

Abstract
The liver is directly or indirectly involved in many essential processes and is affected by numerous inherited diseases. Therefore, many inherited diseases could be effectively treated by targeting the liver using gene transfer approaches. The challenges associated with liver-directed gene therapy are efficient targeting of hepatocytes, stability of the vector genome, and persistent high level expression. Many of these obstacles can be overcome with adeno-associated viral (AAV) gene transfer vectors. The first AAV gene transfer vector developed for in vivo use was based on the AAV2 serotype. AAV2 has a broad tropism and transduces many cell types, including hepatocytes, relatively efficiently in vivo. The capsid protein confers the serological profile and at least 12 primate AAV serotypes have already been characterized. Importantly, pseudotyping a recombinant AAV vector with different capsid proteins can dramatically alter the tropism. Both AAV8 and AAV9 have higher affinities for hepatocytes when compared to AAV2. In particular, AAV8 can transduce 3–4 fold more hepatocytes and deliver 3–4 fold more genomes per transduced cell when compared to AAV2. Depending on the dose, AAV8 can transduce up to 90–95% of hepatocytes in the mouse liver following intraportal vein injection. Interestingly, comparable levels of transduction can be achieved following intravenous injection. Direct intraparenchymal injection of an AAV vector also mediates relatively high level long term expression. Additional specificity can be conferred by using liver-specific promoters in conjunction with AAV8 capsid proteins. In addition to treating primary hepatocyte defects, immune reactions to transgene products can be minimized by circumventing the fixed tissue macrophages of the liver, Kupffer cells, and limiting expression to hepatocytes. The ability to target hepatocytes by virtue of the AAV serotype and the use of liver-specific promoters allows investigators to test novel therapeutic approaches and answer basic clinical and biological questions.
Keywords: Adeno-Associated Virus, Gene Therapy, Liver, Hepatocytes, Inherited Metabolic Disease
1. Introduction
The liver performs a myriad of tasks that are essential to the maintenance and proper function of nearly every organ system (1). An enormous number of proteins are synthesized and metabolized in the liver. These include both intracellular and secreted proteins. Intracellular and integral membrane proteins are responsible for carbohydrate utilization and storage. The liver is also a major site of lipid metabolism. Many secreted proteins are synthesized in the liver. The major serum protein component, albumin, is synthesized in the liver. In addition, most of the circulating clotting factors, α1-antitrypsin, ceruloplasmin, and a host of other secreted proteins are produced in the liver. The liver plays an important role in detoxifying naturally occurring metabolites such as ammonia and bilirubin. Drugs and environmental agents are also detoxified, conjugated, and excreted by the liver.
The enormous number of complex tasks performed by the liver are controlled by proteins (enzymes, integral membrane proteins, signaling molecules, secreted proteins, etc.); all of which are susceptible to mutations in their respective genes. Mutations in any of the genes encoding the proteins involved in these processes typically lead directly to disease or a predisposition to disease. The diseases can range from defects in carbohydrate metabolism, lipid metabolism, bleeding disorders, increased vulnerability to drugs and environmental toxins, and cognitive deficits, just to name a few. Given the vast number of functions the liver carries out and its importance for general health, it is clear that it is an important therapeutic target. The ability to stably and efficiently introduce functional expression cassettes into hepatocytes that can compensate for primary liver defects would be a powerful therapeutic tool. The well developed secretory machinery of the liver could also be exploited in order to express and secrete proteins that are not normally produced in the liver. For example, clotting factor VIII or Von Willebrand factor which are normally produced and secreted from endothelial cells could potentially be expressed and secreted from the liver. Finally, many of the complex interactions and pathways in the liver are not fully understood. Expressing specific transgenes in the livers of intact animals could be an effective method to test various hypotheses and better understand basic mechanisms.
The ability to stably introduce functional genes that mediate persistent high level expression into the liver had been a major challenge. The development of gene transfer vectors based on adeno-associated virus (AAV) overcame many of these obstacles. The first AAV-based gene transfer vector was described by Hermonat and Muzyczka (1984) and utilized the AAV2 serotype (2). However, the first generation vectors were difficult to make and purify in quantities sufficient for in vivo use, even in murine models. Once the technical challenges associated with larger scale production were overcome (3, 4), it quickly became clear that AAV2-based vectors had a relatively broad tropism and mediated persistent high level expression in vivo. Early studies showed that AAV2 appeared to have the greatest tropism for liver and skeletal muscle (5, 6, 7). Intravenous injection of an AAV2-based gene transfer vector in mice resulted in transduction of most tissues with the exception of those rich in hematopoietic-derived cells (6, 7, 8). Importantly, the AAV genome persisted in most tissues, including the liver, for at least one year (Figure 1) and continued to mediate high level expression (8).
Figure 1.
