

Abstract
Autotaxin (ATX) is an autocrine motility factor that promotes cancer cell invasion, cell migration and angiogenesis. ATX, originally discovered as a nucleotide phosphodiesterase, is known now to be responsible for the lysophospholipid-preferring phospholipase D activity in plasma. As such, it catalyzes the production of lysophosphatidic acid (LPA) from lysophophatidylcholine (LPC). ATX is thus an attractive drug target; small molecular inhibitors might be efficacious in slowing the spread of cancers. With this study we have generated a series of beta-keto and beta-hydroxy phosphonate derivatives of LPA, some of which are potent ATX inhibitors.
Keywords: Autotaxin, ATX, Phosphonate, Choline, LPA
The autocrine motility factor autotaxin (ATX) was originally isolated from melanoma cell supernatants as a 125-kD glycoprotein that stimulated tumor cell motility.1 In vivo experiments documented that forced expression of ATX augments tumor cell invasion and metastasis.2 Further, ATX promotes angiogenesis and may act in concert with other angiogenic factors to facilitate new blood vessel formation.3 These biological properties require enzymatic activity.
ATX belongs to the nucleotide pyrophosphatase and phosphodiesterase (NPP) family of enzymes, which hydrolyze phosphodiester and diphosphate bonds, typically found in ATP and ADP.4 Interest in ATX was stimulated by the identification of this enzyme as the long elusive plasma lysophospholipase D activity, which is responsible for the cleavage of choline group of lysophophatidylcholine (LPC) to form lysophosphatidic acid (LPA) (Figure 1).5,6 This is a major pathway of biosynthesis of LPA in plasma.7,8 LPA is an intercellular lipid mediator that influences many biochemical processes including cell proliferation, smooth muscle contraction, platelet aggregation and apoptosis.9–11 For example, LPA is the “ovarian cancer activating factor” in ascitic fluid characteristic of ovarian cancer patients. Elevated levels of LPA are present both at early and late stages in ovarian cancer and may play a role in tumor cell proliferation and invasion.12,13 LPA mediates its effects through the activation of G protein-coupled receptors (GPCR).14 Thus, great efforts have been made on the study of LPA receptor antagonists and agonists due to their therapeutic potential.15–21 In aggregate, these data suggest that ATX is an attractive pharmacological target; blockage of LPA production via ATX inhibition by small molecules could be a useful anticancer chemotherapy.22,23
Figure 1.

Hydrolysis of LPC by lysoPLD/ATX
A lead towards developing ATX inhibitors was provided by the discovery that this enzyme undergoes end product inhibition by, for example, LPA24. Indeed a limited number of ATX inhibitors that are LPA analogs have been reported to date. Recently, a series of fatty alcohol phosphate analogs were identified as LPA receptor ligands.20 Some of the analogs showed ATX inhibition activity. A series of phosphatidic acid derivatives were investigated and only two acyl thiophosphates showed autotaxin inhibition.21 A group of Darmstoff analogs were reported as weak ATX inhibitors recently.25 Most recently 3-carba analogs of cyclic phosphatidic acid were reported.26 Although lacking significant activity at LPA receptors, they were potent inhibitors of ATX activity. In this report, we developed a series of β-hydroxy and β-keto phosphonate derivatives of LPA as ATX inhibitors.
Synthesis of the phosphonate derivatives is described in Scheme 1. It began with the acylation of the ammonium hydrochloride salt of tyrosine O-methyl ester a with appropriate acyl chlorides followed by etherification of the free phenol with appropriate mesylates to afford the fully protected tyrosine c. Next, was the base mediated addition onto the methyl ester with the lithium anion of dimethyl methylphosphonate to achieve β-keto phosphonate dimethyl ester d. A bromotrimethylsaline mediated deprotection of the ester ensued to afford the β-keto phosphonate g.27 Sodium borohydride reduction of d proceeded to give two possible diastereometic β-hydroxy phosphonate dimethyl esters which were separated by column chromatography. The stereochemistry determination is ongoing. The β-hydroxy phosphonate f was obtained by using the same deprotection method (for compounds f41 and f42, pyridine was used in the deprotection).
