

Abstract
Objective:
To comprehensively investigate the relationship between antibodies to single glycolipids and their complexes and Guillain-Barré syndrome subtypes and clinical features.
Methods:
In acute sera from 199 patients with Guillain-Barré syndrome, immunoglobulin G (IgG) antibodies to glycolipids and ganglioside complexes were tested using ELISA against individual antigens from single glycolipids including gangliosides (LM1, GM1, GM1b, GD1a, GalNAc-GD1a, GD1b, GT1a, GT1b, GQ1b) and a neutral glycolipid, asialo-GM1 (GA1), and antigens from the combination of 2 different glycolipids. Based on serial nerve conduction studies, the electrodiagnoses were as follows: 69 demyelinating subtype, 85 axonal subtypes, and 45 unclassified.
Results:
Significant associations were detected between acute motor axonal neuropathy subtype and IgG antibodies to GM1, GalNAc-GD1a, GA1, or LM1/GA1 complex. Reversible conduction failure was significantly associated with IgG antibodies to GM1, GalNAc-GD1a, GD1b, or complex of LM1/GA1. No significant association was demonstrated between acute inflammatory demyelinating polyneuropathy and any of the glycolipids or ganglioside complexes. Anti-ganglioside complex antibodies alone were detected in 7 patients (5 axonal subtype).
Conclusions:
The current study demonstrates that antibodies to single glycolipids and ganglioside complexes are associated with acute motor axonal neuropathy or acute motor conduction block neuropathy but not acute inflammatory demyelinating polyneuropathy.
Classification of evidence:
This study provides Class II evidence that antibodies to glycolipids are increased in patients with acute motor axonal neuropathy and acute motor conduction block neuropathy but not acute inflammatory demyelinating polyneuropathy.
Guillain-Barré syndrome (GBS) is an acute immune-mediated polyneuropathy with 2 major subtypes: acute inflammatory demyelinating polyneuropathy (AIDP) and acute motor axonal neuropathy (AMAN).1 Within the axonal subtype, there are now recognized variants evident on nerve conduction studies (NCS), which demonstrate early reversible conduction failure, referred to as acute motor conduction block neuropathy (AMCBN).2 There is robust evidence that immunoglobulin G (IgG) anti-ganglioside antibodies are associated with the pathogenesis of AMAN, whereas the target antigens in AIDP remain elusive.3
In 2004, antibodies to ganglioside complexes (GSCs) were reported in patients with GBS.4 The patients who were seronegative for antibodies to single gangliosides were found to have anti-GSC antibodies. The authors have since described further associations between anti-GSC antibodies and variants of GBS. This includes antibodies to LM1 and its complexes in AIDP,5 to complex of GM1 and GalNAc-GD1a (GM1/GalNAc-GD1a) in AMCBN,6 and to complexes of GD1a/GD1b and GD1b/GT1b in patients with GBS requiring artificial ventilation.7
In the current study, we aimed to investigate the relationship between anti-GSC antibodies and specific clinical features of GBS as well as the electrodiagnostic subtypes of GBS, the latter based on serial NCS in a large cohort of patients from different geographical locations.
METHODS
Serum samples.
Acute phase sera were collected from patients with GBS presenting consecutively to 5 different centers, namely, University Malaya Medical Centre in Malaysia, National Neuroscience Institute and National University Hospital in Singapore, and Dokkyo Medical University and Chiba University in Japan. Patients from Malaysia and Singapore were prospectively recruited from 2010 to 2012. Patients recruited from the Japanese cohort were consecutively seen between 1998 and 2012. A total of 199 patients (Malaysia, 22; Singapore, 33; Japan, 144) with GBS were recruited. The clinical features in each patient, specifically, the presence of ophthalmoplegia, bulbar palsy, facial palsy, sensory impairment, and respiratory failure necessitating artificial ventilation were documented by the respective neurologists from each center.
Standard protocol approvals and patient consents.
Patients' informed written consents, clinical data, and sera samples were obtained following protocol approved by the respective institution's ethics committee.
Nerve conduction studies.
NCS were performed at presentation and repeated subsequently within a period of 3 to 6 weeks. The electrodiagnosis of GBS was initially defined according to existing criteria.1 However, a final electrodiagnosis was made after the second NCS. The final electrodiagnoses were AIDP, AMAN (which included both AMCBN and acute motor and sensory axonal neuropathy subtypes), and unclassified. In a separate analysis, patients exhibiting the presence of reversible conduction failure defined by a decrease of proximal to distal compound motor action potential amplitude by 50% in intermediate nerve segments without temporal dispersion were considered to have AMCBN, a less severe form of AMAN.8
ELISA.
