

Abstract
Interleukin-33 (IL-33), an IL-1 family cytokine and nuclear alarmin, is constitutively expressed in epithelial barrier tissues and human blood vessels. However, little is known about the induced expression of IL-33 in monocytes and macrophages, which are major cytokine-producing cells of the innate immune system. Here we report the induction of IL-33 expression in both human monocytes and mouse macrophages from C57BL/6 mice by the acute-phase protein serum amyloid A (SAA). SAA induced transcriptional activation of the IL-33 gene, resulting in nuclear accumulation of the IL-33 protein. TLR2, one of the SAA receptors, was primarily responsible for the induction of IL-33. Progressive deletion of the human IL-33 promoter led to the identification of two potential binding sites for interferon regulatory factor 7 (IRF7), one of which (−277/−257) was found to be important for SAA-stimulated IL-33 promoter activity. IRF7 was recruited to the IL-33 promoter upon SAA stimulation, and silencing IRF7 expression in THP-1 cells abrogated SAA-induced IL-33 expression. SAA also promoted an interaction between TRAF6 and IRF7. Taken together, these results identify IRF7 as a critical transcription factor for SAA-induced IL-33 expression in monocytes and macrophages.
Keywords: serum amyloid A, macrophages, IL-33, IRF7
Introduction
Interleukin-33 (IL-33), also known as IL-1F11 and NF-HEV[1], is a recently identified member of the IL-1 cytokine family, which includes IL-1α, IL-1β, IL-18 and IL-1 receptor antagonist [2]. Human IL-33 is a 270-amino acid protein with a predicted N-terminal nuclear localization signal, a helix-turn-helix motif and an IL-1-like C-terminal domain [2]. Similar to IL-1α, IL-33 functions as an intracellular factor with transcriptional regulatory properties and an extracellular protein with cytokine properties [2, 3]. It has been reported that the helix-turn-helix motif of IL-33, not the nuclear localization signal, is responsible for nuclear localization of IL-33, heterochromatin association and targeting to mitotic chromosomes; however, the exact structure for IL-33 nuclear localization has not been confirmed [3]. The C-terminal portion of IL-33 contributes to its biological activity. Unlike IL-1β and IL-18, which require caspase-1 cleavage for maturation and secretion [2], full-length IL-33 is biologically active without caspase-1 processing [4, 5]. Similar to high mobility group box 1, IL-33 is released by necrotic cells in an active form, but it is inactivated during apoptosis through cleavage by caspase-3 and caspase-7 [6]. Outside the cells, IL-33 exerts its biological functions through the ST2/IL-1R accessory protein complex, which signals through its Toll/Interleukin-1 receptor domain [7]. Recent studies have shown that IL-33 is associated with a range of pathological conditions including infection, asthma, anaphylactic shock, allergic inflammation, arthritis, ulcerative colitis, systemic sclerosis and several cardiovascular disorders [8].
Despite increasing awareness of its clinical implications, IL-33 has not been well characterized for its induced expression by environmental stimuli. Moreover, the mode of IL-33 release remains poorly understood. It is also unclear whether IL-33 primarily functions as a cytokine or a nuclear factor. IL-33 is constitutively expressed in epithelial cells of various species, human endothelial cells, fibroblasts, smooth muscle cells and mast cells [8, 9]. The expression of IL-33 in macrophages is modest but can be induced by pro-inflammatory stimuli such as LPS [10]. The purpose of this study was to explore the mechanism of IL-33 induction in monocytes/macrophages. We report that the acute-phase protein serum amyloid A (SAA) provides signals for the transcription and translation of IL-33, which is produced as an intracellular protein with primary localization in the nucleus.
The acute-phase response is a systemic reaction to internal and external changes that alert the body for potential danger. These environmental changes include microbial infection, tissue injury, trauma or surgery, neoplastic growth, and immunological disorders. In acute-phase reactions, SAA is one of the major proteins produced by hepatocytes through induction, and the induced SAA is subsequently released to the blood circulation [11]. Within the first 2–3 days after initiation of an acute-phase response, the plasma level of SAA can often increase by 1000-fold, which follows trauma [12] and infection [13]. Inflammatory cytokines including IL-1β, IL-6, and TNF-α contribute to the induction of SAA [14]. SAA is a biomarker for inflammatory diseases and is associated with the pathogenesis of chronic diseases such as secondary amyloidosis [15], atherosclerosis [16], obesity [17], diabetes [18] and rheumatoid arthritis [19]. In blood circulation, SAA is bound to high density lipoproteins and is related to the loss of protective function of these proteins against the development of atherosclerosis [20]. SAA is also present in a lipid-poor form in various tissues, produced by inflammatory cells such as activated macrophages. Recombinant SAA has cytokine-inducing properties through activation of several receptors including formyl peptide receptor 2 (FPR2), TLR2, TLR4, and scavenger receptor BI [21–24]. Furthermore, previous reports have shown that the lipid-poor SAA is a potent ligand for the induced expression of granulocyte colony-stimulating factor in whole animal [25] and ex vivo [26]. In vitro studies have shown that SAA stimulates the secretion of IL-8 [27], IL-12 and IL-23 [28]. Since IL-33 is known as an alarmin [8], we sought to determine whether SAA has an effect on its induced expression in monocytes and macrophages. In the present study, we report that SAA is a potent inducer of IL-33 expression. We also identified an IRF7-interacting element within the human IL-33 promoter and found that IRF7 is required for IL-33 expression. These results provide new insights into the regulation of IL-33 expression in macrophages activated by an acute-phase protein.
Results
Induction of IL-33 mRNA by SAA in monocytes and macrophages
SAA has been shown to induce the expression of various proinflammatory cytokines in leukocytes. Using cDNA microarrays, we analyzed the gene expression profile in SAA-stimulated macrophages and identified IL33 as one of the up-regulated genes (data not shown). This result was verified in CD14+ monocytes from human blood (Fig. 1A) and in mouse peritoneal macrophages (Fig. 1B and C), as determined by real-time PCR. In human monocytes, optimal induction of the IL-33 transcript was observed 12 hours after stimulation with 0.05 μM of recombinant SAA (Fig. 1A). In mouse peritoneal macrophages, the Il33 transcript appeared 2 hours after SAA stimulation and peaked at about 8 hours (Fig. 1B); maximal induction was observed with 0.05 to 0.5 μM of SAA (equivalent to 0.6 μg/mL to 6 μg/mL of SAA). Next, selected TLR ligands were used to detect their ability to induce IL-33 expression. Consistent with a previous report [10], both the TLR4 ligand LPS (100ng/mL) and the TLR2 ligand Pam3CSK4 (100 ng/mL) induced the expression of IL-33. In contrast, the TLR3 ligand polyI: C (up to 10 μg/mL) had minimal effect on IL-33 mRNA levels (Fig. 1D).
Figure 1.

