

Abstract
Primary established genetic abnormalities in B-cell precursor acute lymphoblastic leukemia include high hyperdiploidy (51–65 chromosomes), the translocations t(12;21)(p13;q22)/ETV6-RUNX1 fusion and t(9;22)(q34;q11)/BCR-ABL1 fusion, MLL rearrangements and intrachromosomal amplification of chromosome 21. These rearrangements are of prognostic and therapeutic relevance and are usually mutually exclusive. We identified 28 patients at diagnosis with both a primary genetic rearrangement and an immunoglobulin heavy chain locus translocation using chromosomal analysis and fluorescence in situ hybridization. Among these patients, the immunoglobulin heavy chain locus translocation partner gene was identified in six (CRLF2, CEBPA, CEBPB, TRA/D@, IGF2BP1 and IGK@). Clonal architecture was investigated in 17 patients using multiple color interphase fluorescence in situ hybridization analysis, which showed that the translocation was acquired as a secondary abnormality in ten patients, in four patients the etiology was undetermined and in three patients it was observed in a separate clone from the primary chromosomal rearrangement. These findings demonstrate the co-existence of immunoglobulin heavy chain locus translocations with other primary chromosomal rearrangements either in the same or separate clones, which may have prognostic significance in B-cell precursor acute lymphoblastic leukemia. Clinical trials: UKALLXII: Study ID n. ISRCTN77346223 and ALL2003: Study ID n. ISRCTN07355119
Introduction
Cytogenetic characterization of acute lymphoblastic leukemia (ALL) indicates prognosis and is paramount for risk stratification for treatment. The main cytogenetic abnormalities used for risk stratification are usually mutually exclusive. They include high hyperdiploidy (51–65 chromosomes with the gain of specific chromosomes) and the ETV6-RUNX1 gene fusion, which are two subtypes associated with a good outcome, whereas BCR-ABL1 fusion, MLL translocations and intrachromosomal amplification of chromosome 21 (iAMP21) are subtypes associated with a poor outcome.1
In a recent study to ascertain the incidence of immunoglobulin heavy chain locus translocations (IGH@-t) in ALL, we identified the co-existence of IGH@-t in association with primary chromosomal rearrangements in 28 patients.7 To investigate these cases in greater detail, we examined the IGH@-t and primary chromosomal rearrangements within the same diagnostic bone marrow cells using multiple color fluorescence in situ hybridization (FISH) and found a range of clonal architecture.
Methods
Patients
A total of 28 patients were included in this study (Online Supplementary Table S1). Of these, 21 were treated on the childhood treatment trial UKALL2003 [including patients with Down syndrome ALL (DS-ALL)] and six on the adult UKALLXII trial. Institutional review board ethical approval was obtained for all patients at each of the collaborating centers. Informed consent was given in accordance with the Declaration of Helsinki.
Fluorescence in situ hybridization
The primary cytogenetic changes were originally identified by cytogenetic analysis; however due to the cryptic nature of the translocation, t(12;21), the presence of the ETV6-RUNX1 fusion was confirmed by FISH or reverse transcriptase polymerase chain reaction analysis. Initial screening for the presence of IGH@-t was carried out using the Vysis LSI IGH Break-Apart Rearrangement Probe (Abbott Molecular, Illinois, USA) (Vysis IGH@ probe). A normal cell with intact IGH@ shows two closely apposed red (R) and green (G) fusion signals (2F). The presence of an IGH@-t is indicated by separation of one pair of red and green signals to give the 1R1G1F pattern. Five control slides made with fixed cells from individuals with no known neoplasia were hybridized with this probe. Both visual and automated scoring was performed to determine the cut-off percentages for false positive results (±3 standard deviations). A patient was classified as IGH@-positive if the percentage of separated red and green signals was ≥5%, as determined independently by two analysts. Due to the cryptic nature and relative frequency of CRLF2 involvement in IGH@-t, patients with this translocation and available material were screened for CRLF2 rearrangements using previously described FISH probes and methods.2 For patients with an IGH@-t visible by cytogenetics, either previously published FISH probes or FISH mapping (if material was available) was used to confirm the partner gene.2–5
Multiple color interphase FISH was carried out to assess the presence of both the primary rearrangement and IGH@-t in the same cells. The IGH Breakapart LPH 014 probe (Cytocell, Cambridge, UK) or a home-made IGH@ single color probe was used for the detection of IGH@-t.5 The FISH probes used to detect aneuploidy in the cohort with high hyperdiploidy were designed to mark those chromosomes most commonly gained (chromosomes 4, 14 and 21) or those rarely gained as a control to exclude tetraploid cells (chromosome 9). The probes used to detect the primary rearrangements are detailed in Table 1.
Table 1.
Multicolour FISH probe combinations, probe details and cut-off values used.
For scoring by eye (independent IGH@ and CRLF2 FISH), results were provided by the regional cytogenetics laboratories or scored using an Olympus BX-61 florescence microscope with a x100 oil objective. A minimum of 100 nuclei were scored for each FISH test by two independent analysts. When combined with probes to mark the co-existing primary rearrangement, automated capture and scoring was carried out using an automated Olympus BX-61 8-bay stage florescence microscope with a x60 oil objective. Images were stored and analyzed using the CytoVision 7.1 SPOT counting system (Leica Microsystems, Gateshead, UK).