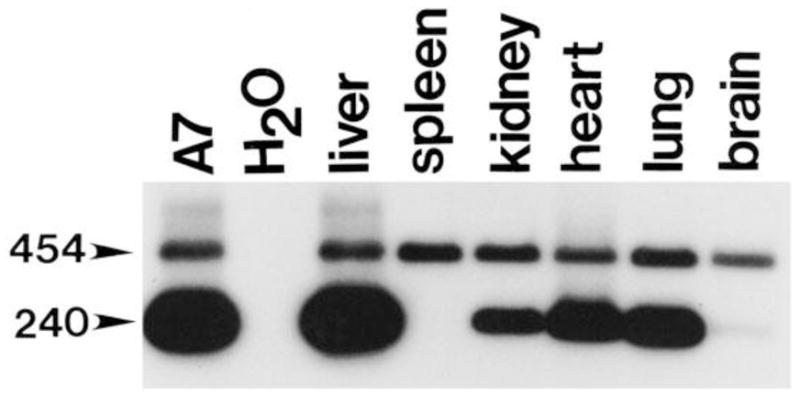
Transduced viral cDNA persists in most tissues for at least 1 year. Tissues from 1-year-old mice were analyzed for the presence of viral cDNA using primers which amplify a 240 bp band from human cDNA, and a 454 bp band from the endogenous murine GUSB gene. A murine fibroblast line (A7) which has been transduced with a single retroviral copy of the human GUSB cDNA is shown as a positive control. Persistence of AAV-tranduced human GUSB cDNA is seen in most tissues at 1 year. The spleen, however, shows no indication of persistent viral transduction at this late time-point. The Southern blot shows the PCR products from a single mouse and is representative of the pattern observed in three separate mice 1 year after injection. Reproduced from Gene Therapy, 2001, with permission from Nature Publishing Group.
These studies were encouraging and represented a major technical advance towards effective in vivo gene therapy. As the field progressed it became clear that the AAV2-based genome was quite versatile and could be efficiently packaged with other AAV capsid proteins. Changing the serotype dramatically affected the tropism. For example, serotypes 1, 5, and 4 were more efficient than AAV2 at transducing cells in the murine central nervous system (9, 10). Likewise, the liver is also more susceptible to transduction by vectors pseudotyped with specific AAV capsid proteins. Although AAV2-based vectors transduced the liver more efficiently than other tissues, serotypes 8 and 9 had an even greater affinity for the liver (11, 12, 13). Portal vein injection of a vector pseudotyped with the AAV8 capsid protein resulted in dramatically increased liver transduction compared to vectors pseudotyped with AAV1, AAV2, AAV5, or AAV7 capsid proteins (Table 1) (11). Interestingly, comparable levels of liver transduction were observed with an AAV8 vector regardless of whether it was delivered by intraportal vein injection or by intravenous (tail vein) injection (Figure 2) (12). It was also demonstrated in the same study that, depending on the dose, greater than 95% of hepatocytes could be transduced with a single injection of an AAV8-pseudotyped vector. The discovery of different AAV capsid proteins and the demonstration that pseudotyped vectors have dramatically different tropisms represents an elegant and effective approach to tissue-specific gene therapy.
Table 1.
Real-time PCR analysis for abundance of AAV vectors in nu/nu mouse liver after injection of 1 × 1011 genome copies of vector. A set of probe and primers targeting the SV40 poly(A) region of the vector genome was used for TagMan PCR. Values shown are means of three individual animals with standard deviations. The animals were sacrificed at day 56 to harvest liver tissues for DNA extraction. Reproduced from the Proceedings of the National Academy of Sciences of the United States of America, 2002, with permission from the National Academy of Sciences of the United States of America.
| AAV vectors/dose Genome copies per cell
| |
|---|---|
| AAV2/1AlbA1AT | 0.6 ± 0.36 |
| AAV2AlbA1AT | 0.003 ± 0.001 |
| AAV2/5AlbA1AT | 0.83 ± 0.64 |
| AAV2/7AlbA1AT | 2.2 ± 1.7 |
| AAV2/8AlbA1AT | 18 ± 11 |
Figure 2.

Comparison of efficiency of rAAV8-mediated liver transduction between tail vein and portal vein injections. (A) Plasma human coagulation factor IX (hF.IX) levels after tail vein (TV) or portal vein (PV) injection of AAV8-hF.IX16 into male C57BL/6 mice. Robust human coagulation factor IX expression with no lag phase was observed with both routes. Expression peaked 4 weeks after injection, followed by a substantial ( 75%) decline. Vertical bars indicate standard deviations. (B) Vector genome copy numbers (ds-vg/dge) in livers transduced with AAV8-EF1 -nlslacZ via tail vein or portal vein injection at 3.0 × 1011 or 7.2 × 1012 vg/mouse. Total liver DNA was extracted 6 weeks postinjection, and 10 μg of DNA was analyzed by Southern blot with BglI digestion and a 2.1-kb lacZ probe (BglI-BglI fragment). The left and right blots were analyzed separately with a different series of vector copy number standards. The double-stranded vector copy number standards (0 to 100 and 0 to 1,000 ds-vg/dge) were prepared by adding the corresponding amount of plasmid, pAAV-EF1 -nlslacZ, to 10 μg of liver DNA extracted from a naïve mouse. Each lane represents an individual mouse. Routes of administration and vector doses are indicated above the lanes. Reprinted with permission of the American Society of Microbiology.