Time and dose-dependent induction of IL-33 transcript by SAA in monocytes and macrophages. (A) Freshly isolated human blood CD14+ monocytes were stimulated with 0.05 μM SAA for 12 and 24 hours (left panel), or with SAA at 0.05 or 1 μM for 12 hours (right panel) prior to qRT-PCR detection of the IL-33 transcript. (B and C) Mouse peritoneal macrophages were treated with (B) 0.5 μM of SAA for the indicated periods of time, or (C) stimulated for 8 hours with various concentrations of SAA prior to qRT-PCR detection of the IL-33 transcript. (D) THP-1 cells were stimulated for 12 hours with LPS (100 ng/mL), Pam3CSK4 (Pam, 100 ng/mL), polyI:C (10 μg/mL), SAA (0.5 μM) or medium control (Con.). Results are expressed as relative levels of the IL-33 transcript, with that of the unstimulated cells set as 1. (A–D) Data are shown as means + SEM of six samples pooled from three independent experiments. Statistical significance was determined using two-tailed paired Student’s t-test. *p < 0.05, **p < 0.01, (SAA-treated cells versus untreated cells).
SAA-induced IL-33 proteins are localized in the nucleus
We sought to determine whether induction of IL-33 mRNA was followed by an increase in IL-33 protein levels. Figure 2A shows a time-dependent induction of the IL-33 protein that peaked at 8 hours after SAA stimulation in THP-1 cells. In the acute-phase response, plasma SAA concentration can easily reach micromolar concentrations. Consistent with the dose required for the induction of IL-33 transcript, the optimal concentration for IL-33 protein induction was 0.05 μM of SAA (Fig. 2B).
Figure 2.

SAA-induced IL-33 proteins are localized in the nucleus. (A) THP-1 cells were stimulated with 0.05 μM SAA for various time periods as indicated, or (B) for 8 hours with SAA at various concentrations. The induced IL-33 protein was detected by western blotting, using β-actin as a loading control. (C) THP-1 cells were treated as in (A) and the induced IL-33 protein in the nuclear and cytosolic fractions were detected by western blotting. In A–C, graphs show protein levels relative to β-actin or HDAC1 quantified by densitometry (means + SEM) from three samples pooled from three independent experiments. *p < 0.05, (SAA-treated cells versus untreated cells), Student’s t-test. (D) HeLa-TLR2 cells were stimulated with 0.05 μM of SAA for the indicated times and analyzed by confocal microscopy (63× objective, scale bar, 20 μm). The IL-33 protein is shown in green and nuclei in blue. (A–D) Results are representative images and blots from one out of at least three independent experiments performed.
Since IL-33 can function as a nuclear factor in an intracrine manner, or as an extracellular alarmin in a “necrocrine” manner [2, 3], we performed ELISA to determine whether IL-33 protein was released from SAA-stimulated cells. Very little IL-33 protein was detected in the culture medium after 48 hours of SAA stimulation in THP-1 cells (Supporting Information Fig. 1A). It is likely that the small amount of IL-33 in the culture medium came from dead cells. Next, we prepared nuclear and cytosolic fractions from the same cell culture to detect IL-33 expression. As shown in Fig. 2C, IL-33 was detected primarily in the nucleus after 4 hours of SAA stimulation. Our previous data showed that TLR2-expressing HeLa cells (HeLa-TLR2) could response to SAA for cytokine production [24]. Thus, HeLa-TLR2 was used to confirm the nuclear localization of IL-33 with immunofluorescent microscopy with a clear cytoskeletal and nuclear morphology. In these stably transfected cells, IL-33 protein (green fluorescence; Fig. 2D) was detected at 4, 8 and 24 hours after SAA stimulation and was co-localized with the blue DAPI stain. Taken together, these results indicate that the SAA-induced IL-33 was predominantly localized in the nucleus.
Identification of SAA receptors that mediate induced expression of IL-33
SAA is known to activate multiple receptors including TLR2 and FPR2, all capable of mediating cytokine induction [24, 27]. To identify the receptors responsible for IL-33 induction, a neutralizing antibody and an antagonist were applied to THP-1 cells prior to SAA stimulation. The SAA-induced IL-33 expression was significantly inhibited by a neutralizing antibody against TLR2 (1μg/mL) (Fig. 3A). Likewise, the antibody also reduced IL-33 induction by Pam3CSK4 (Fig. 3A). In peritoneal macrophages from wild type C57BL/6 mice, SAA induced potent expression of IL-33 (Fig. 3B). However, little induction of Il33 was seen in peritoneal macrophages from Tlr2−/− mice. FPR2, a G protein-coupled receptor known to mediate SAA-induced expression of certain cytokines and chemokines [27, 29], may also contribute to SAA induction of IL-33 as mice lacking Fpr2 showed reduced Il33 expression (Fig. 3B). WKYMVm, a highly potent FPR2 agonist that induces Ca2+ mobilization at low nanomolar concentrations [30], was unable to stimulate IL-33 expression when used alone at 500 nM, but it potentiated IL-33 induction by Pam3CSK4 (Fig. 3C). WKYMVm could potentiate SAA-induced IL-33 only when SAA was used at low concentrations (Supporting Information Fig. 2A). WRW4, an antagonist that blocks FPR2 binding to WKYMVm [31], partially inhibited the SAA-induced expression of IL-33 at a high concentration (20 μM, Supporting Information Fig. 2B). Taken together, these results suggest that the SAA-induced IL-33 is primarily mediated by TLR2, but synergy may be produced with an activated FPR2.
Figure 3.
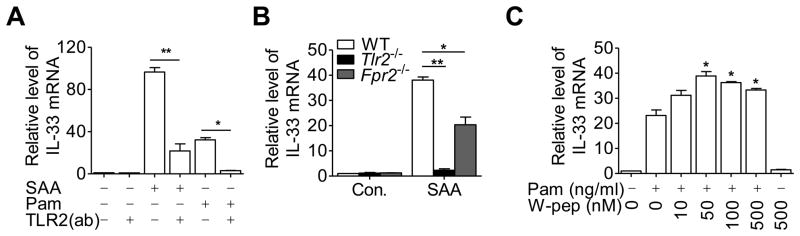
Receptors involved in SAA-induced expression of IL-33. Relative changes in IL-33 mRNA compared with basal level (set as 1) were detected by qRT-PCR, using the following experimental conditions. (A) TLR2-neutralizing antibody (TLR2(ab), 1μg/mL) was applied 1 hour before stimulation with SAA (0.05 μM) or Pam3CSK4 (Pam)(100 ng/mL) in THP-1 cells. (B) Peritoneal macrophages from Tlr2−/− (left) or Fpr2−/− mice and from wildtype littermate controls were stimulated with 0.05 μM of SAA for 8 hours. (C) THP-1 cells were stimulated for 8 hours with Pam3CSK4 (100 ng/mL) in the absence or presence of the indicated concentrations of WKYMVm (W-pep) or W-pep alone. Data shown are means + SEM of six samples pooled from(A, C) three independent experiments or (B) two independent experiments each with n=4 mice/group. *p < 0.05, **p < 0.01; Pam-treated cells versus Pam plus W-pep-treated cells in (C). Differences were analyzed by (A, B) one-way ANOVA followed by Bonderroni post-testing, (C) paired t-test.
