocardial remodeling resulting in ventricular dilation, dysfunction, and infarct rupture that can result in sudden cardiac death and/or heart failure (8, 44, 49). Free wall myocardial rupture is a fatal complication of acute MI and is associated with a high mortality (10, 13). Impaired remodeling of the extracellular matrix (ECM) is a critical contributor to postinfarction dilation, dysfunction, and myocardial rupture (21, 48). ECM integrity is maintained by a balance in the function of matrix metalloproteinases (MMPs), which degrade ECM proteins, and their physiological inhibitors, tissue inhibitors of metalloproteinases (TIMPs). Altered plasma profiles of MMPs/TIMPs correlate with adverse post-MI ventricular remodeling in patients (56). MMPs have been shown to play a key role in post-MI left ventricular (LV) rupture in animal models (18, 33), as well as in patients who died of LV rupture (53). Among the TIMPs, TIMP3 is ECM bound, is highly expressed in the heart (39), can inhibit a broad spectrum of metalloproteinases, and is downregulated in the ischemic myocardium of animals (46), as well as in patients with ischemic heart failure (29). TIMP3 deficiency severely compromises cardiac response to pressure overload, resulting in the early development of dilated cardiomyopathy (25) and excess myocardial fibrosis (24), as well as accelerated adverse LV remodeling post-MI (52). In this study we examine the early molecular events post-MI and demonstrate a region- and time-specific function for TIMP3 in cardiac recovery from myocardial infarction. We further demonstrate that the early rise in MMP activities post-MI is the detrimental factor in a subsequent adverse LV remodeling and dysfunction.
MATERIALS AND METHODS
Experimental model and tissue collection.
Wild-type (WT) C57BL/6 mice were purchased from Jackson Laboratories, and TIMP3-deficient (TIMP3−/−) mice were generated in C57BL/6 background as described previously (28). All animal experiments were carried out in accordance with the Canadian Council on Animal Care Guidelines, and animal protocols were reviewed and approved by the Animal Care and Use Committee at University of Alberta. Male 10- to 12-wk-old TIMP3−/− and WT mice were subjected to MI induced by ligation of the left anterior descending artery (LAD) as described previously (23). Briefly, anesthetized and intubated mice underwent left thoracotomy in the fifth intercostal space. The pericardium was opened to expose the LV of the heart, and then the LAD was encircled and ligated under the tip of the left atrial (LA) appendage using a 7-0 silk suture. Once the LAD ligation was complete, the muscle and skin were closed in layers using a 6-0 silk suture. Sham-operated mice from each genotype underwent the exact same protocol, but their LAD was not ligated. These sham-operated mice were used as baseline controls. At 1 day, 3 days, or 1 wk post-MI, mice were anesthetized, and hearts were quickly excised, dissected into infarct, peri-infarct, and noninfarct regions, and then flash-frozen separately for further protein and RNA analyses. For immunohistochemical analysis, whole hearts were arrested in diastole with 1 M KCl and then fixed in 10% formalin.
Echocardiography and tissue Doppler imaging.
Transthoracic echocardiography was performed noninvasively as described previously using a Vevo 770 high-resolution imaging system equipped with a 30-MHz transducer (RMV-707B; VisualSonics, Toronto, ON, Canada) (5). The temporal resolution for M-mode imaging in this system is a pulse repetition frequency of 8 kHz with an axial resolution of 55 μm, lateral resolution of 115 μm, focal length of 12.7 mm, and depth of field of 2.2 mm. Mice were anesthetized with 0.75% isoflurane for the duration of the recordings. M-mode images were obtained for measurements of LV wall thickness, LV end-diastolic diameter, and LV end-systolic diameter. LV ejection fraction (EF) was calculated as a measure of systolic function using the following equation: EF (%) = [(LV end-diastolic volume − LV end-systolic volume)/LV end-diastolic volume] × 100.
Diastolic function was assessed using pulsed-wave Doppler imaging of the transmitral filling pattern with the early transmitral filling wave (E-wave) followed by the late filling wave due to atrial contraction (A-wave). Isovolumetric relaxation time (IVRT) was calculated as the time from closure of the aortic valve to initiation of the E-wave. The E-wave deceleration time (DT) was determined by measuring the time needed for the down slope of the peak of the E-wave to reach baseline, whereas the E-wave deceleration rate was calculated as the E-wave divided by the DT. Tissue Doppler imaging (TDI) is a novel and validated technique to assess systolic and diastolic function where a reduction in E′ and an elevation in E/E′ are considered markers of elevated LV filling pressure and diastolic dysfunction (5). TDI was made at the inferolateral region in the radial short axis at the base of the LV with the assessment of early (E′) and late diastolic (A′) myocardial velocities. The maximal anteroposterior LA diameter was measured by M-mode in the parasternal long-axis view and used as LA size.
MMP inhibitor administration in vivo.
An MMP-specific inhibitor, PD-166793 (Pfizer) was administered daily by gavage as described previously (25). PD-166793 treatment (30 mg·kg−1·day−1) began 2 days before MI and was continued for 2 days post-MI. Mortality was monitored over 1 wk post-MI, and cardiac structure and function were assessed by echocardiography at 1 wk post-MI.
Autopsy and infarct size measurement.
Autopsy was performed on each mouse found dead throughout the course of the study. Cardiac rupture was confirmed by the presence of a large amount of clotted blood in the chest cavity. Infarct size on surviving mice was measured at 1 wk post-MI by perfusing the hearts at a constant pressure (60 mmHg) with PBS and then with 1% triphenyltetrazolium chloride (Sigma, Oakville, ON, Canada) to stain the live cells. Hearts were then sectioned in 1-mm slices from apex to the base. The infarct area was measured using Image-Pro Plus software and reported as a percentage of the total LV size.
Second harmonic generation and multiphoton imaging.
Collagen fiber density and arrangements were visualized in unfixed and unstained heart tissue by using the novel second harmonic generation and multiphoton excitation fluorescence microscopy imaging technique, as described previously (1).
Immunohistochemistry.
Hearts were fixed in 10% buffered formalin for 48 h and then placed in 80% ethanol. Neutrophil stainings were performed on 5-μm sections using rat anti-mouse neutrophil antibody (MCA771GA; Serotec) as described previously (23).
Protein and RNA analyses and protease activity assays.
Total protein was extracted from frozen tissue by gentle homogenization in EDTA-free RIPA extraction buffer and quantified using Bio-Rad DC protein assay. Gelatin zymography was performed to detect pro- and active MMP2 and MMP9 as described previously (23, 25). Western blot analysis was performed to detect TIMP1 (R&D Systems), TIMP2 (Abcam), TIMP3 (Santa Cruz Biotechnology), and TIMP4 (Novus Biologicals). Total gelatinase and collagenase activities were measured using fluorescence-based activity assays from EnzChek (Molecular Probes) as described previously (23). Total RNA was extracted using TRIzol reagent (Invitrogen), and RNA expression analysis was performed by TaqMan RT-PCR with hypoxanthine phosphoribosyltransferase used as the internal control, as described previously (23, 24).