Although the tropism of AAV-based gene transfer vectors can be shifted towards a specific cell type or tissue by using different capsid proteins, the specificity is not absolute. More restricted expression can be achieved by combining a particular AAV capsid protein with a tissue- or cell-specific promoter. This has particular significance for liver-directed gene therapy. Fixed tissue macrophages in the liver known as Kupffer cells, can act as antigen presenting cells. Expression of foreign proteins in Kupffer cells can elicit an immune response and effectively eliminate any therapeutic effect by developing antibodies to the protein or eliminating the transduced cells through a cytotoxic T cell-mediated mechanism. It has been shown that limiting expression of a foreign transgene to hepatocytes using an AAV8-pseudotyped vector in combination with a hepatocyte-specific promoter dramatically decreases the immune response (14, 15). In fact, recent reports show that liver-specific gene transfer can effectively “tolerize” a mouse model of human disease (16, 17). In one case, the “tolerized” animals can subsequently be treated with intravenous recombinant protein without developing antibodies or lethal hypersensitivity reactions (18).
The thoughtful use of specific AAV capsid proteins is an effective method to enhance liver-specific expression. However, there is an equally effective, if less elegant, means of accomplishing the same goal. It has been shown that direct injection of an AAV2 vector into the liver parenchyma results in relatively widespread transduction throughout the liver (19). This approach mediates persistent expression that is sufficient to reduce the disease burden in several tissues in a murine model of lysosomal storage disease. Although this approach allows some infectious particles to be disseminated through the vasculature and transduce other tissues, the liver remains the predominant target.
An important advantage of AAV gene transfer vectors for human gene therapy applications is that they are believed to persist primarily as extrachromosomal episomes (20, 21). This would effectively eliminate insertional mutagenesis which, in certain circumstances, can lead to serious adverse events when using stably integrating vectors (22, 23). However, several independent studies have shown that rearranged portions of recombinant AAV vectors can stably integrate into the host genome (24, 25). Although there have been no reports of toxicity in most in vivo pre-clinical experiments or in clinical trials, there has been one report of AAV integration being associated with hepatocellular carcinoma (HCC) (26). Toxicity was first observed in a pre-clinical experiment where effective long-term (≥1 year) disease correction was achieved in mice with a lysosomal storage disorder following systemic delivery of an AAV vector during the neonatal period (8, 27). At 18 months of age the AAV-treated animals still appeared healthy, however, three out of five of the remaining animals had HCC (27). It seemed unlikely that over-expression of the transgene product (β-glucuronidase, GUSB) was the cause of the toxicity since several transgenic lines expressing as much as 1,000-fold higher than normal levels of GUSB showed no evidence of toxicity (28). The initial observation of toxicity following systemic neonatal AAV-mediated gene therapy was reproduced in a larger study where 30–50% of both affected and normal animals developed HCC (26). A thorough molecular analysis of four independent tumors from different animals revealed that a portion of the AAV vector had integrated within a six kilobase region near the distal end of mouse chromosome 12. This integration event disrupted the expression of an adjacent micro-RNA-rich region. Interestingly, there have been two additional reports in different disease models using different transgenes of hepatic tumor formation following AAV-mediated gene therapy (29, 30). However, detailed analysis of the proviral structures was not performed in those studies. It remains unclear whether these observations are species- or tissue-specific. Alternatively, these observations could be dependent on the developmental stage at which the vector is delivered (neonatal) or the underlying disease state of the liver. Clearly, the frequency and mechanisms of these observations need to be more precisely elucidated in order for informed risk-vs-benefit determinations to be made.
Progress towards effective liver-directed gene therapy as well as a better understanding of the underlying pathogenesis of primary liver defects will be greatly accelerated by the ability to efficiently and stably transfer functional genes to the livers of intact animal models of disease. The advent of recombinant AAV vectors, the ability to enhance delivery to the liver through pseudotyping with different AAV capsid proteins, and the effective use of liver-specific promoters provides an opportunity to accomplish these goals. Several methods for delivering gene transfer vectors to the livers of murine models of disease are outlined below.
2. Materials
2.1 Liver-Directed Gene Delivery
1 cc Tuberculin syringes.
30G needles.
3X illuminated desktop magnifying glass.
Anesthetic cocktail: 20mg/ml Ketamine (Fort Dodge Animal Health, Fort Dodge, IA), .375mg/ml Xylazine (AGRI Laboratories, St. Joseph, MO), (100μl Anesthetic cocktail/25 gram mouse).
Sterile surgical supplies (scalpel, scissors, forceps, sponges, etc.).
Absorbable gelatin sponge (Ethicon, Somerville, NJ).
4-0 polyester suture (Ethicon, Somerville, NJ).
Topical disinfectant: 70% ethanol
Povidone: topical iodine prep pad (Triad Disposables, Brookfield, WI)
Mouse restrainer.
2.2 Biochemical, Histological and Molecular Analyses
Microhematocrit capillary tubes.
Tissue fixative: 10% neutral-buffered formalin (Sigma, St. Louis, MO).
Standard molecular biology equipment (microcentrifuge, microcentrifuge tubes, micropipettors, agarose gel electrophoresis, transfer membranes, etc.).
DNA isolation kit.
Standard PCR Thermocycler or Real-Time PCR Thermocycler.