SAA stimulates transcription of the IL-33 gene
To investigate the molecular mechanisms of SAA-induced IL-33 expression, we used cycloheximide (CHX), an inhibitor of protein synthesis, and actinomycin D (ActD), an inhibitor of transcription, to determine the level at which IL-33 was induced by SAA. The SAA-induced IL-33 protein expression was significantly inhibited by ActD as well as CHX (Fig. 4A), whereas induction of the IL-33 transcript was inhibited by ActD but not CHX (Fig. 4B). To confirm that SAA induces transcription of the IL-33 gene, a 1.05-kb DNA fragment (−1050/+49) of the human IL-33 promoter was ligated into a pGL3 firefly luciferase reporter plasmid. The resulting plasmid was transfected into the easy-to-transfect HeLa-TLR2 cells [24], which were then stimulated with SAA for 16 hours prior to measurement of luciferase activity. As shown in Fig. 4C, stimulation of the transfected HeLa-TLR2 cells with SAA doubled the IL-33 promoter-driven expression of firefly luciferase, compared with similarly treated cells without TLR2 expression. These results suggest that SAA stimulates transcription of the IL-33 gene.
Figure 4.

SAA regulates IL-33 gene transcription. (A) THP-1 cells were pretreated with cycloheximide (CHX, 100 μM) or actinomycin D (ActD, 10 μg/mL) for 1 hour before stimulation with SAA (0.05 μM). After 8 hours, IL-33 protein in cell lysate was detected by western blotting, using β-actin as a loading control. Densitometry values (means + SEM) from three samples pooled from three independent experiments are shown in the right panel. (B) Real-time PCR results showing changes in IL-33 transcript level using cells from the experiments in (A). (C) The −1050/+49 region of the human IL-33 promoter construct was transiently transfected into HeLa-TLR2 or HeLa cells together with the control plasmid pRL-TK. The transfected cells were treated with SAA for 16 hours. Relative luciferase activity was calculated by normalizing firefly luciferase activity to renilla luciferase activity. Data are shown as mean + SEM of six samples pooled from three independent experiments. (D) Several putative transcription factor binding sites were predicted by MatInspector and are underlined. (E) Schematic drawing of the promoter region with major regulatory elements marked (left). The corresponding changes in SAA-induced luciferase activity (0.05 μM, 16 hours) in HeLa-TLR2 cells are shown to the right side of each construct. (F) HeLa-TLR2 cells were individually transfected with IL-33-pro WT(−662/+49 of IL-33 promoter), IL-33-pro (Mut.A, a mutation in IRF7 #1 site), IL-33-pro (Mut.B, a mutation in IRF7 #2 site) or IL-33-pro (Mut.AB, mutations in IRF7 #1 and IRF7 #2 site). Relative luciferase activity of each construct, as measured by firefly luciferase activity, was normalized against that of the renilla luciferase, encoded by the cotransfected pRL-TK plasmid. Data in (E-F) represent means + SEM of six samples pooled from three independent experiments. *p < 0.05, **p < 0.01, one-way ANOVA followed by Bonderroni post-testing.
Characterization of the IL-33 promoter
To elucidate the molecular basis for IL-33 transcriptional regulation by SAA, the sequence about 1000-bp upstream of the transcription starting site of the human IL-33 gene was analyzed using Mat Inspector (Genomatix Software, Munchen, Germany). The result is shown in Fig. 4D. Several putative cis elements were identified, including four AP-1 sites localized at −971/−968 (AP-1 #1), −937/−930 (AP-1 #2), −806/−803 (AP-1 #3), and −457/−451 (AP-1 #4). There is also one NF-κB site at −510/−498, and two interferon regulatory factor 7 (IRF7) binding sites localized at −277/−257 (IRF7 #1) and −255/−235 (IRF7 #2). The involvement of these cis elements in SAA-induced IL-33 transcription was examined using sequential deletion from the 5′ end (Fig. 4E). The resulting DNA fragments were placed upstream of a luciferase reporter in the pGL3 plasmid and transiently transfected into the HeLa-TLR2 cells, which were then stimulated with SAA for the induction of luciferase activity. As shown in Fig. 4E, sequential deletion from −1000 through −300, including removal of all AP-1 sites and the NF-κB site, only minimally altered the IL-33 promoter activity. However, the response to SAA was significantly reduced in cells transfected with the −160 plasmid construct, which lacked the two putative IRF7 sites. Previous report showed that the consensus sequence for IRF7 binding is 5′-GAA(A/T)N(C/T)GAAAN (T/C)-3′ [32]. The sequence of IRF7 #1 (GAATTTCAAAAT, IRF7 #1) and of IRF7 #2 (GAAGTTGAAGTT) are similar but not identical to the consensus IRF-7 binding sequence (Fig. 4D). To determine whether these sites are indeed essential for IL-33 promoter activation, the first four nucleotides in these binding sites (underlined) were substituted, producing three mutants in the −662 deletion construct: Mut.A (TGCGTTCAAAAT, IRF7 #1), Mut.B (TCGATTGAAGTT), and Mut.AB (combining the above two mutants). These mutants were individually transfected into the HeLa-TLR2 cells, and the SAA-induced luciferase activity was determined. As shown in Fig. 4F, Mut.A but not Mut.B reduced the luciferase activity induced by SAA. Combining Mut.A and Mut.B did not produce any additional effect, suggesting that Mut.B is ineffective for IL-33 promoter activation.
Involvement of IRF7 in SAA-induced IL-33 gene transcription
To identify a role for IRF7 in SAA-induced IL-33 gene transcription, we first detected the expression of IRF7 in THP-1 cells. Both IRF7 transcript and protein were up-regulated upon SAA stimulation (Fig. 5A and B). Moreover, SAA stimulation increased phosphorylation of IRF7, as detected using an anti-phospho-serine antibody in the IRF7 immunoprecipitate (Fig. 5C). Because phophorylation of IRF7 is a prerequisite for its nuclear translocation, we next assessed the effect of SAA on the nuclear localization of IRF7. Nuclear and cytosolic fractions were prepared from unstimulated and SAA-stimulated THP-1 cells, and the presence of IRF7 was detected by western blotting. As shown in Fig. 5D, SAA induced nuclear translocation of IRF7 that that started after 15 min of stimulation and peaked at 60 min.
Figure 5.