Three control slides were set up for all multiple color probe combinations using fixed cells from blood samples of individuals with no known neoplasia (Table 1). For the different probe combinations to mark the IGH@-t and aneuploidy, 15% was used as the cut-off percentage.6 The higher cut-off level is required when analyzing multiple probes in the same cells to allow for interference and obscuring of the many signals.
Results
Patients’ details and primary cytogenetic changes
In an earlier study to determine the incidence of IGH@-t in B-cell precursor-ALL (BCP-ALL), we identified 28 IGH@-t positive patients with co-existing established primary chromosomal rearrangements.7 The clinical, demographic and diagnostic karyotype data of these 28 patients are presented in Online Supplementary Table S1.
Nine IGH@-t patients had chromosomal gains indicative of high hyperdiploidy. While all had >50 chromosomes, two DS-ALL patients had constitutional gain of one copy of chromosome 21. One DS-ALL patient (24390) had only four somatic gains, three of which were commonly observed gains in high hyperdiploidy (chromosomes 14, 21 and X). The second patient with DS-ALL (8447) had somatic gains of eight chromosomes and somatic loss of two. However, only three gains (chromosomes X, 8 and 14) were typical of those observed in high hyperdiploidy. Eight patients with IGH@-t had the ETV6-RUNX1 gene fusion, with additional chromosomal abnormalities in four of them. Six IGH@-t patients had the BCR-ABL1 gene fusion, which was cytogenetically cryptic in two of them. Three IGH@-t patients had iAMP21 and two had MLL translocations, both involving the AFF1 gene at 4q21.
Conventional cytogenetics and dual color fluorescence in situ hybridization
From cytogenetic analysis, eight patients had a visible translocation involving chromosome band 14q32. Interphase and metaphase FISH analysis using the Vysis IGH@ probe and other home-made probes confirmed that IGH@-t was present in all 28 patients and identified the partner genes in six (CRLF2, CEBPA, CEBPB, TRA/D@, IGF2BP1 and IGK@, as shown in Online Supplementary Table S1). The partner genes involved in the remaining 22 patients are unknown.
Multiple color fluorescence in situ hybridization analysis to determine clonal architecture
To assess the presence of the primary rearrangement and the IGH@-t within the same cells, a combination of probes were hybridized simultaneously to the same slide for 17 patients with available material. In all patients a normal population was observed. However, the percentage of nuclei harboring IGH@-t differed according to whether the studies were conducted with the Vysis IGH@ probe, used individually, or with the Cytocell IGH@ probe or home-made IGH@ probe used for the detection of IGH@-t in combination with the primary rearrangements in some patients. This is likely a consequence of hybridization of a single probe scored by eye compared to simultaneous multiple color probe combinations scored by an automated procedure.
IGH@-t is a secondary event
Thirteen patients (8447, 23110, 22105, 11438, 10281, 22420, 20951, 2618, 10285, 21733, 10859, 11061 and 21940) harbored a population of nuclei with the primary rearrangement in the absence of IGH@-t.
Examples of each primary rearrangement with clonal architecture
High hyperdiploidy
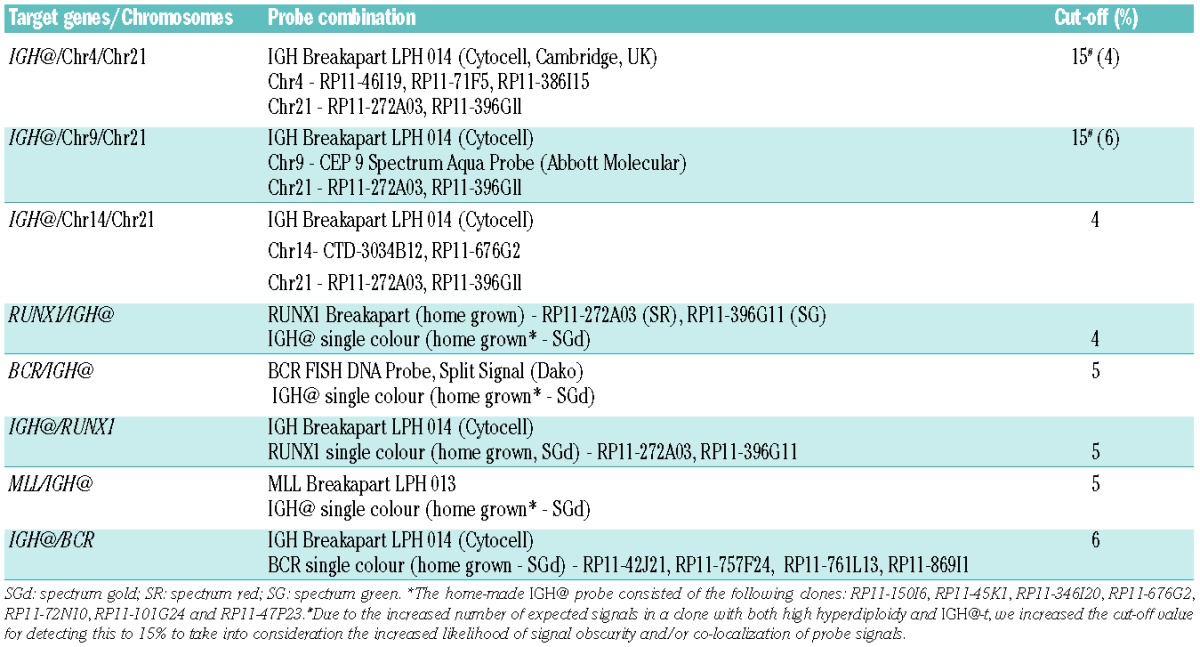
In patient 10281 with high hyperdiploidy (Figure 1i–iii), the Vysis IGH@ probe alone showed a breakpoint within the variable region in 27% of nuclei. By multiple color FISH, 30% of nuclei showed chromosomal gains in the absence of IGH@-t. Two abnormal populations without IGH@-t had gains of chromosomes 4, 14 and 21 (30%). One of these populations acquired IGH@-t on the background of the initial numerical gains (38%), which further evolved with loss of one copy of chromosomes 14 and 21 (11%).
