

In order to comprehensively define the effects of sequence variation on tRNA function, Guy et al. developed a high-throughput in vivo screen to quantify the activity of the nonsense suppressor SUP4oc of Saccharomyces cerevisiae. SUP4oc tolerated numerous sequence variations, accommodated slippage in tertiary and secondary interactions, and exhibited genetic interactions that suggest an alternative functional tRNA conformation. Mutations that sensitized SUP4oc to rapid tRNA decay were found to be located throughout the sequence. This shows that the integrity of the entire tRNA molecule is under surveillance by cellular quality control machinery.
Keywords: tRNA function, tRNA decay, high-throughput analysis, RTD, SUP4oc
Abstract
Sequence variation in tRNA genes influences the structure, modification, and stability of tRNA; affects translation fidelity; impacts the activity of numerous isodecoders in metazoans; and leads to human diseases. To comprehensively define the effects of sequence variation on tRNA function, we developed a high-throughput in vivo screen to quantify the activity of a model tRNA, the nonsense suppressor SUP4oc of Saccharomyces cerevisiae. Using a highly sensitive fluorescent reporter gene with an ochre mutation, fluorescence-activated cell sorting of a library of SUP4oc mutant yeast strains, and deep sequencing, we scored 25,491 variants. Unexpectedly, SUP4oc tolerates numerous sequence variations, accommodates slippage in tertiary and secondary interactions, and exhibits genetic interactions that suggest an alternative functional tRNA conformation. Furthermore, we used this methodology to define tRNA variants subject to rapid tRNA decay (RTD). Even though RTD normally degrades tRNAs with exposed 5′ ends, mutations that sensitize SUP4oc to RTD were found to be located throughout the sequence, including the anti-codon stem. Thus, the integrity of the entire tRNA molecule is under surveillance by cellular quality control machinery. This approach to assess activity at high throughput is widely applicable to many problems in tRNA biology.
tRNAs allow the genetic code to be correctly interpreted for protein synthesis. In consequence, their sequence is under three strong functional constraints. First, tRNAs require similar overall structures to participate equivalently in translation (Kim et al. 1974; Westhof et al. 1985; Basavappa and Sigler 1991; Giege et al. 2012). Second, tRNAs require unique features to ensure specific charging by cognate synthetases and accurately decode mRNA, both of which often require specific modifications (Putz et al. 1994; Giege et al. 1998; Murphy et al. 2004; Agris et al. 2007) and elements outside the anti-codon (Musier-Forsyth et al. 1991; Cochella and Green 2005; Ledoux et al. 2009; Ling et al. 2009; Shepotinovskaya and Uhlenbeck 2013). Third, tRNAs must be stable enough to survive for multiple generations (Gudipati et al. 2012) and avoid turnover (Whipple et al. 2011) yet flexible enough to accommodate conformational changes during ribosome passage (Valle et al. 2003; Schmeing et al. 2009; Zhou et al. 2013).
These sequence constraints on tRNA function suggest that tRNAs would be largely intolerant to mutation. Indeed, numerous tRNA mutations in yeast adversely affect function (Kurjan et al. 1980). Over 230 mitochondrial tRNA mutations have been associated with human diseases (Ruiz-Pesini et al. 2007), including encephalopathy, hearing loss, ataxia, myopathy, diabetes, epilepsy, neuropathy, and gastrointestinal dysfunction (Yarham et al. 2010); these mutations occur in all stems and loops.
Nonetheless, there are also data demonstrating that tRNAs can tolerate variant sequences in the stems and loops. For example, the yeast Saccharomyces cerevisiae tRNAArg(CCG) gene retains function with any of several D-loop or anti-codon loop mutations (Geslain et al. 2003), and the yeast tRNASer(CGA) gene is fully functional with any of seven different base-pair swaps in the acceptor or T stems that retain secondary structure (Whipple et al. 2011). Similarly, several Escherichia coli variants of a partially impaired tRNAAla(CUA) amber suppressor tRNA retain activity with individual mutations in the acceptor stem, the anti-codon stem, or the T stem (Hou and Schimmel 1992).
These seemingly conflicting data make it difficult to predict the effects on tRNA function of the numerous naturally occurring sequence variations in the metazoan tRNA isodecoders, which have the same anti-codon but altered tRNA bodies (Goodenbour and Pan 2006). In addition, most of the numerous disease-associated mitochondrial tRNA variants are poorly understood (Suzuki et al. 2011). Prediction of the function of variants is further complicated by the multiple modifications and quality control pathways that influence tRNA activity (Kadaba et al. 2004; Chernyakov et al. 2008; Hopper 2013; Kramer and Hopper 2013) and by the tRNA internal promoter, which is not quantitatively understood (Koski et al. 1980; Pearson et al. 1985; Kaiser and Brow 1995; Marck et al. 2006; Orioli et al. 2012).
Although there is a wealth of information on the effects of mutating individual tRNA residues on specific steps of tRNA processing and function (Normanly et al. 1986; Schultz and Yarus 1994; Yan and Francklyn 1994; Fechter et al. 2000; Schrader et al. 2009), there has been no quantitative analysis at a large scale of the effects of mutations on tRNA biology. Here we describe the use of a sensitive fluorescent reporter and deep sequencing to quantify the in vivo function of thousands of variants of a tRNA suppressor in the yeast S. cerevisiae and the use of this system to comprehensively define the biological substrates of a prominent tRNA decay pathway (Alexandrov et al. 2006). We identified a large number of mutated tRNAs that are functional, suggesting that tRNA structure is much more flexible than anticipated, and found that the tRNA decay pathway unexpectedly acts on many more classes of variants than previously known or predicted.
Results
Quantification of tRNA function by cell sorting of yeast carrying a library of tRNA variants
To analyze the effect of mutations on tRNA function in vivo, we sought a model system in which we could assay tRNA activity quantitatively, with high sensitivity and on a large scale. In yeast, suppression of a stop codon in the green fluorescent protein gene (GFP) allows fluorescence-activated cell sorting (FACS) of millions of cells based on their level of suppression by a nonsense suppressor tRNA. To test the feasibility of this approach, we integrated the nonsense suppressor SUP4oc (tRNATyr-G34U) into a yeast strain bearing GFPoc in the RNA-ID reporter (Dean and Grayhack 2012); this strain allows a comparison of the expression of GFPoc to the control red fluorescent protein gene (RFP) (Fig. 1A). GFPoc/RFP was minimal without suppression (0.004 of GFP/RFP) but was nearly normal (0.94 of GFP/RFP) with SUP4oc (Fig. 1B), as anticipated for this stop codon because of its poor termination context (Bonetti et al. 1995; Dean and Grayhack 2012). Based on these data, this GFPoc expression assay discriminates with high resolution among tRNA variants, with a 235-fold dynamic range of expression and limited variation in GFP/RFP values for individual cells of a variant (Dean and Grayhack 2012). Moreover, the assay measures the net contribution of all steps of tRNA biogenesis and translation except fidelity.
Figure 1.

High-throughput quantification of tRNA function of SUP4oc variants. (A) Schematic of the RNA-ID reporter used to quantify tRNA function. (B) SUP4oc efficiently suppresses GFPoc. Scatter plot of flow cytometry of cells with integrated RNA-ID reporter expressing GFP (green), GFPoc (red), and GFPoc and SUP4oc (blue). (C) FACS of SUP4oc variant library. Cells were grown in YP galactose medium and sorted. (D) SUP4oc tolerates numerous mutations. Cloverleaf heat map showing GFPSEQ of single-mutant variants. Quadrant color around residues indicates variant activity. Active variants are white (GFPSEQ of 0.026) to blue (GFPSEQ of 1) gradient, and inactive variants are red. Modified bases are indicated in the figure.