SAA regulates the expression and activation of IRF7. THP-1 cells were stimulated with 0.05 μM SAA for up to 16 hours. IRF7 expression was determined by (A) real-time PCR and (B) western blotting, using β-actin as a loading control. (C) Phosphorylation of IRF7 was detected in anti-IRF7 immunoprecipitates (IP) of total lysate with an anti-phosphoserine (p-Ser) antibody, in SAA- stimulated (0.05 μM) THP-1 cells. (D) Western blot analysis of IRF7 in nuclear and cytosolic fractions of THP-1 cell after SAA stimulation. β-actin or HDAC1 was used as a loading control. (A–D) Data are shown as means + SEM of (A) six or (B– D) three samples and are (A–D) pooled from three independent experiments. *p < 0.05, **p < 0.01, (SAA-treated cells versus untreated cells). Differences were analyzed by paired t-test.
Two small interfering RNAs (siRNAs) were prepared for silencing IRF7 expression in THP-1 cells. As shown in Fig. 6A and 6B, both siRNA-1 and siRNA-2 significantly reduced the levels of IRF7 mRNA and protein in THP-1 cell. The more potent siRNA-2 was used in subsequent experiments. Silencing of IRF7 by siRNA-2 significantly inhibited the SAA-induced IL-33 expression at the mRNA and protein levels (Fig. 6C and D).
Figure 6.

A role for IRF7 in IL-33 promoter activation. (A) HEK-293T cells were transfected with an IRF7 expression vector, together with the firefly luciferase reporter constructs IL-33-pro WT, IL-33-pro (Mut.A), IL-33-pro (Mut.B), IL-33-pro (Mut.AB) or the pGL3 vector control, respectively. After 48 hours, the relative luciferase activity of each construct, as assessed by firefly luciferase activity, was normalized against that of the renilla luciferase. (B) real-time PCR and (C) western blot analysis were performed to detect mRNA and protein levels, respectively, of IRF7 in THP-1 cells transfected with the IRF7-specific siRNA-1 and siRNA-2, or a negative control siRNA (NC). Graphs show protein levels relative to β-actin quantified by densitometry. (D) real-time PCR and (E) western blot analysis were performed to detech mRNA and protein levels, respectively, of IRF7 in THP-1 cells stimulated with 0.05 μM SAA for 8 hours after transfection with siRNA-2 for 48 hours. Graphs show protein levels relative to β-actin quantified by densitometry. (F) Nuclear extracts were isolated from THP-1 cells after SAA (0.05 μM) stimulation for the indicated times. EMSA was performed based on chemiluminescence. PF, protein-free; CP, cold competitor probe. Blot is representative of four independent experiments. (G) THP-1 cells were treated with SAA (0.05 μM) or with buffer control for 30 min, followed by ChIP analysis as described in Materials and methods. The relative location of the IRF sites are shown schematically to the left, with the region covered by the real-time PCR primers underlined. (A–E, G) Data are shown as mean + SEM of (A, G) six or (B–E) three samples pooled from three independent experiments. *p < 0.05, **p < 0.01, (siRNA-treated cells versus untreated cells). Differences were analyzed by (A) one-way ANOVA followed by Bonderroni post-testing, (B, C, D, E, G) paired t-test.
The effect of IRF7 over-expression on IL-33 gene transcription was examined using the −662 luciferase reporter in co-transfected HEK-293T cells. IRF7 over-expression produced spontaneous induction of the luciferase reporter without agonist stimulation (Fig. 6E). Substitution of the consensus sequence in IRF7 #1 site, as in Mut.A and Mut.AB, markedly reduced the IRF7-driven expression of the luciferase reporter. In contrast, nucleotide substitution in IRF7 #2 site did not affect luciferase reporter expression in IRF7 co-transfected cells. These results further support a role for IRF7 in transcriptional activation of the IL-33 gene.
To directly measure IRF7 binding to the IL-33 promoter, electrophoretic mobility shift assay (EMSA) was performed using nuclear extracts isolated from SAA-stimulated THP-1 cells. The nuclear extracts were incubated with a probe containing the IRF7 binding site #1 and #2 (WT probe, −277 to −235) (Supporting Information Fig. 3A). A nuclear DNA binding complex appeared 1 and 4 hours after stimulation and was competed off with the unlabeled oligonucleotide probe (cold probe, CP; Fig. 6F). To further test the specificity of the interaction between IRF7 and the binding site within the IL-33 promoter, EMSA assay was performed using nucleotide probes harboring mutations in the IRF7 binding site. As shown in Supporting Information Fig. 3B, IRF7 binding was completely abolished with mutation of the IRF7 #1 site (Mut.A and Mut.AB). In contrast, mutation of the IRF7 #2 site (Mut.B) did not affect IRF7 binding in EMSA.
To determine the interaction of IRF7 with the IL-33 promoter in vivo, chromatin immunoprecipitation (ChIP) was performed using primers encompassing nucleotides −349 to −215 upstream of the transcription starting site in the IL-33 promoter (Fig. 6G). A 30-min stimulation with SAA induced specific binding of IRF7 to the IL-33 promoter in THP-1 cells, as evidenced by an immunoprecipitated complex with the anti-IRF7 antibody but not control IgG. Taken together, these results demonstrate that binding of IRF7 to a proximal site in the IL-33 promoter is a major event in the SAA-induced transcription of the IL-33 gene.
A potential role for TRAF6 in SAA-induced IL-33 expression
Results shown above support a role for IRF7 in SAA-induced expression of IL-33. Because TLR2 is a major receptor for this action of SAA, we sought to identify signaling molecules downstream of this receptor for the mechanism of IRF7 activation. SAA did not affect the phosphorylation of TANK-binding kinase 1 (data not shown), a known activator of IRF7. Therefore, we investigated whether SAA stimulates binding of TRAF6 to IRF7. As shown in Fig. 7A, IRF7 could interact with TRAF6 in vitro, when the individually tagged constructs were expressed in transfected HEK293T cells. In THP-1 cells, which were used as a monocytic cell model throughout this study, SAA stimulation increased TRAF6 interaction with IRF7 (Fig. 7B). Moreover, over-expression of TRAF6 enhanced the activation of IL-33 promoter (Fig. 7C). Taken together, these data suggest a role for TRAF6 as a signaling molecule downstream of TLR2 that interacts and activates IRF7 for SAA-induced expression of IL-33.
Figure 7.
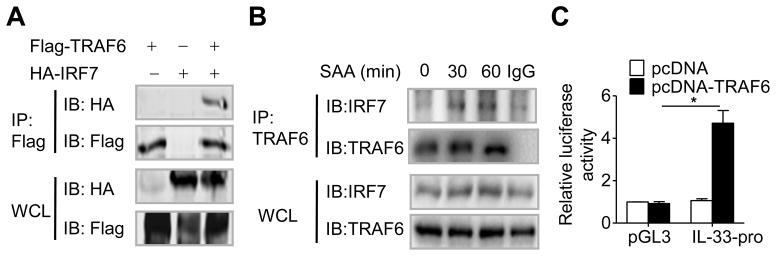
TRAF6 interacts with IRF7 and mediates IL-33 promoter activity. (A) HEK-293T cells were transfected with HA-IRF7 and Flag-TRAF6 expression vector, and analyzed by immunoprecipitation (IP) and/or immunoblotting (IB) as indicated. WCL: whole cell lysate. (B) IP and/or IB analysis of IRF7 and TRAF6 in lysates from THP-1 cells stimulated with SAA for indicated durations. (C) HEK-293T cells were co-transfected with IL-33 promoter luciferase reporter constructs, together with a TRAF6 expression vector. Luciferase activity was measured 48 hours after transfection and normalized against that of the renilla luciferase. IL-33-pro, −662/+49 of IL-33 promoter. (A and B) Data are representative of three experiments. (C) Data are shown as the means + SEM of six samples pooled from three experiments. *p < 0.05, paired t-test.