Scheme 1.

Synthesis of Compounds f and g. Reagents and conditions: (i) appropriate acyl chloride, Et3N, CH2Cl2, 0°C, 3hr, 70–80%; (ii) appropriate mesylate, K2CO3, 18-crown-6, acetone, reflux overnight, 90–95%; (iii) n-BuLi, dimethyl methylphosphonate, then add in ester c, −78°C, 3hr, 50–60%; (iv) NaBH4, THF, EtOH, 0°C, 2hr, 70–80%; (v) bromotrimethylsaline, w/wo pyridine, CH2Cl2, rt, 4hr, then H2O and MeOH, overnight, 90–95%.
The phosphonate derivatives were tested in choline detection assay for ATX inhibition.28 The ATX activity was measured in the presence of the compounds under different concentrations (100µM, 10µM and 1µM). The ATX activity without compounds was used as the standard (100% activity). Most β-hydroxy phosphonate derivatives inhibited ATX activity at only the highest concentration tested. However, f17 and f18 exhibited significant inhibition at 1µM (Table 1). These two compounds were synthesized from protected L-tyrosine and they are diastereomers in terms of the β-hydroxy groups. The less polar isomer, f17, (also known as VPC8a202) was able to inhibit 73% of ATX activity at 1 µM. Compounds f15 and f16, which were synthesized from D-tyrosine, did potently inhibit ATX although they contained the same 4-methoxy-3,5-dimethylpyridyl structure moiety. Other groups including some alkyl chains and aromatic rings were also investigated. However, these compounds were not potent ATX inhibitors. The corresponding β-keto phosphonate derivatives were also tested (Table 2). At the concentration of 100µM, some compounds inhibited 50% – 70% of ATX activity. Further structure optimization was made based on the two lead compounds f17 and f18. We kept the 4-methoxy-3,5-dimethyl-pyridyl moiety and investigated a series of β-hydroxy phosphonate derivatives with a variety of lipophilic tails. These data are presented in Table 3. None of these compounds were as potent as f17 or f18. All of the compounds were tested as potential antagonists at the recombinant LPA1, LPA2 and LPA3 receptors, but none showed significant blockade at concentrations up to 10µM (data not shown).
Table 1.
ATX inhibitory evaluation of compound f1 – f34
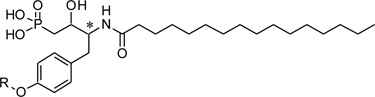 | |||||
|---|---|---|---|---|---|
| Compounds | R | * S/R | ATX activity (%) | ||
| 1 µM | 10 µM | 100 µM | |||
| f1 | R (a1) | 92 | 84 | 55 | |
| f2 | R (b1) | N/D | 84 | 64 | |
| f3 | S (a) | 92 | 80 | 32 | |
| f4 | S (b) | 103 | 83 | 56 | |
| f5 | R (a) | 83 | 82 | 83 | |
| f6 | R (b) | 80 | 81 | 83 | |
| f7 | S (a) | 97 | 77 | 68 | |
| f8 | S (b) | 76 | 83 | 78 | |
| f9 | R (a) | 103 | 76 | 60 | |
| f10 | R (b) | 92 | 78 | 69 | |
| f11 | S (a) | 104 | 81 | 52 | |
| f12 | S (b) | N/D | 74 | 65 | |
| f13 | R (b) | 84 | 80 | 67 | |
| f14 | S (b) | 80 | 80 | 80 | |
| f15 | 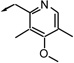 |
R (a) | 108 | 95 | 63 |
| f16 | 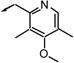 |
R (b) | 69 | 64 | 47 |
| f17 | 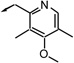 |
S (a) | 27 | 13 | 6 |
| f18 | 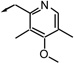 |
S (b) | 63 | 21 | 8 |
| f19 | CH3 | R (a) | 100 | 78 | 57 |
| f20 | CH3 | R (b) | 95 | 84 | 82 |
| f21 | CH3 | S (a) | 77 | 56 | 36 |
| f22 | CH3 | S (b) | 84 | 80 | 69 |
| f23 | n-C3H7 | R (a) | 80 | 79 | 72 |
| f24 | n-C3H7 | R (b) | 108 | 108 | 90 |
| f25 | n-C3H7 | S (a) | 110 | 97 | 77 |
| f26 | n-C3H7 | S (b) | 100 | 100 | 83 |
| f27 | n-C5H11 | R (a) | 81 | 85 | 80 |
| f28 | n-C5H11 | R (b) | 86 | 82 | 70 |
| f29 | n-C5H11 | S (a) | 98 | 83 | 67 |
| f30 | n-C5H11 | S (b) | 94 | 90 | 70 |
| f31 | R (a) | 103 | 104 | 72 | |
| f32 | R (b) | N/D | 104 | 94 | |
| f33 | S (a) | 104 | 104 | 82 | |
| f34 | S (b) | 106 | 101 | 91 | |
a refers to the diastereomer that elutes first, b refers to the diastereomer that elutes second.