We constructed a library of ∼220,000 SUP4oc variants, each integrated into the yeast RNA-ID strain and bearing ∼3% random mutations in nucleotides 1–33 and 38–73 (conventional numbering) (Supplemental Fig. S1A). We grew this library at 28°C, sorted cells into four bins by FACS (Fig. 1C), PCR-amplified the SUP4oc allele from the pooled genomic DNA from each bin, and evaluated the bin distribution of individual variants by sequencing (Supplemental Table S1), similar to an approach used to measure gene expression from thousands of designed promoters (Sharon et al. 2012). The fractional representation of reads for each variant in each bin was converted to a GFP/RFP ratio, which was normalized to the SUP4oc ratio to define relative function (termed GFPSEQ). Filters were then applied to score only those variants with ≥100 reads and enough reads to measure the distribution of ≥30 cells (Supplemental Fig. S1B).
Overall, we scored 25,491 variants (Supplemental Table S2), including all 213 single variants. GFPSEQ was highly reproducible for single mutants of a biological replicate (Supplemental Fig. S1C), with R2 = 0.99. We also confirmed tRNA activity of 60 variants by reconstruction and flow cytometry analysis of the variants; each activity, normalized to the SUP4oc ratio, yielded a GFP/RFP ratio termed GFPFLOW, which correlated highly with the corresponding GFPSEQ up to GFPFLOW of 0.4 (R2 = 0.90) (Supplemental Fig. S1D). To further enhance resolution of highly active variants, we used FACS to subdivide bin 1 into three fractions, extending the linear range of GFPSEQ values to GFPFLOW of 0.55. It is not clear why GFPSEQ is systematically approximately twofold higher than GFPFLOW, resulting in a correlation between GFPSEQ and GFPFLOW that only extends up to 0.55 (Supplemental Fig. S1D). Some of the discrepancy is likely due to the limited resolution of high-fluorescence variants, even in the bin 1 subdivision data set. In addition, PCR chimerism in low-fluorescence bins can lead to spurious wild-type reads, thereby underestimating the function of the wild-type tRNA by the sequencing approach, which in turn leads to overestimation of variant function by GFPSEQ. Finally, minor systematic errors may be introduced by the use of different instruments for GFPSEQ and GFPFLOW measurements and the steps of bin collection and plating, PCR amplification, and sequencing.
SUP4oc is highly tolerant of mutations
To characterize the mutational consequences in SUP4oc, we initially analyzed the 213 single variants, given both their relative simplicity and the previous studies that examined single mutations in this tRNA. SUP4oc is remarkably tolerant of single mutations, with 44 highly functional variants (GFPSEQ ≥ 0.9) (Fig. 1D, dark blue) and 27 substantially functional variants (0.18–0.9) (Fig. 1D, blue), along with nine marginally functional variants (0.026–0.18) (Fig. 1D, light blue). We note that there are minimal consequences due to the higher values of GFPSEQ relative to GFPFLOW. Thus, 26 of 32 nonfunctional or marginally active variants by GFPSEQ were correctly annotated based on reconstruction and GFPFLOW analysis, and six nonfunctional variants had trace amounts of GFPFLOW activity (Supplemental Fig. S1D). Similarly, 13 of 16 of highly functional variants that were tested by reconstruction and flow cytometry had GFPFLOW values >0.7.
The highly or substantially active variants were heavily clustered in specific residues. These included each of the three possible mutations of all five D-loop uridine residues; U4 of the acceptor stem; A9, A13, and A22 of the D loop; residues 44, 45, and 47 of the variable loop; C59 of the T loop; and G62 of the T stem. These results are consistent with, and substantially extend, previous analyses of functional variants of SUP4oc (Kurjan and Hall 1982; Kohalmi and Kunz 1992), yeast tRNAArg(CCG) (Geslain et al. 2003), and an E. coli alanine amber suppressor tRNA (Hou and Schimmel 1992). In contrast, residues that did not tolerate any single mutations included those in conserved tertiary pairs (U8–A14, R15–Y48, G18–U55, G19–C56, and U54–A58), emphasizing the requirement of the L-shaped tertiary fold of the tRNA for activity.
Although our data emphasize that the integrity of the four stems must be intact for tRNA to have full function, flexibility is observed at two locations. Single- and double-mutant variants that preserve canonical pairing were often functional (Fig. 2A), with the notable exceptions of the G53–C61 pair, which is highly conserved as part of the B-box of the internal promoter (Marck et al. 2006); C1–G72, which is a determinant for tyrosine charging (Fechter et al. 2000); and the G10–C25 and C11–G24 pairs of the D stem, which is comprised of only 3 base pairs (bp). In contrast, only eight of 140 stem variants with noncanonical pairing had a GFPSEQ > 0.5, and these eight included four variants of U4–G69 and two variants of C52–G62. Although functional variants with mismatches at U4–G69 might be anticipated because of the weak U–G pair and the known mismatches that occasionally occur among stem base pairs in tRNAs, it is unclear why SUP4oc tolerated mismatches at C52–G62, since this position is rarely occupied by a mismatched pair, G–U, or U–G (Marck and Grosjean 2002).
Figure 2.
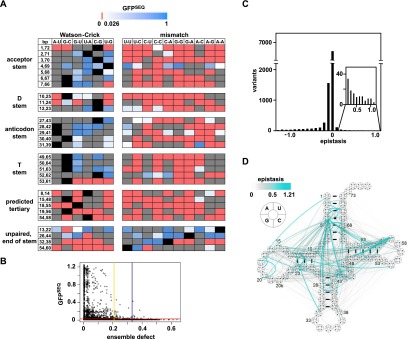
Analysis of single- and double-mutant SUP4oc variants. (A) GFPSEQ of stem base pair and tertiary pair variants. Color-coding as in Figure 1D. (Gray boxes) Variant not scored. (B) Plot of GFPSEQ versus ensemble defect (ED) for all single and double variants. (Red) Undetectable GFPSEQ activity cutoff; (yellow) 95% ED cutoff; (blue) 99% ED cutoff. (C) Epistasis of double-mutant variants. (D) Cloverleaf schematic map of positive epistatic interactions between residues in SUP4oc. Color and width of lines correspond to the strength of the interactions.
Our data also indicate that the tertiary fold must be intact, since little sequence variation is observed in the conserved tertiary pairs. Indeed, 45 of 47 variants with mutations in these pairs resulted in a completely nonfunctional tRNA, and the remaining two had only marginal activity (Fig. 2A).
Among the 9349 double-mutant variants, 1499 were active, including 685 substantially or highly functional variants. One important requirement for activity is a low ensemble defect (ED), which is a parameter that estimates the propensity of a tRNA to misfold (Zadeh et al. 2011a). According to our data, almost all functional variants had an estimated per nucleotide ED < 0.21 (Fig. 2B, 95% cutoff, yellow), which is well within the range of native eukaryotic tRNAs (Supplemental Fig. S2A).
Unexpected positive interactions between residues
To identify previously unappreciated parameters important for tRNA function, we examined double-mutant variants that displayed positive (or negative) epistasis, indicating that they functioned substantially better (or worse) than anticipated from the scores of the corresponding single-mutant variants. Epistasis within a protein or RNA can reveal interactions between residues when the phenotype caused by one mutation is dependent on mutation at another residue. Based on a multiplicative model, we calculated an epistasis score by subtracting the product of the GFPSEQ scores of two single variants from that of the corresponding double variant (Supplemental Tables S3, S4). Most double variants scored close to their predicted values (Fig. 2C), but 6.9% had substantial negative epistasis (defined as a score ≤0.18) (Supplemental Fig. S2B), and 1.5% had positive epistasis (>0.18) (Fig. 2D). As might be expected for a molecule with severe sequence constraints, there was a large excess of negative epistasis over positive epistasis. Indeed, of the double variants that had GFPSEQ scores that allowed the possibility of negative or positive epistasis, 62% were negatively epistatic, whereas only 1.4% were positively epistatic, and this excess was not dependent on the epistasis cutoff score used (Supplemental Fig. S2C). However, the 6.9% of total double variants with negative epistasis includes a remarkably large number (202 of 648, 31%) of completely nonfunctional doubles in which both singles were highly functional, suggesting that while the tRNA tolerates single mutations at multiple locations with little loss of function, it is extremely sensitive to a second mutation.