3. Methods
As mentioned above, liver-directed gene delivery with an AAV vector can be accomplished by several methods. Conceptually, the easiest methods involve the injection of vector into the liver either through the portal vein or directly into the liver parenchyma. Unfortunately, both methods are relatively invasive and require some surgical skill (see below). A more elegant approach involves pseudotyping the viral vector with an AAV capsid that enhances liver tropism, and incorporating a liver-specific promoter into the expression cassette. Liver-directed gene transfer using a liver-specific vector/promoter combination can be accomplished with a simple intravenous injection. This eliminates the need to become proficient in surgical techniques. There are numerous liver-specific expression cassettes, and infectious AAV vector stocks pseudotyped with a variety of capsid proteins can be obtained on a fee-for-service basis from several core laboratories. These techniques are beyond the scope of this chapter and will be discussed elsewhere. The techniques outlined below are intended for use in rodent (rat and mouse) models. The reader should seek assistance from an experienced veterinarian if similar experiments are to be carried out in larger (canine, primate, etc.) animal models.
3.1 Liver-Directed Gene Delivery
Intraportal vein injection, direct intraparenchymal liver injection and intravenous injection techniques have been described in detail elsewhere. Therefore, the methods outlined below represent basic guides for these approaches. The reader is encouraged to seek experienced collaborators to assist with the techniques, in particular the survival surgery techniques.
3.1.1 Intraportal Vein Injection [see Cai, SR, et al., Int. J. Oncol., 2005 (31)]
Place adult mice under surgical anesthesia by intraperitoneal injection of anesthetic cocktail. Surgical anesthesia is confirmed when the animal is unresponsive when the toe is pinched and the blinking reflex is no longer present.
Prepare the abdomen prior to surgery by washing with topical disinfectant followed by Povidone. Establish a sterile field over the abdomen.
Make a 2–3cm long midline incision in the abdomen starting 2–3mm below the xyphiod process.
Eviscerate the intestines and reflect them to the left to expose the portal vein. Note: the portal vein is a relatively large vein that originates in the intestines and drains directly into the liver. The anterior lobes of the liver may have to be lifted in order to see the vein. The intestines are kept moist during the procedure by covering them with sterile gauze soaked in sterile saline.
Introduce a 30G needle directly into the portal vein.
Inject 0.2 ml of viral suspension uniformly and slowly over a period of 1min.
Keep the needle in place for approximately 10–20 sec after the injection.
Remove the needle and gently press a small piece of absorbable sponge over the insertion point to prevent bleeding.
Close the abdomen with two layers (muscle and skin) of running 4-0 polyester suture.
Place the animal under a heating lamp until it has recovered and then house singly until the wound is completely healed.
3.1.2 Intraparenchymal Liver Injection [see Sferra, TJ, et al., Mol. Ther, 2004 (19)]
Up to 200μl of total injectate can be delivered to the adult mouse liver in 4–5 separate injection sites (40–50μl/site).
Place adult mice under surgical anesthesia by intraperitoneal injection of anesthetic cocktail. Surgical anesthesia is confirmed when the animal is unresponsive when the toe is pinched and the blinking reflex is no longer present.
Prepare the abdomen for surgery by washing with topical disinfectant followed by Povidone. Establish a sterile field over the abdomen.
Make a 2–3cm long midline incision in the abdomen starting 2–3mm below the xyphiod process in order to expose the liver.
Inject viral suspension directly into the liver parenchyma through a 30G needle affixed to a tuberculin syringe.
Inject the suspension slowly with intermittent pressure. Note: The injection site is monitored during the injection through a surgical stereoscope to ensure that there is no hemorrhage or extravasation. If either of these situations occurs, the needle is removed, the bleeding stopped with gentle pressure, and a new site is injected.
Remove the needle and stop the bleeding by gentle pressure with a sterile cotton sponge.
Choose a new site and repeat the procedure.
Close the abdomen with two layers (muscle and skin) of running 4-0 polyester suture.
Place the animal under a heating lamp until it has recovered and then house singly until the wound is completely healed.
3.1.3 Adult I.V. Injection
Up to 300–400μl of injectate can be delivered intravenously to an adult mouse by this route.
Place a mouse in a standard mouse restrainer such that the injector has free access to the entire length of tail.
The most accessible veins in the mouse tail are on the lateral sides of the tail. The vein on the dorsal aspect of the mouse tail is difficult to routinely inject. The vessel on the ventral aspect of the tail is an artery and should never be injected.
Insert a 30G needle affixed to a 1cc tuberculin syringe into one of the lateral veins at a site as distal as possible on the tail. Choose a distal site so that if the first attempt is unsuccessful the injector can simply choose another more proximal injection site. Note: a) if the injector experiences difficulty with this approach, keep in mind the lateral veins are EXTREMELY SUPERFICIAL. b) some degree of vasodilation can be achieved if the tail is gently heated with warm water.
Slowly inject the viral suspension once the needle is in place. Note: if the injector has difficulty depressing the plunger due to increased pressure it is likely that the needle is not in the vein. In that case remove the needle and choose a new site.
Remove the needle once the injection is complete and apply pressure to the site until bleeding stops.
Return the mouse to its cage immediately after the injection.
3.1.4 Neonatal I.V. Injection [see Sands, MS and Barker, JE, Lab. Animal Sci., 1999 (32)]
Up to 100 μl of viral suspension can be delivered to a neonatal mouse by this technique.
This procedure is most easily accomplished with two people; one restraining the animal and the other injecting.