Discussion
In the present study, we demonstrated that SAA, an acute-phase protein, promotes the expression of IL-33 in both human monocytes and mouse macrophages. SAA up-regulates IL-33 at the mRNA and protein levels. We further demonstrated that a molecular mechanism by which SAA induces IL-33 expression involves TLR2 signaling and activation of a downstream effector IRF7, which binds to the IL-33 promoter and contributes to IL-33 transcription. Taken together, these results provide a link between an acute-phase protein and monocyte/macrophage expression of IL-33.
IL-33 is a recently described member of the IL-1 family and has attracted attention due to its important roles in modulating inflammation [8]. IL-33 is also known for mediating Th2-type immune responses [9], allergic lung inflammation and asthma, anaphylactic shock, arthritis, and colitis [8]. However, our understanding of the molecular mechanisms for IL-33 induction is very limited. Results from this study show that SAA can increase both the mRNA and cellular protein levels of IL-33. As reported previously, IL-33 functions in two different modes. As a nuclear protein, IL-33 binds to chromatin and has transcriptional repressor activity. A recent report shows that nuclear IL-33 can interact with the transcription factor NF-κB and reduce NF-κB-triggered gene expression to dampen proinflammatory signaling [33]. It is also shown that nuclear IL-33 can induce the expression of intercellular adhesion molecule and vascular cell adhesion molecule for endothelial cell activation though binding to the p65 promoter [34]. IL-33 is also released extracellularly and activates the ST2/ST2/IL-1R accessory protein complex receptor. In the present study, we have successfully detected an increase in the IL-33 mRNA as well as its protein production in monocytes/macrophages challenged by SAA; however, we have not found any significant increase in IL-33 release to the culture medium. Cell fractionation and immunofluorescence studies have also shown an increased nuclear accumulation of IL-33, indicating that SAA promotes intracellular IL-33 expression but not its secretion. The present study focuses on the intracellular production of IL-33 by SAA but not its biological functions, which is an area of considerable interest and requires further exploration.
To date, how IL-33 protein is released from cells remains unclear. Caspase-1 is the first known protease to be involved in the cleavage of this class of cytokines. Early data suggested that pro-IL-33 requires cleavage by caspase-1, much alike the processing of some IL-1 cytokines such as IL-1β and IL-18 which are synthesized as inactive precursor proteins and are activated and secreted following cleavage by caspase-1 [9]. Although SAA is known to activate caspase-1 for IL-1β secretion [35], it does not affect IL-33 secretion in our experiments. These results suggest that caspase-1 activation is either insufficient for or unrelated to IL-33 release. In addition, recent reports show that IL-33 can be released as a biologically active full-length molecule from cells only during necrosis and the release is halted during apoptosis by cleavage of caspase-3 and caspase-7 [6]. However, we found no activation of caspase-3 under SAA stimulation (Supporting Information Fig. 1B). Together, our results indicate that SAA is able to induce intracellular expression of IL-33 but not its release. It is likely that SAA does not provide the necessary signals required for IL-33 release.
Several proteins have been identified as SAA receptors that mediate its diverse functions. Notably, the G protein-coupled FPR2 is responsible for the chemotactic effect of SAA and contributes to its cytokine-inducing property [22, 27]. The scavenger receptor BI mediates the cholesterol efflux function of SAA and also transduces signals leading to MAPK activation [23, 36]. Other receptors such as TLR2 and TLR4 account for the induced expression of IL-23 and granulocyte colony-stimulating factor by SAA [21, 25, 28]. In the present study, we show that TLR2 is primarily responsible for the induced expression of IL-33 by SAA. The role for FPR2 in SAA-induced IL-33 expression is not fully understood. Genetic deletion of Fpr2 reduced IL-33 expression by approximately 50%, and the FPR2 agonist WKYMVm was able to potentiate IL-33 expression induced by Pam3CSK4 and by low concentrations of SAA. At high concentrations of SAA, this effect of WKYMVm is absent probably because the induced expression of IL-33 reaches to a plateau. These findings support the involvement of FPR2 in the SAA-induced IL-33 expression. Published studies have shown that SAA interacts with FPR2 using a binding site that differs from the site used by most other agonists [37], possibly making WRW4 a less effective antagonist for blocking SAA binding to FPR2. Indeed, a blocking effect was only observed at very high concentrations of WRW4 (20 μM), which markedly reduced the Ca2+ flux baseline in a control experiment (Supporting Information Fig. 2C). The Ca2+ flux data also suggested that WRW4 could act as an allosteric modulator, as it could increase the level of Ca2+ at low concentrations. Therefore, due to limited choices for FPR2 antagonists, results obtained with the use of WRW4 should be interpreted with caution. Taken together, although stimulation of FPR2 by WKYMVm alone was insufficient for IL-33 induction, this receptor may work together with TLR2 in SAA-induced expression of IL-33, as suggested by the Fpr2 knockout data. Further investigation will be required for an understanding of the crosstalk between the signaling pathways activated by TLR2 and FPR2.
Given the well-established regulatory effect of IL-33 in allergic lung inflammation, anaphylactic shock, and colitis [8], it is important to understand the mechanism underlying its induction at the transcriptional level, which has not been fully characterized. It was recently reported that activation of TBK-1 leads to activation of the transcription factor IRF3 for IL-33 production [38], thus suggesting the involvement of the IRF proteins in IL-33 induction. Our study identified IRF7 as being critical to the SAA-induced IL-33 gene expression through binding to an IRF7 site in the promoter of IL-33. Of the two potential IRF7 sites identified, only one (IRF7 #1) was used by IRF7 in its induction of IL-33 gene expression despite similarity of both sites to the consensus IRF7 binding sequence. A close examination of the sequence of these cis-acting elements found that IRF7 #1 has higher resemblance to the consensus sequence (5′-GAA(A/T)N(C/T)GAAAN (T/C)-3′) than IRF7 #2, as only the second G in the consensus sequence is replaced by a C in IRF7 #1, and a similar change has been described previously [32]. In comparison, the first A/T in IRF #2 is replaced by a G and the fifth A is replaced by a G, which are changes not described previously [32]. Subsequent analysis by EMSA and chromatin immunoprecipitation confirmed that SAA could induce binding of IRF7 to the −277/−266 IRSE site in the IL-33 promoter. These findings demonstrate for the first time that IRF7 is a critical component for SAA-induced activation of the IL-33 promoter.