Statistical analysis.
All statistical analyses were performed using SPSS software (version 10.1; Chicago, IL). Mortality and rupture incidents were compared using Kaplan Meier survival analysis, followed by the log-rank test. Comparisons between genotypes were performed using multiple ANOVA followed by the Student-Newman-Keuls test for multiple comparison testing. Averaged values are means ± SE. Statistical significance is recognized at P < 0.05.
RESULTS
MI alters TIMP levels in a region- and time-dependent fashion.
The mRNA (Supplemental Fig. 1) and protein levels (Fig. 1) were measured for all TIMPs in the infarct, peri-infarct, and noninfarct regions of WT mice at 3 days (Fig. 1A) and 1 wk post-MI (Fig. 1B). (Supplemental data for this article is available online at the American Journal of Physiology-Heart and Circulatory Physiology website.) At 3 days post-MI, TIMP1 protein levels were significantly increased in the infarct and peri-infarct myocardium, and its levels in the noninfarct region were additionally increased by 1 wk post-MI. TIMP2 levels were decreased in the noninfarct myocardium at both time points. TIMP3 and TIMP4 levels were markedly reduced in the infarct region at both time points, and TIMP3 levels were also reduced in the peri- and noninfarct regions at 3 days post-MI. Changes in the mRNA levels (Supplemental Fig. 1), however, did not directly correspond to the alterations in protein levels, as also reported previously (23, 43). For instance, at 1 wk post-MI, TIMP3 mRNA levels were elevated in the infarct region but unaltered in the peri-infarct and infarct regions, whereas TIMP3 protein levels remained markedly reduced in the infarct and peri-infarct areas. This could represent a compensatory response to the loss of the TIMP3 protein and/or the TIMP3 mRNA synthesized by infiltrating fibroblasts.
Fig. 1.
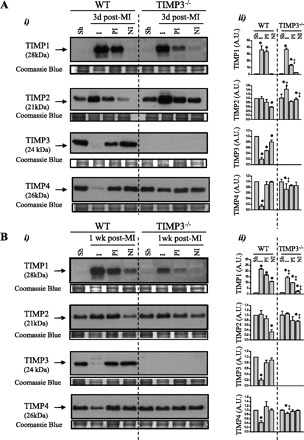
Tissue inhibitor of metalloproteinase (TIMP) levels are altered following myocardial infarction (MI). Representative Western blots (i) and averaged protein levels (ii) for TIMP1, TIMP2, TIMP3, and TIMP4 are shown in sham (Sh), infarct (I), peri-infarct (PI), and noninfarct (NI) myocardium of wild-type (WT) and TIMP3−/− mice at 3 days (A) and 1 wk post-MI (B) (n = 6/genotype). Coomassie blue was used as the internal loading control. A.U., arbitrary units; d, days. Values are presented as Mean ± SEM. *P < 0.05 compared with sham, ‡P < 0.05 compared with WT.
Among all TIMPs, TIMP3 showed a marked reduction in all regions of myocardium early post-MI, with the greatest reduction in the infarct followed by the peri- and noninfarct regions, suggesting a region- and time-sensitive loss of TIMP3 protein during the early cardiac response to MI. Next, we examined whether TIMP3 deficiency impacted the alterations in TIMP levels post-MI (Fig. 1). Compared with WT mice, TIMP3−/− mice showed a markedly smaller increase in TIMP1 levels in the peri-infarct area at 3 days and in all regions at 1 wk post-MI but greater TIMP2 and TIMP4 protein levels in the infarct area at 3 days post-MI, which persisted for TIMP4 at 1 wk post-MI (Fig. 1, A and B).
Suppressed post-MI survival in TIMP3−/− mice due to excess LV rupture.
To determine whether the reduction in TIMP3 post-MI underlies the disease progression and whether the early rise in TIMP2 and TIMP4 in the infarct myocardium of TIMP3−/− mice compensates for the absence of TIMP3, we examined the cardiac response of TIMP3-deficient MI compared with age-matched WT mice over 4 wk. TIMP3−/− mice showed a significantly decreased rate of survival compared with WT mice over 4 wk post-MI (Fig. 2Ai). Autopsy results revealed that a majority of the mice had died due to LV rupture that led to hemopericardium and/or hemothorax. LV rupture occurred in 81% of TIMP3−/− mice at 3–7 days post-MI compared with 32% in WT mice (Fig. 2Aii). Gross histological analysis of WT and TIMP3−/− hearts at 3 days, 1 wk, and 4 wk post-MI showed greater LV dilation and larger infarct expansion in the TIMP3−/− mice (Fig. 2B). Our findings further demonstrate that the increased TIMP2 and TIMP4 protein levels in the infarct area were not sufficient to compensate for the absence of TIMP3 and to improve the rate of post-MI rupture. Further analysis of the infarct size showed that despite comparable initial infarct sizes at 1 day post-MI, TIMP3−/− mice developed a 25% greater infarct expansion compared with WT mice by 1 wk post-MI (Fig. 2C).
Fig. 2.
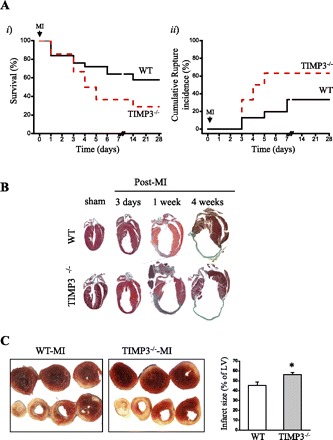
Lack of TIMP3 severely compromises the rate of survival, with increased left ventricular (LV) dilation and infarct expansion post-MI. A: rate of total survival (i) and cumulative rate of rupture incidence (ii) in WT (solid line, n = 35) and TIMP3−/− mice (dashed line, n = 50) over 4 wk following MI. B: gross morphology of WT and TIMP3−/− hearts following sham operation or 3 days, 1 wk, and 4 wk post-MI. C: representative triphenyltetrazolium chloride-stained WT and TIMP3−/− hearts (i) and averaged infarct size (ii; n = 8/genotype) at 1 wk post-MI. *P < 0.05 compared with WT.
TIMP3 deficiency leads to greater LV dilation and exacerbated systolic and diastolic dysfunction post-MI associated with aberrant ECM remodeling.
Echocardiographic imaging at 1 wk (Table 1) and 4 wk post-MI (Table 2) revealed markedly greater LV dilation and lower EF, indicating marked suppression in systolic function in TIMP3−/− compared with WT mice. Wall motion score index (WMSI) is a validated method of evaluating regional LV wall motion abnormalities and correlates with the degree of adverse ventricular remodeling post-MI (23, 35, 60). A WMSI value of 1 indicates intact LV wall motion and contractility, as found in sham-operated mice. At 1 wk post-MI, TIMP3−/− hearts showed significantly higher WMSI, indicating more severe adverse regional remodeling in the ventricles compared with WT mice, due primarily to worsening of midventricular and apical wall motion. TDI further revealed that mice lacking TIMP3 developed a more severe diastolic dysfunction, as indicated by a markedly lower ratio of E′-wave to A′-wave, a larger LA, elevated LV isovolumetric relaxation time, and increased deceleration time, which indicate impaired relaxation (Tables 1 and 2). These results demonstrate that loss of TIMP3 leads to greater LV dilation and worsening of LV wall motion and systolic and diastolic dysfunction following MI.