Figure 1.
Images depicting the emergence of high hyperdiploidy prior to the IGH@-t in patient 10281. (i) Captured interphase nuclei hybridized with FISH probes to track the high hyperdiploidy (chromosome 4; aqua, chromosome 21; gold) and IGH@-t/+14 (red/green). (ii) The same captured interphase nuclei, including colored arrows depicting the signals that were scored: blue arrows highlight the aqua probe marking copies of chromosome 4, the yellow arrows highlight the gold probe marking copies of chromosome 21 and the red/green double arrows highlight the IGH@ breakapart probe that marks both the copy number of chromosome 14 and also detects the presence of a translocation. (iii) Cartoon depicting possible models of evolutionary progression by either gain (green arrows) or loss (red arrows). Solid and dashed lines highlight the possible alternative evolutionary routes taken by the sample. The black circle represents the FISH signal pattern which correlates with the abnormal karyotype observed at metaphase analysis. No cytogenetically visible IGH@-t was observed in these cells.
ETV6-RUNX1 fusion
In patient 20951 with ETV6-RUNX1 (Figure 2), the Vysis IGH@ probe alone showed a breakpoint within the variable region in 59% of nuclei. By multiple color FISH, IGH@-t was detected in 44% of nuclei in association with ETV6-RUNX1, as indicated by the RUNX1 probes. The IGH@-t in this patient appeared to have arisen as a secondary event, as 14% of nuclei harbored the EVT6-RUNX1 translocation and an additional copy of chromosome 21 only. However, as this patient had a variant FISH signal pattern using the Vysis IGH@ probe, this case was classified as having an undetermined etiology because of the difficulties of interpretation when one signal was absent.
Figure 2.

Images of FISH interphase nuclei depicting the apparent emergence of an ETV6-RUNX1 translocation prior to the IGH@-t. (i) Captured interphase nuclei from patient 20951 hybridized with FISH probes to track the IGH@-t (red/green) and the ETV6-RUNX1 translocation (homemade RUNX1 probe; gold). (ii) The same captured interphase nuclei, including colored arrows highlighting the signals that were scored: the yellow arrows highlight the gold probe marking the RUNX1 rearrangement and the red/green double arrows highlight the IGH@ break apart probe that marks both the copy number of chromosome 14 and also detects the presence of a translocation. (iii) Cartoon depicting possible models of evolutionary progression either by gain (green arrows) or loss (red arrows). Forty-three percent of nuclei were normal for both probes (one red/green fusion appears yellow, this is marked with arrows for clarity as shown in (ii) with 14% showing an additional copy of chromosome 21 in the presence of the RUNX1 translocation. On the background of the RUNX1 translocation and additional chromosome 21, a sub-clone acquired an IGH@-t (23%) which subsequently either lost (9%) or gained (11%) a copy of chromosome 21. (iv) Table showing the percentage of positive nuclei when samples were hybridized with either the individual probes for each rearrangement (Individual FISH), or when they were investigated together (Multi-FISH) in the same cell.
BCR-ABL1 fusion
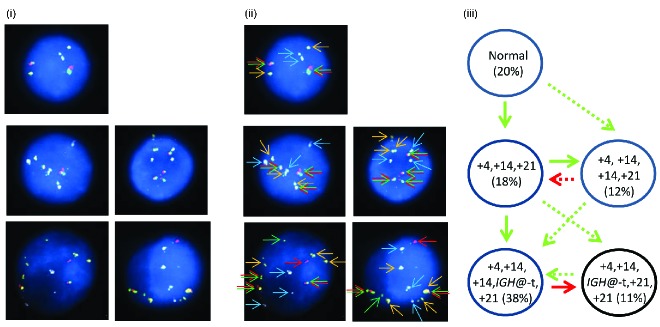
In patient 21733 with BCR-ABL1 (Figure 3), the Vysis IGH@ probe alone showed 9% of interphase nuclei with IGH@-t. Using multiple color FISH, 79% of interphase nuclei showed BCR-ABL1 only with an additional 6% showing both BCR-ABL1 and IGH@-t.