TLR2 is known for its fundamental role in pathogen-associated pattern recognition. Emerging evidence also suggests that TLR2 plays a role in inflammation resulting from endogenous ligands [39]. Our work provided preliminary data showing a potential role for TRAF6 in the interaction with IRF7. This interaction is believed to be important for IL-33 transcriptional activation downstream of a SAA-activated TLR2. Our work suggests the possible existence of a TLR2-TRAF6-IRF7 axis that leads to IL-33 induction.
In summary, the present study shows that the acute-phase protein SAA can induce the nuclear expression of IL-33 at the transcription level. We have also identified an IRF7-interacting element in the IL-33 promoter region and demonstrated that IRF7 is required for IL-33 expression in SAA-activated macrophages. These results lead to the speculation that SAA produced during the acute-phase response may extend its function as an inflammatory modulator through the induction of IL-33. The pathological roles as well as detailed mechanism underlying the induced IL-33 expression during inflammatory disease are potentially interesting areas for further investigation.
Materials and methods
Mice
All knockout mice were obtained from the Jackson Laboratory except the Fpr2−/− mice [40], which were generously provided by Dr. Ji Ming Wang (NCI-Frederick, MD). C57BL/6 mice were purchased from Shanghai Laboratory Animal Center (SLAC, Shanghai, China). Age- and sex-matched littermates were used in the experiments. The procedures involving mice were carried out using protocols approved by the institutional Animal Care and Use Committee at the Shanghai Jiao Tong University.
Plasmids and Reagents
The IL-33-pGL3 reporters (−1050 to +49, −662 to +49, −520 to +49, −460 to +49, −300 to +49, −160 to +49) were generated by cloning the PCR products from THP-1 cell genomic DNA into the pGL3-basic vector (Promega). The IRF7 binding site mutants (Mut.A, Mut.B and Mut.AB) were generated by GeneArt® Site-Directed Mutagenesis System from Life Technologies. Human IRF7 sequence was amplified by PCR with total cDNA from THP-1 cells, which was cloned into pcDNA3.0-HA vector. All primers used were shown in the Supporting Information Table 1. Recombinant human SAA was obtained from PeproTech (Rocky Hill, NJ). cycloheximide (CHX) and actinomycin D (ActD) was purchased from Sigma-Aldrich (St Louis, MO). The TRAF6 plasmid, Pam3CSK4, polyI: C, TLR2 neutralizing antibody and its isotype control IgA were obtained from InVivoGen (San Diego, CA). Anti-IL-33 (ab118503) and anti-IRF7 were obtained from Abcam (Cambridge, MA). Anti-pSer/Thr was obstained from BD Biosciences (San Jose, CA). Antibodies for caspase-3, β-actin, HDAC1, HRP-conjugated anti-rabbit IgG, HRP-conjugated anti-mouse IgG were obtained from Cell Signaling Technology (Danvers, MA).
Cells preparation and culture
Monocytes from healthy blood donors were isolated and purified from buffy coats, following a protocol approved by the Institutional Review Board of Shanghai Jiao Tong University. Briefly, human peripheral blood mononuclear cells (PBMCs) were separated by centrifugation on Ficoll-Paque gradient. Monocytes were then isolated from PBMCs by positive sorting using anti-CD14-conjugated magnetic microbeads (Miltenyi, Germany). Monocyte purity was greater than 90% as assessed by flow cytometry. Mouse peritoneal macrophages were prepared from WT or knockout mice with C57BL/6 background as described [41]. Human monocytic leukemia cell line THP-1 (TIB-202) was purchased from ATCC (Manassas, VA). Cells were maintained in RPMI 1640 supplemented with 10% FBS, 1% streptomycin-penicillin and 10 μM β-mercaptoethanol. All cell cultures were kept in a humidified atmosphere with 5% CO2 at 37°C.
RNA extraction, RT-PCR and real-time PCR
Total RNA was extracted from the cells using an RNeasy Mini Kit (Qiagen, Germany) according to the manufacturer’s instructions. First-strand cDNA was prepared using AMV reverse transcriptase (Promega) and oligo(dT)15. Gene-specific real-time quantitative PCR was performed using primers and probes from SYBR Green® Real-time PCR Master Mix (TOYOBO, Japan) with a Mastercycler Realplex system (Eppendorf, Germany). The results for each gene were normalized to that for GAPDH expression in the sample. The primer sequences were shown in Supporting Information Table 1. The ΔΔCt method was used for calculation of the mRNA levels.
Transfection and reporter assay
TLR2-HeLa and HEK-293T cells were transiently transfected with plasmids containing different fragments of the human IL-33 promoter fused to a luciferase reporter and the pRL-TK plasmid (a Renilla luciferase control plasmid, Promega), using Lipofectamine™ 2000 reagent (Invitrogen). Promoter activities were analyzed using a Dual-Luciferase Reporter Assay Kit (Promega) and were normalized to Renilla luciferase activities.
Electrophoretic mobility shift assay (EMSA)
Cells were treated with 0.05 μM of SAA or buffer only, and nuclear proteins were collected using the NE-PER Nuclear and Cytosolic Extraction Reagents (Thermo scientific). EMSA was performed with a LightShift Chemiluminescent EMSA Kit (Thermo scientific) according to the manufacturer’s instructions. For EMSA, WT and mutant IRF7 oligonucleotide probes labeled with biotin were synthetized (Invitrogen). The probe sequences were shown in Supporting Information Fig. 3A.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) was performed using a ChIP assay kit (Millipore, Billerica, MA) according to the manufacturer’s instruction with minor modifications. The primer sequences were shown in Supporting Information Table 1. Data are presented as the amount of DNA recovered relative to the input control.
Immunofluorecence
Cells were grown on Microscope cover glass (Thermo-Fisher) and fixed in cold 4% paraformaldehyde for 15 min. Cells were permeabilized with 0.1% Triton X-100/PBS for 15 min, and blocked with 1% BSA/PBS for 15 min. Then the cells were incubated with an anti-IL-33 (10 μg/mL) overnight at 4°C. The cells were then repeatedly washed and incubated with 20μg/mL of Alexa-Fluor 488 goat anti-rabbit IgG (H+L) fluorescein isothiocyanate (FITC, Invitrogen) for 60 min. Nuclei were stained with DAPI (10μg/mL) for 5 min. The cover glasses were washed in PBS and examined on a Leica TCS SP UV confocal laser scanning microscope (Leica, Wetzlar, Germany).
siRNA interference
The human monocytic THP-1 cells were transfected with specific siRNA using Silencer® siRNA Transfection II Kit (Ambion) according to the manufacturer’s instructions. The cells were then recovered for 48 hours before stimulation. The siRNA oligonucleotides were designed and synthesized by Shanghai RIBOBIO Co., LTD (Guangzhou, China). IRF7-specific siRNA-2 (5′-CGAGCUGCACGUUCCUAUA dTdT-3′) was used to suppress endogenous IRF7 expression. ‘Nonsense’ sequence (5′-UUCUCCGAACGUGUCACGUdTdT-3′) was used as a control siRNA.