Table 1.
Echocardiographic parameters show more severe structural and functional deterioration in TIMP3−/− compared with WT mice at 1 wk post-MI
| WT-Sham | WT-MI | TIMP3−/−-Sham | TIMP3−/−-MI | |
|---|---|---|---|---|
| Sample size | 6 | 8 | 6 | 8 |
| HR, beats/min | 489 ± 17 | 494 ± 20 | 482 ± 18 | 499 ± 20 |
| LVEDV, μl | 65.9 ± 2.1 | 133.1 ± 14.5* | 66.1 ± 4.6 | 165.4 ± 20.2*† |
| LVESV, μl | 28.4 ± 3.0 | 95.4 ± 14.4* | 30.8 ± 2.6 | 135.8 ± 23.9*† |
| LVPWTd, mm | 0.75 ± 0.02 | 0.64 ± 0.06* | 0.73 ± 0.01 | 0.52 ± 0.02* |
| EF, % | 66.7 ± 0.2 | 37.1 ± 6.2* | 60.4 ± 0.3 | 17.4 ± 2.6*† |
| WMSI | 1 | 1.67 ± 0.09* | 1 | 2.48 ± 0.06*† |
| E′/A′ | 1.3 ± 0.2 | 0.9 ± 0.1* | 1.1 ± 0.1 | 0.7 ± 0.1*† |
| LA size, mm | 1.5 ± 0.1 | 2.2 ± 0.2* | 1.6 ± 0.1 | 2.5 ± 0.1*† |
| IVRT, ms | 14.7 ± 0.5 | 17.5 ± 1.1* | 14.2 ± 0.7 | 21.9 ± 2.1*† |
| DT, ms | 22.7 ± 0.6 | 24.4 ± 2.3 | 23.3 ± 0.8 | 29.3 ± 3.6*† |
HR, heart rate; LVEDV, left ventricular end-diastolic volume; LVESV, LV end-systolic volume; LVPWTd, LV posterior wall thickness during diastole; EF, ejection fraction; WMSI, wall motion score index; E′/A′, ratio of early tissue Doppler velocity (E′) to tissue Doppler velocity due to atrial contraction (A′); LA, left atrial; IVRT, isovolumetric relaxation time (of LV); DT, deceleration time of E-wave. Values are means ± SE.
P < 0.05 compared with corresponding sham-operated group.
P < 0.05 compared with WT-MI group.
Table 2.
Echocardiographic parameters show greater structural and functional deterioration in TIMP3−/− compared with WT mice at 4 wk post-MI
| WT-Sham | WT-MI | TIMP3−/−-Sham | TIMP3−/−-MI | |
|---|---|---|---|---|
| Sample size | 6 | 10 | 6 | 10 |
| HR, beats/min | 489 ± 17 | 492 ± 14 | 490 ± 9 | 499 ± 20 |
| LVEDV, μl | 68.5 ± 5.4 | 167.5 ± 15.2* | 66.8 ± 3.7 | 190.4 ± 15.5*† |
| LVESV, μl | 31.8 ± 2.1 | 138.3 ± 14.2* | 29.4 ± 3.1 | 165.2 ± 5.5*† |
| EF, % | 61.4 ± 1.3 | 25.5 ± 3.2* | 63.7 ± 2.1 | 12.2 ± 4.3*† |
| E′/A′ | 1.1 ± 0.1 | 0.8 ± 0.1* | 1.2 ± 0.1 | 0.6 ± 0.1* |
| LA size, mm | 1.5 ± 0.2 | 2.3 ± 0.2* | 1.6 ± 0.1 | 2.6 ± 0.3* |
| IVRT, ms | 14.2 ± 0.7 | 19.2 ± 1.2* | 14.7 ± 0.5 | 25.1 ± 2.3*† |
| DT, ms | 23.3 ± 0.9 | 25.9 ± 1.3 | 25.5 ± 2.1 | 32.4 ± 1.9*† |
Values are means ± SE.
P < 0.05 compared with corresponding sham-operated group.
P < 0.05 compared with WT-MI group.
TIMP3 is a potent inhibitor of a number of MMPs that degrade the ECM structural proteins. In addition, the severe LV dilation in TIMP3−/− mice indicates structural instability that could result from disrupted ECM structure. We assessed ECM structure in the infarct and noninfarct myocardium of WT and TIMP3−/− mice at 3 days post-MI by second harmonic generation imaging of unfixed and unstained hearts as described previously (23). We found that TIMP3-deficient mice exhibited lower density and greater disarray of collagen fibers in the infarct area compared with WT mice (Fig. 3B), whereas expression of procollagen types Iα and IIIα were comparable between the genotypes (Fig. 3C), suggesting excess MMP activity and collagen degradation in TIMP3−/− mice.
Fig. 3.
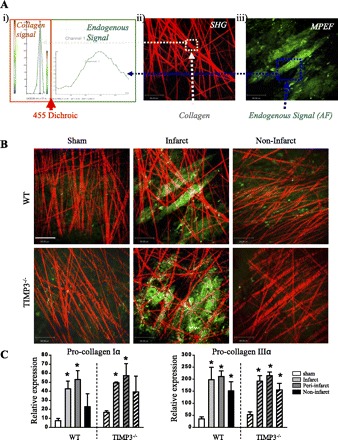
Lack of TIMP3 results in aberrant degradation of the fibrillar structure of the extracellular matrix. A: second harmonic generation (SHG; red) (i) and multiphoton excitation fluorescence (MPEF; green) imaging (ii) were used to visualize the collagen fibers and endogenous autofluorescence, respectively, and SHG and MPEF images were superimposed (iii). B: representative images showing the density and organization of the collagen fibers in the infarct and noninfarct myocardium of WT and TIMP3−/− mice at 3 days post-MI. Scale bar, 80 μm. C: TaqMan mRNA expression of procollagen types Iα (i) and IIIα (ii) in WT and TIMP3−/− mice at 3 days post-MI (n = 5/group). *P < 0.05 compared with sham.
Early post-MI proteolysis is markedly elevated in the absence of TIMP3.