Figure 3.
Images of FISH interphase nuclei depicting the apparent emergence of a BCR-ABL1 translocation prior to the IGH@-t. (i) Captured interphase nuclei from patient 21733 hybridized with FISH probes to track BCR-ABL1 (DAKO BCR split signal probe; red/green) and a home-made IGH@ probe (gold). (ii) The same captured inter-phase nuclei, including colored arrows highlighting the signals that were scored: the yellow arrows highlight the gold probe marking the home-made IGH@ probe that detects both the copy number of chromosome 14 and also the presence of a translocation. The red/green double arrows highlight the BCR split signal probe that marks the BCR-ABL1 translocation. (iii) Cartoon depicting a possible model of evolutionary progression by gain (green arrows). A normal population of cells was observed (15%) with the emergence of a clone harboring only the BCR-ABL1 translocation in 79% of nuclei. This clone subsequently acquired an IGH@-t in 6%. (iv) Table showing the percentages of positive nuclei when samples were hybridized with either the individual probes for each rearrangement (Individual FISH), or when they were investigated together (Multi-FISH) in the same cell.
Intrachromosomal amplification of chromosome 21
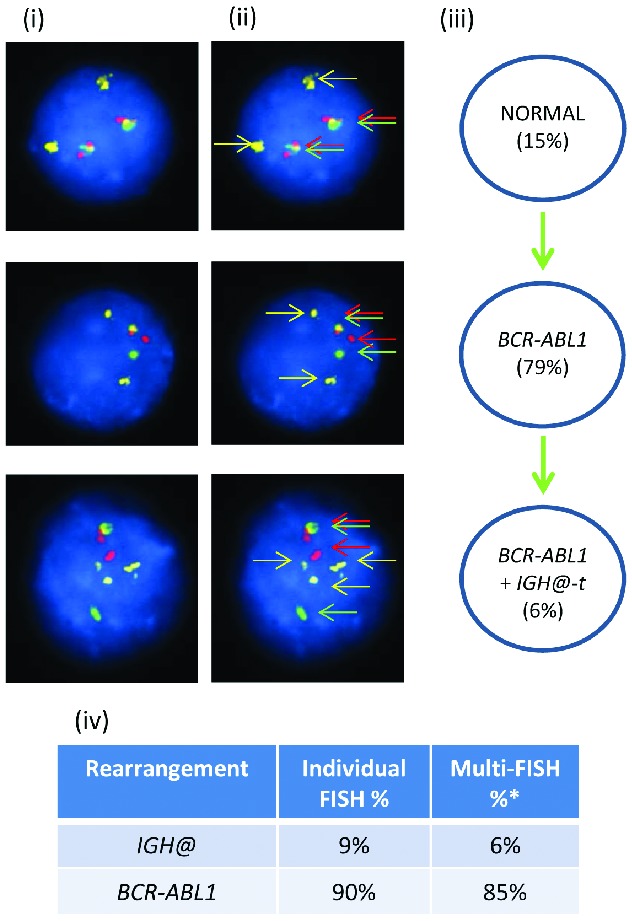
In patient 11061 with iAMP21, the Vysis IGH@ probe showed 47% interphase nuclei with IGH@-t (Figure 4), while multiple color FISH showed 97% of cells with iAMP21, of which only 21% had IGH@-t in addition to iAMP21.
Figure 4.
Images of FISH interphase nuclei depicting the apparent emergence of an iAMP21 prior to the IGH@-t. (i) Captured interphase nuclei from patient 11061 hybridized with FISH probes to track iAMP21 using a home-made RUNX1 breakapart probe (red/green) and a home-made IGH@ single color probe (gold). (ii) The same captured interphase nuclei, including colored arrows highlighting the signals that were scored: the yellow arrows highlight the gold probe marking the home-made IGH@ probe that detects both the copy number of chromosome 14 and also the presence of a translocation. The red/green double arrows highlight the home home-made RUNX1 breakapart probe that marks the iAMP21 aberration. (iii) Cartoon depicting a possible model of evolutionary progression. Seventy-six percentage of nuclei showed multiple copies of RUNX1 with 21% of these nuclei also harboring IGH@-t. (iv) Table showing the percentages of positive nuclei when samples were hybridised with either the individual probes for each rearrangement (Individual FISH), or when they were investigated together (Multi-FISH) in the same cell.
MLL translocation
In patient 21940 with an MLL translocation, the Vysis IGH@ probe alone showed a rearrangement in 13% of inter-phase nuclei (Figure 5). Multiple color FISH showed the pres ence of IGH@-t in addition to the MLL translocation in 8% of cells with 82% having only MLL translocation with no evidence of IGH@-t.
Figure 5.
