

Abstract
Of the five G-protein-coupled muscarinic acetylcholine receptors (mAChRs or M1-M5), M5 is the least explored and understood due to a lack of mAChR subtype selective ligands. We recently performed a high-throughput functional screen and identified a number of weak antagonist hits that were selective for M5. An iterative parallel synthesis and detailed molecular pharmacologic profiling effort, led to the discovery of the first highly selective, CNS penetrant M5 orthosteric antagonist tool compound, with submicromolar potency (hM5 IC50 = 450 nM, hM5 Ki = 340 nM, M1-M4 IC50s >30 μM), enantiospecific inhibition and an acceptable DMPK profile for in vitro and electrophysiology studies.
Keywords: muscarinic, M5, antagonist, orthosteric, acetylcholine
Numerous studies have suggested that subtype-selective muscarinic acetylcholine receptor (mAChR) antagonists may provide novel approaches to treat a number of central nervous system (CNS) disorders;[1–7] however, the majority of existing antagonists are not selective for specific mAChR subtypes, forcing the field to rely on biochemical and or genetic approaches to discern therapeutic potential.[1–7] Of the central mAChRs, M5 expression is the lowest (<2%), and M5 is predominantly localized to dopaminergic neurons originating in the substantia nigra pars compacta (SNc) and the ventral tegmental area (VTA).[4–10] Studies with M5−/− mice have confirmed the hypothesis that M5 modulates dopaminergic neurotransmission and that M5 functions in addiction/reward mechanisms, with M5−/− mice displaying reduced cocaine place preference and self-administration without affecting food intake.[4–10] Further biochemical studies with M5-targeted antisense oligonucleotides, or scopolamine, infused into the VTA have recapitulated the genetic findings,[11,12] strongly suggesting that selective M5 inhibition may provide a novel therapeutic approach for the treatment of addiction.[1–12]
Only recently have M5-preferring and M5-selective ligands begun to emerge. By targeting allosteric sites[13,14] on mAChRs, we recently described the first M5-selective ligands, a series of isatin-based M5 positive allosteric modulators (PAMs) (1-3, Figure 1).[15–17] Shortly thereafter, Dwoskin and co-workers described an M5-preferring orthosteric antagonist (4, Ki= 2.2 μM) with 11-fold selectivity for M5 over M1, but no functional (IC50) or DMPK data was reported.[18] Last year, we reported the first results from a functional M5 high-throughput screen (HTS), where we identified and optimized the first M5 selective negative allosteric modulator (NAM), 5, with submicromolar potency (hM5 IC50 = 300 nM, hM1-M4 IC50s >30 μM).[19] Based on the Dwoskin work,[18] and our earlier studies that had yielded a highly (>75-fold versus M2-M5) subtype-selective M1 orthosteric antagonist 6,[20] we revisited our HTS hits in an attempt to identify and optimize a highly M5 selective orthosteric antagonist to complete our tool kit of M5 probes to dissect the role of M5 in the CNS.
Figure 1.

Structures of recently reported M5 PAMs (1-3), an M5-preferring orthosteric antagonist 4, the first highly selective M5 NAM 5 and a highly M1 selective orthosteric antagonist 6.
Under the auspice of the Molecular Libraries Probe Center Network (MLPCN),[21] a triple-add, functional HTS was performed evaluating 360,000 compounds for M5 modulatory activity. In parallel, we screened the same collection in triple-add, functional mode against M1, M4 and the parental Chinese hamster ovary (CHO) cell line, resulting in nine confirmed and selective inhibitors of M5, from which the M5 NAM 5 resulted.[19] Evaluation of the eight remaining hits led us to quickly focus on a weak, yet selective, isoxazole-based amide 7 (Figure 2), which confirmed in the HTS center with an M5 IC50 of 9.3 μM and selectivity versus M1 and M4 (IC50s > 30 μM). Resynthesis of 7 (VU0480131), and screening fresh powder significantly increased our enthusiasm for the hit, as 7 displayed improved potency for both human and rat M5 (hM5 IC50 = 1.12 μM (pIC50 = 5.94±0.03), rM5 IC50 = 3.47 μM (pIC50 = 5.46±0.07)) and high selectivity versus human M1-M4 (IC50s > 30 μM). We then assessed the mechanism of action of 6 and performed competition binding studies with [3H]-N-methylscopolamine (NMS) and an atropine control, and observed that 7 displaced [3H]-NMS in a concentration dependent manner with a Ki of 1.3 μM, pKi = 5.88±0.05, (comparable to its IC50 of 1.1 μM), indicating competitive, and thus, orthosteric antagonism, reminiscent of the highly selective M1 orthosteric antagonist 6.[19,20]
Figure 2.