Neonatal mice can be injected by this route between 1 and 4 days of life. It becomes increasingly difficult to visualize the vein beyond the first four days of life.
Place neonatal mice on a dry towel at 4°C for several minutes prior to injection to minimize their movement and reduce trauma.
One person gently immobilizes the forelimbs and holds the head such that the lateral aspect is facing up. This exposes the superficial temporal vein that extends from behind the eye into the neck.
Position the mouse under the 3X illuminated magnifying glass.
The other person inserts the 30G needle affixed to a 1 cc tuberculin syringe under the skin next to the vein. The approach should be from the head towards the neck.
Move the needle over the vein and advance slowly into the vein until the bevel is obscured by blood. This indicates that the needle is in the vessel. Note: the skin is translucent at this age and the bevel of the needle can easily be seen through the skin.
Do not advance further.
Inject the viral suspension slowly. Note: Typically the vein distal to the injection site will blanch if the injection is successful. If the needle is not in the vein, an easily visible bulge will immediately form with as little as 10 microliters injected subcutaneously.
Once the injection is complete remove the needle and apply gentle pressure to the vein. Bleeding typically stops within 2–3 minutes.
Place the mouse under a heating lamp before returning to the female.
If the injection is unsuccessful, allow the mouse to recover for 5–10 minutes and then attempt a second injection on the other side.
3.2 Liver-Specific Analyses
Two important and disease-independent considerations when evaluating liver-directed gene transfer are the presence of the transgene and toxicity. Determining the presence of a specific transgene in liver tissue can be accomplished by several standard molecular biology methods. However, the exact methods and conditions will vary depending on the transgene used. Therefore, the methods will be discussed in general terms. Although the techniques for toxicity determinations are standard, it is inefficient for most small independent research laboratories to incorporate those technologies into their laboratory. Therefore, the assays necessary for an initial determination of toxicity will be outlined and examples (not an exhaustive list) of representative service providers will be listed. If toxicity is suspected after an initial screen, the investigator is encouraged to seek expert (Pathologist, etc.) advice to determine the appropriate course of action to better understand the findings.
3.2.1 Southern Blot (see Sambrook and Russel, Molecular Cloning: A Laboratory Manual, 2001 (33)]
Isolate high molecular weight genomic DNA or lower molecular weight “Hirt” DNA from liver tissue by standard techniques.
Digest the DNA to completion with appropriate restriction endonucleases. Note: Restriction sites within the transgene or expression cassette will liberate the entire transgene or internal fragments of the transgene. A single restriction site within the transgene or expression cassette will liberate transgene/genomic junction fragments from integrated proviral forms.
Separate the restriction fragments by agarose gel electrophoresis. Note: The appropriate agarose concentration will depend on the expected size of the fragments. In general, low concentration gels will be used to identify large junction fragments and higher agarose concentrations will be used to identify internal fragments of known size.
Transfer the DNA fragments by passive diffusion or by electro-transfer to a charged synthetic membrane.
Prepare a suitable radiolabeled or fluorescently labeled DNA fragment, being mindful of where the restriction sites are located.
Equilibrate the membrane containing the bound DNA fragments in buffer, probe with the labeled DNA fragment, then washe extensively to remove any unbound DNA probe.
Visualize the labeled membrane (blot) by exposure to film or by an alternate imaging system.
Incorporate known molecular weight markers or an internal standard in order to: 1) identify the position of the transgene, 2) estimate the copy number of the transgene, and 3) estimate the number of unique integration sites of the transgene.
3.2.2 Standard PCR (see Sambrook and Russel, Molecular Cloning: A Laboratory Manual, 2001 (33)]
Real-Time (RTPCT) or Quantitative (QPCR) PCR is perhaps the easiest and most reliable method to quantify the amount of transgene in the liver of animals following viral-mediated gene transfer. However, this requires specialized equipment that may be beyond the capabilities of small independent laboratories. Alternate, less expensive PCR-based methods have been established that allow investigators to detect the presence and estimate the level of gene transfer. These methods are briefly outlined below:
Isolate total DNA from liver tissue samples by standard techniques.
Design single stranded DNA primers that are complementary to either the transgene or the expression cassette within the gene transfer vector.
PCR primers can be developed for several purposes. A single pair of primers can be devised that will detect the presence of the transgene or expression cassette. This is useful when using a transgene that does not naturally exist in the host genome [bacterial β-galactosidase, green fluorescent protein (GFP), etc.]. When amplifying a therapeutic transgene that has a corresponding full length endogenous gene, an internal control can be incorporated into this type of analysis. A single set of primers that amplifies both the transgene (usually a cDNA) and the corresponding full length gene within the host genome can be devised (see Figure 1). Regions of the cDNA that have exact, or nearly exact sequence homology to the host gene but are separated by one or more introns are required for this analysis. Typically, the PCR products from the transgene and the full length gene should differ by 50 to several hundred base pairs in length in order to easily separate the two fragments by gel electrophoresis. Alternatively, a second set of PCR primers can be developed that amplifies a PCR product of distinct size from a different host gene. This type of analysis can be used to simultaneously detect any transgene and an independent host gene.
Perform PCR on the total DNA isolated from liver (the exact PCR protocol will vary depending on the transgene and primer configurations).
Separate the PCR products by gel electrophoresis. Visualize the PCR products by ethidium-bromide fluorescence or by Southern blot analysis (see above).