Many of the 1.5% of double variants displaying positive epistasis can be explained simply, such as by restoration of a base pair that was lost in both of the corresponding single variants. However, there were several striking examples of unexpected positive epistasis, four classes of which are highlighted below because they suggest structural rearrangements.
First, an alternative tRNA conformation appears to form in variants with mutations in the 26- to 44-nucleotide (nt) pair. The nucleotides at residues 26 and 44 are mismatched ∼65% of the time in eukaryotes and, in known structures, often form a propeller-twisted noncanonical base pair in a Watson-Crick-like orientation (Fig. 3A; Kim et al. 1974); however, these nucleotides are also frequently canonically paired, with Watson-Crick (17%) or G–U (18%) pairings (Marck and Grosjean 2002). We found that the A44U mutation (opposite G26) had nearly opposite effects on the function of double variants, dependent on the identity of the other mutation. Thus, the A44U mutation substantially rescued the function of variants with the destabilizing anti-codon stem mutations A29C, A29U, and A28U; in contrast, the A44U mutation had large negative epistatic effects with A9U, A9C, A22U, and G57A (Fig. 3B,C; Supplemental Table S4; Supplemental Fig. S3A), all of which often participate in the tertiary fold (Giege et al. 2012). One likely interpretation of these results is that A44U alters tRNA conformation by pairing with G26, strengthening the anti-codon stem and thereby countering other destabilizing anti-codon stem mutations while simultaneously causing structural shifts that impair the function of variants with otherwise benign mutations affecting the tRNA fold. It is notable that the 26–44 pair is in the “hinge” region of tRNA, which undergoes substantial conformational changes during ribosome passage in the A/T state with EF-Tu (Valle et al. 2003; Schmeing et al. 2009) and in the pe*/E state during translocation (Zhou et al. 2013). Flexibility in this region of the tRNA may also explain why the inactive G26U variant (opposite A44) was substantially rescued by mutation of G45 (Supplemental Table S4; Supplemental Fig. S3B–D). G45 sometimes interacts with the 10–25 pair (Westhof et al. 1985; Gautheret et al. 1995; Giege et al. 2012), suggesting that mutating G45 could alter or break this tertiary interaction, thus adding more flexibility to the hinge region and allowing for a Watson-Crick pair at 26–44.
Figure 3.

Evidence for an alternative conformation in SUP4oc 26–44 variants. (A) G26–A44 structure in tRNAPhe. Data from Protein Data Bank (PDB) ID 1EHZ. (B) An A44U mutation confers both positive and negative epistasis on variants. Flow cytometry of cells expressing A44U and/or A29C (top) and/or A9U (bottom). (C) Cloverleaf map of epistatic interactions involving A44U. (Cyan) Positive epistasis; (amber) negative epistasis.
Second, the virtually universally conserved U8–A14 pair (Randau et al. 2009) could be replaced by A8–G14, resulting in substantial function (Supplemental Table S4; Supplemental Fig. S4A–C), whereas none of eight other substitutions of this pair resulted in a tRNA that was functional (Supplemental Table S3). Since U8–A14 forms a critical reverse Hoogsteen pair to help position the D stem, it seems plausible that A8–G14 is functional in part because it maintains this geometry (Sterner et al. 1995), perhaps with N1 of A8 protonated (Supplemental Fig. S4D; Leontis et al. 2002). However, it is not clear why only the A8–G14 variant had function, since five of the other eight 8–14 pairs that we scored are predicted to accommodate this geometry, albeit with slightly differing spacing (Leontis et al. 2002).
Third, tertiary interactions involving the D loop appear to shift to adjacent residues. For example, although G18 and G19 in the D loop are virtually universally conserved and interact with U55 and C56, respectively, the inactive G19U variant was completely rescued by U17G but not by U16G, U20aG, or other mutations (Fig. 4A; Supplemental Table S4; Supplemental Fig. S5A–C), and the G18A variant was rescued only by U20G (Supplemental Table S4). A plausible explanation for these data is that the positions of the guanosines can be altered while retaining critical tertiary interactions with Ψ55 and C56, presumably by physical displacement of D-loop residues. This mechanism is consistent with the known variability of D-loop size (Giege et al. 2012), but since the crystal structure of tRNATyr is not known, epistasis at these residues may be due to another mechanism. We note that tRNAAsp lacks the G19–C56 interaction (Westhof et al. 1985).
Figure 4.

Positive epistasis due to shift of interactions to neighboring residues in SUP4oc variants. (A) Cloverleaf map of epistatic interactions involving U17G. (Cyan) Positive epistasis; (amber) negative epistasis. (B) A C27U mutation restores activity to an A28C variant. Flow cytometry of cells expressing C27U and/or A28C (top) and Δ42 derivatives of C27U A28C and SUP4oc (bottom). (C) Predicted base pair rearrangement of the C27A U28A variant. (Red) Mutations; (underlined) proposed rearranged bases.
Fourth, flexibility in the anti-codon stem and V loop may accommodate a bulged base. Although it was puzzling that a destabilizing A28C variant (opposite U42) was substantially rescued by a destabilizing C27U mutation (opposite G43) (Fig. 4B; Supplemental Table S4; Supplemental Fig. S5D,E), a plausible explanation is that U42 bulges out of the anti-codon stem helix, allowing U27 to form a Watson-Crick pair with A44 of the V loop and allowing C28 to pair with G43 while reducing the V-loop size by 1 nt (Fig. 4C). Consistent with this interpretation, activity was retained if the putative bulged U42 was deleted from the C27U A28C variant (Fig. 4B; Supplemental Table S4).
The rapid tRNA decay (RTD) pathway monitors the integrity of the entire tRNA molecule
Our high-throughput screening approach to quantify tRNA function allows us to vary the parameters of the assay to define how mutations affect many distinct aspects of tRNA biology. One critical process modulating tRNA turnover is the RTD pathway, which targets specific mature tRNAs for degradation due to lack of one or more body modifications or to a destabilized acceptor or T stem, resulting in attack by the 5′–3′ exonucleases Rat1 and Xrn1 (Fig. 5A; Alexandrov et al. 2006; Chernyakov et al. 2008; Whipple et al. 2011; Dewe et al. 2012). However, the full scope of sequence variants subject to RTD is not clear, as only the tRNASer family has been examined in any detail; the roles of the anti-codon stem–loop, the D stem–loop, and the T loop have been only minimally examined; tRNASer family members are in the minority class II of tRNAs that have a long variable stem; and acceptor stem/T-stem stability estimates do not always accurately predict RTD susceptibility for other tRNA species (Whipple et al. 2011).
Figure 5.

Analysis of SUP4oc RTD substrates. (A) Schematic of RTD. (B) The RTD substrate SUP4-3oc (SUP4-G62C) has increased GFPFLOW in the met22∆ strain. (C) Mutations throughout the tRNA appear to trigger RTD. Cloverleaf heat map of SUP4oc single variants analyzed for RTD based on GFPSEQ RTD ratios [GFPSEQ(met22∆)/GFPSEQ(wild type)]. (Green shades) RTD substrate; (purple) nonsubstrate; (wedge border thickness) GFPSEQ(met22∆) − GFPSEQ(wild type). Gray hatches indicate variants not scored by sequencing or those with a GFPSEQ < 0.052 in met22Δ cells (the minimum score in met22Δ cells to observe an RTD ratio >2.0). (D) Analysis of SUP4oc levels of putative RTD variants in met22∆ and wild-type strains. Bulk RNA from the indicated strains was analyzed using poison primer extension with ddCTP. (E) ROC (receiver operating characteristic) curves for RTD prediction based on estimated ΔΔG°28 for single variants for which the RTD ratio could be scored (see the Materials and Methods).
