

Abstract
A total synthesis of the unusual ent-kaurane maoecrystal V is described. The synthesis strategy features a counterintuitive early disconnection of the lactone subunit to a polycyclic enol ether intermediate in order to preserve the central tetrahydrofuran ring until the beginning stages of the synthesis. This strategy enables an application of C–H functionalization at the early phase of the synthesis during the construction of a dihydrobenzo-furan intermediate.
Among many known ent-kauranoids,1 maoecrystal V stands out for its atypical molecular architecture in this class of natural products. Isolated and characterized by Sun and co-workers in 2004,2 this unique C19 diterpenoid displayed potent (IC50 = 20 ng/mL) and remarkably selective cytotoxicity against HeLa cells. The pentacyclic framework of maoecrystal V integrates three contiguous quaternary stereocenters (two all-carbon), a bicyclo[2.2.2]octan-2-one subunit, and a strained central tetrahydrofuran flanked by trans-fused six-membered rings. Collectively, these structural characteristics amount to an exquisite challenge for chemical synthesis. Numerous research groups initiated programs directed at the synthesis of maoecrystal V,3 identifying the construction of the quaternary stereogenic centers as one of the main strategic goals and developing many effective solutions. The successful completion of the first total synthesis of maoecrystal V in racemic form, reported by Yang and co-workers in 2010,4 relied on a concise strategy centered on an intramolecular Diels–Alder reaction (IMDA). The reaction enables a rapid assembly of the bicyclooctanone and tetrahydrofuran ring systems in one event, albeit with low facial selectivity for the diene counterpart. The second of the two completed total syntheses of (±)-maoecrystal V reported to date was described by Peng and Danishefsky in 2012.5 Other research groups have also developed creative and efficient approaches based on IMDA chemistry that is envisioned to precede tetrahydrofuran formation.3
Our strategy for the total synthesis of maoecrystal V has emerged from the decision to pursue an early installation of the central oxolane ring. We postulated that an early introduction of the heterocycle would be beneficial for controlling stereoselectivity in several key transformations. Conversely, its construction late in the synthesis would carry undesired risks due to its strained nature. Following this line of analysis, a plan summarized in Scheme 1 was developed.
Scheme 1.
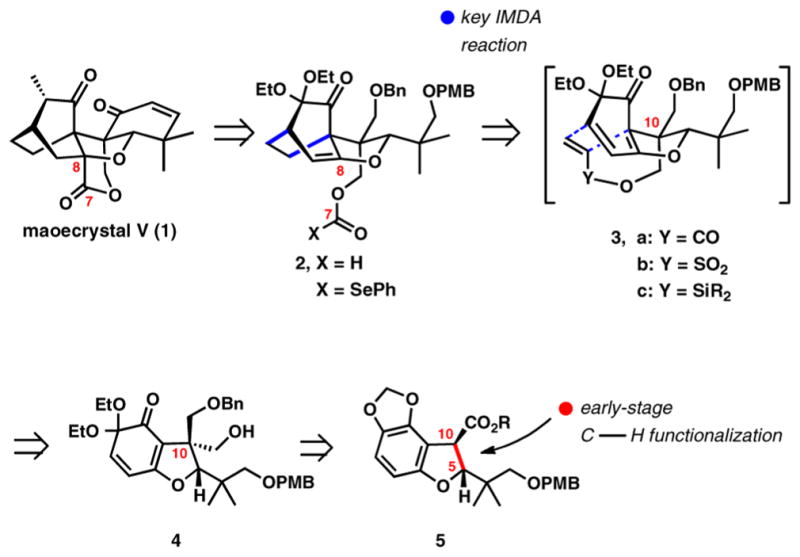
Synthesis Design for Maoecrystal V
The strategic constrains of early THF installation suggested a somewhat counterintuitive initial disassembly of the lactone along the C7—C8 bond (1⇒2, Scheme 1). We envisioned that the target lactone subunit could be accessed from an appropriately functionalized formate 2 (X=H) by an intra-molecular hydroformylation or radical cyclization of selenocarbonate (2, X=SePh) onto the enol ether. The bicyclo[2.2.2]-octanone is unraveled further by an IMDA disconnection (2⇒3). The hydroxymethyl group at C10 used previously to deliver the lactone carbonyl group is now refunctionalized to direct a synthetic equivalent of ethylene for the IMDA reaction, thus achieving the requisite facial selectivity. In our preliminary investigations,8 we validated the efficiency of the IMDA reaction and stability of the enol ether in the case of acrylate and vinylsulfonate adducts 3a (Y=CO) and 3b (Y=SO2). However, when faced with removal of the linking functional groups, we found them to be less than ideal, necessitating lengthy and cumbersome detours for their deletion. In search for an alternative, we found that a silyl group serves the purpose of enabling both an efficient [4 + 2] cycloaddition and an effective tether removal.9 This modification provides a direct path from 4 to 2. Further analysis led to functionalized dihydrobenzofuran intermediate 5, which we planned to prepare by a C–H-insertion process along the highlighted C5—C10 bond. In the long term, the C–H-insertion reaction can potentially be carried out using asymmetric catalysis,10 paving the way for the enantioselective synthesis of maoecrystal V.
