

Abstract
The cellular prion protein (PrPC) has been implicated in several neurodegenerative diseases as a result of protein misfolding. In humans, prion disease occurs typically with a sporadic origin where uncharacterized mechanisms induce spontaneous PrPC misfolding leading to neurotoxic PrP-scrapie formation (PrPSC). The consequences of misfolded PrPC signalling are well characterized but little is known about the physiological roles of PrPC and its involvement in disease. Here we investigated wild-type PrPC signalling in synaptic function as well as the effects of a disease-relevant mutation within PrPC (proline-to-leucine mutation at codon 101). Expression of wild-type PrPC at the Drosophila neuromuscular junction leads to enhanced synaptic responses as detected in larger miniature synaptic currents which are caused by enlarged presynaptic vesicles. The expression of the mutated PrPC leads to reduction of both parameters compared with wild-type PrPC. Wild-type PrPC enhances synaptic release probability and quantal content but reduces the size of the ready-releasable vesicle pool. Partially, these changes are not detectable following expression of the mutant PrPC. A behavioural test revealed that expression of either protein caused an increase in locomotor activities consistent with enhanced synaptic release and stronger muscle contractions. Both proteins were sensitive to proteinase digestion. These data uncover new functions of wild-type PrPC at the synapse with a disease-relevant mutation in PrPC leading to diminished functional phenotypes. Thus, our data present essential new information possibly related to prion pathogenesis in which a functional synaptic role of PrPC is compromised due to its advanced conversion into PrPSC thereby creating a lack-of-function scenario.
INTRODUCTION
The cellular prion protein (PrPC) is a cell membrane-anchored glycoprotein which plays an important role in a variety of neuronal processes including circadian rhythm, neuroprotection and neuroplasticity (1,2). Although the physiological role of PrPC remains elusive, the conversion of PrPC into the neurotoxic PrPSC during prion disease and its signalling are well documented (2–4). As a consequence of protein misfolding, several mammalian species develop neurodegenerative conditions best known as scrapie in sheep, bovine spongiform encephalopathy in cattle or Creutzfeldt–Jacob disease (CJD) and Gerstmann–Sträussler–Scheinker Syndrome (GSS) in human. The unique feature of these conditions is that, in addition to sporadic and inherited forms, it can be transmitted by infectious agents according to the ‘protein only’ hypothesis. The early onset of disease may now be caused by either loss-of-function of PrPC or gain-of-function of cytotoxic PrPSC, or both.
PrPC is present in all mammalian cortico-cerebellar, deep nuclei neurons and neuromuscular junctions (NMJs) (5). Morphological studies suggest that PrPC is preferentially located along axons and in presynaptic terminals (6) but postsynaptic localization and signalling has also been reported (7,8). Evidence accumulates that neuroprotective roles of PrPC are essential (9,10) as loss-of-function in PrPC knock-out (KO)/mutant models leads to neuronal dysfunction (11–13). Interestingly, KO animals for the gene encoding PrPC exhibit phenotypes with impaired long-term potentiation (14–16), abnormal circadian rhythm (17) or effects on glutamatergic transmission (18,19) but also more severe characteristics such as Purkinje cell degeneration and demyelination of peripheral nerves leading to ataxia (11,20). As the exact cellular functions of PrPC remain unknown, it is essential to characterize the physiological and neuroprotective roles of PrPC in order to better understand the changes which occur during early onset prion disease.
Recently, several non-mammalian neurodegeneration models have been employed (21–23) and in particular, expression of PrPC and PrPSC in Drosophila or Caenorhabditis elegans allows investigations of prion function in host organisms that do not have a direct prion ortholog (24–29). PrPC can convert into PrPSC in adult Drosophila causing neurodegeneration and expression of a mutated PrPC (PrPP101L) is sufficient to mimic neurodegenerative phenotypes in adult Drosophila (25,30). PrPC can modulate synaptic transmission (31) including potentiation of acetylcholine release at the mouse NMJ (32), whereas PrPC-KO mice exhibit reduced inhibitory release (14). Research suggests that synaptic dysfunction precedes the cell death that occurs at later stages during prion pathogenesis (33,34) but studies have yet to define the exact physiological mechanisms of PrPC in order to explain the underpinning synaptic loss and/or dysfunction before disease onset.
In the current study, presynaptic expression in Drosophila of mouse wild-type PrPC (PrP3F4) and a mutated form of PrPC [PrPP101L, which induces a GSS-like disease in mice and is related to a human GSS-associated mutation (P102L) (35)] was investigated to elucidate potential effects on synaptic release before manifestation of neurodegeneration thereby contributing to our understanding of PrPC function. The data show that endogenous PrP3F4 facilitates synaptic release and this function is partially compromised following expression of PrPP101L indicating a pivotal role of PrPC (PrP3F4) signalling.
RESULTS
Expressed wild-type and mutated murine prion proteins are sensitive to proteinase digestion
Expression of wild-type murine PrPC (PrP3F4) in Drosophila causes spongiform degeneration in adult fly brains (26) and importantly this degeneration is accelerated following expression of a mutated PrPC (P101L) [PrPP101L], a mutation which is linked to the human prion disease GSS. In initial experiments we aimed to validate expression of either PrP3F4 or the mutated prion protein (PrPP101L) in transgenic Drosophila larvae by performing immunohistochemistry (IHC) which confirmed strong and specific expression of either protein within all boutons of the NMJ and lack of expression in UAS controls [Fig. 1A, co-stained for vesicular glutamate transporter (vGlut)]. Western blot analysis further confirmed expression of either prion protein (Fig. 1B). Assessing expression levels of both prion proteins revealed no differences between both lines (prion protein/α-tubulin ratio: PrP3F4: 1.2 ± 0.4 (n = 10), PrPP101L: 1.0 ± 0.2 (n = 7), P = 0.35, Student's t-test). As PrPC expression induces a neurodegenerative phenotype in older adult flies (26,30) we next tested whether the expressed prion proteins were sensitive to proteinase digestion at these developmentally younger larval stages. In order to test this we employed the Proteinase K (PK)-digestion protocol. An increased resistance to PK digestion has been shown in older flies (30 days) expressing PrP3F4 where digestion occurs above ∼5–7 μg/ml (30,36) but it is unknown whether this resistance to PK digestion, an indicator of PrPSC misfolding, is evident in third instar larvae. Any digestion occurring below ∼5–7 μg/ml could suggest a lack of misfolding and that the putative function of these proteins may not be affected (12). Incubation of larval preparations with various concentrations of PK (Fig. 1C, 0–1 μg/ml) did not indicate any formation of PK-resistant prion in either PrP3F4 or PrPP101L transgenic lines as PrP3F4 and PrPP101L digestion starts at ∼1 μg/ml (so is α-tubulin being digested by this concentration of PK, Fig. 1C). Thus, protein misfolding and aggregation is unlikely to occur at this developmental stage (12). It is established that synaptic loss in murine prion disease models precedes degeneration of the cell soma (34) but it remains to be investigated what the underlying mechanisms for this dysfunction are and whether pre- or postsynaptic prion protein signalling is involved. The current study aims to investigate prion protein-mediated effects on presynaptic release mechanisms to elucidate its physiological roles.