We applied this library-based approach to comprehensively define SUP4oc variants that are substrates for RTD. RTD is readily detected with the RNA-ID reporter, since the known substrate SUP4oc-G62C (Whipple et al. 2011) had reduced GFPFLOW in MET22+ (wild-type) cells compared with that in met22∆ cells (Fig. 5B, Supplemental Table S5), in which RTD is inactivated (Chernyakov et al. 2008). We made a SUP4oc library in the met22Δ strain, analyzed variants by FACS and sequencing (Supplemental Fig. S6A), and compared GFPSEQ of variants with that from wild-type cells. GFPSEQ from the met22Δ strain was highly reproducible and correlated with GFPFLOW (Supplemental Fig. S6B,C).
This analysis revealed many single variants that were putative RTD substrates, with mutations surprisingly located throughout the tRNA body. In the met22Δ strain, 70 single variants were highly functional, including all 44 that we identified in the wild type (Supplemental Fig. S6D). Overall, 38 single variants were more than twofold more active in met22Δ cells than in wild-type cells (GFPSEQ RTD ratio >2), suggesting that they are RTD substrates (Fig. 5C, green; Supplemental Table S6), and for 16 of these, the increase in activity was >0.3 (Fig. 5C, dark wedge outlines). Eleven of these 38 RTD candidates have mutations in the acceptor or T stem, as expected for RTD substrates (Whipple et al. 2011). Remarkably, the other 27 RTD candidates have mutations in regions not previously associated with RTD, including 17 in the anti-codon stem and loop, six in the D stem, and one each in the D loop, V loop, and T loop and at N8 (Fig. 5C).
We determined that a number of these variants are RTD substrates by two approaches. First, we reconstructed individual RTD candidate variants with mutations in different regions of the tRNA and tested them by flow cytometry when integrated into met22Δ and wild-type reporter strains. Nineteen of 21 putative RTD substrates with GFPSEQ RTD ratios ranging from 24.4 to 2.3 had GFPFLOW RTD ratios >2.0 (Supplemental Fig. S7A; Supplemental Table S5), whereas 26 of 30 putative non-RTD substrates with GFPSEQ RTD ratios ranging from 1.4 to 0.9 had GFPFLOW RTD ratios <2.0. We therefore conclude that RTD ratios determined by GFPSEQ scores have high predictive value for potential RTD substrates as measured by GFPFLOW. Second, a primer extension assay with ddCTP instead of dCTP (which results in a G34 stop for tRNATyr and a G30 stop for SUP4oc) showed that tRNA levels of RTD candidates were increased in the met22Δ strain relative to the wild-type strain (Fig. 5D). As expected, SUP4oc levels did not change in met22Δ compared with wild-type cells; however, tRNA levels were substantially increased in the met22Δ mutant for the acceptor stem U2C variant, the D-stem C25U variant, the T-stem G62C variant, and the anti-codon stem C27A, A29U, and A31U variants, providing strong evidence that these are all RTD substrates. Based on these data, we estimate that the vast majority of the 38 single-mutant and 605 double-mutant variants that are candidate RTD substrates are authentic (Supplemental Table S6), suggesting that RTD places a significant constraint on tRNA sequences.
Previous analysis of determinants for RTD in the tRNASer family demonstrated that the predicted folding stability of the combined acceptor and T stem correlated inversely with susceptibility to RTD (Whipple et al. 2011). However, since mutations throughout the tRNA elicited RTD, we examined the relationship of RTD to the predicted stability of the entire molecule, as quantified by ΔΔG°28 (Reuter and Mathews 2010). Consistent with the importance of stability in RTD, a threshold of ΔΔG°28 of 2.65 kcal/mol has good predictive value, since 28 of 38 qualified single-mutant variants with ΔΔG°28 >2.65 kcal/mol are RTD substrates (Supplemental Table S6). Since these 28 variants occur in all of the stems of SUP4oc, we conclude that the influence of stability on RTD extends to the entire molecule. In contrast, only 10 of 65 variants with ΔΔG°28 <2.65 kcal/mol were RTD substrates. Since a number of these 10 variants have mutations in loop residues that participate in tertiary interactions, we presume that stability is affected here, too, but is not captured by calculated ΔΔG°28, which only measures secondary structure contributions. Overall, a ΔΔG°28 cutoff of 2.65 kcal/mol results in a true positive rate of 0.74 and a false positive rate of 0.15 (Fig. 5E). We also found that ΔΔG°28 is predictive of RTD for double-mutant variants (Supplemental Fig. S7B).
The numerous examples of positive epistasis involving the stabilizing U4C acceptor stem mutation (opposite G69) may be due to protection from RTD. A large number of variants that were rescued by the U4C mutation are themselves RTD substrates, including several variants with mutations in the anti-codon stem (Fig. 6A,B; Supplemental Fig. S8A,B). Moreover, for each of three variants examined, the U4C double variants had similar tRNA levels in wild-type and met22∆ cells (Fig. 6C; Supplemental Fig. S8C). This result suggests that U4C protects the 5′ end of variants subject to RTD from exonucleolytic attack, presumably by stabilizing the 4–69 base pair.
Figure 6.

A U4C stabilizing mutation rescues variants that are RTD substrates. (A) Cloverleaf map of epistatic interactions involving U4C. (B, top) Flow cytometry of cells expressing the indicated SUP4oc variants. (Bottom) Flow cytometry of cells expressing the indicated SUP4oc variant in wild-type and met22∆ cells. (C) A U4C mutation restores SUP4oc levels of RTD substrate variants in wild-type cells to those in the met22∆ strain. Levels of SUP4oc were determined as described in Figure 5D.
Discussion
Although tRNAs have evolved for their efficient transcription, processing, and modification, high structural stability, and accurate and efficient usage in the translation cycle, the analysis of >25,000 variants of the model yeast tRNA SUP4oc demonstrates that it is highly robust to mutation. This robustness was unexpected based on the numerous constraints on tRNA sequences but was consistent with models of RNA evolution in which sequences converge to those that are robust to mutation (van Nimwegen et al. 1999). Nonetheless, although many single-base changes are tolerated in SUP4oc, a second mutation is much more likely to abolish function than to rescue it (Supplemental Fig. S2C). Since this excess of negative epistasis was present also in met22∆ cells (Supplemental Table S3), RTD is not the primary cause of negative epistasis, although it is likely one contributing factor. Part of the reason that SUP4oc has a preponderance of negative epistasis may be that the multiple constraints on structure and function are too great to accommodate most double mutations.
The analysis of positive epistasis suggests a remarkable amount of flexibility allowed in the sequence of SUP4oc. The suggestion that an alternative tRNA conformation is provoked by mutation of the 26–44 pair in the hinge region (Fig. 3; Supplemental Fig. S3A) may be compatible with conformational changes during translation (Valle et al. 2003; Schmeing et al. 2009; Zhou et al. 2013), as previously proposed for a D-stem variant (Cochella and Green 2005). In the met22Δ mutant, there is even more pronounced evidence for this alternative conformation based on additional examples of negative and positive epistasis for mutations affecting the 26–44 pair (Supplemental Table S3) and more extreme epistasis values. These data suggest that the large fraction (35%) of 1984 surveyed eukaryotic tRNAs with canonical 26–44 pairing (Marck and Grosjean 2002) may have common compensatory features that distinguish them from tRNAs with unmatched residues at this position. The epistasis data derived from the met22Δ mutant underscore the flexibility of tRNA, since increased positive epistasis was observed for the C27U A28C variant and the U8A A14G variant. In addition, there are several examples of epistasis to preserve adjacent guanosine residues in the D loop (normally located at positions 18 and 19) (see Fig. 4A), including one variant that was not scored in wild-type cells (Supplemental Table S3).