The goal of initial studies was to define a synthetic path to maoecrystal V in racemic form according to the aforementioned synthesis design. The preparation of the substrate for the early C–H functionalization reaction began with alkylation of sesamol using 3-(4-methoxybenzyloxy)-2,2-dimethyl-1-propanol (6) in the presence of diisopropyl azodicarboxylate and triphenylphosphine,11 which afforded aryl ether 7 in 79% yield (Scheme 2). Its directed ortho-metalation with n-butyllithium in THF, followed by transmetalation to the arylzinc reagent and coupling with methyl chlorooxaloacetate, afforded α-keto ester 8,12 which was converted directly to α-diazoester 9.13 The essential C–H-insertion was achieved by treatment of 9 with rhodium acetate that provided the benzofuran product 10 in 74% yield and >10:1 diastereoselectivity in favor of the trans-substituted isomer.14
Scheme 2.

Further elaboration included stereoselective installation of the quaternary carbon center at C10 using zincate enolate generated from 10 and benzyl chloromethyl ether (76% yield, dr 9:1).15 Subsequent reduction of the ester group with lithium aluminum hydride and opening of the formyl acetal of catechol with methylmagnesium bromide,16 achieved upon heating in benzene, afforded 13. Oxidation of the substituted phenol with iodobenzene diacetate/EtOH and vinylsilylation of the primary hydroxy group in the resulting protected ortho-quinone intermediate provided the substrate for the intramolecular Diels–Alder reaction (14) in high yield.
As with other related substrates, the IMDA reaction occurred cleanly upon heating a solution of 14 in toluene at 110 °C, giving bicyclo[2.2.2]octane cycloadduct 15 in a nearly quantitative yield. The enol ether within the ring system of 15 was found to be rather stable and generally suitable for subsequent transformations.
Our next major goal was the appendage of the lactone ring. We planned to accomplish this task by a rare formyl radical endo-cyclization onto the enol ether double bond (Scheme 3a).17 This approach first required desilylation of 15, which was found to be rather challenging. The substrate for desilylation reaction was accessed after an efficient reductive removal of the gem-diethoxy substituents with samarium(II) iodide (90% yield, Scheme 3b).18 Under extensively optimized reaction conditions, the silyl group could be removed in a reproducible 50% yield upon treatment with tetra-n-butylammonium fluoride in DMPU at 0 °C for 1 h. The unusually high reactivity of the trialkylsilyl ether is due to its position within the bicyclo[2.2.2]-octanone ring system, and the presence of the carbonyl group is essential for high reactivity. For example, when the carbonyl group is replaced with a hydroxyl, the resulting compound is substantially more resistant to desilylation.19
Scheme 3.

After straightforward derivatization to selenocarbonate20 18, our efforts concentrated on the lactone closure by radical cyclization. Multiple attempts with tri-n-butyltin hydride as the reagent using several initiators (AIBN, ACHN, V70) under a variety of addition protocols and temperature regimes resulted only in reduction to formate 23 (Scheme 3c), with none of the desired lactone 19 observed. Clearly, the reaction of the initially generated formyl radical with the hydrogen atom donor, n-Bu3SnH,21 was too rapid relative to the desired cyclization (step a, Scheme 3a). We hypothesized that using a less efficient hydrogen atom donor would result in a more effective ring closure. Tris(trimethylsilyl)silane ((Me3Si)3SiH) was the reagent of choice.21 To our delight, slow addition of a mixture of (Me3Si)3SiH and AIBN to a solution of phenylselenocarbonate 18 in benzene at 80 °C resulted in the successful formation of lactone 19 (55% yield), along with a minor amount of byproduct 24 resulting from radical fragmentation (12%).22
Formation of the last six-membered ring, the gem-dimethyl-substituted cyclic enone, was required to complete the total synthesis of maoecrystal V. Toward that goal, removal of the p-methoxybenzyl group,23 oxidation of the exposed primary hydroxyl with Dess-Martin periodinane (DMP),24 and the Wittig methylenation25 delivered 20. Our synthetic studies revealed that ketone 20 is the optimal substrate for the introduction of the C17 methyl group, an operation that proved to be difficult to achieve with any level of stereocontrol in the previous total synthesis efforts. In this instance, addition of iodomethane to the enolate derived from 20 and LiN(SiMe3)2 afforded the desired product as the major component in a 7:1 mixture of diastereomers (90% combined yield). After debenzylation (DDQ, wet CH2Cl2, 50 °C, 12 h), the diastereomers were separated, and the major isomer was advanced to intermediate 22 by oxidation to the aldehyde with DMP and chemoselective addition of vinylmagnesium bromide in the presence of anhydrous cerium(III) chloride.26 Maoecrystal V was obtained in two additional steps, which included ring-closing metathesis27 and oxidation to enone with Dess-Martin periodinane.