Structure and pharmacological profile of HTS hit 7 (VU0480131). A) Structure and pharmacology of 7 as an HTS stock solution (hM5 IC50 = 9.3 μM) and as resynthesized, fresh powder (hM5 IC50 = 1.12 μM). B) Concentration-response curves (CRCs) of 7 for human and rat M5, as well as human M1-M4 (IC50s >30 μM, n=3). C) [3H]NMS competition binding [n = 3] in membranes prepped from human M5 cells, showing competitive displacement (Ki = 1.3 μM).
Figure 3 highlights the chemical optimization strategy for 7, wherein we evaluated multiple dimensions of the ligand simultaneously through iterative parallel synthesis, as SAR for the analogous 6 was very steep. The first round of library synthesis surveyed alternate amides; thus, commercial acid 8 was acylated under HATU conditions to afford analogs 9 in yields ranging from 56–90% (Scheme 1). SAR was extremely steep, with all analogs tested, even regioisomeric pyridines, possessing hM5 IC50s >10 μM. This effort only afforded one weakly active compound 9a (hM5 IC50 = 5.6 μM, rM5 IC50 >10 μM, an N-ethyl congener of 7 (Figure 4).
Figure 3.
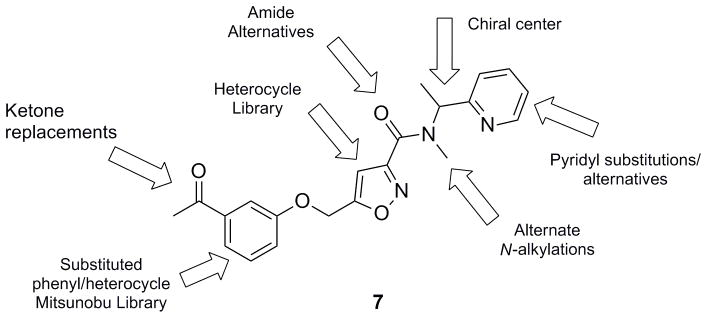
Chemical optimization plan for HTS hit 7 (VU0480131) via multidimensional, iterative parallel synthesis.
Scheme 1.
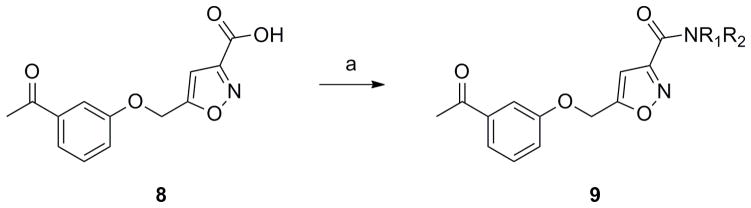
Preparation of amide analogs 9. Reagents and conditions: a) R1R2NH, HATU, CH2Cl2, rt, 56–90%.
Figure 4.

Structures and activities of amide analogs 9. All were uniformly inactive, except for the N-ethyl analog 9a.
With the amide region of 7 intolerant to modification, we next prepared a library of diverse aryl ethers to assess SAR and potentially identify a replacement for the ketone (Scheme 2). Alcohol 10 smoothly reacted with a variety of phenols under Mitsunobu conditions to provide a library of ether analogs 11, in yields ranging from 35–70%.[22] Nothing in this collection showed activity (hM5 IC50s >10 μM), despite surveying nitro, halogen, alkoxy and alkyl moieties around the pendant phenyl ring (See experimental section for full listing of analogs). Replacement of the ether oxygen with an NH, or acylated congener, also led to a complete loss of M5 inhibitory activity, as did amide analogs in place of the aryl ether moiety. Replacement of the central isoxazole with a phenyl ring also led to inactive compounds. Again, akin to the steep SAR noted for the highly selective M1 orthosteric antagonist 6, very little modification to 7 was tolerated. Thus, we elected to next synthesize the single enantiomers of 7 and assess if enantioselective inhibition was observed.
Scheme 2.
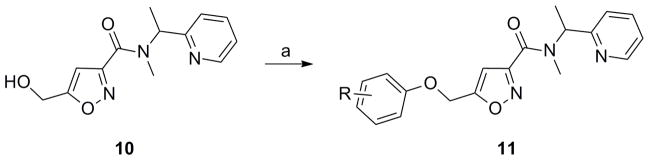
Preparation of aryl ether analogs 11. Reagents and conditions: a) ArOH, PPh3, DIAD, CH2Cl2, rt, 2h, 35–70%.