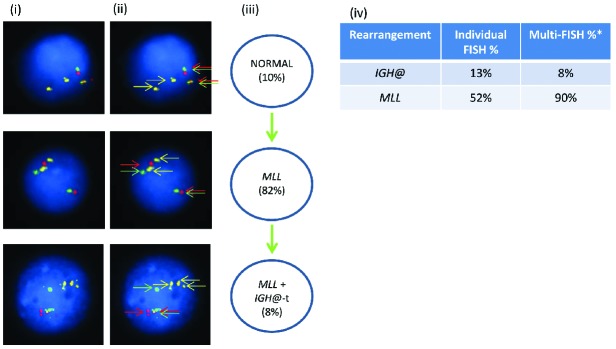
Images of FISH interphase nuclei depicting the apparent emergence of an MLL translocation prior to the IGH@-t. (i) Captured inter-phase nuclei from patient 21940 hybridized with FISH probes to track the MLL translocation using a dual color MLL breakapart probe (Vysis, Abbott Molecular) (red/green) and home-made IGH@ probe (gold). (ii) The same captured interphase nuclei, including colored arrows highlighting the signals that were scored: the yellow arrows highlight the gold probe marking the home-made IGH@ probe that detects both the copy number of chromosome 14 and also the presence of a translocation. The red/green double arrows highlight the dual color MLL breakapart probe that marks the MLL translocation. (iii) Cartoon depicting a possible model of evolutionary progression. An MLL rearrangement was observed in 82% of cells and an additional IGH@-t in 8% of cells. (iv) Table showing the percentages of positive nuclei when samples were hybridized with either the individual probes for each rearrangement (Individual FISH), or when they were investigated together (Multi-FISH) in the same cell. *percentages may vary between the FISH results obtained when using an individual probe, and probes in combination, likely reflecting scoring by automation versus scoring by eye and increased interference and obscuring of signals when there are many of them.
IGH@-t and primary rearrangements are independent clones
In three patients (24390, 11520 and 3737), independent abnormal clones were identified. In patient 24390, cytogenetics and FISH identified independent high hyperdiploidy and IGH@-t abnormal clones. Chromosomal analysis showed a high hyperdiploidy karyotype with 51 chromosomes including constitutional gain of chromosome 21 (+21c), consistent with this patient having DS-ALL. Interphase and metaphase FISH using the Vysis IGH@ probe showed a normal population with two intact IGH@ signals (28% interphase nuclei) (Figure 6Ai). Three abnormal popu lations were also observed: (ii) an additional chromosome 14 with intact IGH@ signals observed on all three chromosomes 14 in 10/23 metaphases and 29% interphase nuclei; (iii) an IGH@-t in 2/23 metaphases and 11% interphase nuclei but with no evidence of gain of chromosome 14; (iv) a rearrangement of both copies of the IGH@ locus in 9/23 metaphases and 28% interphase nuclei but no evidence of gain of chromosome 14. The quality of the metaphase cells with IGH@-t was too poor to establish the number of chromosomes present. The FISH and cytogenetic results indicated two independent abnormal clones with either high hyperdiploidy or a cytogenetically cryptic IGH@-t. Further supporting evidence for two independent abnormal clones included a significant difference in the nuclear morphology of the interphase cells. The nuclei with IGH@ rearrangements (Figure 6Aiii–iv) were significantly smaller than those with chromosomal gains (Figure 6Ai–ii). In this patient, CRLF2 FISH indicated that this IGH@-t was IGH@-CRLF2.
Figure 6.

Dual color and multiple color FISH showing independent aneuploid and IGH@-t clones in patient 24390. (A) Dual color IGH@ FISH showing the presence of a normal clone (i) and three abnormal clones (ii,iii,iv). The red/green colored arrows highlight the signals that were scored. The first abnormal clone (ii) shows an additional copy of the IGH@ intact fusion, the second abnormal clone (iii) shows the separation of one fusion indicating the presence of a translocation involving one IGH@ locus and the third abnormal clone (iv) shows the separation of both IGH@ fusions suggesting the presence of two IGH@-t. (B) Cartoon depicting the potential clonal evolution by either gain (green arrows) or loss (red arrows). Solid and dashed lines highlight the possible alternative evolutionary routes taken by the sample. Two intact IGH@ signals with three copies of chromosome 21 were observed in 48% of nuclei. This clone evolved into two independent sub-clones. The first showed evolution within the high hyperdiploidy clone with additional copies of chromosome 21 (4%), both chromosome 14 and 21 (20%) and loss of one copy of chromosome 21 (8%). The second sub-clone harbored a single IGH@-t followed by rearrangement of the second IGH@ locus (18%). The black circles represent the FISH signal patterns which correlate with the karyotypes observed at metaphase analysis. (C) Table showing the percentages of positive nuclei when samples were hybridized with either the individual probes for each rearrangement (Individual FISH), or when they were investigated together (Multi-FISH) in the same cell.
Multiple color FISH for IGH@ and chromosomes 14 and 21 provided additional evidence that the translocation and the copy number abnormalities were present in different clones (Figure 6B). Two intact IGH@ signals with three copies of chromosome 21, indicating constitutional gain (+21c), were observed in 48% of nuclei. This clone evolved into two independent sub-clones. The first acquired an additional copy of chromosome 21 (4%) which further evolved with gain of chromosome 14 (20%). This clone appeared to lose one copy of chromosome 21 in a further 8% of nuclei. There is a possibility that the latter clone may have evolved from the normal clone, however the hypothesis that high hyperdiploidy arises in one cell division makes loss of chromosomes at segregation more likely.8 The second sub-clone harbored a single IGH@-t in the presence of +21c. Although this rearrangement was observed in only 2% of nuclei, the metaphase and interphase FISH results described above, using only the Vysis IGH@ probe, confirmed this result. This clone appeared to have evolved further, with a translocation involving the other IGH@ locus in 18% of nuclei. CRLF2 FISH identified a rearrangement, suggesting IGH@-CRLF2. The partner gene involved in the second IGH@-t remains unknown.