Figure 1.

Prion protein expression does not result in PK resistance at the larval NMJ. (A) IHC staining of third instar NMJs show strong prion protein labelling in elav-Gal4/UAS-MoPrP3F4 and elav-Gal4/UAS-MoPrPP101L larvae but no prion expression in respective UAS control NMJs. (B) Protein extracts of Tg-PrP lines (elav-Gal4/UAS-MoPrP3F4 and elav-Gal4/UAS-MoPrPP101L) with appropriate controls (UAS-MoPrP3F4/+ and UAS-MoPrPP101L/+) were probed for PrP (Ab: AH6 anti-PrP) and α-tubulin. Note, PrPC can undergo glycosylation leading to multiple PrP bands (25,98) which are not detectable in larvae. n = 3 animals per lane. (C) Third instar larval extracts from Tg-PrP lines (elav-Gal4/UAS-MoPrP3F4 and elav-Gal4/UAS-MoPrPP101L) treated with a mild PK gradient (0–1 μg/ml) showed no PK resistance. PK completely digested prion protein (∼25 kDa bands) at relatively low concentrations (∼1 μg/ml) and so is α-tubulin being digested. n = 5 larval heads per lane.
Murine prion protein enlarges synaptic vesicles
To test the effects of PrP3F4 (pan-neuronal expression) on the synaptic physiology at Drosophila NMJs, Two-Electrode Voltage Clamp (TEVC) experiments were conducted to record miniature excitatory junctional currents (mEJCs). PrP3F4 expression induced a 38% increase in mean mEJC amplitudes compared with wild-type w1118 (WT) controls [WT: 0.74 ± 0.03 nA (n = 48), PrP3F4: 1.02 ± 0.04 nA (n = 35), analysis of variance (ANOVA), P < 0.001, Fig. 2A and B]. The use of w1118 as controls in addition to respective UAS controls was justified as all lines were backcrossed to w1118 for at least six generations. This augmentation in mean mEJC amplitudes was also observed at NMJs expressing PrPP101L (23%), although to a lesser degree then caused by the PrP3F4 expression (0.91 ± 0.04 nA (n = 36), P < 0.001 versus WT controls and P < 0.05 versus PrP3F4, ANOVA, n-number of NMJs indicated in bars). As additional controls, male larvae containing one copy of either UAS-PrP3F4 or UAS-P101LD PrPC [PrPP101L] were used (from now on referred to as UAS controls). Expression of either prion protein induced larger mEJCs relative to their respective UAS controls (P < 0.05 each, ANOVA). Each UAS control for PrP3F4 or PrPP101L did not differ from WT (w1118) controls (UAS-PrP3F4: 0.84 ± 0.04 nA; UAS-PrPP101L: 0.71 ± 0.07 nA, P > 0.05, ANOVA). Comparison of mEJC amplitudes in WT controls of male and female larvae did not reveal any difference (male: 0.80 ± 0.07 nA (n = 8) versus female: 0.82 ± 0.07 nA (n = 5), P = 0.36, Student's t-test) consistent with no sex-dependence. The increase in mEJC amplitudes is due to a shift in the distribution of single quantal size as depicted in the normalized histograms and cumulative histograms for the three different genotypes. As shown in Figure 2C and D, normalized relative histograms for mEJC amplitudes and cumulative frequency histograms demonstrated an increased probability of larger amplitude events and hence a right shift in mEJC amplitude distributions in PrP3F4 but to a lesser degree in PrPP101L expressing larvae. Using the more sensitive Kolmogorov–Smirnov test (K–S test) a difference between the distributions became evident (PrP3F4 versus Ctrl: D = 0.22, P = 0.024, PrPP101L versus Ctrl: D = 0.30, P = 0.00023, PrP3F4 versus PrPP101L: D = 0.42, P < 0.00001, K–S test) suggesting that functional properties between both prion proteins within the presynaptic terminal are different.
Figure 2.

Prion causes enlarged miniature EJCs. TEVC recordings were performed at the NMJ to investigate the effects of neuronal PrP3F4 or PrPP101L expression. (A) Sample traces showing mEJCs in control (Ctrl [w1118]) and PrP3F4 expressing larvae. (B) Mean mEJC amplitudes are increased following PrP3F4 and PrPP101L expression with PrPP101L showing a reduction relative to PrP3F4 (n—number of NMJs is indicated in bars. ***P < 0.001, *P < 0.05, n = 6–48 NMJs, [n = 19 animals for w1118, n = 15 animals for PrPP101L, n = 16 animals for PrP PrP3F4, n = 4–8 animals for UAS controls]). Data denote mean ± SEM, ANOVA with Tukey–Kramer post hoc test. (C) Normalized amplitude histograms for mEJCs show a right-shift of amplitudes following PrP3F4 expression. PrPP101L expression left-shifted amplitudes relative to PrPC but still caused an increase versus control (w1118). There is no evidence for multi-quantal release as distributions do not exhibit multiple peaks. (D) Relative cumulative amplitude histograms illustrate the right-shift in amplitudes for PrP3F4 expressing larvae. PrPP101L expression induced a left-shift relative to PrP3F4 (K–S test, PrP3F4 versus w1118: D = 0.22, P = 0.024, PrPP101L versus w1118: D = 0.30, P = 0.00023, PrP3F4 versus PrPP101L: D = 0.42, P < 0.00001). Distributions for UAS controls are not shown for simplicity.