Starting with commercial (S)- or (R)-1-(pyridin-2-yl)ethanamine 12, treatment with Nosyl chloride provides sulphonamides 13 in good yield (Scheme 3). The N-methyl moiety was installed via a Mitsunobu reaction with MeOH to afford tertiary sulphonamides 14. Deprotection was accomplished by treatment with 3-chlorothiophenol in a modest 20% yield to provide chiral secondary amines 15. Finally, a HATU mediated coupling with acid 8 afforded the single enantiomers of 7, (S)-16 and (R)-16, in overall yields of ~15%.[22]
Scheme 3.

Preparation of (S)-16 and (R)-16, single enantiomers of racemic 7. Reagents and conditions: a) NosCl, DIEPA, CH2Cl2, rt, 78–90% b) MeOH, PPh3, DIAD, CH2Cl2, rt, 74–90% c) 3-ClPhSH, K2CO3, MeCN, rt, 20% d) 8, HATU, DIEPA, CH2Cl2, rt, 89–96%. Enantiopurity >99% ee as analysed by chiral SFC.
When screened in the human M5 cell-based assay, enantiospecific inhibition was observed, with (R)-16 possessing no activity (hM5 IC50 >10 μM), yet (S)-16 displayed (Figure 5) submicromolar potency (hM5 IC50 = 450 nM, pIC50 = 6.34±0.05) and exceptional mAChR selectivity (hM1-M4, IC50s >30 μM). These findings led us to then profile (S)-16 against the rat mAChRs, and once again, (S)-16 was selective (rM1-rM4 >30 μM), but a 4-fold diminution in potency was noted for rat M5 (rM5 IC50 = 1.65 μM, pIC50 = 5.78+0.04). Based on the potency and selectivity of (S)-16, it was approved as an MLPCN probe and given the identifier ML381 (VU0488130).[21] As expected, competition binding with [3H]NMS indicated that ML381 was an orthosteric antagonist, with a Ki of 341 nM, pKi = 6.47±0.03, (comparable to its IC50 of 450 nM, Figure 5B).
Figure 5.

Structure and pharmacological profile of (S)-16, ML381 (VU0480131). A) Structure and pharmacology of ML381. B) [3H]NMS competition binding [n = 3] in membranes prepared from human M5 cells, showing competitive displacement (Ki = 340 nM). C) CRCs of ML381 for human M5 (hM5 IC50 = 450 nM) and human M1-M4 (hM1-M4 IC50s >30 μM). D) CRCs of ML381 for rat M5 (rM5 IC50 = 1.65 μM) and rat M1-M4 (rM1-M4 IC50s >30 μM).
Next, we began to assess the physiochemical and DMPK profile of ML381. ML381 is a low molecular weight probe (MW = 379) with favourable lipophilicity (cLogP = 2.74) and an acceptable TPSA of 86. Moreover, ML381 was found be both soluble in phosphate buffered saline, PBS, (96 μM@ pH = 7.4 at 23 °C) and stable (99% parent remaining at 48 hours in PBS at 23 °C). In rat and human hepatic microsomes, ML381 exhibited high intrinsic clearance (rat CLint: 770 mL/min/kg, human CLint: 93 mL/min/kg) with predicted hepatic clearance values (rat CLhep: 64 mL/min/kg, human CLhep: 17 mL/min/kg) near the respective rates of hepatic blood flow in each species (Table 1). In plasma protein binding experiments, the compound exhibited exceptionally high fraction unbound (fuplasma) in rat (58.4%) and moderately high fuplasma in human (11%), which prompted investigation into the compound’s stability over time (4 hours; 37 °C) in plasma of both species, ultimately revealing a pronounced instability in rat plasma (approximately 1 % of parent remaining) and a moderate instability in human plasma (approximately 76 % of parent remaining), which ultimately prevented accurate determination of fuplasma. A high fraction unbound in brain (fubrain: 0.14) was observed for ML381 in rat whole brain homogenate. In order to determine the relevant biotransformation pathway(s) contributing to the poor metabolic stability observed in vitro, metabolite identification (Met ID) experiments were performed using hepatic microsomes and plasma from both species (Figure 6). These analyses revealed hydrolytic cleavage of the amide as the principle pathway of biotransformation in microsomes of both species as well as in rat plasma. Additionally, a number of NADPH-dependent mono-oxidation pathways were also identified in rat and human microsomes, including O-dearylation (M2), phenyl hydroxylation (M3), keto-reduction (M4), and N-dealkylations at the amide (M5, M6; Figure 6).