MMPs have been linked to LV dilation, rupture, and inflammation post-MI (18, 27, 33, 53). We therefore performed gelatin zymography to determine the levels of MMP2 and MMP9, the two MMPs that have been associated with LV rupture post-MI (27, 33). We assessed the total proteolytic activity in the infarct, peri-infarct, and noninfarct myocardial tissue by using gelatinase and collagenase activity assays from EnzChek. At 1 day post-MI, MMP9 and active MMP2 were detectable in the infarct and peri-infarct regions of TIMP3−/− but not WT hearts (Fig. 4A, i and ii). Consistently, total gelatinase (Fig. 4Aiii) and collagenase activities (Fig. 4Aiv) were significantly greater in TIMP3−/− compared with WT myocardium. Interestingly, by 3 days post-MI, gelatin zymography showed elevated MMP9 and active MMP2 only in the noninfarct myocardium of TIMP3−/− compared with WT hearts (Fig. 4B, i and ii). Total gelatinase activity (Fig. 4Biii), but not collagenase activity (Fig. 4Biv), was higher in the peri-infarct TIMP3−/− myocardium. A similar pattern was observed at 1 wk post-MI (Fig. 4C). These data indicate that MI results in time- and region-specific alterations, including a very early rise in MMP activities, which could be a key determinant of the subsequent LV remodeling and dysfunction.
Fig. 4.
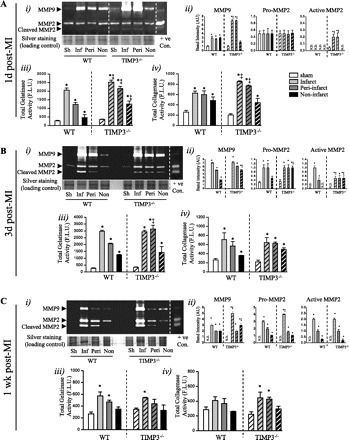
TIMP3 deficiency triggers a rapid and transient increase in MMP levels and activity. Representative gelatin zymography (i), averaged band intensities for MMP9, pro-MMP2, and active MMP2 (ii), total gelatinase activity (iii), and total collagenase activity (iv) are shown in WT and TIMP3−/− hearts at 1 day (A), 3 days (B), and 1 wk post-MI (C) (n = 6/group per genotype). Sh, sham; Inf, infarct; peri, peri-infarct; non, noninfarct; F.L.U., fluorescent light unit. *P < 0.05 compared with sham. ‡P < 0.05 compared with WT.
Increased rate of LV rupture in TIMP3−/−-MI mice is associated with heightened inflammation.
MI triggers a rapid inflammatory response that involves the influx of leukocytes in the infarct and peri-infarct regions of the myocardium (14, 15). Among the leukocytes, neutrophils are the first responders (27), and neutrophil influx in the infarct area of myocardium has been linked to LV rupture post-MI (12, 16). TIMP3 deficiency has been shown to result in increased inflammation in other disease models (34, 47). Staining for neutrophils revealed a significantly greater population of these cells in the infarct and peri-infarct regions of TIMP3−/− compared with WT hearts at 3 days post-MI (Fig. 5, A and B). In addition, expression of inflammatory MMPs was increased in these hearts. MMP8 mRNA levels were significantly higher in all regions, and MMP12 in the infarct area, of TIMP3−/− hearts (Fig. 5C, i and ii). The severe inflammation and expression of inflammatory MMPs in TIMP3-deficient mice at 3 days post-MI coincides with the peak of LV rupture post-MI. In addition, MT1-MMP, a major collagenase in the heart, was significantly elevated in TIMP3−/− myocardium (Fig. 5Ciii). By 1 wk post-MI, the inflammatory response was dissipated in both genotypes (data not shown).
Fig. 5.
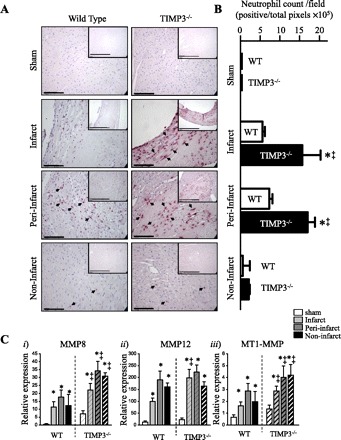
Enhanced neutrophil infiltration in the TIMP3-deficient infarct and peri-infarct myocardium at 3 days post-MI. A: immunostaining for mouse neutrophils in WT and TIMP3−/− hearts after sham or MI. Scale bar, 100 μm. Inset shows a lower magnification; scale bar, 200 μm. B: averaged neutrophil counts in sham, infarct, peri-infarct, and noninfarct myocardium in WT and TIMP3−/− mice. Neutrophil counts were performed on 6 fields per cross section, 3 cross sections per heart, and 5 hearts per genotype. C: expression levels of inflammatory MMPs MMP8 (i) and MMP12 (ii) and collagenase MT1-MMP (iii) in WT and TIMP3−/− hearts (n = 6/group per genotype). *P < 0.05 compared with sham. ‡P < 0.05 compared with WT.
Early inhibition of MMPs blunts the adverse outcomes of TIMP3 deficiency.
Next, we aimed to determine whether the early rise in total MMP activities is in fact the underlying mechanism for the more severe LV remodeling, dysfunction, and rupture in TIMP3−/− mice. We treated TIMP3−/− mice with PD-166793, a broad-spectrum MMP inhibitor (MMPi) that does not inhibit ADAM17 (a disintegrin and metalloproteinase-17), also known as TNF-α-converting enzyme (TACE) (42), 2 days before and 2 days after induction of MI (Fig. 6A). This treatment significantly improved total survival (Fig. 6B) and the rate of LV rupture incidence (Fig. 6C) in TIMP3−/− mice. In addition, this early MMPi treatment decreased the infarct expansion (Fig. 6D), reduced LV dilation (Fig. 7, A and Ci), improved the systolic dysfunction as indicated by increased EF (Fig. 7Cii) and lowered WMSI (Fig. 7Ciii), and improved diastolic dysfunction as shown by increased E′/A′ (Fig. 7Di) in TIMP3−/− mice. The decreases in DT and IVRT showed a trend (P = 0.055) but were not statistically significant (Fig. 7D, ii and iii). This experiment illustrates that MMP activation very early post-MI is a key determinant of the subsequent outcome and that the beneficial effects of MMP inhibition outlast the treatment.
Fig. 6.
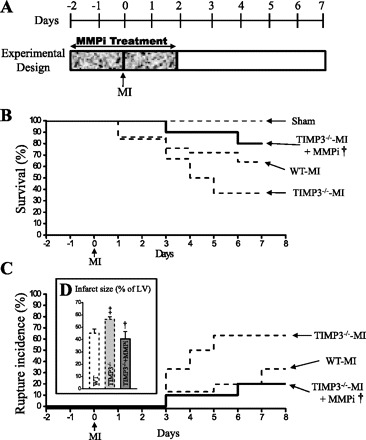
Early treatment with an MMP inhibitor (MMPi; PD-166793) improved post-MI survival in TIMP3−/− mice. A: experimental design for MMPi treatment (30 mg·kg−1·day−1). Total survival (B) and cumulative rate of LV rupture (C) are shown in sham-WT/TIMP3−/−, WT-MI, TIMP3−/−-MI, and TIMP3−/−-MI+MMPi groups (n = 10/sham per genotype, 35/WT, 50/TIMP3−/−, 20/MI+MMPi). D: infarct size at 1 wk post-MI for WT (n = 8), TIMP3−/− (n = 8), and TIMP3−/−+MMPi (n = 6) groups. ‡P < 0.05 compared with WT-MI. †P < 0.05 compared with TIMP3−/−-MI.