Serologic analyses were performed for IgG antibodies to single glycolipids including gangliosides (LM1, GM1, GM1b, GD1a, GalNAc-GD1a, GD1b, GT1a, GT1b, and GQ1b) and a neutral glycolipid, asialo-GM1 (GA1), using ELISA.9 Patients' sera were also assessed for IgG antibodies to GSC, which were tested with a mixture of individual glycolipids at 5 pmol/well each. Anti-glycolipid and -GSC antibodies were considered positive when the optical density was greater than 0.5 of the sum of antibodies to individual antigens. The tests were performed in quadruplicate and a mean of the optical density value was measured.
Statistical analysis.
Comparative analyses of categorical outcomes were performed with the Fisher exact test or χ2 test. A p value <0.05 was considered statistically significant.
Classification of evidence.
The primary objectives of our study were to describe the relationship between antibodies against single glycolipids and glycolipid complexes and GBS subtypes. The study provides Class II evidence that antibodies to single glycolipids and glycolipid complexes are increased in AMAN and AMCBN but not AIDP.
RESULTS
Comparison between the Malaysian-Singaporean and Japanese cohorts.
The presence of ophthalmoplegia, facial palsy, bulbar weakness, sensory impairment, and need for artificial ventilation were significantly more frequent in the Malaysian-Singaporean (n = 55) than the Japanese cohort (n = 144) (table 1). Electrodiagnosis between the 2 cohorts reached no significant difference in AIDP and AMAN, but there were significantly more cases that were unclassified in the Japanese cohort. In contrast, more patients were seen to have reversible conduction failure in keeping with AMCBN in the Malaysian-Singaporean cohort. Despite the differences in the clinical patterns, there were no significant differences between seropositivity for either anti-ganglioside alone or anti-GSC alone between the cohorts.
Table 1.
Comparison of clinical features, electrodiagnosis, and serologic analyses
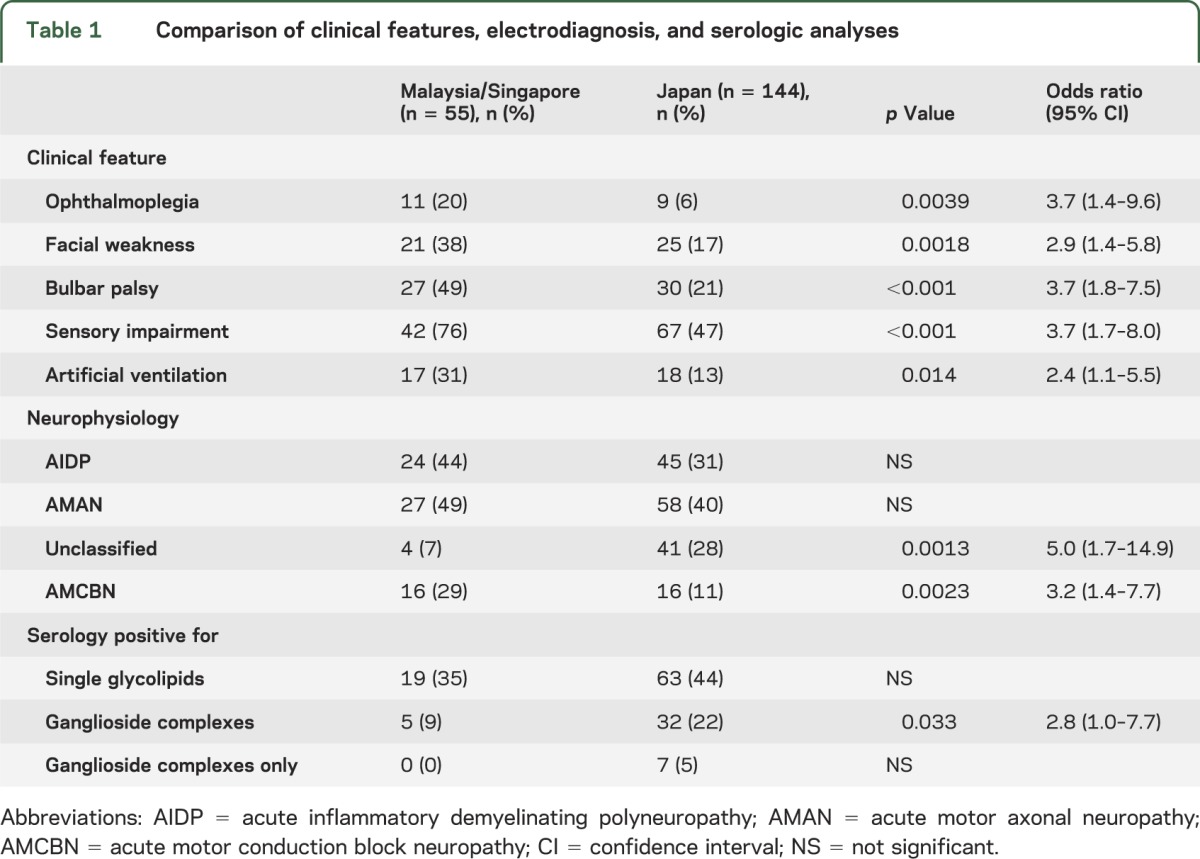
In both cohorts, there was a significant association between the presence of antibodies to single glycolipids and AMAN as well as the absence of anti-ganglioside antibodies and AIDP (table 2). The same pattern was also observed with anti-GSC antibodies, but only in the Japanese cohort. The relationships among anti-glycolipid or -GSC antibodies, the GBS subtypes, and various clinical features were further analyzed in the entire group (tables 3 and 4).