Using individual FISH probes, patient 11520 (Figure 7Ai–iii) showed IGH@-t in 32% of cells and ETV6-RUNX1 in 15% of cells (Figure 7Aiii). Patient 3737 (Figure 7Bi–iii) had IGH@-t in 76% of cells and the BCR-ABL1 gene fusion in 12% of cells (Figure 7Biii). However, in both patients, multiple color FISH showed that these clones were independent; no cells were observed with either the ETV6-RUNX1 or BCR-ABL1 rearrangement in association with IGH@-t (Figures 7Aii and 7Bii). The IGH@-t partner gene in these patients remains unknown.
Figure 7.
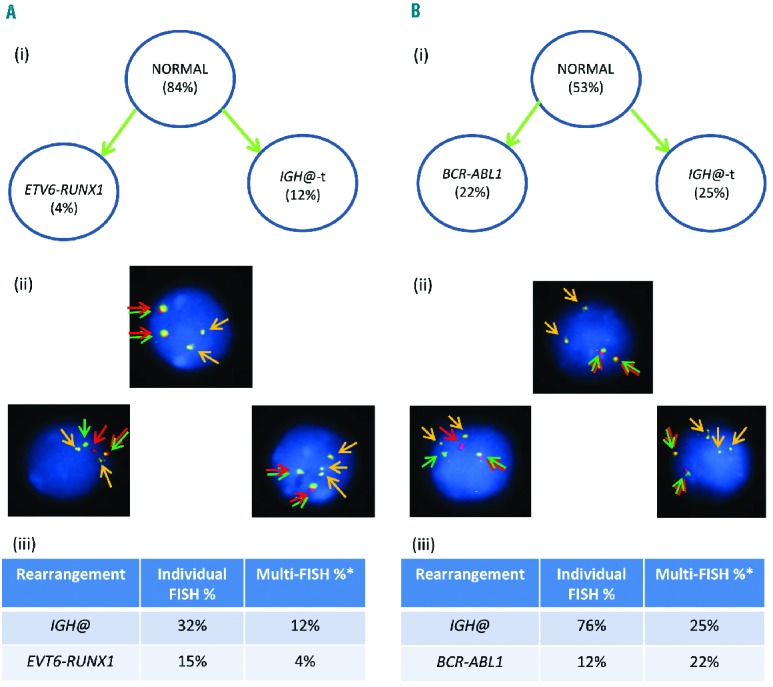
Images of FISH interphase nuclei depicting an independent primary rearrangement and IGH@-t clones. (A) Patient 11520 with ETV6-RUNX1 demonstrated using a RUNX1 breakapart probe (Vysis, Abbott Molecular) (red/green) and home-made IGH@ probe (gold). (i) Cartoon depicting the possible model of evolutionary progression with both aberrations being observed in separate cells. (ii) Captured interphase nuclei, including colored arrows highlighting the signals that were scored: the yellow arrows highlight the gold probe marking the home-made IGH@ probe that detects both the copy number of chromosome 14 and also the presence of a translocation. The red/green double arrows highlight the RUNX1 breakapart probe that marks the ETV6-RUNX1 fusion. (iii) Table showing the percentages of positive nuclei when samples were hybridized with either the individual probes for each rearrangement (Individual FISH), or when they were investigated together (Multi-FISH) in the same cell. (B) Patient 3737 with BCR-ABL1 demonstrated using the dual color BCR-ABL1 breakapart probe (Vysis, Abbott Molecular) (red/green) and home-made IGH@ probe (gold). (i) Cartoon depicting the possible model of evolutionary progression with both aberrations being observed in separate cells. (ii) Captured interphase nuclei, including colored arrows highlighting the signals that were scored: the yellow arrows highlight the gold probe marking the home-made IGH@ probe that detects both the copy number of chromosome 14 and also the presence of a translocation. The red/green double arrows highlight the BCR-ABL1 breakapart probe that marks the BCR-ABL1 fusion. (iii) Table showing the percentages of positive nuclei when samples were hybridized with either the individual probes for each rearrangement (Individual FISH), or when they were investigated together (Multi-FISH) in the same cell. *percentages vary between the FISH results obtained when using an individual probe, or a probe in combination with others.
Excluding the three patients with independent primary and IGH@-t clones, ten patients had IGH@-t at the time of presentation of the disease and also at the time when the IGH@-t positive clone represented the dominant abnormal clone (largest clone), eight presented when the IGH@-t clone was either at a comparable level to that of the primary rearrangement or a significant minor sub-clone (≥15% of cells), and seven presented with a minor sub-clone (those close to the false positive level and <15% of cells) (Online Supplementary Table S1).