Fig. 7.
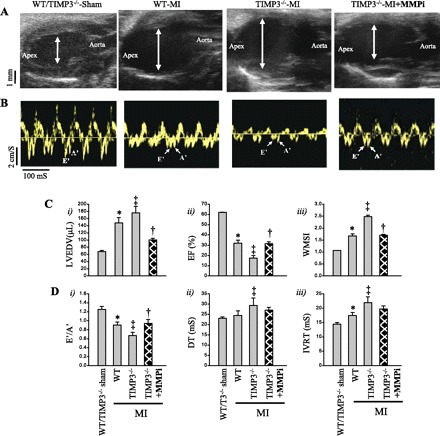
Early MMP inhibition markedly improved LV dilation and dysfunction in TIMP3−/− mice post-MI. Representative parasternal long-axis view (A) and tissue Doppler images (B) are shown in sham of either genotype, WT-MI, TIMP3−/−-MI, and TIMP3−/−-MI hearts with MMPi treatment at 1 wk post-MI. C: averaged left ventricular end-diastolic volume (LVEDV; i), ejection fraction (EF; ii), and wall motion score index (WMSI; iii) are shown in sham-operated (n = 5/group), WT-MI (n = 8/group), TIMP3−/−-MI (n = 8/group), and TIMP3−/−-MI+MMPi (n = 8/group) groups. D: averaged diastolic parameters E′/A′ (i), deceleration time of the E-wave (DT; ii), and isovolumetric relaxation time of LV (IVRT; iii) are shown (n = 8/group). *P < 0.05 compared with sham. ‡P < 0.05 compared with WT-MI. †P < 0.05 compared with TIMP3−/−-MI.
DISCUSSION
Myocardial remodeling following infarction is a complex process orchestrated by a number of cellular and extracellular events (49, 59). Hence, identifying the early events that lead to activation of multiple factors in mediating adverse cardiac response to MI can provide new potential therapies to lessen the resulting complications. We (25) have reported previously that TIMP3-deficient mice are more susceptible to pressure overload induced by aortic constriction. TIMP3-deficient mice have also been reported to develop accelerated remodeling following MI (52); however, the molecular analyses in this study were performed at or after 1 wk post-MI. We and others (16, 23, 26, 58) have found that post-MI LV rupture peaks at 3 days with no incidence of LV rupture after 7 days post-MI. In the current study we have demonstrated that LAD ligation triggers early downregulation of TIMP3 in WT mice and that TIMP3-deficient mice show a very early increase in MMP activities post-MI, aberrant degradation of ECM fibrillar structure, and enhanced inflammation, leading to exacerbated LV dilation and systolic and diastolic dysfunction at 1 and 4 wk post-MI. Diastolic dysfunction is an important and independent prognostic marker of adverse outcome in patients with acute MI (36, 37). The worsened diastolic dysfunction in TIMP3−/−-MI mice is consistent with the more severe LV remodeling and systolic dysfunction in these mice. These data provide a thorough analysis of the early molecular events that lead to deteriorated cardiac remodeling, morphometry, and dysfunction by 1 wk post-MI. This study was performed on male mice, and the impact of TIMP3 deficiency in post-MI remodeling in female mice needs to be evaluated.
The markedly greater rate of LV rupture in TIMP3-deficient mice is associated with a greater infarct expansion in these mice, which is consistent with a recent report that infarct size determines the risk of LV rupture post-MI (16). We used second harmonic generation imaging on unfixed and unstained heart tissue to visualize the ECM fibrillar structure. The advantage of this technique is that it excludes the tissue alterations resulting from manipulations such as fixing or staining. The greater disarray and lower density of the collagen fibers in TIMP3−/− infarct myocardium despite comparable collagen synthesis are consistent with the enhanced inflammation products of matrix proteins in these mice (15). Fragments of ECM proteins can attract inflammatory cells (2), and initial degradation of collagen fibers has been shown to precede inflammation post-MI (48). Degradation products of collagen type I, such as the Pro-Gly-Pro (PGP) peptide that is produced by MMP9 (32) and MMP8 (30), can serve as neutrophil chemoattractants in vivo (55). In addition, TIMP3 was recently reported to constrain neutrophil influx in lung through inhibiting MMPs (17). Hence, the severe inflammation in TIMP3−/− myocardium further indicates excess damage to the ECM structural components in these mice post-MI. Recent studies have shown that a high neutrophil count is correlated with a higher incidence of adverse cardiac events in patients with acute MI (51) and that the neutrophil-to-lymphocyte ratio serves as an independent predictor of cardiac death in patients with stable coronary artery disease (41). Inflammatory cells can produce a number of MMPs (including MMP8, MMP9, and MMP12) that will further mediate ECM degradation and overall adverse tissue remodeling (9, 40, 54). The greater neutrophil infiltration in TIMP3−/− hearts is consistent with elevated levels of MMP8, MMP9, and MMP12 in these hearts. Inflammation and elevated MMP9 levels have also been linked to LV rupture in patients and in animal models (18, 27, 33, 53). In addition, mice lacking MMP9 are protected against LV rupture post-MI (18) and exhibit improved LV dilation and dysfunction post-MI (11).
We examined the time- and region-specific MMP activities and found that MMP9 and MMP2 levels, as well as total collagenase and gelatinase activities, were elevated in the infarct and peri-infarct regions by 1 day post-MI in TIMP3−/− mice. This is also consistent with reports showing that MMP9 increases early post-MI in patients (22) and in experimental models (31). An increase in MMP activities and collagen degradation within hours of MI also has been reported in rat hearts (50). Tian et al. (52) reported increased MMP2 activity in TIMP3−/− mice at 1 wk post-MI, whereas we found the rise in MMP2 activity only at 1 day post-MI but not at later time points. The finding of Tian et al. most likely resulted because activity was measured in the whole LV, and the region-dependent (infarct, peri-infarct vs. noninfarct) alterations in MMP2 activation were not taken into consideration. A region-specific MMP expression and TIMP downregulation in sheep heart post-MI has also been reported (57). Mice lacking MMP2 are protected against LV rupture (33), whereas mice overexpressing MMP2 develop LV dilation and dysfunction (6). The early increase in proteolytic activities in the infarct and peri-infarct regions could explain the greater rate of LV rupture in TIMP3−/− mice, whereas the increased MMP levels and proteolytic activity in the noninfarct myocardium could explain the exacerbated LV dilation and dysfunction in these mice compared with the parallel WT group. We recently reported that TIMP2-deficient mice exhibited comparable LV rupture incidence compared with the WT mice despite a far more severe LV dilation and dysfunction (23). In addition, TIMP2 upregulation in the infarct area of TIMP3-deficient hearts was not sufficient to decrease the rate of LV rupture in these mice. These findings indicate that whereas TIMP2 deficiency primarily affected the remodeling and function of the noninfarct myocardium, lack of TIMP3 exerted a global effect influencing the infarct as well as the noninfarct myocardium. This could result from a number of factors. In WT mice, myocardial infarction results in reduction of TIMP2 protein primarily in the noninfarct region, whereas TIMP3 is reduced in the infarct as well as the noninfarct myocardium. In addition, TIMP2 deficiency results in lack of MMP2 activation (23), as opposed to increased MMP2 activation within 1 day post-MI in TIMP3−/− mice. MMP2 has been reported to be a key factor in post-MI LV rupture (33) and as such could partly explain the differential post-MI rupture in TIMP2- vs. TIMP3-deficient mice. In patients with MI, the adverse ventricular remodeling has been linked to an imbalance in MMP/TIMP activity (56). Our current study and previous report (23) demonstrate that TIMP2 and TIMP3 play different roles in cardiac response to MI. TIMP2 is more critical in the function and remodeling of the peri- and noninfarct myocardium, whereas TIMP3 globally impacts all myocardium, influencing the rate of LV rupture as well as the remodeling and function of the noninfarct myocardium.