Table 2.
Antibodies to glycolipids and ganglioside complexes in Malaysian/Singaporean and Japanese populations
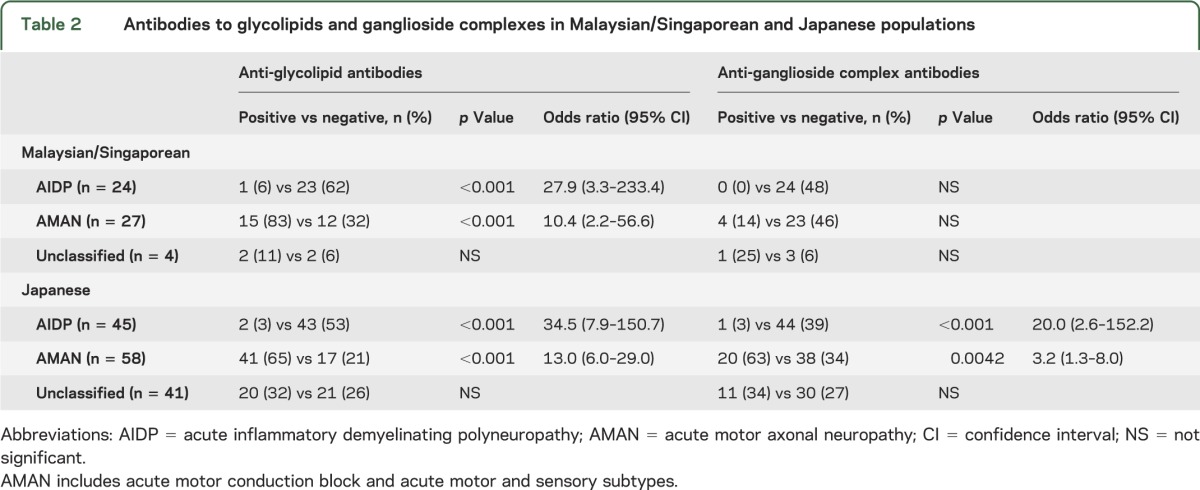
Table 3.
Association of electrodiagnostic subtypes with antibodies to glycolipids and ganglioside complexes
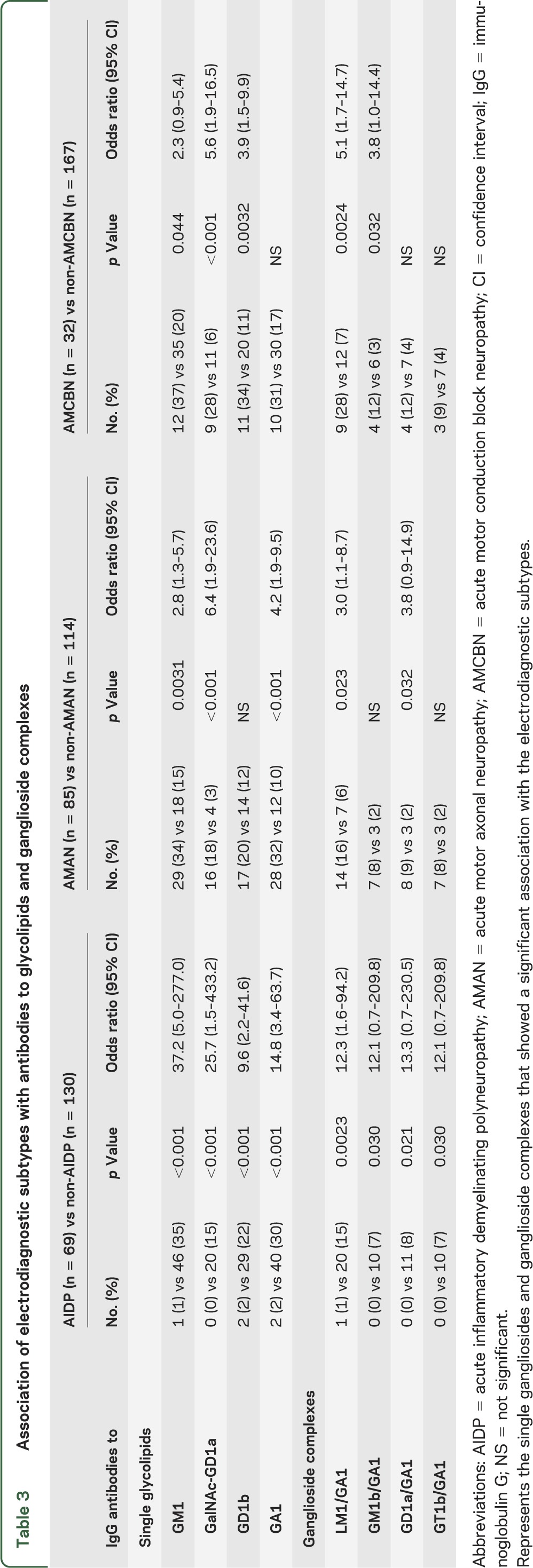
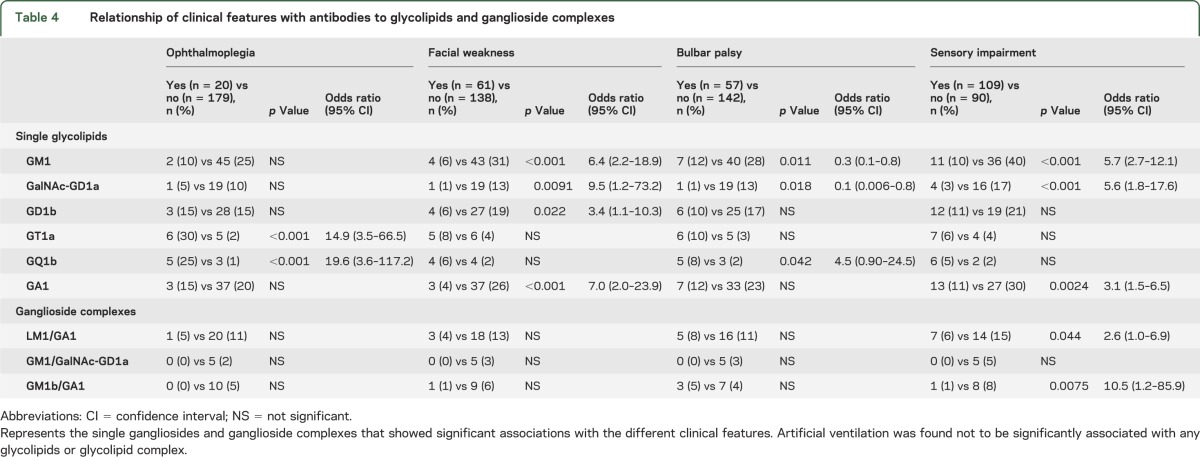
Table 4.
Relationship of clinical features with antibodies to glycolipids and ganglioside complexes
Relationships among anti-ganglioside or -GSC antibodies, electrodiagnoses, and clinical features.
The final electrodiagnoses based on serial studies for the entire group (n = 199) were as follows: AIDP = 69 patients, AMAN = 85, and unclassified = 45. The serologic analyses revealed 88 patients (44%) with positive serology. The results are shown in table 1. Analyses of IgG antibodies to individual single glycolipid and GSC revealed significant associations between AMAN and anti-GM1, -GalNAc-GD1a, -GA1, and -LM1/GA1 antibodies (table 3). Figure e-1 on the Neurology® Web site at Neurology.org depicts an example of seropositive findings in a patient with AMAN. AMCBN was associated with anti-GM1, -GalNAc-GD1a, and -GD1b antibodies as well as anti-LM1/GA1 antibodies. In contrast, AIDP was not significantly associated with any of the glycolipids or GSCs.
Regarding the clinical features, significant associations were detected between IgG anti-GT1a and -GQ1b antibodies and ophthalmoplegia (table 4). Patients with IgG anti-GM1, -GalNAc-GD1a, -GD1a, and -GA1 antibodies were less likely to have facial palsy, and those with IgG anti-GalNAc-GD1a antibodies were also less likely to have bulbar palsy. In addition, sensory impairment was less likely to be demonstrated in patients who had IgG anti-GM1, -GalNAc-GD1a, -GA1, -LM1/GA1, -GM1/GalNAc-GD1a, and -GM1b/GA1 antibodies. The need for artificial ventilation showed no significant association with the presence of IgG antibodies to glycolipids or GSCs.