This PrP3F4-induced increase in mEJCs could either result from larger presynaptic vesicles or altered postsynaptic responses to a released vesicle. To test whether postsynaptic Drosophila glutamate receptor (DGluR) have functionally changed, kinetics of mEJC decays [indicative for DGluRIIA versus DGluRIIB contributions (37,38)] were compared. Mean decay tau (τ) values did not differ between genotypes nor did the τ distributions between tested conditions, indicating that DGluR composition had not changed (τCtrl: 6.1 ± 0.1 ms, τPrP3F4: 6.5 ± 0.1 ms, τPrPP101L: 6.2 ± 0.1 ms, P > 0.05, ANOVA). Further to this, the frequency (f) of mEJCs was not different between the three genotypes (fCtrl = 1.4 ± 0.1 s−1, fPrP3F4 = 1.7 ± 0.2 s−1, fPrPP101L = 1.6 ± 0.2 s−1, P > 0.05, ANOVA) and comparable with published data (39,40) suggesting that the number of functional synapses responsible for Ca2+-independent release or basal release probability have not changed in response to prion protein expression. These data suggest that the prion protein-mediated changes are not due to postsynaptic alterations but most likely due to presynaptic changes in vesicular release.
To further investigate synaptic vesicles at NMJ synapses we used electron microscopy (EM) to measure presynaptic vesicle sizes. As the Drosophila NMJ harbours two different kinds of boutons, namely 1s and 1b, both exhibiting different vesicular sizes, a subdivision of EM images into 1s and 1b boutons was made as shown before (41). The images in Figure 3A illustrate representative 1s and 1b boutons with release sites [long arrows: active zones (AZs)] and vesicles and higher magnification images for genotypes indicated. Bouton types were identified by size, number of synapses and AZs and size of the sub-synaptic reticulum (41) (Fig. 3A). Figure 3B shows average relative cumulative frequency histograms and histograms for vesicle diameters demonstrating an increased probability of larger diameters and hence can explain a right-shift in mEJC amplitude distributions in PrP3F4 and PrPP101L expressing larvae relative to their respective controls (PrP3F4 versus UAS Ctrl: D = 0.131, P < 0.0001, PrPP101L versus UAS Ctrl: D = 0.191, P < 0.0001, K–S test, n = 9–17 boutons). Furthermore, the difference between vesicle diameters from PrP3F4 and PrPP101L boutons shows also significant differences (PrPP101L versus PrP3F4: D = 0.137, P < 0.0001). Mean total vesicular sizes provide the best comparison to the mean mEJC amplitudes shown above (Fig. 2). Figure 3C illustrates the increase in mean total diameters in PrPP101L (n = 16–17 boutons) and PrP3F4 larvae (n = 9–17 boutons) relative to their UAS controls but also the difference between PrPP101L and PrP3F4 expressing larvae (*P < 0.05, **P < 0.01, Student's t-test). As these values are composed of 1s and 1b type boutons (equal number of each bouton type), we decided next to test whether prion protein effects could be observed at both bouton types. We subdivided the boutons according to previously published characteristics (number of synapses and AZs and size of the sub-synaptic reticulum) and vesicle diameter values for 1s and 1b boutons of ∼45 and ∼38 nm, respectively (41). Expression of either prion protein led to an increase in mean vesicle diameter in both types of boutons compared with their UAS controls (Fig. 3D, *P < 0.05, Student's t-test, n = 4–10 boutons). Interestingly, the number of AZs per bouton type did not differ between the genotypes (data not shown). The overall shift in the distribution of synaptic vesicle diameter in 1s and 1b boutons towards larger values suggests that PrP3F4/PrPP101L is directly or indirectly involved in regulating vesicle size. Based on the changes in vesicular diameter we calculated the increase in volume which increased in PrP3F4 expressing larvae by 60 and 50% in 1s and 1b boutons, respectively, whereas PrPP101L mutants showed an increase of 30 and 41% in 1s and 1b boutons, respectively. These increases are comparable with data from electrophysiological recordings although one has to consider an overestimation of measured mEJC amplitudes due to non-detectable smaller mEJCs (42). The data also imply that the mutated form of prion protein (PrPP101L) exhibits a diminished and altered function compared with PrP3F4 signalling. Together, this data suggest that PrP3F4 has an endogenous function in vesicle biogenesis and positively controls vesicle size and transmitter release at the Drosophila NMJ.
Figure 3.

Prion causes enlarged presynaptic vesicles. (A) EM images for genotypes indicated. Images of synaptic boutons showing synaptic active zones (AZ—arrows with T-bars in left two images) and synaptic vesicles at higher magnification (SV—right three images for genotypes indicated). The boutons have been identified by the following features: 1b boutons are larger and possess more synapses, active zones and mitochondria compared with 1s. The enveloping sub-synaptic reticulum is more voluminous around type 1b boutons. (B) Relative average and cumulative histograms of total vesicle diameter counts including both 1s and 1b boutons (with similar percentage of both bouton types) in elav-Gal4/UAS-MoPrPP101L (left) and elav-Gal4/UAS-MoPrP3F4 (right) larvae with their respective UAS controls. No difference in the mean number of AZ and T-bars between genotypes was detected. (C) Mean diameters of total vesicle counts including both 1s and 1b boutons (with similar percentage of both bouton types) for genotypes indicated showing increases of mean vesicle diameters following PrPP101L and PrP3F4 expression. Note, PrP3F4 caused a greater increase relative to PrPP101L. (D) Left, mean vesicle diameters for 1s and 1b boutons from elav-Gal4/UAS-MoPrPP101L and control UAS-MoPrPP101L/+ larvae. Right, mean vesicle diameters for 1s and 1b boutons from elav-Gal4/UAS-MoPrP3F4 and control UAS-MoPrP3F4/+ larvae. (PrP3F4: 46.2 ± 0.7 to 54.0 ± 1.1 nm [1 s]* and 37.7 ± 0.5 to 43.3 ± 0.6 nm [1b]*; PrPP101L: 43.7 ± 0.7 to 47.5 ± 0.8 nm [1s]* and 38.4 ± 0.4 to 42.1 ± 0.4 nm [1b]*; *P < 0.05, **P < 0.01, Student's t-test). Data denote mean ± SEM, n—number of boutons is indicated in bars [n = 3 animals for each genotype].