The ability to simultaneously amplify a distinct sized PCR product using either a single set of primers or two sets of primers allows the investigator to estimate the amount of the transgene relative to a known host gene. Note: Numerous controls must be run with this type of analysis to determine the appropriate PCR protocol, the relative rates of amplification from the primers, and the relative affinity of the probes for the PCR products if Southern blot analysis is to be performed.
3.2.3 Real-Time (RTPCR) or Quantitative (QPCR) PCR (see Sambrook and Russel, Molecular Cloning: A Laboratory Manual, 2001 (33)]
Isolate total DNA from liver tissue samples by standard techniques.
Synthesize PCR primers specific for the transgene. PCR primers used for QPCR typically amplify a fragment less than 100 bp in length.
Determine the PCR protocol (annealing time, extension time, etc.) empirically in test runs using standard techniques. Perform the amplification using either a fluorescently labeled probe or fluorescently labeled nucleotides.
PCR products are detected and quantified by two methods: 1) Synthesize a fluorescently labeled primer that binds a region of DNA between the two original primers. As the internal primer is incorporated into the PCR products, the fluorescent label is liberated and detected by the Real-Time PCR instrument. This is a method that indirectly measures the amplification of the product. However, this method provides additional specificity since all three primers must bind their respective sites in order for the fluorescent probe to be liberated. 2) Use fluorescently labeled nucleotides that will be incorporated into the amplified product. This method provides a direct measure of the amount of amplified product and is typically more versatile since a gene-specific fluorescently labeled primer is not required. However, this method may not be as specific since it only requires two primers to faithfully bind their respective complementary sequences. Any off-target amplification cannot be distinguished from the desired PCR product.
Perform the quantitative PCR reaction and compare to a known internal standard. In the case of transgene copy number the standard is typically a single copy gene [eg. glyceraldehydes-3-phosphate dehydrogenase (GAPDH)]. When quantifying mRNA levels by QPCR, the internal standard is typically mRNA from a house-keeping gene such as β-actin or GAPDH. However, care must be taken when quantifying mRNA since the levels of certain housekeeping genes can change depending on the disease state or the transgene that is expressed.
3.2.4 Liver Toxicity
An important consideration for the development of liver-directed gene therapy is safety. Toxic effects of gene transfer to the liver can be determined in animal models of disease either indirectly (serum chemistry) or directly (histology). The advantage of determining changes in liver enzymes by serum chemistry is that this can be performed repeatedly in the same animal using survival techniques. In contrast, performing histology to directly determine liver toxicity typically involves sacrificing the animal, this is especially true for small (rodent) animal models of disease. In most cases, a thorough evaluation of toxicity involves both serial serum chemistry analyses and histology at a terminal time point.
3.2.5 Serum Chemistries
Anesthetize animals (see above) prior to blood collection to minimize unnecessary stress.
Collect blood from the lateral saphenous vein which proceeds dorsally then laterally over the tarsal joint.
Shave the lateral aspect of the hind leg to expose the skin. Cleanse the area with a topical disinfectant.
Knick the vein with a sterile scalpel.
Collect up to 150μl of blood into either a heparinized or non-heparinized microhematocrit capillary tube, depending on whether plasma or serum is needed, respectively. Apply gentle pressure until bleeding is stopped. Note: This procedure can be performed once every two weeks without altering normal hematologic parameters.
Alternatively, blood can be obtained from the tail.
Cleanse the tail thoroughly with topical disinfectant and cut two to three mm of the tail tip off with sharp sterile scissors.
Collect up to 150μl of blood from the tail tip. Apply gentle pressure to stop the bleeding. Note: Unlike blood collection from the saphenous vein, blood collection from the tail tip can only be performed one or two times. One advantage of collecting blood from the tail tip is that DNA can be isolated from the piece of tissue for genotyping.
The most common serum enzymes that serve as indicators of liver damage are: Serum Glutamic-Oxaloacetic Transaminase (SGOT or AST), Serum Glutamic-Pyruvic Transaminase (SGPT or ALT), Alkaline Phosphatase (ALP), Gamma-Glutamyl Transpeptidase (GGT), Lactic Acid Dehydrogenase (LDH), and Bilirubin. Note: Elevations or reductions in these circulating enzymes are non-specific indicators of liver disease or damage. If the levels of these enzymes deviate from normal a liver specialist should be consulted so more specific liver tests can be performed. It is not cost-effective for most independent research laboratories to establish these techniques in their laboratories. There are numerous contract laboratories that will perform these assays on a fee-for-service basis [National Toxicology Program, Department of Health and Human Services (http://ntp.niehs.nih.gov/index.cfm?objectid=070C9D76-D1D1-84FD-998ECE4408785E21), Quest Diagnostics, Madison, NJ (http://www.questdiagnostics.com)]. Finally, it is critical for the investigator to provide serum from normal control animals since the standards and normal values used by contract laboratories are typically human-specific.
3.2.6 Histology
An initial histological screen for liver toxicity can be performed by staining fixed tissue with Hematoxylin and Eosin (H&E).
Sacrifice animals by anesthetic overdose or by CO2 asphyxiation.
Make a midline abdominal incision to expose the liver.