DISCUSSION
In the current study, we investigated the relationship between anti-glycolipid or -GSC antibodies with electrophysiologic subtypes or specific clinical features of GBS. Patients were recruited from 2 geographical locations: Southeast Asia (represented by Malaysia and Singapore) and Japan. Although a comparison between the 2 cohorts revealed differences in the frequencies of certain clinical features, neither the electrodiagnostic classification of AIDP and AMAN nor the serologic analyses were significantly different. Analyses of the entire cohort revealed that significant associations of antibodies to certain single glycolipids and GSCs were evident in patients with an electrodiagnosis of AMAN but not AIDP. There were also specific antibodies that were significantly associated with reversible conduction failure as well as certain clinical characteristics such as ophthalmoplegia and bulbar palsy.
In a previous comparative study between Japanese and Italian cohorts, no significant differences were found in the final GBS electrodiagnosis (also based on serial studies) and anti-ganglioside antibodies.10 The current study also demonstrates that both GBS cohorts from Southeast Asia and Japan were not significantly different regarding the final electrodiagnoses of AIDP and AMAN or their serologic reactivities. The majority of seropositive patients had IgG antibodies to single glycolipids (with some also reacting to GSCs). In seronegative patients, analysis of IgG antibodies to GSC identified a further 7 patients. GSCs are thought to represent new clustered epitopes that are recognized by antibodies that would normally not recognize epitopes of a single glycolipid.
LM1 is a predominant peripheral nerve ganglioside, localized in motor nerve myelin,11,12 and thus it is possible that antibodies to LM1 and its complexes are involved in the development of AIDP. Several studies have investigated the presence of both IgG and IgM anti-LM1 antibodies in GBS, and the results have been variable. The frequencies range from 43%13 and 23% of patients with GBS14 to less than 10% of GBS in other series.15–17 In a more recent study, a significant association of AIDP with antibodies to LM1 and its complexes was reported.5 However, in the current study, we did not detect as strong an association of AIDP with IgG antibodies to LM1 and its complexes making it less likely that LM1 or its complexes are target antigens, at least in AIDP. Instead, we found that the LM1/GA1 complex was significantly associated with AMAN, reversible conduction failure or AMCBN, and the absence of sensory impairment. In previous studies, the electrodiagnosis of GBS was based on a single study. Given our current understanding that the neurophysiologic findings in GBS can rapidly change in the early stages of the disease, we believe that the diagnosis of AIDP was likely to have been overestimated. Based on our findings, pathogenic autoantibodies involved in AIDP remain elusive.
Certain electrophysiologic features, such as reversible conduction failure, have previously been associated with the presence of IgG antibodies to specific gangliosides, namely, GM1.18–21 In the current study, we found significant associations of reversible conduction failure with IgG anti-GM1, -GalNAc-GD1a, -GD1b, and -LM1/GA1 antibodies. Reversible conduction failure was first described in 1998 and this was followed by reports of similar findings in other cohorts.18 Some authors have referred to patients with such features as having AMCBN, associated with a better prognosis in comparison to AMAN. AMCBN is a predominantly motor neuropathy and thus it is not surprising to find associations with IgG anti-GM1 and -GalNAc-GD1a antibodies, both of which have been described in AMAN. Notably, there were significantly more patients with AMCBN in the Southeast Asian cohort compared with the Japanese cohort, and the significance of this merits further study in a larger cohort.