IGH@-t etiology is unknown
One patient (7143) showed the presence of both high hyperdiploidy and IGH@-t in the same nuclei in all abnormal cells and therefore the point at which IGH@-t arose was unknown. In this cases the etiology was classified as undetermined (Figure 8i–iii). Two models of evolution are possible (Figure 8iii). Clonal evolution occurred with either 25% of nuclei gaining a third additional copy of chromosome 21, or 71% of nuclei losing the third additional copy of 21. The IGH@-t was cytogenetically visible in this patient and sub sequent interphase FISH with a breakapart probe to CEBPA confirmed IGH@-CEBPA.
Figure 8.
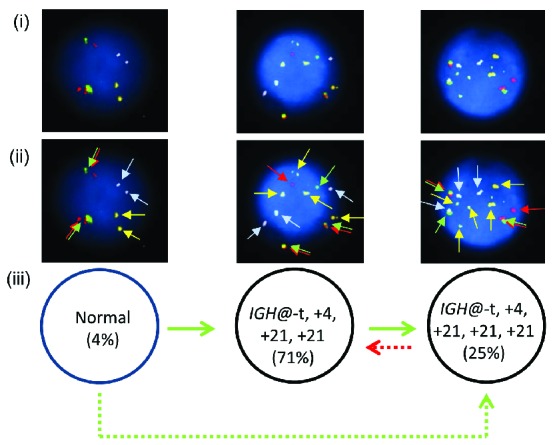
Multiple color FISH for patient 7143 showing co-existing high hyperdiploidy and IGH@-t. (i) Captured interphase nuclei from patient 7143 hybridized with FISH probes to track the IGH@-t (red/green) (Cytocell), chromosome 4 (aqua) and chromosome 21 (gold). (ii) The same captured interphase nuclei, including colored arrows highlighting the signals that were scored: the red/green double arrows highlight the IGH@ breakapart probe that marks both the copy number of chromosome 14 and also detects the presence of a translocation, the blue arrows highlight chromosome 4 and the gold arrows chromosome 21. (iii) Cartoon depicting possible models of evolutionary progression by either gain (green arrows) or loss (red arrows). Solid and dashed lines highlight the possible alternative evolutionary routes taken by the sample. The black circle represents the FISH signal pattern which correlates with the abnormal karyotype observed at metaphase analysis.
Discussion
High hyperdiploidy, t(12;21)/ETV6-RUNX1, t(9;22)/BCR-ABL1, iAMP21 and MLL translocations are all established recurrent cytogenetic abnormalities in BCP-ALL.9 This study has shown that these rearrangements and IGH@-t may occur together in both childhood and adult BCP-ALL. IGH@-t did not appear to be associated with any particular primary rearrangement, as the frequency observed correlated with the frequency observed in BCP-ALL in general. High hyperdiploidy and ETV6-RUNX1 are the two most common subtypes of childhood BCP-ALL (~25% each), BCR-ABL1 is observed in around 9% (3% in children and 15% in adults), whereas iAMP21 and MLL-t are two of the relatively rarer subtypes (~2% each). The median age and white cell count at presentation in these patients compared well to those typically observed in BCP-ALL.9
This study is the first to look at the existence and evolution of a primary abnormality and a structural rearrangement (IGH@-t) in the same cells. Although the number of cases is small, our results support the literature showing that high hyperdiploidy, ETV6-RUNX1, iAMP21 and MLL-t are primary events.10,11 IGH@-t did not arise as primary events in the presence of these established changes. The models proposed for the four patients described (10281 21733, 11061 and 21940), and confirmed in six others (8447, 22105, 11438, 22420, 2618 and 10859), suggest that in the majority of patients harboring both a primary rearrangement and IGH@-t, the IGH@-t is a secondary event in leukemia development. In three patients they were apparently independent events and in four the etiology was undetermined.
The partner genes identified in this cohort of patients with both a primary rearrangement and IGH@-t were CRLF2, CEBPA, CEBPB, TRA/D@, IGF2BP1 and IGK@. These genes and others have previously been identified as being deregulated in ALL due to juxtaposition with the IGH@ enhancer.2–5,12–14 The translocation, t(14;14)(q11;q32), involving both the IGH@ and TRA/D@ loci is typically observed in T-lineage ALL. However, the presence of rearrangements involving IGH@ and TRA/D@ in B-lineage neoplasia has been previously reported in the literature, albeit rarely.7,15 Of the patients with apparently independent clones, the DS-ALL (patient 24390) showed unusual results with different clones having either one or two IGH@-t. FISH studies identified one CRLF2 gene rearrangement, while the other remains unknown. These results suggest the occurrence of one IGH@-t with subsequent translocation of the second IGH@ locus. The apparently unrelated abnormal clones in this patient and two others are likely to have arisen in either a common precursor clone with an unknown rearrangement and/or gene mutation, or both could have arisen as independent leukemias.
Whether these rearrangements represent drivers or passengers may be dependent on which partner gene is aberrantly expressed. Those present as a low level population may be passenger abnormalities with no leukemogenic advantage.
None of the patients in the favorable risk cohort showed any evidence of minimal residual disease after induction therapy. All but two are alive at this time and are in complete remission which has lasted a median of 43 months; the two patients who died had relapsed at 13 and 24 months. It remains to be determined whether IGH@-t in the presence of a favorable or intermediate marker alters these risks.