The protective effects of MMPi treatment early post-MI in TIMP3−/− mice indicate that the early rise in MMP activities post-MI is a major determinant of subsequent LV remodeling and dysfunction. In addition to inhibiting a large number of MMPs, TIMP3 also inhibits TACE, which is responsible for cell surface shedding of TNF and activation of its downstream signaling (3, 7). We recently reported that activation of the TNF pathway induces activation of a number of MMPs in cardiomyocytes within 1 h of treatment (4). Moreover, the current study was aimed to examine the beneficial effects of early MMPi treatment. Continuous MMPi treatment attenuates the adverse LV remodeling in mice (45) and in pigs (38) following MI. We demonstrate that the beneficial effects of MMPi treatment during the first 2 days post-MI persist to prevent adverse LV remodeling, infarct expansion, and LV rupture up to 1 wk post-MI. Targeting the early rise in MMP activities post-MI is particularly important, since we have demonstrated that levels of TIMPs (TIMP2, TIMP3, and TIMP4) are also reduced early post-MI. In addition, this could also explain the unsuccessful outcome of the PREMIER trial, where MI patients received PG-116800, an MMPi, 48–72 h after the onset of MI (19). Perhaps an earlier treatment window could have been more beneficial.
In summary, in this study we have provided definitive evidence that the MMP inhibitory function of TIMP3 is critical during early stages of recovery from myocardial infarction. Since TIMP3 levels are significantly reduced in the hearts of patients with ischemic cardiomyopathy, overexpressing or supplementing TIMP3 in myocardial ischemic injury may prove to be a promising therapeutic approach.
GRANTS
This study was funded by Canadian Institute of Health Research Operating Grants 84279 (to Z. Kassiri) and 86602 (to G. Y. Oudit). Z. Kassiri is a New Investigator of the Heart and Stroke Foundation of Canada and an Alberta Heritage Foundation for Medical Research (AHFMR) scholar, G. Y. Oudit is an AHFMR Clinician Investigator, and G. D. Lopaschuk is an AHFMR Medical Scientist.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge technical support from the core facility at the Cardiovascular Research Centre at the University of Alberta and an Alberta HEART interdisciplinary team grant.
REFERENCES
- 1. Abraham T, Carthy J, McManus B. Collagen matrix remodeling in 3-dimensional cellular space resolved using second harmonic generation and multi photon excitation fluorescence. J Struct Biol 169: 36–44, 2010. [DOI] [PubMed] [Google Scholar]
- 2. Adair-Kirk TL, Senior RM. Fragments of extracellular matrix as mediators of inflammation. Int J Biochem Cell Biol 40: 1101–1110, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Amour A, Slocombe PM, Webster A, Butler M, Knight CG, Smith BJ, Stephens PE, Shelley C, Hutton M, Knauper V, Docherty AJ, Murphy G. TNF-α converting enzyme (TACE) is inhibited by TIMP-3. FEBS Lett 435: 39–44, 1998. [DOI] [PubMed] [Google Scholar]
- 4. Awad AE, Kandalam V, Chakrabarti S, Wang X, Penninger JM, Davidge ST, Oudit GY, Kassiri Z. Tumor necrosis factor induces matrix metalloproteinases in cardiomyocytes and cardiofibroblasts differentially via superoxide production in a PI3Kγ-dependent manner. Am J Physiol Cell Physiol 298: C679–C692, 2010. [DOI] [PubMed] [Google Scholar]
- 5. Basu R, Oudit GY, Wang X, Zhang L, Ussher JR, Lopaschuk GD, Kassiri Z. Type 1 diabetic cardiomyopathy in the Akita (Ins2WT/C96Y) mouse model is characterized by lipotoxicity and diastolic dysfunction with preserved systolic function. Am J Physiol Heart Circ Physiol 297: H2096–H2108, 2009. [DOI] [PubMed] [Google Scholar]
- 6. Bergman MR, Teerlink JR, Mahimkar R, Li L, Zhu BQ, Nguyen A, Dahi S, Karliner JS, Lovett DH. Cardiac matrix metalloproteinase-2 expression independently induces marked ventricular remodeling and systolic dysfunction. Am J Physiol Heart Circ Physiol 292: H1847–H1860, 2007. [DOI] [PubMed] [Google Scholar]
- 7. Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385: 729–733, 1997. [DOI] [PubMed] [Google Scholar]
- 8. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling—concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol 35: 569–582, 2000. [DOI] [PubMed] [Google Scholar]
- 9. Cuadrado E, Ortega L, Hernández-Guillamon M, Penalba A, Fernández-Cadenas I, Rosell A, Montaner J. Tissue plasminogen activator (t-PA) promotes neutrophil degranulation and MMP-9 release. J Leukoc Biol 84: 207–214, 2008. [DOI] [PubMed] [Google Scholar]
- 10. Davis N, Sistino JJ. Review of ventricular rupture: key concepts and diagnostic tools for success. Perfusion 17: 63–67, 2002. [DOI] [PubMed] [Google Scholar]
- 11. Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest 106: 55–62, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fang L, Gao XM, Moore XL, Kiriazis H, Su Y, Ming Z, Lim YL, Dart AM, Du XJ. Differences in inflammation, MMP activation and collagen damage account for gender difference in murine cardiac rupture following myocardial infarction. J Mol Cell Cardiol 43: 535–544, 2007. [DOI] [PubMed] [Google Scholar]
- 13. Figueras J, Cortadellas J, Soler-Soler J. Left ventricular free wall rupture: clinical presentation and management. Heart 83: 499–504, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Frangogiannis NG. Targeting the inflammatory response in healing myocardial infarcts. Curr Med Chem 13: 1877–1893, 2006. [DOI] [PubMed] [Google Scholar]
- 15. Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res 53: 31–47, 2002. [DOI] [PubMed] [Google Scholar]
- 16. Gao XM, Ming Z, Su Y, Fang L, Kiriazis H, Xu Q, Dart AM, Du XJ. Infarct size and post-infarct inflammation determine the risk of cardiac rupture in mice. Int J Cardiol 143: 20–28, 2010. [DOI] [PubMed] [Google Scholar]