Ca2+-dependent evoked release is enhanced by prion protein expression
The above data suggest a functional role of prion protein in synaptic function and vesicular release. To further investigate these effects on release mechanisms we measured evoked release at NMJs with presynaptic expression of PrP3F4 or PrPP101L. Evoked EJC (eEJC) amplitudes at low frequency stimulation (0.2 Hz) were enhanced following PrP3F4 (134 ± 5 nA*** (n = 19); P < 0.001, ANOVA) but not PrPP101L expression (110 ± 9 nA (n = 7)) compared with w1118 Ctrl (101 ± 5 nA (n = 18)) and UAS-PrP3F4 controls (103 ± 2 nA (n = 10)) suggesting a differential effect on synaptic transmission (Fig. 4A and B). eEJCs did not differ in their decay kinetics (data not shown) again confirming no alterations in postsynaptic receptor composition (37). Assuming that single quanta summate linearly in eEJCs and mEJCs and evoked responses arise from the same pool of vesicles at these low stimulus frequencies, quantal content [QC, the number of vesicles released per action potential (AP)] was estimated as the quotient of eEJC and mEJC amplitude per NMJ. The QC was strongly increased in PrP3F4 NMJs (w1118 Ctrl: 119 ± 6 (n = 12); PrPP101L: 140 ± 8 (n = 5); PrP3F4: 162 ± 11* (n = 18); UAS-PrP3F4: 121 ± 5 (n = 10); P < 0.05, ANOVA, Fig. 4B). The enhanced QC could be attributable to an increase in either the number of release-ready vesicles or the release probability per vesicle.
Figure 4.

Evoked synaptic release and QC are enhanced by PrP3F4 expression. (A) representative eEJC recordings by stimulating the motor nerve in genotypes indicated at 1.5 mm [Ca2+]e. (B) Summary of mean eEJC amplitudes and QC for different genotypes at 1.5 mm [Ca2+]e, *P < 0.05, ***P < 0.001, ANOVA with Tukey–Kramer post hoc test, n = 5–19 NMJs (indicated in bars [n = 10 animals for controls, n = 5 animals for PrPP101L, n = 15 animals for PrP3F4, n = 6 animals for UAS-PrP3F4]). (C) Left, example traces for PPR for a Ctrl (w1118) and PrPC NMJ at 20 ms inter-stimulus-intervals (ISI) at 1.5 mm [Ca2+]e. Black arrows indicate measurements of eEJC amplitudes. Right, summary of mean PPR versus ISI between 20 to 200 ms for three different genotypes. The dotted line indicates a PPR of 1 (20 ms: PPRctrl: 1.07 ± 0.03, PPRPrP3F4: 0.89 ± 0.03*, PPRPrPP101L: 0.98 ± 0.04; 40 ms: PPRctrl: 1.04 ± 0.03, PPRPrP3F4: 0.88 ± 0.02*, PPRPrPP101L: 0.94 ± 0.05; 100 ms: PPRctrl: 1.04 ± 0.02, PPRPrP3F4: 0.91 ± 0.01*, PPRPrPP101L: 0.92 ± 0.03; 200 ms: PPRctrl: 0.97 ± 0.02, PPRPrP3F4: 0.92 ± 0.01, PPRPrPP101L: 0.91 ± 0.01; ANOVA (n = 21 [Ctrl], n = 7 [PrPP101L], n = 13 [PrP3F4] NMJs [n = 10 animals for controls, n = 4 animals for PrPP101L, n = 7 animals for PrPC]), PrP3F4 versus Ctrl: *P < 0.05 at different ISI. ANOVA with Tukey–Kramer post hoc test. Data denote mean ± SEM.
To discriminate between these two options we initially analyzed paired pulse ratios (PPR) which critically depend on the initial release probability (pvr) (43) but also on the degree of facilitation (44) or on receptor saturation and desensitization (45,46). Receptor desensitization was excluded as eEJCs decay kinetics were identical between the first and last eEJC within a 1 s train (data not shown).
PPR provide a measure of pvr, in that the amplitude ratio of two closely spaced EJCs (EJC2/EJC1) increases as pvr decreases (45). Here, the ratio of two eEJCs with inter-spike-intervals (ISI) varying from 20 to 200 ms was assessed at 1.5 mm [Ca2+]e. In all further experiments we used only w1118 larvae as controls since responses in UAS-PrP3F4 controls and UAS-PrPP101L controls did not differ from w1118 (compare Figs 2 and 4B) and all strains were back-crossed for at least six generations to w1118. In agreement with previous data, control larvae showed weak paired-pulse facilitation at 100, 40 and 20 ms ISI (Fig. 4C) (42), whereas PrP3F4 expressing larvae showed significant depression at various ISI tested (ANOVA, P < 0.05, Fig. 4C) suggesting an increase in pvr in PrP3F4 larvae and consistent with increases in QC following PrP3F4 expression (Fig. 4B). PrPP101L again showed a non-significant tendency towards an increased pvr.