In closing, a concise total synthesis of maoecrystal V has been accomplished (24 steps, ~1.5% overall yield from sesamol). The strategic focus on the central strained tetrahydrofuran ring resulted in an initial disassembly of the lactone ring to a polycyclic enol ether. The enol ether was constructed by an IMDA reaction of a tethered CH2=CH2 equivalent with a 2,4-cyclohexadienone fragment obtained by oxidative dearomatization of a dihydrobenzofuran intermediate. This intermediate, in turn, was prepared by an effective rhodium-catalyzed C–H functionalization reaction which can potentially be modified to access enantioenriched products using chiral rhodium catalysts.10
Supplementary Material
Acknowledgments
We dedicate this manuscript to Professor Larry E. Overman in honor of his 70th birthday. Financial support for this work was provided by the NSF (CHE-0836757), NIH (R01 GM077379), and additional kind gifts from Amgen and Eli Lilly. We would like to thank Professor Alexei Novikov (University of North Dakota) for helpful discussions. Dr. Hongjun Zhou is thanked for continued assistance with NMR spectroscopy. We are grateful to Dr. James Pavlovich for performing the high-resolution mass-spectroscopic analysis.
Footnotes
The authors declare no competing financial interest.
Experimental procedures and characterization data for all reactions and products. Copies of 1H, 13C, HSQC, and NOESY NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Sun HD, Huang SX, Han QB. Nat Prod Rep. 2006;23:673–698. doi: 10.1039/b604174d. [DOI] [PubMed] [Google Scholar]
- 2.Li SH, Niu XM, Shen YH, Zhang HJ, Sun HD, Li ML, Tian QE, Lu Y, Cao P, Zhang QT. Org Lett. 2004;6:4327–4330. doi: 10.1021/ol0481535. [DOI] [PubMed] [Google Scholar]
- 3.Synthetic studies toward maocrystal V: Gong J, Lin G, Li CC, Yang Z. Org Lett. 2009;11:4770–4773. doi: 10.1021/ol9014392.Krawczuk PJ, Schone N, Baran PS. Org Lett. 2009;11:4774–4776. doi: 10.1021/ol901963v.Peng F, Yu M, Danishefsky SJ. Tetrahedron Lett. 2009;50:6586–6587. doi: 10.1016/j.tetlet.2009.09.050.Nicolaou KC, Dong L, Deng LJ, Talbot AC, Chen DYK. Chem Commun. 2010;46:70–72. doi: 10.1039/b917045f.Lazarski KE, Hu DX, Sterm CL, Thomson RJ. Org Lett. 2010;12:3010–3013. doi: 10.1021/ol101025r.Singh V, Bhalerao P, Mobin SM. Tetrahedron Lett. 2010;51:3337–3339.Baitinger I, Mayer P, Trauner D. Org Lett. 2010;12:5656–5659. doi: 10.1021/ol102446u.Dong L, Deng L, Lim YH, Leugn GYC, Chen DYK. Chem—Eur J. 2011;17:5778–5781. doi: 10.1002/chem.201100232.Peng F, Danishefsky SJ. Tetrahedron Lett. 2011;52:2104–2106. doi: 10.1016/j.tetlet.2010.11.029.Lazarski KE, Akpinar B, Thomson RJ. Tetrahedron Lett. 2013;54:635–637. doi: 10.1016/j.tetlet.2012.11.135.Carberry P, Viernes DR, Choi LB, Fegley MW, Chisholm JD. Tetrahedron Lett. 2013;54:1734–1737.Total synthesis of (−)-maoecrystal Z: Cha JY, Yeoman JTS, Reisman SE. J Am Chem Soc. 2011;133:14964–14967. doi: 10.1021/ja2073356.Recent total synthesis of related ent-kauranoids: Yeoman JTS, Mak VW, Reisman SE. J Am Chem Soc. 2013;135:11764–11767. doi: 10.1021/ja406599a.