Several other positive epistatic interactions cannot be explained easily by structural alterations (Supplemental Table S3). Since our approach measures the overall function of the tRNA, which includes all steps from its transcription by RNA polymerase III through its role in translation, such positive epistasis could arise because of altered function in the double mutant due to any combination of steps during the biogenesis of tRNA or its deployment in translation.
Our results suggest that RTD monitors the integrity of the entire tRNA molecule, greatly expanding the scope of variants subject to this pathway. Indeed, since 446 of 838 substantially functional double variants are likely subject to RTD, RTD is a major factor in determining the sequence limits to tRNA function (Supplemental Table S6). Since our analysis suggests that increased ΔΔG°28 correlates with susceptibility to RTD regardless of the stem that is affected (Fig. 5C; Supplemental Table S5, S6; Supplemental Fig. S7), this brings up the question of how overall tRNA stability influences RTD. Our suggestion that degradation occurs through the 5′ end is supported by widespread U4C suppression of RTD (Fig. 6; Supplemental Fig. S8). Mutations to SUP4oc in the D stem, D loop, and T loop may trigger RTD by altering the tertiary fold, allowing increased 5′ end attack. However, it is difficult to rationalize how mutations in the anti-codon stem expose the 5′ end, since residues in this stem do not interact with other regions. These anti-codon stem mutations might trigger RTD due to cooperative unfolding of the acceptor stem, altered stacking with the D stem and consequent destabilization, or another sensing mechanism, perhaps an element of the translation machinery.
Our results provide a framework for understanding how sequence variation influences many aspects of tRNA biology, including the role and function of tRNA isodecoders in metazoans and the molecular basis of diseases caused by mitochondrial tRNA mutations (Yarham et al. 2010). An analysis of ED shows that tRNA structure prediction software may be useful for giving an upper bound on tolerated defects, but other parameters need to be incorporated for these programs to predict function successfully. Application of the high-throughput approach described here to define functional determinants of other tRNA species should lead to large improvements in our ability to predict function of variants.
The approach described here is generally applicable to many problems in tRNA biology. FACS followed by deep sequencing of tRNA genes can be used to score the effect of mutations that affect tRNA processing, modification, or translation by comparing the scores with those in a wild-type background. A prerequisite for this approach is that the tRNA can be made into a suppressor or that cells carrying the tRNA can be scored for growth or another activity. By the use of appropriate screens, this approach can also measure tRNA charging fidelity (Kramer et al. 2010). Furthermore, this overall approach can be adapted to study many problems in the biology of noncoding RNAs.
Materials and methods
Yeast strains
The BY4741 can1∷PGAL1-GFPoc-PGAL10-RFP strain (YK380-1) was constructed by PCR amplification of the PGAL1-GFPoc-PGAL10-RFP reporter and its adjacent MET15 marker from plasmid EKD1302 (Dean and Grayhack 2012), using primers with sequence complementary to the 5′ and 3′ ends of CAN1, followed by linear transformation of the DNA into BY4741. A met22∆ derivative of the YK380-1 (YK391-1) was generated by PCR amplification of the met22∷kanMX strain (Open Biosystems) followed by linear transformation. SUP4oc and SUP4oc variant derivatives of strains YK380-1 and YK391-1 were generated by linear transformation to integrate the StuI fragment of the plasmid containing SUP4oc (derived from AB230-1) into the ADE2 locus, followed by selection on S-His dropout medium. Since the StuI fragment has different sequences of ADE2 DNA at each end flanking the DNA containing SUP4oc and Schizosaccharomyces pombe his5+, linear transformation should not generate multiple integrants at this locus. For each variant strain analyzed, three individual transformants were constructed and used.
Plasmids
AB230-1was constructed by replacement of the MET15 marker of JW132 (Whipple et al. 2011) with a fragment of DNA from pUG27 expressing S. pombe his5+ (which complements S. cerevisiae his3) (Gueldener et al. 2002) followed by insertion of a 1508-base-pair (bp) fragment of Fluc DNA into the BglII and XhoI restriction sites to facilitate detection of inserts when SUP4oc variants were inserted into these sites.
Variant tRNAs were constructed by insertion of the appropriate SUP4oc tRNA sequence, flanked by the 22 bp 5′ of the +1 site and the 7 bp 3′ of residue 73 of mature tRNAHis(GUG) [tH(GUG)G2], into the BglII XhoI site of AB230-1, essentially as described previously (Whipple et al. 2011). The final sequence inserted was as follows: 5′-AACAAAGTTCATAAAGAAATTACTCTCGGTAGCCAAGTTGGTTTAAGGCGCAAGACTTTAATTTATCACTACGAAATCTTGAGATCGGGCGTTCGACTCGCCCCCGGGAGATTTTTTCCTCGAG-3′, with the SUP4oc exon sequence underlined, the anti-codon in bold, and the intron in italics.
Analytical flow cytometry
Strains were grown overnight at 28°C in S-His liquid dropout medium containing 2% raffinose and 2% galactose supplemented with 80 mg/L adenine, followed by growth for 24 h in YP medium containing 2% raffinose and 2% galactose supplemented with 80 mg/L adenine. Dilutions were made as necessary to maintain log phase growth. Cells were then diluted in the same medium to an OD600 of 0.3 and grown to an OD600 between 0.8 and 1.2. Samples were prepared and analyzed essentially as described previously. Briefly, 10,000 events were recorded after analysis on an LSR-II flow cytometer (BD Biosciences) using laser and fluorescence detection filter parameters as described previously, with filter voltages set so that both GFP and RFP fluorescence intensities were ∼26,000 and with only those cells that passed an RFP cutoff of 5 × 103 analyzed (Dean and Grayhack 2012). Data analysis was performed using FlowJo software (Tree Star). GFPFLOW scores for a culture of a given SUP4oc variant represent the ratio of the median GFP divided by the median RFP, normalized to the median GFP/median RFP for wild-type SUP4oc. Biological triplicates were used to obtain standard deviations.
SUP4oc library construction and analysis
The SUP4oc library was generated by annealing two partially complementary oligonucleotides (MPG P346 and MPG P347) with 3% random mutations in SUP4oc residues 1–33 and 39–73 (IDT) followed by filling in the unpaired overhangs (Supplemental Fig. S1) and cloning. The sequence of P346 was 5′-TTTTGAGATCTAACAAAGTTCATAAAGAAATTACTCTCGGTAGCCAAGTTGGTTTAAGGCGCAAGACTTTAATTTATCACTACGAA-3′, and that of P347 was 5′-AGTTGCTCGAGGAAAAAATCTCCCGGGGGCGAGTCGAACGCCCGATCTCAAGATTTCGTAGTGATAAATTAA-3′, with the residues containing mutations underlined and the complementary sequence (comprising residues 34–38 and the intron) in bold. Annealing of the two oligonucleotides was done by heating for 5 min to 100°C followed by slow cooling to 30°C and then immediate placement on ice. The unpaired overhangs were then filled using the Klenow fragment of DNA polymerase at 37°C, and the reaction product was digested with BglII and XhoI, purified by gel extraction, and ligated into AB230-1, giving ∼325,000 E. coli transformants. An aliquot of these transformants containing ∼2.7 × 1010 cells was amplified by ∼4.3 generations of growth, and then plasmid DNA was extracted and digested with StuI for integration into yeast as described above. The yeast transformants were scraped, pooled, and frozen in aliquots for subsequent use.