Table 2.
ATX inhibitory evaluation of compound g1 – g15
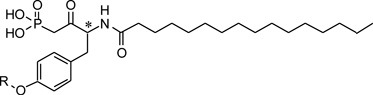 | |||||
|---|---|---|---|---|---|
| Compounds | R | *S/R | ATX activity (%) | ||
| 1 µM | 10 µM | 100 µM | |||
| g1 | R | 85 | 70 | 57 | |
| g2 | S | 79 | 68 | 44 | |
| g3 | R | N/D | 82 | 76 | |
| g4 | S | N/D | 71 | 61 | |
| g5 | R | 78 | 68 | 52 | |
| g6 | S | 102 | 72 | 50 | |
| g7 | 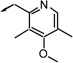 |
S | 71 | 74 | 26 |
| g8 | CH3 | R | 84 | 71 | 32 |
| g9 | CH3 | S | 96 | 68 | 45 |
| g10 | n-C3H7 | R | 104 | 97 | 75 |
| g11 | n-C3H7 | S | N/D | 97 | 72 |
| g12 | n-C5H11 | R | 93 | 84 | N/D |
| g13 | n-C5H11 | S | 95 | 83 | N/D |
| g14 | R | 97 | 101 | 76 | |
| g15 | S | 104 | 104 | 85 | |
Table 3.
ATX inhibitory evaluation of compound f35 – f44
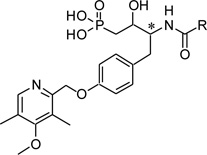 | |||||
|---|---|---|---|---|---|
| Compounds | R | * S/R | ATX activity (%) | ||
| 1 µM | 10 µM | 10 µM | |||
| f35 | n-C9H19 | R (a1) | N/D | 86 | 30 |
| f36 | n-C9H19 | R (b1) | N/D | N/D | N/D |
| f37 | n-C13H27 | R (a) | N/D | 99 | 67 |
| f38 | n-C13H27 | R (b) | N/D | N/D | 35 |
| f39 | n-C17H35 | S (a) | 101 | 91 | 81 |
| f40 | n-C17H35 | S (b) | N/D | 90 | 62 |
| f41 | n-C17H332 | S (a) | N/D | 45 | 15 |
| f42 | n-C17H332 | S (b) | 77 | 68 | 58 |
| f43 | n-C19H39 | S (a) | 64 | 45 | 10 |
| f44 | n-C19H39 | S (b) | N/D | N/D | N/D |
a refers to the diastereomer that elutes first, b refers to the diastereomer that elutes second.
cis double bond located between C9 and C10 from the carbonyl.
A potential complication of our studies is the reported inhibition of ATX by its product, LPA,24 which is generated in our assay using LPC as a substrate.28 There, we measured the inhibition of ATX using the artificial substrate, para-nitrophenol-thymidylic acid.5,6 Using this substrate, we found the same rank order potency of our most potent inhibitors, including f17 and f18 (data not shown).