- 17. Gill SE, Huizar I, Bench EM, Sussman SW, Wang Y, Khokha R, Parks WC. Tissue inhibitor of metalloproteinases 3 regulates resolution of inflammation following acute lung injury. Am J Pathol 176: 64–73, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nube O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med 5: 1135–1142, 1999. [DOI] [PubMed] [Google Scholar]
- 19. Hudson MP, Armstrong PW, Ruzyllo W, Brum J, Cusmano L, Krzeski P, Lyon R, Quinones M, Theroux P, Sydlowski D, Kim HE, Garcia MJ, Jaber WA, Weaver WD. Effects of selective matrix metalloproteinase inhibitor (PG-116800) to prevent ventricular remodeling after myocardial infarction: results of the PREMIER (Prevention of Myocardial Infarction Early Remodeling) trial. J Am Coll Cardiol 48: 15–20, 2006. [DOI] [PubMed] [Google Scholar]
- 20. Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, Oates JA, Rahko PS, Silver MA, Stevenson LW, Yancy CW, Antman EM, Smith SC, Jr, Adams CD, Anderson JL, Faxon DP, Fuster V, Halperin JL, Hiratzka LF, Jacobs AK, Nishimura R, Ornato JP, Page RL, Riegel B. ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 112: e154–e235, 2005. [DOI] [PubMed] [Google Scholar]
- 21. Jugdutt BI. Ventricular remodeling after infarction and the extracellular collagen matrix: when is enough enough? Circulation 108: 1395–1403, 2003. [DOI] [PubMed] [Google Scholar]
- 22. Kaden JJ, Dempfle CE, Sueselbeck T, Brueckmann M, Poerner TC, Haghi D, Haase KK, Borggrefe M. Time-dependent changes in the plasma concentration of matrix metalloproteinase 9 after acute myocardial infarction. Cardiology 99: 140–144, 2003. [DOI] [PubMed] [Google Scholar]
- 23. Kandalam V, Basu R, Abraham T, Wang X, Soloway PD, Jaworski DM, Oudit GY, Kassiri Z. TIMP2 deficiency accelerates adverse post-myocardial infarction remodeling because of enhanced MT1-MMP activity despite lack of MMP2 activation. Circ Res 106: 796–808, 2010. [DOI] [PubMed] [Google Scholar]
- 24. Kassiri Z, Defamie V, Hariri M, Oudit GY, Anthwal S, Dawood F, Liu P, Khokha R. Simultaneous transforming growth factor beta-tumor necrosis factor activation and cross-talk cause aberrant remodeling response and myocardial fibrosis in Timp3-deficient heart. J Biol Chem 284: 29893–29904, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kassiri Z, Oudit GY, Sanchez O, Dawood F, Mohammed FF, Nuttall RK, Edwards DR, Liu PP, Backx PH, Khokha R. Combination of tumor necrosis factor-alpha ablation and matrix metalloproteinase inhibition prevents heart failure after pressure overload in tissue inhibitor of metalloproteinase-3 knock-out mice. Circ Res 97: 380–390, 2005. [DOI] [PubMed] [Google Scholar]
- 26. Kassiri Z, Zhong J, Guo D, Basu R, Wang X, Liu PP, Scholey JW, Penninger JM, Oudit GY. Loss of angiotensin-converting enzyme 2 accelerates maladaptive left ventricular remodeling in response to myocardial infarction. Circ Heart Fail 2: 446–455, 2009. [DOI] [PubMed] [Google Scholar]
- 27. Kawakami R, Saito Y, Kishimoto I, Harada M, Kuwahara K, Takahashi N, Nakagawa Y, Nakanishi M, Tanimoto K, Usami S, Yasuno S, Kinoshita H, Chusho H, Tamura N, Ogawa Y, Nakao K. Overexpression of brain natriuretic peptide facilitates neutrophil infiltration and cardiac matrix metalloproteinase-9 expression after acute myocardial infarction. Circulation 110: 3306–3312, 2004. [DOI] [PubMed] [Google Scholar]
- 28. Kawamoto H, Yasuda O, Suzuki T, Ozaki T, Yotsui T, Higuchi M, Rakugi H, Fukuo K, Ogihara T, Maeda N. Tissue inhibitor of metalloproteinase-3 plays important roles in the kidney following unilateral ureteral obstruction. Hypertens Res 29: 285–294, 2006. [DOI] [PubMed] [Google Scholar]
- 29. Li YY, Feldman AM, Sun Y, McTiernan CF. Differential expression of tissue inhibitors of metalloproteinases in the failing human heart. Circulation 98: 1728–1734, 1998. [DOI] [PubMed] [Google Scholar]
- 30. Lin M, Jackson P, Tester AM, Diaconu E, Overall CM, Blalock JE, Pearlman E. Matrix metalloproteinase-8 facilitates neutrophil migration through the corneal stromal matrix by collagen degradation and production of the chemotactic peptide Pro-Gly-Pro. Am J Pathol 173: 144–153, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lindsey M, Wedin K, Brown MD, Keller C, Evans AJ, Smolen J, Burns AR, Rossen RD, Michael L, Entman M. Matrix-dependent mechanism of neutrophil-mediated release and activation of matrix metalloproteinase 9 in myocardial ischemia/reperfusion. Circulation 103: 2181–2187, 2001. [DOI] [PubMed] [Google Scholar]
- 32. Malik M, Bakshi CS, McCabe K, Catlett SV, Shah A, Singh R, Jackson PL, Gaggar A, Metzger DW, Melendez JA, Blalock JE, Sellati TJ. Matrix metalloproteinase 9 activity enhances host susceptibility to pulmonary infection with type A and B strains of Francisella tularensis. J Immunol 178: 1013–1020, 2007. [DOI] [PubMed] [Google Scholar]
- 33. Matsumura S, Iwanaga S, Mochizuki S, Okamoto H, Ogawa S, Okada Y. Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J Clin Invest 115: 599–609, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mohammed FF, Smookler DS, Taylor SE, Fingleton B, Kassiri Z, Sanchez OH, English JL, Matrisian LM, Au B, Yeh WC, Khokha R. Abnormal TNF activity in Timp3−/− mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat Genet 36: 969–977, 2004. [DOI] [PubMed] [Google Scholar]
- 35. Moller JE, Hillis GS, Oh JK, Reeder GS, Gersh BJ, Pellikka PA. Wall motion score index and ejection fraction for risk stratification after acute myocardial infarction. Am Heart J 151: 419–425, 2006. [DOI] [PubMed] [Google Scholar]
- 36. Moller JE, Hillis GS, Oh JK, Seward JB, Reeder GS, Wright RS, Park SW, Bailey KR, Pellikka PA. Left atrial volume: a powerful predictor of survival after acute myocardial infarction. Circulation 107: 2207–2212, 2003. [DOI] [PubMed] [Google Scholar]