Previous studies have provided evidence that IgG anti-GM1 or -GD1a antibodies are pathogenic in the development of AMAN.22 Several clinical patterns have since been described in association with certain anti-ganglioside antibodies. This includes the association of pure motor GBS with IgG anti-GM1/GalNAc-GD1a antibodies6 and IgG antibodies to GD1a/GD1b and GD1b/GT1b with the need for artificial ventilation.7 We investigated the various clinical features and their associations with seropositivity in our cohort. We found that patients who lacked sensory impairment (indicating a predominant motor form of GBS) were significantly associated with IgG antibodies to GM1, GalNAc-GD1a, and GA1 as well as IgG antibodies to LM1/GA1, GM1/GalNAc-GD1a, and GM1b/GA1. In contrast, none of the patients who required mechanical ventilation had significant associations with anti-GQ1b antibodies, which has previously been reported to be predictive of mechanical ventilation.23 Of note, in the current study, only one patient was seropositive for anti-GD1a/GD1b and none for GD1b/GT1b, both of which have also been postulated to have associations with severity of GBS.7
The presence of ophthalmoplegia and bulbar palsy was associated with IgG anti-GQ1b antibodies, in keeping with previous reports.24,25 Ophthalmoplegia is a key feature of Fisher syndrome, which has a strong association with anti-GQ1b antibodies,25 whereas bulbar palsy is typically seen in patients with the pharyngeal-cervical-brachial variant of GBS, which is associated with monospecific anti-GT1a antibodies.26,27 In the current study, the association of bulbar palsy with anti-GT1a antibodies did not reach significance. Instead, anti-GQ1b antibodies, which are recognized to crossreact with GT1a, were significantly associated with bulbar palsy.25 This association has been demonstrated in previous studies comparing GBS with and without bulbar palsy.28 None of the patients in the current cohort had monospecific anti-GT1a antibodies. Contrary to previous reports, antibodies to GSCs were not significantly higher in either group of patients with ophthalmoplegia or bulbar palsy.7 In our cohort, the presence of facial palsy was associated with a diagnosis of AIDP without significant serologic associations. Instead, the presence of IgG anti-GM1, -GalNAc-GD1a, -GD1b, and -GA1 antibodies was less likely to result in the development of facial palsy. Facial palsy in GBS has been described to occur in almost 60% of patients with GBS,29 and there are reports that recognize the presence of “bifacial weakness and paraesthesia” as a variant of AIDP.30,31 Our studies would support this hypothesis and that there are as yet no specific antigens that can be associated with facial palsy.
Before the current work, the majority of the literature on GSCs and the clinical characteristics associated with them has originated from a different Japanese cohort.4 Although our findings share similarities to their cohort, there were also discrepancies such as the lack of association of antibodies to LM1 and LM1 complexes in AIDP. The most apparent reason for the differences is the different methodologies in serologic analyses by ELISA and GBS electrodiagnosis. In comparison to previous studies, our serologic analyses utilized a reduced amount of antigen (e.g., GM1, 7.5 vs 200 ng) and a higher serum and secondary antibody dilution (1:500 and 1:2,000 vs 1:40 and 1:500, respectively). The optical density value for seropositivity also differed (≥0.5 in the current study vs >0.1 in single gangliosides and >0.2 in GSCs in other studies). We believe that the methodology adopted in our study would result in more specific findings. The final electrodiagnostic criteria in the current study were based on serial studies, taking into account the existing limitations of a single study.2 In contrast, other studies have used different criteria based on one study, which may overestimate AIDP.32
In a more recent study, antibodies to glycolipid complexes were assessed in sera from a Western European cohort utilizing the combinatorial glycoarray method.33 The method differs from traditional ELISA, and discrepancies of results obtained from ELISA were noted by the authors. In the study, a large number of heterodimeric glycolipid complexes were assessed (n = 162) and the authors found an increase in seropositivity to the glycolipid complexes of patients with “demyelinating” GBS or unclassified. Similar to previous studies, the GBS electrodiagnoses were based on a single NCS. These are some of the limitations of the current study and highlight the importance of standardizing methodology of serology and electrophysiology among investigators to allow for improved and more valid comparisons of GBS patterns between cohorts. One likely platform for such work to be done could be the ongoing multicentered International GBS Outcome Study, recently initiated by the Inflammatory Neuropathy Consortium.
The current study of a large multicentered GBS population suggests that antibodies to glycolipids and GSCs are associated with classical AMAN and AMCBN but not demyelinating GBS. Future work incorporating standardized methodology, including reliable electrodiagnostic criteria for classifying GBS subtypes, is required to better clarify the true relationship between antibodies to glycolipids and GSCs and the clinical and electrophysiologic patterns.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Yuki Fukami for preparing the tables and figure e-1 and Madams Saiko Koike and Chiaki Yanaka for their help in the serologic analyses of the Japanese sera.
GLOSSARY
- AIDP
acute inflammatory demyelinating polyneuropathy
- AMAN
acute motor axonal neuropathy
- AMCBN
acute motor conduction block neuropathy
- GBS
Guillain-Barré syndrome
- GSC
ganglioside complex
- Ig
immunoglobulin
- NCS
nerve conduction study
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Design or conceptualization of the study (N.S., N.Y.), analysis or interpretation of the data (all authors), and drafting or revising the manuscript for intellectual content (N.S., N.Y.).
STUDY FUNDING
Dr. Shahrizaila receives research funding from University of Malaya (RG491/13HTM). Dr. Umapathi receives grant support from the Singapore National Medical Research Council (IRG 10nov086). Dr. Yuki receives grant support from the Singapore National Medical Research Council (IRG 10nov086 and CSA/047/2012) and a start-up grant (R-172-000-264-733) from Yong Loo Lin School of Medicine.