To grow yeast SUP4oc libraries, ∼4.9 million cells were thawed, inoculated into S-His medium containing 2% raffinose supplemented with 80 mg/L adenine, and grown for 24 h at 28°C followed by dilution to OD600 of 0.04 and growth for 24 h in YP medium containing 2% raffinose and 2% galactose supplemented with 80 mg/L adenine. Dilutions were made as necessary to maintain log phase and ensure that at least 4.9 million cells were propagated at each step. Cells were then diluted in the same medium to an OD600 of 0.4 and grown to an OD600 of 1.1 prior to FACS into four bins on an Aria-ll cell sorter (BD Biosciences) at the University of Rochester Medical Center Flow Cytometry Core facility. Laser and fluorescence detection filter parameters were set as previously described, and only those cells with a RFP > 5 × 103 were collected (Dean and Grayhack 2012). Bin borders were set at a GFPFLOW of 0.007 (the lowest activity readily distinguished for a strain containing SUP4oc variants as compared with strains with no SUP4oc, corresponding to GFPSEQ of 0.026), with successive borders at 0.038 and 0.384. At least 2 million cells were collected (Supplemental Table S1) and then plated on YPD medium. After incubation for 3 d at 25°C, cells were scraped, pooled, and stored at −80°C. Genomic DNA was then directly isolated from frozen aliquots of the stored cells in each bin. Libraries WT1 and WT2 are replicates of the SUP4oc library analyzed in wild-type cells, and libraries ∆1 and ∆2 are replicates of the SUP4oc library analyzed in met22∆ cells. The GFPSEQ RTD analysis was based on comparing the WT2 and ∆2 libraries.
To enhance resolution of highly functional variants, aliquots of stored bin 1 cells (cells with high GFP expression) collected from the first sorting of the WT2 library were thawed, grown, and further sorted by FACS into four bins, three of which were subdivisions of the original bin1, and one of which was the original bin 2. Pooled cells were treated as described above prior to sequence analysis. Data from this analysis are referred to as WT2 6 bins and are the data set used for single, double, and epistasis analysis of SUP4oc in wild-type cells.
Sequencing
The SUP4oc construct, including 27 5′ and 16 3′ nucleotides, was amplified for 20 cycles (10 sec at 98°C, 30 sec at 52°C, and 30 sec at 72°C) from 1–3 μg of genomic DNA using Phusion polymerase and one of four sets of primers. Y19 (5′-AATGATACGGCGACCACCGAGATCTACACCTCCGCCTAACCCGAGTCCACCCGTCCCNNNNGATCTAACAAAGTTCATAAAGAAATTA-3′), used in all four primer sets, contains the Illumina adaptor sequence (1–29), a sequencing primer (30–56), and four Ns to mitigate low-complexity library cluster registration problems on the MiSeq. Y22 (5′-CAAGCAGAAGACGGCATACGAGATATTCCTTTCTTCCCTGCCCACCACCAGCTCCGTTGCTCGAGGAAAAAA-3′), Y23 (5′-CAAGCAGAAGACGGCATACGAGATCGGGTAAACTTCCCTGCCCACCACCAGCTCCGTTGCTCGAGGAAAAAA-3′), Y24 (5′-CAAGCAGAAGACGGCATACGAGATGGATATAGCTTCCCTGCCCACCACCAGCTCCGTTGCTCGAGGAAAAAA-3′), and Y25 (5′-CAAGCAGAAGACGGCATACGAGATTCCAGCCCCTTCCCTGCCCACCACCAGCTCCGTTGCTCGAGGAAAAAA-3′) each contain the Illumina adaptor, a sequencing primer, and a different index to discriminate between the bins (ATTCCTTT, CGGGTAAA, GGATATAG, and TCCAGCCC). Amplicons were gel-purified, quantitated with the qbit and Kapa quantitative PCR (qPCR) quantification kits, and sequenced on either the MiSeq V2 (using the 2x150 kit) or the HiSeq 2500 (using the 2x101 kit) using the manufacturer’s instructions.
Sequence assembly and quality filtering
Sequences were trimmed and split into bins using a custom python script. Forward and reverse reads were combined and filtered for quality using Enrich version 0.2 with a minimum phred score of 30 for any given cycle (Fowler et al. 2011).
Calculation of GFPSEQ
The number of reads corresponding to each unique variant for each bin was tabulated using Enrich version 0.2 (Fowler et al. 2011). The reads were then normalized to the sequencing depth of each bin, and the frequency of a given variant in a given bin was converted to an estimated number of collected cells by multiplying by the total number of cells collected for that bin during the FACS analysis. To calculate an approximate cellular fluorescence score for a given variant, the fraction of that variant’s cells in each bin was multiplied by the median GFP/median RFP value for each bin, and the results were summed, resulting in a weighted average fluorescence for that variant. This score was then normalized to the wild-type score (labeled NA-NA in the ID column of Supplemental Table S2) to give the normalized weighted average fluorescence referred to as GFPSEQ.
Data quality control
To focus analysis on validated sequences for which there were sufficient data, two sets of filters were applied: read counts per variant from sequencing and estimated cell counts per variant. A comparison of GFPSEQ values between WT1 and WT2 was taken as the gauge of quality in order to select the thresholds for filtering. Fifty-six combinations of both filtering parameters derived from seven different values from zero to 60 cell counts and eight different values from zero to 200 sequencing reads were applied to the data set, and a manual selection was made to reduce spurious correlations while not reducing the number of sequences needlessly. Scatter plots of comparisons of GFPSEQ values between the WT1 and WT2 libraries are shown in Supplemental Figure S1B, for nine representative combinations. The thresholds chosen were total read counts ≥100 and total cell counts ≥30.
Prediction of ED
The EDcalculator from RNAstructure (Reuter and Mathews 2010) was applied to calculate each mutant’s ED (Zadeh et al. 2011b) using the secondary structure of SUP4oc as reference. Modified bases dihydrouridine (D), N2,N2-dimethylguanosine (m2,2G), and 1-methyladenosine (m1A) were forced to be unpaired in the ED prediction, since these bases are known to block canonical base-pairing, and the assumption was also made that only mutation of the modified base would remove the modification. For the assessment of the ED of natural tRNAs, we used all eukaryotic tRNA sequences for which the modification status is known from the following species: Bombyx mori, Bos taurus, Candida cylindracea, Drosophila melanogaster, Homo sapiens, Leishmania tarentolae, Lupinus albus, Lupinus luteus, Mus musculus, Nicotiana rustica, Nicotiana tabacum, Oryctolagus cuniculus, Pichia jadinii, Rattus norvegicus, S. cerevisiae, S. pombe, Tetrahymena thermophila, Triticum aestivum, and Xenopus laevis. Sequences were obtained from the Modomics database (http://modomics.genesilico.pl).
Calculation of ΔΔG°28
The ∆∆G°28 is the folding free energy change difference between the mutant and wild-type tRNA structure at 28°C. The predicted ∆∆G°28 for each tRNA variant was computed with a custom program using the C2+ classes from the RNAstructure package (Reuter and Mathews 2010). ∆∆G°s were calculated using nearest neighbor rules (Mathews et al. 2004), where base pairs were disrupted if a nucleotide mutation prevented canonical pairing.
Epistasis analysis
The formula to determine epistasis for double mutants that passed the read count filter was
The product of GFPSEQ of the two single mutants was correlated with the actual double-mutant GFPSEQ with an R2 of 0.53 for ∆1, 0.56 for ∆2, 0.545 for WT1, and 0.534 for WT2. Epistasis can be >1 or less than −1, since some double-mutant variants have GFPSEQ values >1, because all sequencing reads for that variant were in bin 1, whereas some wild-type SUP4oc sequence reads occurred outside of bin 1.
Parameters that can be manipulated on the interactive SUP4oc Web site (http://depts.washington.edu/sfields/tRNA_supplemental/tRNA_interactive.html) are as follows: predicted fitness cutoff, only display epistatic interactions (links) in which the product of the GFPSEQ for the constituent single mutant variants is greater than this number; cell count cutoff, only display interactions for double mutants with more total estimated cell counts than this number; read count cutoff, only display interactions for double mutants with more total sequencing reads than this number; epistasis color control, controls the point at which the negative and positive epistasis values switch from gray to color for links; opacity cutoff, the epistasis value below which link opacity is set to “minimum opacity”; and minimum opacity, a value between 0 (transparent) and 1 (opaque) that defines the transparency of epistatic links with values below the “opacity cutoff.” The default is 0.3.