Immunoprecipitation
For immunoprecipitation, cell lysates were prepared in RIPA buffer (Cell Signaling Technology) and precleared with Protein G Agarose (Millipore, Billerica, MA). The lysates were immunoprecipitated with the addition of protein G-Sepharose, the M2 FLAG antibody (Sigma-Aldrich, St. Louis, MO), anti-IRF7 or anti-TRAF6 (Santa Cruz, Dallas, TX), respectively, and the mixture was incubated with gentle shaking for 4 hours at 4 °C. The samples were resolved on SDS-PAGE, and underwent immunoblotting analysis using anti-HA (Sigma-Aldrich, St. Louis, MO) and the M2 FLAG antibody, respectively.
Statistical analysis
Each experiment was performed independently for at least three times. For comparison between two groups, two-tailed paired Student’s t-test was used. For comparison between two or more groups one-way ANOVA was applied. A p-value < 0.05 considered statistically significant. The Prism software (version 5.0, GraphPad, San Diego, CA) was used for statistical analysis.
Supplementary Material
Acknowledgments
The authors thank Dr. Ji Ming Wang for providing the Fpr2 knockout mice and Dr. Runsheng Li for helpful discussions. We also thank the staff of the Experimental Animal Center at Shanghai Jiao Tong University for excellent husbandry and animal care. This work was supported by grants from National Natural Science Foundation of China (Grants 81202316 and 31270941), from National Basic Research Program of China (973 Program, Grant 2012CB518001), and from the Specialized Research Fund for the Doctoral Program of Higher Education of China (Grants 20120073110069 and 20120073120092). This work was also supported in part by U.S. National Institutes of Health grant AI 033503 and AI 040176.
Abbreviations
- SAA
serum amyloid A
- FPR2
formyl peptide receptor 2, FPRL1/ALX
- NF-HEV
nuclear factor from high endothelial venules
- HeLa-TLR2
TLR2-expressing HeLa cells
- CHX
cycloheximide
- ActD
actinomycin D
- siRNAs
small interfering RNAs
- EMSA
electrophoretic mobility shift assay
- IRF7
Interferon regulatory factor 7
- TRAF6
TNF receptor-associated factor 6
- ChIP
Chromatin immunoprecipitation
Footnotes
Conflict of Interest:
The authors declare no financial or commercial conflict of interest.
References
- 1.Baekkevold ES, Roussigne M, Yamanaka T, Johansen FE, Jahnsen FL, Amalric F, Brandtzaeg P, et al. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am J Pathol. 2003;163:69–79. doi: 10.1016/S0002-9440(10)63631-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev. 2008;223:20–38. doi: 10.1111/j.1600-065X.2008.00624.x. [DOI] [PubMed] [Google Scholar]
- 3.Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, Bouche G, Girard JP. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci U S A. 2007;104:282–287. doi: 10.1073/pnas.0606854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohno T, Oboki K, Kajiwara N, Morii E, Aozasa K, Flavell RA, Okumura K, et al. Caspase-1, caspase-8, and calpain are dispensable for IL-33 release by macrophages. J Immunol. 2009;183:7890–7897. doi: 10.4049/jimmunol.0802449. [DOI] [PubMed] [Google Scholar]
- 5.Talabot-Ayer D, Lamacchia C, Gabay C, Palmer G. Interleukin-33 is biologically active independently of caspase-1 cleavage. J Biol Chem. 2009;284:19420–19426. doi: 10.1074/jbc.M901744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, Brumatti G, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31:84–98. doi: 10.1016/j.immuni.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Chackerian AA, Oldham ER, Murphy EE, Schmitz J, Pflanz S, Kastelein RA. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. J Immunol. 2007;179:2551–2555. doi: 10.4049/jimmunol.179.4.2551. [DOI] [PubMed] [Google Scholar]
- 8.Liew FY, Pitman NI, McInnes IB. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nat Rev Immunol. 2010;10:103–110. doi: 10.1038/nri2692. [DOI] [PubMed] [Google Scholar]
- 9.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 10.Nile CJ, Barksby E, Jitprasertwong P, Preshaw PM, Taylor JJ. Expression and regulation of interleukin-33 in human monocytes. Immunology. 2010;130:172–180. doi: 10.1111/j.1365-2567.2009.03221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem. 1999;265:501–523. doi: 10.1046/j.1432-1327.1999.00657.x. [DOI] [PubMed] [Google Scholar]
- 12.Jabor A, Holub Z, Franekova J, Pavlisova M, Boril P, Fenclova E, Kliment L. Serum amyloid A as an effective marker for the assessment of surgical trauma and risk of post-operative complications. Ceska Gynekol. 2006;71:131–136. [PubMed] [Google Scholar]
- 13.Lannergard A, Viberg A, Cars O, Karlsson MO, Sandstrom M, Larsson A. The time course of body temperature, serum amyloid A protein, C-reactive protein and interleukin-6 in patients with bacterial infection during the initial 3 days of antibiotic therapy. Scand J Infect Dis. 2009;41:663–671. doi: 10.1080/00365540903127417. [DOI] [PubMed] [Google Scholar]
- 14.Uhlar CM, Grehan S, Steel DM, Steinkasserer A, Whitehead AS. Use of the acute phase serum amyloid A2 (SAA2) gene promoter in the analysis of pro- and anti-inflammatory mediators: differential kinetics of SAA2 promoter induction by IL-1 beta and TNF-alpha compared to IL-6. J Immunol Methods. 1997;203:123–130. doi: 10.1016/s0022-1759(96)00220-7. [DOI] [PubMed] [Google Scholar]
- 15.Ishii W, Matsuda M, Nakamura A, Nakamura N, Suzuki A, Ikeda S. Abdominal fat aspiration biopsy and genotyping of serum amyloid A contribute to early diagnosis of reactive AA amyloidosis secondary to rheumatoid arthritis. Intern Med. 2003;42:800–805. doi: 10.2169/internalmedicine.42.800. [DOI] [PubMed] [Google Scholar]
- 16.King VL, Thompson J, Tannock LR. Serum amyloid A in atherosclerosis. Curr Opin Lipidol. 2011;22:302–307. doi: 10.1097/MOL.0b013e3283488c39. [DOI] [PubMed] [Google Scholar]
- 17.Zhao Y, He X, Shi X, Huang C, Liu J, Zhou S, Heng CK. Association between serum amyloid A and obesity: a meta-analysis and systematic review. Inflamm Res. 2010;59:323–334. doi: 10.1007/s00011-010-0163-y. [DOI] [PubMed] [Google Scholar]