DISCLOSURE
N. Shahrizaila, N. Kokubun, S. Sawai, T. Umapathi, and Y. Chan report no disclosures relevant to the manuscript. S. Kuwabara receives research support from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and Grants-in-Aid from the Ministry of Health, Labour and Welfare of Japan, and serves as an associate editor of the Journal of Neurology, Neurosurgery & Psychiatry, and as an editorial board member of the Journal of the Neurological Sciences and Internal Medicine. K. Hirata reports no disclosures relevant to the manuscript. N. Yuki serves as an editorial board member of Expert Review of Neurotherapeutics, Journal of the Neurological Sciences, Journal of the Peripheral Nervous System, ISRN Neurology, and Journal of Neurology, Neurosurgery & Psychiatry. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Ho TW, Mishu B, Li CY, et al. Guillain-Barré syndrome in northern China: relationship to Campylobacter jejuni infection and anti-glycolipid antibodies. Brain 1995;118:597–605 [DOI] [PubMed] [Google Scholar]
- 2.Uncini A, Kuwabara S. Electrodiagnostic criteria for Guillain-Barré syndrome: a critical revision and the need for an update. Clin Neurophysiol 2012;123:1487–1495 [DOI] [PubMed] [Google Scholar]
- 3.Shahrizaila N, Yuki N. Antiganglioside antibodies in Guillain-Barré syndrome and its related conditions. Expert Rev Neurother 2011;11:1305–1313 [DOI] [PubMed] [Google Scholar]
- 4.Kaida K, Morita D, Kanzaki M, et al. Ganglioside complexes as new target antigens in Guillain-Barré syndrome. Ann Neurol 2004;56:567–571 [DOI] [PubMed] [Google Scholar]
- 5.Kuwahara M, Suzuki S, Takada K, Kusunoki S. Antibodies to LM1 and LM1-containing ganglioside complexes in Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy. J Neuroimmunol 2011;239:87–90 [DOI] [PubMed] [Google Scholar]
- 6.Kaida K, Sonoo M, Ogawa G, et al. GM1/GalNAc-GD1a complex: a target for pure motor Guillain-Barré syndrome. Neurology 2008;71:1683–1690 [DOI] [PubMed] [Google Scholar]
- 7.Kaida K, Morita D, Kanzaki M, et al. Anti-ganglioside complex antibodies associated with severe disability in GBS. J Neuroimmunol 2007;182:212–218 [DOI] [PubMed] [Google Scholar]
- 8.Yuki N, Shahrizaila N. Keeping it simple: is there a need for the various subtyping of axonal forms of Guillain-Barré syndrome? J Neurol Neurosurg Psychiatry 2011;82:592. [DOI] [PubMed] [Google Scholar]
- 9.Shahrizaila N, Goh KJ, Kokubun N, Abdullah S, Yuki N. Serial nerve conduction studies provide insight into the pathophysiology of Guillain-Barré and Fisher syndromes. J Neurol Sci 2011;309:26–30 [DOI] [PubMed] [Google Scholar]
- 10.Sekiguchi Y, Uncini A, Yuki N, et al. Antiganglioside antibodies are associated with axonal Guillain-Barré syndrome: a Japanese-Italian collaborative study. J Neurol Neurosurg Psychiatry 2012;83:23–28 [DOI] [PubMed] [Google Scholar]
- 11.Ogawa-Goto K, Funamoto N, Ohta Y, Abe T, Nagashima K. Myelin gangliosides of human peripheral nervous system: an enrichment of GM1 in the motor nerve myelin isolated from cauda equina. J Neurochem 1992;59:1844–1849 [DOI] [PubMed] [Google Scholar]
- 12.Svennerholm L, Bostrom K, Fredman P, et al. Gangliosides and allied glycosphingolipids in human peripheral nerve and spinal cord. Biochim Biophys Acta 1994;1214:115–123 [DOI] [PubMed] [Google Scholar]
- 13.Fredman P, Vedeler CA, Nyland H, Aarli JA, Svennerholm L. Antibodies in sera from patients with inflammatory demyelinating polyradiculoneuropathy react with ganglioside LM1 and sulfatide of peripheral-nerve myelin. J Neurol 1991;238:75–79 [DOI] [PubMed] [Google Scholar]
- 14.Ilyas AA, Mithen FA, Dalakas MC, Chen ZW, Cook SD. Antibodies to acidic glycolipids in Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy. J Neurol Sci 1992;107:111–121 [DOI] [PubMed] [Google Scholar]