Table 1.
DMPK profile of ML381.
| Species | Property | Value1 |
|---|---|---|
| Sprague Dawley Rat | Hepatic Microsomal CLint | 770 mL/min/kg |
| Predicted CLhep | 64 mL/min/kg | |
| fuplasma; fubrain | 0.582; 0.14 | |
| Cbrain:Cplasma (Kp) | 0.583 | |
|
| ||
| Human | Hepatic Microsomal CLint | 93 mL/min/kg |
| Predicted CLhep | 17 mL/min/kg | |
| fuplasma | 0.112 | |
| MDCK-MDR1 ER | 1.6 | |
| P450 1A2 IC50 | > 30 μM | |
| P450 2C9 IC50 | 6.3 μM | |
| P450 2D6 IC50 | > 30 μM | |
| P450 3A4 IC50 | 27 μM | |
values represent means of at least two replicates with similar results
fuplasma values not accurate due to instability in rat plasma
Kp determined at 0.25 hr following a 0.2 mg/kg IV dose (n = 2)
Figure 6.

Metabolite identification for 7, ML381 (VU0480131), in rat and human, indicating six major routes of metabolism (M1-M6).
In order to gauge distribution to the central nervous system (CNS), concentrations of ML381 in whole brain and plasma at a single time point (0.25 hr) were measured following a single intravenous (IV) administration (0.2 mg/kg) to male, Sprague Dawley rats (n = 2). This study revealed a brain:plasma partition coefficient (Kp) of 0.58, suggesting moderate distribution to the CNS (Table 1); determination of an unbound brain:plasma partition coefficient (Kp,uu) was precluded by an absence of accurate fuplasma data due to the aforementioned plasma instability. In a bidirectional MDCK-MDR1 transwell assay, ML381 (5 μM) exhibited an efflux ratio (ER) of 1.6, suggesting an absence of P-glycoprotein (P-gp)-mediated active efflux liabilities at the blood-brain barrier (BBB; Table 1). Furthermore, favorably low inhibition of four major human cytochrome P450 enzymes by ML381 was observed in human hepatic microsomes, with inhibition of 2C9 (6.3 μM) representing the predominant liability (Table 1). Moreover, ML381 was screened in a Eurofins radioligand binding panel of 68 GPCRs, ion channels and transporters at a concentration of 10 μM,[23] and no significant activity was noted (no inhibition >50%@10 μM). Thus, in addition to unprecedented selectivity versus M1-M4, ML381 possessed clean ancillary pharmacology against a diverse array of discrete molecular targets. Together, these findings suggest that ML381 possesses an overall acceptable drug metabolism and pharmacokinetic (DMPK) profile for pharmacodynamic studies in rat,[24] with the exception of poor metabolic stability and a potential for amide hydrolysis in plasma; however, ML381 is best suited as an in vitro/electrophysiological probe.
In summary, we have developed (S)-16 (also known as ML381 or VU0480131), the most potent and selective M5 orthosteric antagonist (hM5 IC50 = 450 nM, M5 Ki = 340 nM, hM1-hM4 IC50s >30 μM) reported to date. Moreover, ML381 possesses a favourable DMPK profile and CNS penetration; however, instability in rat plasma indicates the utility of ML381 will be primarily as a molecular probe for in vitro and electrophysiology studies. SAR was extremely steep for this series, with subtle modifications leading to complete loss of M5 inhibitory activity, reminiscent of the M1 ligand 6; however, enantiospecific inhibition was observed, with all of the activity residing in the (S)-enantiomer. Current efforts are focused on further chemical optimization to address the plasma instability identified in Met ID studies and to improve PK and CNS penetration. Studies are underway and will be reported in due course. ML381 is an MLPCN probe and is freely available upon request.
Experimental Section
Experimental procedures for medicinal chemistry, pharmacology and drug metabolism as well as characterization of compounds are provided in the Supporting information, available at http://dx.doi.org/10.1002/cmdc.20xxxxxxx.
Acknowledgments
This work was generously supported by the NIH/MLPCN grant U54 MH084659 (C.W.L.) and grant U54 MH084512 (Scripps). Dr. Lindsley acknowledges the Warren Family and Foundation for funding the William K. Warren, Jr. Chair in Medicine.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cmdc.20xxxxxxx.