Prion protein reduces synaptic vesicle pool size
As changes in the observed PPR between control and PrP3F4 expressing synapses could be caused by alterations of two different parameters, namely: (i) the initial pvr of the synapse but also (ii) the size of the ready-releasable pool (RRP) (43,47,48), it is important to estimate both of these synaptic parameters. A method to determine pool size was successfully applied at Drosophila NMJs by analyzing cumulative amplitudes within trains of higher frequency stimulation (42,49). Figure 5A shows 50 Hz trains (500 ms) of a control and PrP3F4 expressing NMJ. Control recordings show a strong initial potentiation followed by depletion which plateaued after ∼300 ms. In contrast, PrP3F4 expression did not allow potentiation within the train. Under these conditions, receptor desensitization was excluded as eEJCs decayed with identical time constants at the beginning and end of the train (data not sown). The average data of peak amplitudes revealed the difference between the genotypes as shown in Figure 5B. To check whether the observed differences are due to changes in pvr and/or size of RRP we estimated these parameters (Fig. 5C–F). Stimulation at 50 Hz for 500 ms revealed a smaller cumulative eEJC amplitude (extrapolation to time point zero) of 162 ± 32 nA*** (n = 8) in PrPP101L and 133 ± 20 nA*** (n = 13) in PrP3F4 expressing larvae compared with 324 ± 45 nA (n = 7) in control larvae (ANOVA, Fig. 5C and E) suggesting a strong reduction in pool size. The pool size was subsequently calculated by multiplying the cumulative amplitude by the mean quantal size of each muscle, confirming a strong reduction following PrP3F4 but also PrPP101L expression (Fig. 5E). The number of release-ready vesicles was calculated by dividing the cumulative amplitude by the quantal size and was therefore reduced from control of 382 ± 44 (n = 7) to 174 ± 30** (n = 13) in PrP3F4 expressing larvae but not in PrPP101L (198 ± 32 (n = 8), Fig. 5D and F; ANOVA). The possibility that recovery from depression was interfering with this data analysis could be excluded as it was shown previously that recovery occurs earliest at times >20 s (42).
Figure 5.

Number of readily releasable vesicles is reduced following PrP3F4 expression. (A) Example traces of 50 Hz trains (500 ms) from a Ctrl (black) and PrP3F4 expressing (red) larva at 1.5 mm [Ca2+]e. (B) Mean eEJC amplitudes from synaptic trains for the three different genotypes. (C) Cumulative eEJC amplitudes for the three genotypes with linear fits to the last 200 ms for each condition. Line fits to the cumulative eEJCs were back-extrapolated to time zero (indicated in italics, see Materials and Methods). (D) Average cumulative QCs with back-extrapolation of linear fits to the last 200 ms yielded estimates for the readily releasable pool (indicated in italics) for each genotype. Inset, mean QC during the 50 Hz stimulus train in the three genotypes indicated. (E) Mean values for cumulative eEJC amplitudes (black bars, ***P < 0.001) and vesicle pool sizes (open bars, *P < 0.05, **P < 0.01) are reduced compared with control. Pool size was determined by multiplying the cumulative eEJC amplitudes with the mean quantal size for this cell. (F) Mean number of ready-releasable vesicles (black bars, **P < 0.01) and the release probability for each genotype (open bars, **P < 0.001). The number of vesicles was estimated by dividing pool size by mean quantal size. The release probability was estimated by dividing eEJC0 by the pool size. ANOVA with Tukey–Kramer post hoc test (n = 7–13 NMJs as indicated in bars [n = 4 animals for controls, n = 5 animals for PrPP101L, n = 9 animals for PrP3F4]). Data denote mean ± SEM.
Using this method, we further estimated the initial pvr which can be calculated based on the cumulative amplitude analysis by dividing EJC0 of the train by the size of the RRP as described previously (50). The comparison between the three genotypes revealed that pvr is increased following expression of PrP3F4 but not PrPP101L in agreement with above data (Ctrl: 0.34 ± 0.04 (n = 7); PrPP101L: 0.74 ± 0.12 (n = 8); PrP3F4: 1.01 ± 0.14** (n = 13); Fig. 5F, ANOVA).
The above data consistently show that PrP3F4 enhanced pvr and decreased the size of the RRP, whereas PrPP101L only induced a decrease in the RRP size without affecting pvr.
We next decided to apply a different and independent approach to estimate the synaptic parameters by which prion protein modulates synaptic release: fluctuation analysis (51–53) estimates the quantal parameters: N, pvr and quantal size q. eEJCs were elicited at varying calcium concentration (0.5–3 mm, 0.2 Hz, Fig. 6A and B) and N, pvr and q were estimated from parabolic fits to the variance–mean plots for each cell (Fig. 6D). By plotting the mean QC over various [Ca2+]e (Fig. 6C) it became apparent that PrP3F4 expression led to enhanced release at 0.5–2 mm [Ca2+]e, whereas PrPP101L expression only enhanced QC at 1 mm [Ca2+]e (*P < 0.05, **P < 0.01, ANOVA). Data were fitted with a Hill equation yielding the Hill slope as a measure of Ca2+ co-operativity for prion protein expressing and control larvae. Interestingly, the Hill slope was reduced in PrP3F4 versus PrPP101L and control NMJs from 3.3 ± 0.5 [Ctrl] and 4.3 ± 0.3 [PrPP101L] to 2.4 ± 0.3 [PrP3F4] (PrP3F4 versus Ctrl: *P < 0.05, PrP3F4 versus PrPP101L: ***P < 0.001, mean ± SD, ANOVA, Fig. 6) without affecting the half-maximal effective Ca2+ concentrations (EC50, ANOVA, P > 0.05, n = 8–11 NMJs) indicating that sensitivity to Ca2+ was not altered (54). A lower Hill slope has been associated with a tightening of the release site/Ca2+ channel complex (55,56) opening up the possibility for an interaction of PrP3F4 with Ca2+ signalling.
Figure 6.