- 37. Moller JE, Pellikka PA, Hillis GS, Oh JK. Prognostic importance of diastolic function and filling pressure in patients with acute myocardial infarction. Circulation 114: 438–444, 2006. [DOI] [PubMed] [Google Scholar]
- 38. Mukherjee R, Brinsa TA, Dowdy KB, Scott AA, Baskin JM, Deschamps AM, Lowry AS, Escobar GP, Lucas DG, Yarbrough WM, Zile MR, Spinale FG. Myocardial infarct expansion and matrix metalloproteinase inhibition. Circulation 107: 618–625, 2003. [DOI] [PubMed] [Google Scholar]
- 39. Nuttall RK, Sampieri CL, Pennington CJ, Gill SE, Schultz GA, Edwards DR. Expression analysis of the entire MMP and TIMP gene families during mouse tissue development. FEBS Lett 563: 129–134, 2004. [DOI] [PubMed] [Google Scholar]
- 40. Owen CA, Hu Z, Lopez-Otin C, Shapiro SD. Membrane-bound matrix metalloproteinase-8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase-resistant collagenase and serpinase. J Immunol 172: 7791–7803, 2004. [DOI] [PubMed] [Google Scholar]
- 41. Papa A, Emdin M, Passino C, Michelassi C, Battaglia D, Cocci F. Predictive value of elevated neutrophil-lymphocyte ratio on cardiac mortality in patients with stable coronary artery disease. Clin Chim Acta 395: 27–31, 2008. [DOI] [PubMed] [Google Scholar]
- 42. Peterson JT, Hallak H, Johnson L, Li H, O'Brien PM, Sliskovic DR, Bocan TM, Coker ML, Etoh T, Spinale FG. Matrix metalloproteinase inhibition attenuates left ventricular remodeling and dysfunction in a rat model of progressive heart failure. Circulation 103: 2303–2309, 2001. [DOI] [PubMed] [Google Scholar]
- 43. Peterson JT, Li H, Dillon L, Bryant JW. Evolution of matrix metalloprotease and tissue inhibitor expression during heart failure progression in the infarcted rat. Cardiovasc Res 46: 307–315, 2000. [DOI] [PubMed] [Google Scholar]
- 44. Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 81: 1161–1172, 1990. [DOI] [PubMed] [Google Scholar]
- 45. Rohde LE, Ducharme A, Arroyo LH, Aikawa M, Sukhova GH, Lopez-Anaya A, McClure KF, Mitchell PG, Libby P, Lee RT. Matrix metalloproteinase inhibition attenuates early left ventricular enlargement after experimental myocardial infarction in mice. Circulation 99: 3063–3070, 1999. [DOI] [PubMed] [Google Scholar]
- 46. Sawicki G, Menon V, Jugdutt BI. Improved balance between TIMP-3 and MMP-9 after regional myocardial ischemia-reperfusion during AT1 receptor blockade. J Card Fail 10: 442–449, 2004. [DOI] [PubMed] [Google Scholar]
- 47. Smookler DS, Mohammed FF, Kassiri Z, Duncan GS, Mak TW, Khokha R. Tissue inhibitor of metalloproteinase 3 regulates TNF-dependent systemic inflammation. J Immunol 176: 721–725, 2006. [DOI] [PubMed] [Google Scholar]
- 48. Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev 87: 1285–1342, 2007. [DOI] [PubMed] [Google Scholar]
- 49. Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation 101: 2981–2988, 2000. [DOI] [PubMed] [Google Scholar]
- 50. Takahashi S, Barry AC, Factor SM. Collagen degradation in ischaemic rat hearts. Biochem J 265: 233–241, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Takahashi T, Hiasa Y, Ohara Y, Miyazaki S, Ogura R, Suzuki N, Hosokawa S, Kishi K, Ohtani R. Relationship of admission neutrophil count to microvascular injury, left ventricular dilation, and long-term outcome in patients treated with primary angioplasty for acute myocardial infarction. Circ J 72: 867–872, 2008. [DOI] [PubMed] [Google Scholar]
- 52. Tian H, Cimini M, Fedak PW, Altamentova S, Fazel S, Huang ML, Weisel RD, Li RK. TIMP-3 deficiency accelerates cardiac remodeling after myocardial infarction. J Mol Cell Cardiol 43: 733–743, 2007. [DOI] [PubMed] [Google Scholar]
- 53. van den Borne SW, Cleutjens JP, Hanemaaijer R, Creemers EE, Smits JF, Daemen MJ, Blankesteijn WM. Increased matrix metalloproteinase-8 and -9 activity in patients with infarct rupture after myocardial infarction. Cardiovasc Pathol 18: 37–43, 2008. [DOI] [PubMed] [Google Scholar]
- 54. Van Lint P, Libert C. Matrix metalloproteinase-8: cleavage can be decisive. Cytokine Growth Factor Rev 17: 217–223, 2006. [DOI] [PubMed] [Google Scholar]
- 55. Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, Folkerts G, Nijkamp FP, Blalock JE. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med 12: 317–323, 2006. [DOI] [PubMed] [Google Scholar]
- 56. Webb CS, Bonnema DD, Ahmed SH, Leonardi AH, McClure CD, Clark LL, Stroud RE, Corn WC, Finklea L, Zile MR, Spinale FG. Specific temporal profile of matrix metalloproteinase release occurs in patients after myocardial infarction: relation to left ventricular remodeling. Circulation 114: 1020–1027, 2006. [DOI] [PubMed] [Google Scholar]
- 57. Wilson EM, Moainie SL, Baskin JM, Lowry AS, Deschamps AM, Mukherjee R, Guy TS, St John-Sutton MG, Gorman JH, 3rd, Edmunds LH, Jr, Gorman RC, Spinale FG. Region- and type-specific induction of matrix metalloproteinases in post-myocardial infarction remodeling. Circulation 107: 2857–2863, 2003. [DOI] [PubMed] [Google Scholar]
- 58. Yang Y, Ma Y, Han W, Li J, Xiang Y, Liu F, Ma X, Zhang J, Fu Z, Su YD, Du XJ, Gao XM. Age-related differences in postinfarct left ventricular rupture and remodeling. Am J Physiol Heart Circ Physiol 294: H1815–H1822, 2008. [DOI] [PubMed] [Google Scholar]
- 59. Zamilpa R, Lindsey ML. Extracellular matrix turnover and signaling during cardiac remodeling following MI: causes and consequences. J Mol Cell Cardiol 48: 558–563, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang Y, Takagawa J, Sievers RE, Khan MF, Viswanathan MN, Springer ML, Foster E, Yeghiazarians Y. Validation of the wall motion score and myocardial performance indexes as novel techniques to assess cardiac function in mice after myocardial infarction. Am J Physiol Heart Circ Physiol 292: H1187–H1192, 2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.