References
- 1.Smythies J. Int Rev Neurobiol. 2005;64:1–122. doi: 10.1016/S0074-7742(05)64001-9. [DOI] [PubMed] [Google Scholar]
- 2.Wess J, Eglen RM, Gautam D. Nat Rev Drug Discov. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]
- 3.Langmead CJ, Watson J, Reavill C. Pharmacol Ther. 2008;117:232–243. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 4.Denker D, Thomsen M, Wortwein G, Weikop G, Cui Y, Jeon J, Wess J, Fink-Jensen A. Muscarinic acetylcholine receptor subtypes as potential drug targets for the treatment of schizophrenia, drug abuse and Parkinson’s disease. ACS Chem Neurosci. 2012;3:80–89. doi: 10.1021/cn200110q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caulfield MP, Birdsall NJM. Pharmacol Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- 6.Weiner DM, Levey AI, Brann MR. Proc Natl Acad Sci USA. 1990;87:7050–7054. doi: 10.1073/pnas.87.18.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yasuda RP, Ciesla W, Flores LR, Wall SJ, Li M, Satkus SA, Weisstein JS, Spagnola BV, Wolfe BB. Mol Pharmacol. 1993;43:149–157. [PubMed] [Google Scholar]
- 8.Basile AS, Fedorova I, Zapata A, Liu X, Shippenberg T, Duttaroy A, Yamada M, Wess J. Proc Natl Acad Sci USA. 2002;99(17):11452–11457. doi: 10.1073/pnas.162371899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fink-Jensen A, Fedorova I, Wörtwein G, Woldbye DP, Rasmussen T, Thomsen M, Bolwig TG, Knitowski KM, McKinzie DL, Yamada M, Wess J, Basile A. J Neurosci Res. 2003;74(1):91–96. doi: 10.1002/jnr.10728. [DOI] [PubMed] [Google Scholar]
- 10.Thomsen M, Woldbye DP, Wörtwein G, Fink-Jensen A, Wess J, Caine SB. J Neurosci. 2005;25:8141–8149. doi: 10.1523/JNEUROSCI.2077-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yeomans JS, Takeuchi J, Baptista M, Flynn DD, Lepik K, Nobrega J, Fulton J, Ralph MR. J Neurosci. 2000;20:8861–8867. doi: 10.1523/JNEUROSCI.20-23-08861.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lester DB, Miller AD, Blaha CD. Synapse. 2010;64:216–223. doi: 10.1002/syn.20717. [DOI] [PubMed] [Google Scholar]
- 13.Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. J Med Chem. 2012;55:1445–1464. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conn PJ, Jones C, Lindsley CW. Trends in Pharm Sci. 2009;30:148–156. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bridges TM, Marlo JE, Niswender CM, Jones JK, Jadhav SB, Gentry PR, Weaver CD, Conn PJ, Lindsley CW. J Med Chem. 2009;52:3445–3448. doi: 10.1021/jm900286j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bridges TM, Kennedy JP, Cho HP, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2010;20:558–562. doi: 10.1016/j.bmcl.2009.11.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gentry PR, Bridges TM, Lamsal A, Vinson PN, Smith E, Chase P, Hodder PS, Engers JL, Niswender CM, Daniels JS, Conn PJ, Wood MR, Lindsley CW. Bioorg Med Chem Lett. 2013;23:2996–3000. doi: 10.1016/j.bmcl.2013.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng G, Smith AM, Huang X, Subramanian KL, Siripuarpu KB, Deaciuc A, Zhan CG, Dwoskin LP. J Med Chem. 2013;56:1693–1703. doi: 10.1021/jm301774u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gentry PR, Kokubo M, Bridges TM, Kett NR, Harp JM, Cho HP, Smith E, Chase P, Hodder PS, Niswender CM, Daniels SJ, Conn PJ, Wood MR, Lindsley CW. J Med Chem. 2013;56:9351–9355. doi: 10.1021/jm4013246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheffler DJ, Williams R, Bridges TM, Lewis LM, Xiang Z, Zheng F, Kane AS, Byum NE, Jadhav S, Mock MM, Zheng F, Lewis LM, Jones CK, Niswender CM, Weaver CD, Conn PJ, Lindsley CW, Conn PJ. Mol Pharmacol. 2009;76:356–368. doi: 10.1124/mol.109.056531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.For the MLPCN see: http://mli.nih.gov/mli/mlpcn/; ML381 is an MLPCN probe and freely available upon request.
- 22.See Supporting Information for full details and Supplemental SAR tables.
- 23.For information on the Eurofins Lead Profiling Screen, please see: www.eurfins.com
- 24.Wenthur CJ, Niswender CM, Morrison R, Daniels JS, Conn PJ, Lindsley CW. J Med Chem. 2013;56:5208–5212. doi: 10.1021/jm400439t. [DOI] [PMC free article] [PubMed] [Google Scholar]