Prion expression induced changes in synaptic release parameters. (A) Group of eEJCs recorded from one NMJ at different indicated [Ca2+]e in mm. (B) eEJC amplitudes of a PrP3F4 expressing NMJ elicited at 0.2 Hz at different [Ca2+]e. The lines indicate regions used for analysis. (C) Mean QC plotted versus different [Ca2+]e for the genotypes indicated and fitted with a Hill function according to QC([Ca2+]) = QCmax[1+(EC50/[Ca2+])slope]−1 which yielded QCmax = 136 ± 6, 171 ± 5 and 194 ± 15; EC50 = 1.0 ± 0.02 mm, 0.9 ± 0.01 mm and 0.9 ± 0.06 mm and Hill slope = 3.3 ± 0.5, 4.3 ± 0.3 and 2.4 ± 0.3 for Ctrl, PrPP101L and PrPC NMJs, respectively (Hill slope: PrP3F4 versus Ctrl: *P < 0.05, PrP3F4 versus PrPP101L: ***P < 0.001, data denote mean ± SD, ANOVA with Tukey–Kramer post hoc test, n = 8–11 NMJs [n = 5 animals for control, n = 6 animals for PrPP101L, n = 5 animals for PrP3F4]). Note, PrP3F4 causes a left-shifted [Ca2+]e—QC relationship but also and change in the slope of the fitted curves. (D) Parabolic fits to the variance–mean relationships for the three genotypes indicated. Note data are from a different subset of experiments as in C (data denote mean ± SEM). (E) Mean quantal parameters quantal size q, vesicular release probabilities (pvr) at different [Ca2+]e and number of release-ready vesicles (N) estimated from fluctuation analysis in all three genotypes (*P < 0.05, **P < 0.01, ANOVA with Tukey–Kramer post hoc test, with n—number of NMJs indicated in bars [n = 4 animals for control, n = 4 animals for PrPP101L, n = 4 animals for PrP3F4], data denote mean ± SEM). (F) Double-logarithmic plot of the QC as a function of [Ca2+]e for all three genotypes as indicated. Ca2+ dependency is decreased in PrP3F4 but not PrPP101L: controls (3.7 ± 0.4), PrPC (2.7 ± 0.3*, P = 0.036), PrPP101L (4.0 ± 0.9, P = 0.23), n = 20–27 NMJs [n = 12 animals for control, n = 11 animals for PrPP101L, n = 14 animals for PrPC]. Data denote mean ± SD, Student's t-test, ANOVA did not reveal any significance.
The fluctuation analysis further revealed, in agreement with data from mEJC measurements (Fig. 2), that q was enlarged following PrPP101L (731 ± 8 pA, n = 7, P < 0.01) expression and even further increased in PrP3F4 (886 ± 24 pA, n = 5, P < 0.01 versus Ctrl and P < 0.05 versus PrPP101L) expressing larvae compared with controls (347 ± 27 pA, n = 5; Fig. 6E left, ANOVA). Furthermore, PrP3F4 had a stronger phenotype then PrPP101L also in agreement with above data (Figs 2–4). The estimations further revealed an increase in pvr at 2 and 3 mm [Ca2+]e in PrP3F4 but not PrPP101L expressing larvae although once again, PrPP101L showed a tendency towards enhanced pvr values (Fig. 6E middle) consistent with the enhanced QC and decreased PPR in PrP3F4 expressing larvae (Figs 4 and 5). On the other hand, the analysis showed that the number of release-ready vesicles decreased from 630 ± 104 (n = 5) in controls to 288 ± 91 (n = 7) in PrPP101L and 246 ± 30 (n = 5) in PrP3F4 expressing larvae (Fig. 6E right, P < 0.05, ANOVA). Therefore, both, cumulative postsynaptic current and fluctuation analysis showed roughly a 50% reduction in those parameters following prion protein expression. The estimation of N from fluctuation analysis (∼600) in controls is in accordance with previously reported EM data showing a number of synapses onto muscle 6 of ∼500 (57). Together, these estimations are consistent with the data from above experiments where cumulative eEJC amplitude measurements (Fig. 5) or direct eEJC and mEJC measurements (Figs 2, 4 and 6) were conducted.
The enhanced QC seen in PrP3F4 expressing larvae can also be attributable to a change in Ca2+ dependence of release so we determined whether the elevated transmitter release is due to an altered Ca2+ co-operativity of release (58–60). The changes in the Hill slope shown above (Fig. 6C) indicate a reduction in Ca2+ co-operativity in PrP3F4 larvae but to corroborate this we analyzed the slopes of the linear regression lines to the double log QC–[Ca2+]e relationship at lower non-saturating [Ca2+]e (Fig. 6F). The data show that the Ca2+ co-operativity was reduced following PrP3F4 expression (P < 0.05, Student's t-test, Fig. 6) but not versus PrPP101L (P > 0.05, Student's t-test, Fig. 6).
The detected reduction in the number of effective release sites as a consequence of prion protein expression could result from either functional or morphological alterations as developmental morphological changes at the NMJ are associated with various signalling pathways (61,62). To distinguish between these two possibilities we performed confocal imaging of NMJs and analyzed the number of NC82 puncta [Bruchpilot (Brp)—an essential component of the active zone cytomatrix T-bar] per NMJ and the NMJ size by calculating the area of vGlut staining. Figure 7 shows that both parameters are unchanged between the three genotypes (three NMJs from three larvae per genotype, P > 0.05, ANOVA) suggesting that prion protein expression does not induce any morphological changes and the reduction in release sites is due to functional rather than morphological changes.
Figure 7.

NMJ morphology is not affected by prion proteins. (A) Confocal images were taken from control, PrPP101L and PrP3F4 expressing NMJs co-stained with anti-vGlut C-terminus and NC82 (anti-Brp) (Muscle 6). The number of NC82 puncta and vGlut area was calculated using Volocity software. (B) Mean data for counted NC82 puncta and vGlut area for the three genotypes indicated (n = 3 NMJs from three animals for each genotype, P > 0.05, data denote mean ± SEM. ANOVA with Tukey–Kramer post hoc test).
Prion protein mediates enhanced Drosophila larval locomotor activities
The outcome of the changes in synaptic transmission induced by prion protein expression would be a stronger depolarization of the postsynaptic muscle as long as the reduction in pool sizes does not become the limiting factor. On the behavioural level, a measure of low frequency muscle contraction would be the locomotor activity. The expectation would be that a stronger depolarisation leads to a stronger contraction and further locomotor distances. To test this behaviour, larvae were placed on a crawling device allowing on-line monitoring of individual larval activities (63). Locomotive impairment is a commonly used diagnostic for characterization of models for neurodegeneration where foraging behaviours are affected (64) and this test was employed to investigate locomotor activities of prion protein expressing larvae. Expression of either prion protein caused an increase in activity as detected in greater crawling distances per 30 min (P < 0.001, ANOVA, Fig. 8) relative to WT control larvae suggesting that the synaptic effects of prion protein expression also altered behavioural activities.