In summary, we developed a series of β-hydroxy and β-keto phosphonate derivatives. ATX inhibitory activity was determined for these compounds. Two β-hydroxy phosphonates, which originated from protected Ltyrosine, were identified as the lead compounds. The stereochemistry of original tyrosines and 4-methoxy-3,5-dimethyl-pyridyl moiety proved to be important to the activity. Further SAR studies are ongoing.
Acknowledgements
This work was supported by a grant from the NIH (R01 GM052722).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Stracke ML, Clair T, Liotta LA. Adv. Enzyme Regul. 1997;37:135. doi: 10.1016/s0065-2571(96)00017-9. [DOI] [PubMed] [Google Scholar]
- 2.Nam SW, Clair T, Campo CK, Lee HY, Liotta LA, Stracke ML. Oncogene. 2000;19:241. doi: 10.1038/sj.onc.1203263. [DOI] [PubMed] [Google Scholar]
- 3.Nam SW, Clair T, Kim YS, McMarlin A, Schiffmann E, Liotta LA, Stracke ML. Cancer Res. 2001;61:6938. [PubMed] [Google Scholar]
- 4.Goding JW, Terkeltaub R, Maurice M, Deterre P, Sali A, Bell SI. Immunol. Rev. 1998;161:11. doi: 10.1111/j.1600-065x.1998.tb01568.x. [DOI] [PubMed] [Google Scholar]
- 5.Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, Fukuzawa K. J. Biol. Chem. 2002;277:39436. doi: 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- 6.Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, Yamori T, Mills GB, Inoue K, Aoki J, Arai H. J. Cell Biol. 2002;158:227. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sano T, Baker D, Virag T, Wada A, Yatomi Y, Kobayashi T, Igarashi Y, Tigyi G. J. Biol. Chem. 2002;277:21197. doi: 10.1074/jbc.M201289200. [DOI] [PubMed] [Google Scholar]
- 8.Aoki J, Taira A, Takanezawa Y, Kishi Y, Hama K, Kishimoto T, Mizuno K, Saku K, Taguchi R, Arai H. J. Biol. Chem. 2002;277:48737. doi: 10.1074/jbc.M206812200. [DOI] [PubMed] [Google Scholar]
- 9.Tigyi G, Parrill AL. Prog. Lipid Res. 2003;42:498. doi: 10.1016/s0163-7827(03)00035-3. [DOI] [PubMed] [Google Scholar]
- 10.Mills GB, Moolenaar WH. Nat. Rev. Cancer. 2003;3:582. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 11.Lynch KR, Macdonald TL. Prost. Lipid Med. 2001;64:33. doi: 10.1016/s0090-6980(01)00106-x. [DOI] [PubMed] [Google Scholar]
- 12.Xu Y, Gaudette DC, Boynton JD, Frankel A, Fang X-J, Sharma A, Hurteau J, Casey G, Goodbody A, Mellors A, Holub BJ, Mills GB. Clin. Cancer Res. 1995;1:1223. [PubMed] [Google Scholar]
- 13.Xu Y, Fang XJ, Casey G, Mills GB. Biochem J. 1995;309:933. doi: 10.1042/bj3090933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Contos JJA, Ishii I, Chun J. Mol. Pharm. 2000;58:1188. doi: 10.1124/mol.58.6.1188. [DOI] [PubMed] [Google Scholar]
- 15.Heasley BH, Jarosz R, Carter KM, Jenny Van S, Lynch KR, Macdonald TL. Bioorg. Med. Chem. Lett. 2004;14:4069. doi: 10.1016/j.bmcl.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 16.Webster LS, Heasley BH, Jarosz R, Carter KM, Lynch KR, Macdonald TL. Bioorg. Med. Chem. Lett. 2004;14:3473. doi: 10.1016/j.bmcl.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 17.Heasley BH, Jarosz R, Lynch KR, Macdonald TL. Bioorg. Med. Chem. Lett. 2004;14:2735. doi: 10.1016/j.bmcl.2004.03.076. [DOI] [PubMed] [Google Scholar]