ROC (receiver operating characteristic) analysis
To assess the extent to which ΔΔG°28 correlates with RTD, we performed ROC analysis. All of the single and double variants were filtered with the criterion requiring met22∆2 GFPSEQ ≥ 0.052 (twice the cutoff for a variant to be considered active). With that, all of the variants with a ratio of met22∆2 GFPSEQ to WT2 GFPSEQ ≥2.0 were classified as RTD substrates. ΔΔG°28 was taken as a predictor to compute the false positive rate and the true positive rate for ROC analysis. The plot was generated, and thresholds were computed by the R package pROC (http://www.R-project.org) (Robin et al. 2011).
Isolation of bulk RNA and tRNA purification
Wild-type and met22∆ cells containing integrated SUP4oc variants were grown as described for analytical flow cytometry. Bulk low-molecular-weight RNA was extracted from 300 OD-mL pellets by hot phenol extraction followed by two ethanol precipitations and resuspension in ddH2O, as previously described (Jackman et al. 2003). tRNATyr was purified using biotinylated oligomer MP129, which is complementary to residues 76–52 of endogenous tRNATyr, SUP4oc, and the variants analyzed (Jackman et al. 2003).
Primer extension of SUP4oc variants
Bulk low-molecular-weight RNA was subjected to a poison primer extension assay using a primer from nucleotides 57–37 or 62–43 of mature tRNATyr that was 5′ end-labeled with T4 polynucleotide kinase and [γ-32P]ATP. Two-hundred nanograms of bulk low-molecular-weight RNA (or 7 ng of purified tRNA) was annealed to ∼1 pmol of 5′ radiolabeled primer after incubation for 3 min at 95°C before slow cooling and incubation for 30 at 50°C. Annealed RNA was then incubated for 1 h at 50°C in the presence of 1 mM each ddCTP, dATP, dGTP, and dTTP and 2 U of AMV reverse transcriptase (Promega). After completion, the reaction was resolved on a 7 M urea and 15% polyacrylamide gel for ∼4 h. The resulting gel was then dried and exposed to a phosphorimager plate for analysis, as previously described (Jackman et al. 2003).
An interactive Web site for analysis of GFPSEQ, epistasis, and RTD on tRNA cloverleaf maps is also available at http://depts.washington.edu/sfields/tRNA_supplemental/tRNA_interactive.html.
Acknowledgments
We thank John Abelson, Anita Hopper, and Yi-Tao Yu for comments. This work was supported by National Institutes of Health (NIH) grants GM052347 to E.M.P., GM076485 to D.H.M., and 1P41 GM103533 to S.F., and National Science Foundation grant MCB-1329545 to E.J.G. M.J.P. was partially supported by NIH Training Grant in Cellular, Biochemical, and Molecular Sciences 5T32 GM068411. S.F. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.245936.114.
References
- Agris PF, Vendeix FA, Graham WD. 2007. tRNA’s wobble decoding of the genome: 40 years of modification. J Mol Biol 366: 1–13 [DOI] [PubMed] [Google Scholar]
- Alexandrov A, Chernyakov I, Gu W, Hiley SL, Hughes TR, Grayhack EJ, Phizicky EM. 2006. Rapid tRNA decay can result from lack of nonessential modifications. Mol Cell 21: 87–96 [DOI] [PubMed] [Google Scholar]
- Basavappa R, Sigler PB. 1991. The 3 A crystal structure of yeast initiator tRNA: functional implications in initiator/elongator discrimination. EMBO J 10: 3105–3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetti B, Fu L, Moon J, Bedwell DM. 1995. The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. J Mol Biol 251: 334–345 [DOI] [PubMed] [Google Scholar]
- Chernyakov I, Whipple JM, Kotelawala L, Grayhack EJ, Phizicky EM. 2008. Degradation of several hypomodified mature tRNA species in Saccharomyces cerevisiae is mediated by Met22 and the 5′–3′ exonucleases Rat1 and Xrn1. Genes Dev 22: 1369–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochella L, Green R. 2005. An active role for tRNA in decoding beyond codon:anticodon pairing. Science 308: 1178–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean KM, Grayhack EJ. 2012. RNA-ID, a highly sensitive and robust method to identify cis-regulatory sequences using superfolder GFP and a fluorescence-based assay. RNA 18: 2335–2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewe JM, Whipple JM, Chernyakov I, Jaramillo LN, Phizicky EM. 2012. The yeast rapid tRNA decay pathway competes with elongation factor 1A for substrate tRNAs and acts on tRNAs lacking one or more of several modifications. RNA 18: 1886–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fechter P, Rudinger-Thirion J, Theobald-Dietrich A, Giege R. 2000. Identity of tRNA for yeast tyrosyl-tRNA synthetase: tyrosylation is more sensitive to identity nucleotides than to structural features. Biochemistry 39: 1725–1733 [DOI] [PubMed] [Google Scholar]
- Fowler DM, Araya CL, Gerard W, Fields S. 2011. Enrich: software for analysis of protein function by enrichment and depletion of variants. Bioinformatics 27: 3430–3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautheret D, Damberger SH, Gutell RR. 1995. Identification of base-triples in RNA using comparative sequence analysis. J Mol Biol 248: 27–43 [DOI] [PubMed] [Google Scholar]
- Geslain R, Martin F, Camasses A, Eriani G. 2003. A yeast knockout strain to discriminate between active and inactive tRNA molecules. Nucleic Acids Res 31: 4729–4737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giege R, Sissler M, Florentz C. 1998. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res 26: 5017–5035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giege R, Juhling F, Putz J, Stadler P, Sauter C, Florentz C. 2012. Structure of transfer RNAs: similarity and variability. RNA 3: 37–61 [DOI] [PubMed] [Google Scholar]
- Goodenbour JM, Pan T. 2006. Diversity of tRNA genes in eukaryotes. Nucleic Acids Res 34: 6137–6146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudipati RK, Xu Z, Lebreton A, Seraphin B, Steinmetz LM, Jacquier A, Libri D. 2012. Extensive degradation of RNA precursors by the exosome in wild-type cells. Mol Cell 48: 409–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH. 2002. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 30: e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper AK 2013. Transfer RNA post-transcriptional processing, turnover, and subcellular dynamics in the yeast Saccharomyces cerevisiae. Genetics 194: 43–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou YM, Schimmel P. 1992. Novel transfer RNAs that are active in Escherichia coli. Biochemistry 31: 4157–4160 [DOI] [PubMed] [Google Scholar]
- Jackman JE, Montange RK, Malik HS, Phizicky EM. 2003. Identification of the yeast gene encoding the tRNA m1G methyltransferase responsible for modification at position 9. RNA 9: 574–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadaba S, Krueger A, Trice T, Krecic AM, Hinnebusch AG, Anderson J. 2004. Nuclear surveillance and degradation of hypomodified initiator tRNA(Met) in S. cerevisiae. Genes Dev 18: 1227–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser MW, Brow DA. 1995. Lethal mutations in a yeast U6 RNA gene B block promoter element identify essential contacts with transcription factor-IIIC. J Biol Chem 270: 11398–11405 [DOI] [PubMed] [Google Scholar]
- Kim SH, Suddath FL, Quigley GJ, McPherson A, Sussman JL, Wang AH, Seeman NC, Rich A. 1974. Three-dimensional tertiary structure of yeast phenylalanine transfer RNA. Science 185: 435–440 [DOI] [PubMed] [Google Scholar]