Figure 8.
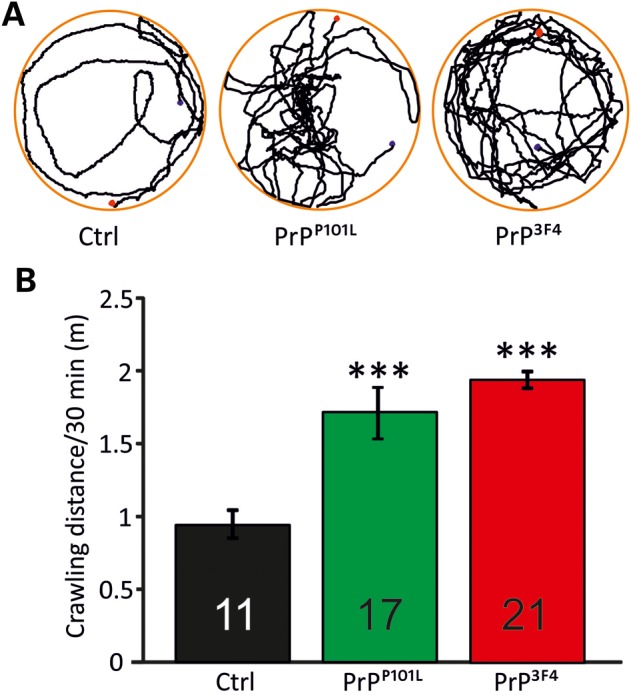
Prion proteins induce greater locomotor activities. Larval locomotor behaviour was recorded over a period of 30 min. Third instar larvae were put on a moist, food-free and temperature controlled (20°C) surface. Larval tracks were monitored (A, blue dot—starting point; red dot—end point of tracking) and data for each genotype were expressed as crawling distance (m) per 30 min (B) (n = 11 animals for control, n = 17 animals for PrPP101L, n = 21 animals for PrP3F4]). Data denote mean ± SEM (***P < 0.001, ANOVA with Tukey–Kramer post hoc test).
DISCUSSION
Our data demonstrate a novel physiological function of prion protein at the synapse. Although many studies have reported neurodegenerative effects of misfolded prion protein, its physiological roles are much less understood. Initial data from mouse studies suggest that prion protein is required for synaptic transmission and mice lacking PrPC show reduced excitatory and inhibitory synaptic currents. Various studies demonstrated a positive correlation between prion protein expression and synaptic transmission (14,18,32) suggesting that PrPC is involved in neurotransmission (16,27), neurite outgrowth (65,66), circadian rhythm (17) and neuronal excitability (67). In the present study we have shown that PrP3F4 not only leads to larger vesicles but also increases pvr with simultaneous reduction in the number of functional release sites equivalent to fewer ready-releasable vesicles. A mutated PrPC (PrPP101L) exhibited diminished phenotypes illustrating a dysfunctional signalling.
As PrPC expression causes an increase in vesicle diameter (gain-of-function), conversely, one would predict that deletion of prion protein signalling would induce the opposite effect. As the Drosophila genome does not contain an ortholog of mammalian PrPC, expression of the PrP3F4/PrPP101L transgene induced the observed gain-of-function effect. In mammalian tissue, PrPC is widely expressed and a loss-of-function would be observed in PrPC-KO mutants. In fact, PrPC-KO mice show reduced transmitter release at inhibitory synapses (14) and conversely, addition of PrPC induces a potentiation of end-plate currents at mouse NMJs, most likely due to presynaptic mechanisms (32) supporting a physiological role of endogenous PrPC in synaptic function. Recent evidence shows that PrPC has physiological roles and hence loss-of-function effects due to conversion into PrPSC or in PrPC-KO/mutant models were observed (11–13). PrPC can interact with synapsin Ib, metabotropic glutamate receptors, Ca2+ channels or laminin, all of which represent potential binding partners for PrPC (13,31,68,69). The role for PrPC–laminin interaction is consistent with scaffolding properties of PrPC and its role in regulation of signal transduction (70) with altered PrPC signalling leading to synaptic dysfunction.
The neurotoxic gain-of-function during prion disease is believed to be due to accumulation of PrPSC but little evidence is available that loss of PrPC plays a role during the pathogenesis. However, the possibility that prion diseases have loss-of-function components remains open (71,72) and a critical examination of this hypothesis depends on determining the elusive neuronal functions of PrPC. This current study took advantage of the Drosophila model by using the UAS/Gal4 bipartite expression system with pan-neuronal PrP3F4/PrPP101L expression. The advantages of the Drosophila model to investigate neurodegenerative signalling pathways are well-recognized and several prion-related disease models in flies investigate ovine- (28) and mouse-prion protein (26,36) or GSS-mediated (25,30) neurodegeneration.
Prion protein interaction with vesicle formation
Previous studies in mouse NMJs and hippocampal CA1 neurons suggested that PrPC potentiates synaptic release (18,32) consistent with PrPC expression at presynaptic terminals (73). However, pre- but also postsynaptic PrPC signalling precludes a precise determination of its function. We have confirmed presynaptic actions of PrP3F4 showing an increase in the size of synaptic vesicles. An interaction between synaptic vesicles and PrPC has been proposed at mammalian NMJs based on morphological studies (74) and our functional data support a role for PrPC in vesicular homeostasis.
As PrPC interacts with synapsin (31) and its internalization is mediated via clathrin-coated pits (75) in a dynamin-dependent process (76), one could speculate that PrPC may play a role in endocytosis, vesicle replenishment and release. This interaction would offer a new functional explanation of how PrPC modulates release processes at the synapse and how a consequent conversion of PrPC into PrPSC could induce synaptic dysfunction.