Our study has shown that IGH@-t can occur together with primary rearrangements and, when present, IGH@-t usually arise as secondary events, likely contributing to the development of the leukemia phenotype. Due to the cryptic nature of some IGH@-t we would recommend their exclusion by FISH. Whether IGH@-t in the presence of other primary rearrangements associated with a good or intermediate risk has an impact on outcome has yet to be determined.
Acknowledgments
We thank the Kay Kendall Leukaemia Fund and Leukaemia and Lymphoma Research for financial support and member laboratories of the United Kingdom Cancer Cytogenetic Group (UKCCG) for providing cytogenetic data and material. We also thank the Central England Haemato-oncology Research Biobank for providing some of the material. Finally, we thank all the clinicians who entered patients into the trial and the children and families who agreed to take part.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Moorman AV, Ensor HM, Richards SM, Chilton L, Schwab C, Kinsey SE, et al. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010;11(5):429–38 [DOI] [PubMed] [Google Scholar]
- 2.Russell LJ, Capasso M, Vater I, Akasaka T, Bernard OA, Calasanz MJ, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114(13):2688–98 [DOI] [PubMed] [Google Scholar]
- 3.Chapiro E, Russell L, Radford-Weiss I, Bastard C, Lessard M, Struski S, et al. Overexpression of CEBPA resulting from the translocation t(14;19)(q32;q13) of human precursor B acute lymphoblastic leukemia. Blood. 2006;108(10):3560–3 [DOI] [PubMed] [Google Scholar]
- 4.Akasaka T, Balasas T, Russell LJ, Sugimoto KJ, Majid A, Walewska R, et al. Five members of the CEBP transcription factor family are targeted by recurrent IGH translocations in B-cell precursor acute lymphoblastic leukemia (BCP-ALL). Blood. 2007;109(8):3451–61 [DOI] [PubMed] [Google Scholar]
- 5.Russell LJ, Akasaka T, Majid A, Sugimoto KJ, Loraine KE, Nagel I, et al. t(6;14)(p22;q32): a new recurrent IGH@ translocation involving ID4 in B-cell precursor acute lymphoblastic leukemia (BCP-ALL). Blood. 2008;111(1):387–91 [DOI] [PubMed] [Google Scholar]
- 6.Eastmond DA, Schuler M, Rupa DS. Advantages and limitations of using fluorescence in situ hybridization for the detection of aneuploidy in interphase human cells. Mutat Res Letters. 1995;348(4):153–62 [DOI] [PubMed] [Google Scholar]
- 7.Russell LJ, Enshaei A, Jones L, Erhorn A, Masic D, Bentley H, et al. IGH@ translocations are prevalent in teenagers and young adults with ALL and are associated with a poor outcome. J Clin Oncol. 2014;32(14):1453–62 [DOI] [PubMed] [Google Scholar]
- 8.Paulsson K, Mörse H, Fioretos T, Behrendtz M, Strömbeck B, Johansson B. Evidence for a single-step mechanism in the origin of hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromos Canc. 2005;44(2):113–22 [DOI] [PubMed] [Google Scholar]
- 9.Moorman AV. The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood Rev. 2012;26(3):123–35 [DOI] [PubMed] [Google Scholar]
- 10.Rand V, Parker H, Russell LJ, Schwab C, Ensor H, Irving J, et al. Genomic characterization implicates iAMP21 as a likely primary genetic event in childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2011;117(25):6848–55 [DOI] [PubMed] [Google Scholar]
- 11.Maia AT, van der Velden VHJ, Harrison CJ, Szczepanski T, Williams MD, Griffiths MJ, et al. Prenatal origin of hyperdiploid acute lymphoblastic leukemia in identical twins. Leukemia. 2003;17(11):2202–6 [DOI] [PubMed] [Google Scholar]
- 12.Russell LJ, De Castro DG, Griffiths M, Telford N, Bernard O, Panzer-Grumayer R, et al. A novel translocation, t(14;19)(q32;p13), involving IGH@ and the cytokine receptor for erythropoietin. Leukemia. 2008;23(3):614–7 [DOI] [PubMed] [Google Scholar]
- 13.Chapiro E, Russell LJ, Struski S, Cave H, Radford-Weiss I, Valle VD, et al. A new recurrent translocation t(11;14)(q24;q32) involving IGH@ and miR-125b-1 in B-cell progenitor acute lymphoblastic leukemia. Leukemia. 2010;24(7):1362–4 [DOI] [PubMed] [Google Scholar]
- 14.Aventín A, Sánchez J, Nomdedéu JF, Estany C, Forcada P, La Starza R, et al. Novel IGH translocations, t(2;14)(q14.3;q32) and t(14;17)(q32;q21), in B-cell precursor acute lymphoblastic leukemia. Canc Genet Cytogen. 2008;185(1):57–9 [DOI] [PubMed] [Google Scholar]
- 15.Wong KF, Kwong YL, Wong TK. Inversion 14q in acute lymphoblastic leukemia of B-lineage. Cancer Genet Cytogen. 1995;80(1):72–4 [DOI] [PubMed] [Google Scholar]