- 18.Du JL, Liu JF, Men LL, Yao JJ, Sun LP, Sun GH, Song GR, et al. Effects of five-year intensive multifactorial intervention on the serum amyloid A and macroangiopathy in patients with short-duration type 2 diabetes mellitus. Chin Med J (Engl) 2009;122:2560–2566. [PubMed] [Google Scholar]
- 19.Connolly M, Marrelli A, Blades M, McCormick J, Maderna P, Godson C, Mullan R, et al. Acute serum amyloid A induces migration, angiogenesis, and inflammation in synovial cells in vitro and in a human rheumatoid arthritis/SCID mouse chimera model. J Immunol. 2010;184:6427–6437. doi: 10.4049/jimmunol.0902941. [DOI] [PubMed] [Google Scholar]
- 20.Artl A, Marsche G, Lestavel S, Sattler W, Malle E. Role of serum amyloid A during metabolism of acute-phase HDL by macrophages. Arterioscler Thromb Vasc Biol. 2000;20:763–772. doi: 10.1161/01.atv.20.3.763. [DOI] [PubMed] [Google Scholar]
- 21.Sandri S, Rodriguez D, Gomes E, Monteiro HP, Russo M, Campa A. Is serum amyloid A an endogenous TLR4 agonist? J Leukoc Biol. 2008;83:1174–1180. doi: 10.1189/jlb.0407203. [DOI] [PubMed] [Google Scholar]
- 22.Su SB, Gong W, Gao JL, Shen W, Murphy PM, Oppenheim JJ, Wang JM. A seven-transmembrane, G protein-coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J Exp Med. 1999;189:395–402. doi: 10.1084/jem.189.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baranova IN, Vishnyakova TG, Bocharov AV, Kurlander R, Chen Z, Kimelman ML, Remaley AT, et al. Serum amyloid A binding to CLA-1 (CD36 and LIMPII analogous-1) mediates serum amyloid A protein-induced activation of ERK1/2 and p38 mitogen-activated protein kinases. J Biol Chem. 2005;280:8031–8040. doi: 10.1074/jbc.M405009200. [DOI] [PubMed] [Google Scholar]
- 24.Cheng N, He R, Tian J, Ye PP, Ye RD. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J Immunol. 2008;181:22–26. doi: 10.4049/jimmunol.181.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He RL, Zhou J, Hanson CZ, Chen J, Cheng N, Ye RD. Serum amyloid A induces G-CSF expression and neutrophilia via Toll-like receptor 2. Blood. 2009;113:429–437. doi: 10.1182/blood-2008-03-139923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim JS, Ryoo SB, Heo K, Kim JG, Son TG, Moon C, Yang K. Attenuating effects of granulocyte-colony stimulating factor (G-CSF) in radiation induced intestinal injury in mice. Food Chem Toxicol. 2012;50:3174–3180. doi: 10.1016/j.fct.2012.05.059. [DOI] [PubMed] [Google Scholar]
- 27.He R, Sang H, Ye RD. Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood. 2003;101:1572–1581. doi: 10.1182/blood-2002-05-1431. [DOI] [PubMed] [Google Scholar]
- 28.He R, Shepard LW, Chen J, Pan ZK, Ye RD. Serum amyloid A is an endogenous ligand that differentially induces IL-12 and IL-23. J Immunol. 2006;177:4072–4079. doi: 10.4049/jimmunol.177.6.4072. [DOI] [PubMed] [Google Scholar]
- 29.Lee HY, Kim MK, Park KS, Shin EH, Jo SH, Kim SD, Jo EJ, et al. Serum amyloid A induces contrary immune responses via formyl peptide receptor-like 1 in human monocytes. Mol Pharmacol. 2006;70:241–248. doi: 10.1124/mol.105.022103. [DOI] [PubMed] [Google Scholar]
- 30.Le Y, Gong W, Li B, Dunlop NM, Shen W, Su SB, Ye RD, Wang JM. Utilization of two seven-transmembrane, G protein-coupled receptors, formyl peptide receptor-like 1 and formyl peptide receptor, by the synthetic hexapeptide WKYMVm for human phagocyte activation. J Immunol. 1999;163:6777–6784. [PubMed] [Google Scholar]
- 31.Bae YS, Lee HY, Jo EJ, Kim JI, Kang HK, Ye RD, Kwak JY, Ryu SH. Identification of peptides that antagonize formyl peptide receptor-like 1-mediated signaling. J Immunol. 2004;173:607–614. doi: 10.4049/jimmunol.173.1.607. [DOI] [PubMed] [Google Scholar]
- 32.Lin R, Génin P, Mamane Y, Hiscott J. Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Molesular and Cellular Biology. 2000;20:6342–6353. doi: 10.1128/mcb.20.17.6342-6353.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ali S, Mohs A, Thomas M, Klare J, Ross R, Schmitz ML, Martin MU. The dual function cytokine IL-33 interacts with the transcription factor NF-kappaB to dampen NF-kappaB-stimulated gene transcription. J Immunol. 2011;187:1609–1616. doi: 10.4049/jimmunol.1003080. [DOI] [PubMed] [Google Scholar]
- 34.Choi YS, Park JA, Kim J, Rho SS, Park H, Kim YM, Kwon YG. Nuclear IL-33 is a transcriptional regulator of NF-kappaB p65 and induces endothelial cell activation. Biochem Biophys Res Commun. 2012;421:305–311. doi: 10.1016/j.bbrc.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 35.Niemi K, Teirila L, Lappalainen J, Rajamaki K, Baumann MH, Oorni K, Wolff H, et al. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J Immunol. 2011;186:6119–6128. doi: 10.4049/jimmunol.1002843. [DOI] [PubMed] [Google Scholar]
- 36.Baranova IN, Bocharov AV, Vishnyakova TG, Kurlander R, Chen Z, Fu D, Arias IM, et al. CD36 is a novel serum amyloid A (SAA) receptor mediating SAA binding and SAA-induced signaling in human and rodent cells. J Biol Chem. 2010;285:8492–8506. doi: 10.1074/jbc.M109.007526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiang N, Fierro IM, Gronert K, Serhan CN. Activation of lipoxin A(4) receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J Exp Med. 2000;191:1197–1208. doi: 10.1084/jem.191.7.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Polumuri SK, Jayakar GG, Shirey KA, Roberts ZJ, Perkins DJ, Pitha PM, Vogel SN. Transcriptional Regulation of Murine IL-33 by TLR and Non-TLR Agonists. J Immunol. 2012;189:50–60. doi: 10.4049/jimmunol.1003554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–179. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 40.Chen K, Le Y, Liu Y, Gong W, Ying G, Huang J, Yoshimura T, et al. A critical role for the g protein-coupled receptor mFPR2 in airway inflammation and immune responses. J Immunol. 2010;184:3331–3335. doi: 10.4049/jimmunol.0903022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008;Chapter 14(Unit 14):11. doi: 10.1002/0471142735.im1401s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.