- 15.Susuki K, Yuki N, Hirata K, Kuwabara S. Fine specificities of anti-LM1 IgG antibodies in Guillain-Barré syndrome. J Neurol Sci 2002;195:145–148 [DOI] [PubMed] [Google Scholar]
- 16.Yuki N, Tagawa Y, Handa S. Autoantibodies to peripheral nerve glycosphingolipids SPG, SLPG, and SGPG in Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy. J Neuroimmunol 1996;70:1–6 [DOI] [PubMed] [Google Scholar]
- 17.Yako K, Kusunoki S, Kanazawa I. Serum antibody against a peripheral nerve myelin ganglioside, LM1, in Guillain-Barré syndrome. J Neurol Sci 1999;168:85–89 [DOI] [PubMed] [Google Scholar]
- 18.Kuwabara S, Yuki N, Koga M, et al. IgG anti-GM1 antibody is associated with reversible conduction failure and axonal degeneration in Guillain-Barré syndrome. Ann Neurol 1998;44:202–208 [DOI] [PubMed] [Google Scholar]
- 19.Kokubun N, Nishibayashi M, Uncini A, Odaka M, Hirata K, Yuki N. Conduction block in acute motor axonal neuropathy. Brain 2010;133:2897–2908 [DOI] [PubMed] [Google Scholar]
- 20.Susuki K, Johkura K, Yuki N, Hasegawa O, Kuroiwa Y. Rapid resolution of nerve conduction blocks after plasmapheresis in Guillain-Barré syndrome associated with anti-GM1b IgG antibody. J Neurol 2001;248:148–150 [DOI] [PubMed] [Google Scholar]
- 21.Capasso M, Caporale CM, Pomilio F, Gandolfi P, Lugaresi A, Uncini A. Acute motor conduction block neuropathy: another Guillain-Barré syndrome variant. Neurology 2003;61:617–622 [DOI] [PubMed] [Google Scholar]
- 22.Shahrizaila N, Yuki N. Guillain-Barré syndrome animal model: the first proof of molecular mimicry in human autoimmune disorder. J Biomed Biotechnol 2011;2011:829129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaida K, Kusunoki S, Kanzaki M, Kamakura K, Motoyoshi K, Kanazawa I. Anti-GQ1b antibody as a factor predictive of mechanical ventilation in Guillain-Barré syndrome. Neurology 2004;62:821–824 [DOI] [PubMed] [Google Scholar]
- 24.Nagashima T, Koga M, Odaka M, Hirata K, Yuki N. Continuous spectrum of pharyngeal-cervical-brachial variant of Guillain-Barré syndrome. Arch Neurol 2007;64:1519–1523 [DOI] [PubMed] [Google Scholar]
- 25.Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barré syndrome: clinical and immunohistochemical studies. Neurology 1993;43:1911–1917 [DOI] [PubMed] [Google Scholar]
- 26.Mizoguchi K, Hase A, Obi T, et al. Two species of antiganglioside antibodies in a patient with a pharyngeal-cervical-brachial variant of Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry 1994;57:1121–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koga M, Yoshino H, Morimatsu M, Yuki N. Anti-GT1a IgG in Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry 2002;72:767–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshino H, Harukawa H, Asano A. IgG antiganglioside antibodies in Guillain-Barré syndrome with bulbar palsy. J Neuroimmunol 2000;105:195–201 [DOI] [PubMed] [Google Scholar]
- 29.Ropper AH, Wijdicks EFM, Truax BT. Guillain-Barré Syndrome. Philadelphia: F.A. Davis; 1991 [Google Scholar]
- 30.Ropper AH. Further regional variants of acute immune polyneuropathy: bifacial weakness or sixth nerve paresis with paresthesias, lumbar polyradiculopathy, and ataxia with pharyngeal-cervical-brachial weakness. Arch Neurol 1994;51:671–675 [DOI] [PubMed] [Google Scholar]
- 31.Susuki K, Atsumi M, Koga M, Hirata K, Yuki N. Acute facial diplegia and hyperreflexia: a Guillain-Barré syndrome variant. Neurology 2004;62:825–827 [DOI] [PubMed] [Google Scholar]
- 32.Hadden RDM, Cornblath DR, Hughes RAC, et al. Electrophysiological classification of Guillain-Barré syndrome: clinical associations and outcome. Ann Neurol 1998;44:780–788 [DOI] [PubMed] [Google Scholar]
- 33.Rinaldi S, Brennan KM, Kalna G, et al. Antibodies to heteromeric glycolipid complexes in Guillain-Barré syndrome. PloS One 2013;8:e82337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.