- 18.Tamaruya Y, Suzuki M, Kamura G, Kanai M, Hama K, Shimizu K, Aoki J, Arai H, Shibasaki M. Angew. Chem. Int. Ed. Engl. 2004;43:2834. doi: 10.1002/anie.200454065. [DOI] [PubMed] [Google Scholar]
- 19.Qian L, Xu Y, Simper T, Jiang G, Aoki J, Umezu-Goto M, Arai H, Yu S, Mills GB, Tsukahara R, Makarova N, Fujiwara Y, Tigyi G, Prestwich GD. Chem Med Chem. 2006;1:376. doi: 10.1002/cmdc.200500042. [DOI] [PubMed] [Google Scholar]
- 20.Durgam GG, Virag T, Walker MD, Tsukahara R, Yasuda S, Liliom K, van Meeteren LA, Moolenaar WH, Wilke N, Siess W, Tigyi G, Miller DD. J. Med. Chem. 2005;48:4919. doi: 10.1021/jm049609r. [DOI] [PubMed] [Google Scholar]
- 21.Durgam GG, Tsukahara R, Makarova N, Walker MD, Fujiwara Y, Pigg KR, Baker DL, Sardar VM, Parrill AL, Tigyi G, Miller DD. Bioorg. Med. Chem. Lett. 2006;16:633. doi: 10.1016/j.bmcl.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 22.Mills GB, Moolenaar WH. Nat. Rev. Cancer. 2003;3:582. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 23.Goto M, Tanyi J, Lahad J, Liu S, Lapushin R, Hasegawa Y, Lu Y, Trost R, Bevers T, Jonasch E, Aldape K, Liu J, James RD, Ferguson CG, Xu Y, Prestwich GD, Mills GB. J. Cell. Biochem. 2004;92:1115. doi: 10.1002/jcb.20113. [DOI] [PubMed] [Google Scholar]
- 24.van Meeteren LA, Ruurs P, Christodoulou E, Goding H, Takakusa K, Kikuchi K, Perrakis A, Nagano T, Moolenaar WH. J Biol. Chem. 2005;280:21155. doi: 10.1074/jbc.M413183200. [DOI] [PubMed] [Google Scholar]
- 25.Gududuru V, Zeng K, Tsukahara R, Makarova N, Fujiwara Y, Pigg K, Baker D, Tigyi G, Miller D. Bioorg. Med. Chem. Lett. 2006;16:451. doi: 10.1016/j.bmcl.2005.08.096. [DOI] [PubMed] [Google Scholar]
- 26.Baker DL, Fujiwara Y, Pigg KR, Tsukahara R, Kobayashi S, Murofushi H, Uchiyama A, Murakami-Murofushi K, Koh E, Bandle RW, Byun H, Bittman R, Fan D, Murph M, Mills GB, Tigyi G. J. Bio. Chem. 2006;281:22786. doi: 10.1074/jbc.M512486200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rabinowitz R. J. Org. Chem. 1963;28:2975. [Google Scholar]
- 28.ATX activity assay: in 0.1ml, buffer: (in mM: Tris-HCl 100, pH9, NaCl 150, MgCl2 5, CoSO4 0.03, 0.05% Triton X-100), 1mM 18:1 LPC, recombinant human ATX with or without inhibitors; incubation was for 16 hours at 37°C. Released choline was detected as follows: Samples were mixed with 0.15 ml of 50mM TrisHCl containing 2.7 mM TOOS (N-ethyl-N-2-hydroxy-3-sulfopropyl)-m-toluidine,2.7mM 4-AAP (4-aminoantipyrine), 47.7 U/ml horseradish peroxidase, 18U/ml choline oxidase, and 5mM MgCl2. After 30 min incubation at 37°C, light absorbance (550nm) was determined, and the amount of choline release was calculated from a standard curve.