Remove a piece of liver ranging from several mm3 to 1cm3 from one of the liver lobes and place in one to five ml of tissue fixative for at least 48hr.
Dehydrate the piece of liver and embed in paraffin for sectioning. The sections are then stained with H&E and evaluated by a pathologist with experience examining rodent tissue. Note: Normal control animals will aid in this evaluation. As with serum chemistry analyses, it is inefficient for most independent laboratories to bring this expertise into the laboratory. There are numerous laboratories that will perform histology on a fee-for-service basis [eg. HistoTox Labs, Boulder, CO, (www.histotoxlabs.com), Wax-it, Vancouver, CA, (www.waxitinc.com)].
Specialized histological analyses (eg. electron microscopy, immunohistochemistry, etc.) may be warranted if abnormalities are detected on the H&E-stained sections. The most appropriate analysis can be determined after discussions with an experienced pathologist and is beyond the scope of this review.
References
- 1.Goldman L, Ausiello D, editors. Cecil Textbook of Medicine. 22. Saunders; Philadelphia, PA: 2004. [Google Scholar]
- 2.Hermonat PL, Muzyczka N. Use of adeno-associated virus as a mammalian DNA cloning vector: Transduction of neomycin resistance into mammalian tissue culture cells. Proc Natl Acad Sci. 1984;81:6466–70. doi: 10.1073/pnas.81.20.6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grimm D, Kern A, Rittner K, Kleinschmidt JA. Novel tools for production and purification of recombinant adeno-associated virus vectors. Hum Gene Ther. 1998;9:2745–60. doi: 10.1089/hum.1998.9.18-2745. [DOI] [PubMed] [Google Scholar]
- 4.Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, Summerford C, Samulski RJ, Muzyzcka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–85. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 5.Xiao X, Li J, Samulski RJ. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J Virol. 1996;70:8098–108. doi: 10.1128/jvi.70.11.8098-8108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ponnazhagan S, Mukherjee P, Yoder MC, Wang XS, Zhou SZ, Kaplan J, Wadsworth S, Srivastava A. Adeno-associated virus 2-mediated gene transfer in vivo: Organ-tropism and expression of transduced sequences in mice. Gene. 1997;190:203–10. doi: 10.1016/s0378-1119(96)00576-8. [DOI] [PubMed] [Google Scholar]
- 7.Daly TM, Vogler C, Levy B, Haskins ME, Sands MS. Intravenous injection of recombinant AAV into neonatal mice with mucopolysaccharidosis type VII results in persistent β-glucuronidase expression and widespread reduction of lysosomal storage. Proc Natl Acad Sci. 1999;96:2296–300. doi: 10.1073/pnas.96.5.2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daly TM, Ohlemiller KK, Roberts MS, Vogler CA, Sands MS. Prevention of systemic clinical disease in MPS VII mice following AAV-mediated neonatal gene transfer. Gene Ther. 2001;8:1291–8. doi: 10.1038/sj.gt.3301420. [DOI] [PubMed] [Google Scholar]
- 9.Davidson BL, Stein CS, Heth JA, Martins I, Kotin RM, Derksen TA, Zabner J, Ghodsi A, Chiorini JA. Recombinant adeno-associated virus type 2, 4, and 5 vectors: Transduction of variant cell types and regions in the mammalian central nervous system. Proc Natl Acad Sci. 2000;97:3428–32. doi: 10.1073/pnas.050581197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burger C, Gorbatyuk OS, Velardo MJ, Peden CS, Williams P, Zolotukhin S, Reier PJ, Mandel RJ, Muzyczka N. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol Ther. 2004;10:302–17. doi: 10.1016/j.ymthe.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 11.Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci. 2002;99:11854–9. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakai H, Fuess S, Storm TA, Muramatsu S, Nara Y, Kay MA. Unrestricted hepatocyte transduction with adeno-associated virus serotype 8 vectors in mice. J Virol. 2005;79:214–24. doi: 10.1128/JVI.79.1.214-224.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inagaki K, Fuess S, Storm TA, Gibson GA, McTiernan CF, Kay MA, Nakai H. Robust systemic transduction with AAV9 vectors in mice: Efficient global cardiac gene transfer superior to that of AAV8. Mol Ther. 2006;14:45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Nichols TC, Read MS, Bellinger DA, Verma IM. Sustained expression of therapeutic levels of factor IX in hemophilia B dogs by AAV-mediated gene therapy in liver. Mol Ther. 2000;1:154–8. doi: 10.1006/mthe.2000.0031. [DOI] [PubMed] [Google Scholar]
- 15.Franco LM, Sun B, Yang X, Bird A, Zhang H, Schneider A, Brown T, Young SP, Clay TM, Amalfitano A, Chen YT, Koeberl DD. Evasion of immune responses to introduced human acid alpha-glucosidase by liver-restricted expression in glycogen storage disease type II. Mol Ther. 2005;12:876–84. doi: 10.1016/j.ymthe.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 16.Cooper M, Nayak S, Hoffman BE, Terhorst C, Cao O, Herzog RW. Improved induction of immune tolerance to factor IX by hepatic AAV8 gene transfer. Hum Gene Ther. 2009;20:767–76. doi: 10.1089/hum.2008.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishiwata A, Mimuro J, Mizukami H, Kashiwakura Y, Takano K, Ohmori T, Madoiwa S, Ozawa Sakata Y. Liver-restricted expression of the canine factor VIII gene facilitates prevention of inhibitor formation in factor VIII-deficient mice. J Gene Med. 2009;11:1020–9. doi: 10.1002/jgm.1391. [DOI] [PubMed] [Google Scholar]