- 4.Gong J, Lin G, Sun W, Li C, Yang Z. J Am Chem Soc. 2010;132:16745–16746. doi: 10.1021/ja108907x. [DOI] [PubMed] [Google Scholar]
- 5.Peng F, Danishefsky SJ. J Am Chem Soc. 2012;134:18860–18867. doi: 10.1021/ja309905j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For reviews on the intramolecular Diels–Alder reaction, see: Roush WR. Intramolecular Diels–Alder Reactions. In: Trost BM, Fleming I, editors. Comprehensive Organic Synthesis. Vol. 5. Pergamon Press; New York: 1991. pp. 513–550.Takao K-i, Munakata R, Tadano K-i. Chem Rev. 2005;105:4779–4807. doi: 10.1021/cr040632u.Tadano K-i. Eur J Org Chem. 2009:4381–4394.Juhl M, Tanner D. Chem Soc Rev. 2009;38:2983–2992. doi: 10.1039/b816703f.. Selected applications in synthesis: Xiao Q, Young K, Zakarian A. Org Lett. 2013;15:3314–3317. doi: 10.1021/ol401354a.Tortosa M, Yakelis NA, Roush WR. J Am Chem Soc. 2008;130:2722–2723. doi: 10.1021/ja710238h.
- 7.Reviews on dearomatization-Diels–Alder strategies in total synthesis: Roche SP, Porco JA. Angew Chem, Int Ed. 2011;50:4068–4093. doi: 10.1002/anie.201006017.Magdziak D, Meek SJ, Pettus TRR. Chem Rev. 2004;104:1383–1429. doi: 10.1021/cr0306900.. Recent examples of masked ortho-quinones as substrates for Diels–Alder reactions: Chu CS, Lee TH, Rao PD, Song LD, Liao CC. J Org Chem. 1999;64:4111–4118.Wenderski TA, Marsini MA, Pettus TRR. Org Lett. 2011;13:118–121. doi: 10.1021/ol102652t.Kobayakawa Y, Nakada M. Angew Chem, Int Ed. 2013;52:7569–7573. doi: 10.1002/anie.201303224.Snyder SA, Kontes F. J Am Chem Soc. 2009;131:1745–1752. doi: 10.1021/ja806865u.
- 8.Gu Z, Zakarian A. Org Lett. 2011;13:1080–1082. doi: 10.1021/ol1031238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Review on silicon-tethered reactions: Bols M, Skydstrup T. Chem Rev. 1995;95:1253–1277.. For seminal examples of silicon-tethered Diels–Alder reactions: Stork G, Chan TY, Breault GA. J Am Chem Soc. 1992;114:7578–7579.
- 10.Review: Davies HML, Manning JR. Nature. 2008;451:417–424. doi: 10.1038/nature06485.For recent examples of enantioselective synthesis of dihydrobenzofurans by C—H-insertion, see: Wang HB, Li G, Engle KM, Yu JQ, Davies HML. J Am Chem Soc. 2013;135:6774–6777. doi: 10.1021/ja401731d.Koizumi Y, Kobayshi H, Wakimoto T, Furuta T, Fukuyama T, Kan T. J Am Chem Soc. 2008;130:16854–16855. doi: 10.1021/ja807676v.Reddy RP, Lee GH, Davies HML. Org Lett. 2006;8:3437–3440. doi: 10.1021/ol060893l.Saito H, Oishi H, Kitagaki S, Nakamura S, Anada M, Hashimoto S. Org Lett. 2002;4:3887–3890. doi: 10.1021/ol0267127.
- 11.Mitsunobu O. Synthesis. 1981:1–28. [Google Scholar]
- 12.Velkov J, Mincheva Z, Bary J, Boireau G, Fujier C. Synth Commun. 1997;27:375–378. [Google Scholar]
- 13.Xu HD, Zhang W, Shu DX, Werness JB, Tang WP. Angew Chem, Int Ed. 2008;47:8933–8936. doi: 10.1002/anie.200803910. [DOI] [PubMed] [Google Scholar]
- 14.The assignment of relative stereochemistry is tentative and is based on literature precedent; see refs 10b–e.
- 15.Morita Y, Suzuki M, Noyori R. J Org Chem. 1989;54:1785–1787.. Recent applications: Ilardi EA, Isaacman MJ, Qin YC, Shelly SA, Zakarian A. Tetrahedron. 2009;65:3261–3269.Iimira S, Overman LE, Paulini R, Zakarian A. J Am Chem Soc. 2006;128:13095–13101. doi: 10.1021/ja0650504.Fisher EL, Wilkerson-Hill SM, Sarpong R. J Am Chem Soc. 2012;134:9946–9949. doi: 10.1021/ja3045647.