- Kohalmi L, Kunz BA. 1992. In vitro mutagenesis of the yeast SUP4-o gene to identify all substitutions that can be detected in vivo with the SUP4-o system. Environ Mol Mutagen 19: 282–287 [DOI] [PubMed] [Google Scholar]
- Koski RA, Clarkson SG, Kurjan J, Hall BD, Smith M. 1980. Mutations of the yeast SUP4 tRNA(Tyr) locus: transcription of the mutant genes in vitro. Cell 22: 415–425 [DOI] [PubMed] [Google Scholar]
- Kramer EB, Hopper AK. 2013. Retrograde transfer RNA nuclear import provides a new level of tRNA quality control in Saccharomyces cerevisiae. Proc Natl Acad Sci 110: 21042–21047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer EB, Vallabhaneni H, Mayer LM, Farabaugh PJ. 2010. A comprehensive analysis of translational missense errors in the yeast Saccharomyces cerevisiae. RNA 16: 1797–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurjan J, Hall BD. 1982. Mutations at the Saccharomyces cerevisiae SUP4 tRNA(Tyr) locus: isolation, genetic fine-structure mapping, and correlation with physical structure. Mol Cell Biol 2: 1501–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurjan J, Hall BD, Gillam S, Smith M. 1980. Mutations at the yeast SUP4 tRNA(Tyr) locus: DNA sequence changes in mutants lacking suppressor activity. Cell 20: 701–709 [DOI] [PubMed] [Google Scholar]
- Ledoux S, Olejniczak M, Uhlenbeck OC. 2009. A sequence element that tunes Escherichia coli tRNA(Ala)(GGC) to ensure accurate decoding. Nat Struct Mol Biol 16: 359–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leontis NB, Stombaugh J, Westhof E. 2002. The non-Watson-Crick base pairs and their associated isostericity matrices. Nucleic Acids Res 30: 3497–3531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J, Reynolds N, Ibba M. 2009. Aminoacyl-tRNA synthesis and translational quality control. Annu Rev Microbiol 63: 61–78 [DOI] [PubMed] [Google Scholar]
- Marck C, Grosjean H. 2002. tRNomics: analysis of tRNA genes from 50 genomes of eukarya, archaea, and bacteria reveals anticodon-sparing strategies and domain-specific features. RNA 8: 1189–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marck C, Kachouri-Lafond R, Lafontaine I, Westhof E, Dujon B, Grosjean H. 2006. The RNA polymerase III-dependent family of genes in hemiascomycetes: comparative RNomics, decoding strategies, transcription and evolutionary implications. Nucleic Acids Res 34: 1816–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. 2004. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci 101: 7287–7292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy FV 4th, Ramakrishnan V, Malkiewicz A, Agris PF. 2004. The role of modifications in codon discrimination by tRNA(Lys)UUU. Nat Struct Mol Biol 11: 1186–1191 [DOI] [PubMed] [Google Scholar]
- Musier-Forsyth K, Usman N, Scaringe S, Doudna J, Green R, Schimmel P. 1991. Specificity for aminoacylation of an RNA helix: an unpaired, exocyclic amino group in the minor groove. Science 253: 784–786 [DOI] [PubMed] [Google Scholar]
- Normanly J, Ogden RC, Horvath SJ, Abelson J. 1986. Changing the identity of a transfer RNA. Nature 321: 213–219 [DOI] [PubMed] [Google Scholar]
- Orioli A, Pascali C, Pagano A, Teichmann M, Dieci G. 2012. RNA polymerase III transcription control elements: themes and variations. Gene 493: 185–194 [DOI] [PubMed] [Google Scholar]
- Pearson D, Willis I, Hottinger H, Bell J, Kumar A, Leupold U, Soll D. 1985. Mutations preventing expression of sup3 tRNA(Ser) nonsense suppressors of Schizosaccharomyces pombe. Mol Cell Biol 5: 808–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putz J, Florentz C, Benseler F, Giege R. 1994. A single methyl group prevents the mischarging of a tRNA. Nat Struct Biol 1: 580–582 [DOI] [PubMed] [Google Scholar]
- Randau L, Stanley BJ, Kohlway A, Mechta S, Xiong Y, Soll D. 2009. A cytidine deaminase edits C to U in transfer RNAs in archaea. Science 324: 657–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter JS, Mathews DH. 2010. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Muller M. 2011. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Pesini E, Lott MT, Procaccio V, Poole JC, Brandon MC, Mishmar D, Yi C, Kreuziger J, Baldi P, Wallace DC. 2007. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res 35: D823–D828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeing TM, Voorhees RM, Kelley AC, Gao YG, Murphy FV 4th, Weir JR, Ramakrishnan V. 2009. The crystal structure of the ribosome bound to EF-Tu and aminoacyl-tRNA. Science 326: 688–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader JM, Chapman SJ, Uhlenbeck OC. 2009. Understanding the sequence specificity of tRNA binding to elongation factor Tu using tRNA mutagenesis. J Mol Biol 386: 1255–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz DW, Yarus M. 1994. tRNA structure and ribosomal function. I. tRNA nucleotide 27-43 mutations enhance first position wobble. J Mol Biol 235: 1381–1394 [DOI] [PubMed] [Google Scholar]
- Sharon E, Kalma Y, Sharp A, Raveh-Sadka T, Levo M, Zeevi D, Keren L, Yakhini Z, Weinberger A, Segal E. 2012. Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat Biotechnol 30: 521–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepotinovskaya I, Uhlenbeck OC. 2013. tRNA residues evolved to promote translational accuracy. RNA 19: 510–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner T, Jansen M, Hou YM. 1995. Structural and functional accommodation of nucleotide variations at a conserved tRNA tertiary base pair. RNA 1: 841–851 [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Nagao A, Suzuki T. 2011. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu Rev Genet 45: 299–329 [DOI] [PubMed] [Google Scholar]
- Valle M, Zavialov A, Li W, Stagg SM, Sengupta J, Nielsen RC, Nissen P, Harvey SC, Ehrenberg M, Frank J. 2003. Incorporation of aminoacyl-tRNA into the ribosome as seen by cryo-electron microscopy. Nat Struct Biol 10: 899–906 [DOI] [PubMed] [Google Scholar]
- van Nimwegen E, Crutchfield JP, Huynen M. 1999. Neutral evolution of mutational robustness. Proc Natl Acad Sci 96: 9716–9720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhof E, Dumas P, Moras D, Romby P. 1985. Crystallographic refinement of yeast aspartic acid transfer RNA. J Mol Biol 184: 119–145 [DOI] [PubMed] [Google Scholar]
- Whipple JM, Lane EA, Chernyakov I, D’Silva S, Phizicky EM. 2011. The yeast rapid tRNA decay pathway primarily monitors the structural integrity of the acceptor and T-stems of mature tRNA. Genes Dev 25: 1173–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Francklyn C. 1994. Cytosine 73 is a discriminator nucleotide in vivo for histidyl-tRNA in Escherichia coli. J Biol Chem 269: 10022–10027 [PubMed] [Google Scholar]
- Yarham JW, Elson JL, Blakely EL, McFarland R, Taylor RW. 2010. Mitochondrial tRNA mutations and disease. RNA 1: 304–324 [DOI] [PubMed] [Google Scholar]
- Zadeh JN, Steenberg CD, Bois JS, Wolfe BR, Pierce MB, Khan AR, Dirks RM, Pierce NA. 2011a. NUPACK: analysis and design of nucleic acid systems. J Comput Chem 32: 170–173 [DOI] [PubMed] [Google Scholar]
- Zadeh JN, Wolfe BR, Pierce NA. 2011b. Nucleic acid sequence design via efficient ensemble defect optimization. J Comput Chem 32: 439–452 [DOI] [PubMed] [Google Scholar]
- Zhou J, Lancaster L, Donohue JP, Noller HF. 2013. Crystal structures of EF-G-ribosome complexes trapped in intermediate states of translocation. Science 340: 1236086. [DOI] [PMC free article] [PubMed] [Google Scholar]