- 18.Sun B, Kulis MD, Young SP, Hobeika AC, Li S, Bird A, Zhang H, Li Y, Clay TM, Burks W, Kishnani PS, Koeberl DD. Immunomodulatory gene therapy prevents antibody formation and lethal hypersensitivity reactions in murine Pompe disease. Mol Ther. 2010;18:353–60. doi: 10.1038/mt.2009.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sferra TJ, Backstrom K, Wang C, Rennard R, Miller M, Hu Y. Widespread correction of lysosomal storage following intrahepatic injection of a recombinant adeno-associated virus in the adult MPS VII mouse. Mol Ther. 2004;10:478–90. doi: 10.1016/j.ymthe.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 20.Duan D, Sharma P, Yang J, Yue Y, Dudas L, Zhang Y, Fisher KJ, Engelhardt JF. Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue. J Virol. 1998;72:8568–77. doi: 10.1128/jvi.72.11.8568-8577.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakai H, Yant SR, Storm TA, Fuess S, Meuse L, Kay MA. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J Virol. 2001;75:6969–76. doi: 10.1128/JVI.75.15.6969-6976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hacein-Bey-Abina S, Von Kolle C, Schmidt M, McCormack MP, Wulffrat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, Sorensen R, Forster A, Fraser P, Cohen JI, de Saint Basile G, Alexander I, Wintergerst U, Freborg T, Aurias A, Stoppa-Lyonnet D, Romana S, Radford-Weiss I, Gross F, Valensi F, Delabesse E, Macintyre E, Sigaux F, Soulier J, Leiva LE, Wissler M, Prinz C, Rabbitts TH, Le Deist F, Fischer A, Cavazzana-Calvo M. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–9. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 23.Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, Schmidt M, Kramer A, Schwable J, Glimm H, Koehl U, Preiss C, Ball C, Martin H, Gohring G, Schwarzwaelder K, Hofmann WK, Karakaya K, Tchatchou S, Yang R, Reinecke P, Kuhlcke K, Schlegelberger B, Thrasher AJ, Hoelzer D, Seger R, von Kalle C, Grez M. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198–205. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 24.Nakai H, Wu X, Fuess S, Storm TA, Munroe D, Montini E, Burgess SM, Grompe M, Kay MA. Lareg-scale molecular characterization of adeno-associated virus vector integration in mouse liver. J Virol. 2005;79:3606–14. doi: 10.1128/JVI.79.6.3606-3614.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller DG, Trobridge GG, Petek LM, Jacobs MA, Kaul R, Russell DW. Large-scale analysis of adeno-associated virus vector integration sites in normal human cells. J Virol. 2005;79:11434–42. doi: 10.1128/JVI.79.17.11434-11442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donsante A, Miller DG, Li Y, Vogler C, Brundt EM, Russell DW, Sands MS. AAV vector integration sites in mouse hepatocellular carcinoma. Science. 317:477. doi: 10.1126/science.1142658. [DOI] [PubMed] [Google Scholar]
- 27.Donsante A, Vogler C, Muzyczka N, Crawford JM, Barker J, Flotte T, Campbell-Thompson M, Daly T, Sands MS. Observed incidence of tumorigenesis in long-term rodent studies of rAAV vectors. Gene Ther. 2001;8:1343–46. doi: 10.1038/sj.gt.3301541. [DOI] [PubMed] [Google Scholar]
- 28.Vogler C, Galvin N, Levy B, Grubb J, Jiang J, Zhou XY, Sly WS. Transgene produces massive overexpression of human β-glucuronidase in mice, lysosomal storage of enzyme, and strain-dependent tumors. Proc Natl Acad Sci. 2003;100:2669–73. doi: 10.1073/pnas.0437941100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Embury JE, Charron CC, Poirier AE, Zori A, Carmichael R, Flotte TR, Laipis PJ. Long term portal vein administration of AAV-WPRE vector results in increased incidence of neoplastic disease and hepatic pathology. Mol Ther. 2006;13:S83. [Google Scholar]
- 30.Bell P, Moscioni D, McCarter RJ, Wu D, Gao G, Hoang A, Sanmiguel JC, Sun X, Wivel NA, Raper SE, Furth EE, Batshaw ML, Wilson JM. Analysis of tumors arising in male B6C3F1 mice with and without AAV vector delivery to liver. Mol Ther. 2006;14:34–44. doi: 10.1016/j.ymthe.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 31.Cai SR, Garbow JR, Culverhouse R, Church RD, Zhang W, Shannon WD, McLeod HL. A mouse miodel for developing treatment for secondary liver tumors. Int J Oncol. 2005;27:113–20. [PubMed] [Google Scholar]
- 32.Sands MS, Barker JE. Percutaneous intravenous injection into neonatal mice. Lab Animal Sci. 1999;49:328–31. [PubMed] [Google Scholar]
- 33.Sambrook J, Russell D, editors. Molecular Cloning: A laboratory manual. 3 . Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]