- 16.Beltrame P, Gelli G, Lai A, Monduzzi M. J Org Chem. 1976;41:580–581. [Google Scholar]
- 17.For selective examples on radical cyclizations with enol ethers as acceptors, see: Begley MJ, Ladlow M, Pattenden G. J Chem Soc, Perkin Trans 1. 1988:1095–1100.Hori N, Matsukura H, Nakata T. Org Lett. 1999;1:1099–1101.Davies KA, Wulff JE. Org Lett. 2011;13:5552–5555. doi: 10.1021/ol202286q.
- 18.For recent reviews on samarium iodide mediated reactions, see: Nicolaou KC, Ellery SP, Chen JS. Angew Chem, Int Ed. 2009;48:7140–7165. doi: 10.1002/anie.200902151.Edmonds DJ, Johnston D, Procter DJ. Chem Rev. 2004;104:3371–3403. doi: 10.1021/cr030017a.Gopalaiah K, Kagan HB. New J Chem. 2008;32:607–637.Nicolaou KC, Toh QY, Chen DYK. J Am Chem Soc. 2008;130:11292–11293. doi: 10.1021/ja804588r.
-
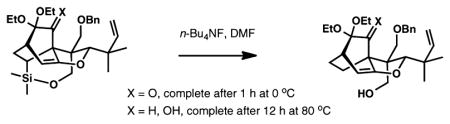
19.In a closely related system, the desilylation of the reduced substrate was sluggish at 23 °C with only a trace amount of product formed. A complete reaction was achieved upon heating at 80 °C for 12 h. Desylilation of the corresponding ketone was complete within 1 h at 0 °C:
- 20.For recent examples of applications of radical cyclizations with selenocarbonates or selenoesters in total synthesis, see: Trost BM, Waser J, Meyer A. J Am Chem Soc. 2007;129:14556–14557. doi: 10.1021/ja076165q.Zaimoku H, Taniguchi T, Ishibashi H. Org Lett. 2012;14:1656–1658. doi: 10.1021/ol300280s.
- 21.Bond dissociation energies: (a) 74 kcal/mol for n-Bu3Sn—H: Burkey TJ, Majewski M, Griller D. J Am Chem Soc. 1986;108:2218–2219. doi: 10.1021/ja00269a017.. (b) 79.0 kcal/mol for (Me3Si)3Si—H: Kanabus-Kaminska JM, Hawari JA, Griller D, Chatgilialoglu C. J Am Chem Soc. 1987;109:5267–5268.Chatgilialoglu C, Griller D, Lesage M. J Org Chem. 1988;53:3642–3644.
-
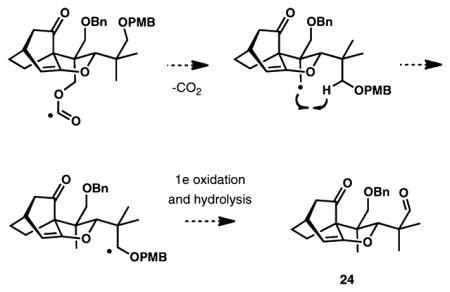
22.A proposed pathway for the formation of compound 24 is illustrated in the graphic below. Virtually no formyl radical reduction product was observed with (Me3Si)3SiH.
- 23.Horita K, Yoshioka T, Tanaka T, Oikawa Y, Yonemitsu O. Tetrahedron. 1986;42:3021–3028. [Google Scholar]
- 24.Dess DB, Martin JC. J Org Chem. 1983;48:4155–4156. [Google Scholar]
- 25.Wittig G, Schoöllkopf U. Chem Ber. 1954;87:1318–1330. [Google Scholar]
- 26.Imamoto T, Takiyama N, Nakamura K, Hatajima T, Kamiya Y. J Am Chem Soc. 1989;111:4392–4398. [Google Scholar]
- 27.Reviews on applications of ring-closing metathesis reactions in total synthesis: Nicolaou KC, Bulger PG, Sarlah D. Angew Chem, Int Ed. 2005;44:4490–4527. doi: 10.1002/anie.200500369.Cossy J, Arsenyadis S, Meyer C, editors. Metathesis in Natural Product Synthesis. Wiley-VCH; Weinheim, Germany: 2010. Fürstner A. Chem Commun. 2011;47:6505–6511. doi: 10.1039/c1cc10464k.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.