Prion protein modulates release probability and vesicle pool size
Our findings that PrP3F4 expression leads to an increase in pvr and decrease in the size of the functional readily releasable pool suggests further actions of PrPC in regulating synaptic transmission. Changes in pvr could be caused by alterations in Ca2+ channel density at release sites which occurs in mutants of Rab3 and Rab3-interacting molecule (RIM) (39,49,77), or by differential coupling of voltage-gated Ca2+ channels (VGCC) with synaptic vesicles (55,56). Furthermore, RIM-binding protein family members and the cytomatrix-associated protein Brp are essential in binding Ca2+ channels and loss of RIM or mutations in Brp lead to a loss of Ca2+ channels (clustering) within the AZ (78,79) illustrating an important connection between the architecture of AZs and Ca2+-mediated vesicle release.
In this context, a mutation in PrPC leads to impaired membrane delivery of the α2δ-1 subunit of VGCC in cerebellar granule neurons (13) suggesting that PrPC is required for intact synaptic Ca2+ signalling which may explain our results with respect to the increased pvr.
The number of release sites determined by fluctuation analysis (∼600) and postsynaptic current analysis (∼380) in our study is similar to the number of AZs reported previously using serial EM images (∼500) (57), current analysis (∼380) (42) or fluctuation analysis (∼500) (39). The fact that one AZ harbours not only one but two release-ready vesicles as shown at mouse NMJs (80) and following synaptic strengthening at Drosophila NMJs (42), allows for the variability of reported numbers of AZs. Importantly, in our study PrP3F4 expression caused a reduction in the number of release-ready vesicles with simultaneously enhanced pvr as determined by independent methods demonstrating a synaptic role of PrPC.
Functional differences between single release sites have been reported at Drosophila NMJs indicating a specific and heterogeneous identity of each release site which determines its pvr (81,82). This identity depends on clustering and the number and activation of VGCC as shown at the calyx of Held (83,84) or the structure and architecture of the cytomatrix (57,81). The organization of AZs is not static but undergoes plastic changes (85) and PrPC could provide a potential candidate to interact with this structure. Our data point towards a model where prion protein expression results in fewer release sites with those remaining having greater pvr possibly due to tighter coupling of Ca2+ channel/release site complexes by interacting with members of the AZ architecture. Our morphological studies indicate that PrP3F4 modulates the functionality rather than the number of AZs (Fig. 7).
Is prion protein modulating calcium-mediated release?
Our findings that PrP3F4 expression alters the Ca2+ co-operativity as shown by a reduced Hill coefficient and slope of the log–log QC–[Ca2+]e relationship (Fig. 6) supports the notion of an increased clustering of synaptic Ca2+ channels. At the mouse calyx of Held, synaptic strength increases with development (86,87) and this occurs in parallel with decreased Ca2+ co-operativity. The classical Ca2+ co-operativity usually refers to the co-operative interaction of 3–4 Ca2+ ions with the release machinery (58). Our data show similar co-operativity values in controls (∼3.3), whereas PrP3F4 exhibits a reduced value of ∼2.4. In contrast, the increase in co-operativity in PrPP101L could reflect a crucial function of PrPC in modulating Ca2+ signalling where this mutation shows a malfunctional phenotype. A reduced co-operativity and Hill slope (as seen with PrP3F4 expression) and increased synaptic strength are associated with tightening of VGCC to release sites in developmentally older compared with higher co-operativities and loosely coupled VGCC in younger synapses (55) suggesting that lower co-operativity, in a dynamic fashion, is associated with faster and more Ca2+-efficient synaptic responses (56). Physiologically, this lower co-operativity associated with higher pvr may also reduce the effects of residual Ca2+ build-up and thus prevent depletion of synaptic vesicles allowing a reduction of release-ready vesicles whilst preserving the synaptic strength as shown in our data.
CONCLUSION
In summary, based on the fact that PrPC interacts with several proteins involved in synaptic release (31,76) and a range of ion channels, including VGCC (13), thereby modulating neuronal transmission, we investigated synaptic PrPC signalling in more detail. Our data provide new evidence for a functional role of PrPC in synaptic transmission where a modulation of vesicles and release properties by PrPC increases synaptic strength. In relation to our findings, PrPC-mediated trafficking of VGCC subunits to the membrane which provides functional glutamatergic transmission (13), may play a crucial role in PrPC-mediated increases in synaptic release.
The molecular mechanisms of AZ organization are important determinants of synaptic function, in particular, proteins such as laminin β2 (scaffolding protein and PrPC binding partner), Bassoon, CAST/Erc2/ELKS2α, Piccolo and RIMs (78,88) are essential components. In this context, it is worth highlighting the functions of Rab GTPases and RIM, which are crucially involved in synaptic transmission (22,77,78), and the facts that Rab3a activity is compromised in CJD (89) and Rab7a is a PrPC interacting partner (90). This connection may present a signalling pathway by which PrPC is required for intact Rab signalling, facilitating release, which will subsequently be diminished in prion disease resulting in synaptic dysfunction. Thus, the complex relationship between synaptic proteins and PrPC, with our data pointing towards an interaction of PrPC with AZ function, suggests a novel physiological role. We propose a model, where prion protein signalling may lead to an optimization and increased efficiency of release, leading to higher Ca2+-dependent release probabilities with concurrent reduction in the number of enlarged release-ready vesicles. Further experiments to elucidate the exact interaction of PrPC with the release machinery and synaptic ion channels, specifically molecules of the cytomatrix and Ca2+-mediated release, will allow a better understanding of the physiological roles of PrPC signalling.
MATERIALS AND METHODS
Drosophila stocks
Flies were raised on standard maize media at 25°C. The elav-Gal4 [c155] driver was obtained from the Bloomington Stock Center (Indiana, USA). The transgenic flies contain a UAS construct of either wild-type mouse prion protein on the third chromosome (UAS-Mo-PrP3F4) or mutant mouse prion protein (UAS-Mo-PrP3F4 containing a proline-to-leucine substitution at residue 101) on the second chromosome (P101LD, referred to as P101LD PrPC [PrPP101L]) (25). Flies homozygous for the UAS-PrP3F4 or UAS-P101LD constructs were crossed with flies homozygous for the elav-Gal4 driver to produce offspring expressing either PrP3F4 or mutant prion protein PrPP101L. The 3F4 epitope tag on Mo-PrP does not distort the normal topology or functions of PrPC (91,92). The use of the UAS/Gal4 bipartite expression system to drive pan-neuronal expression excludes potential postsynaptic effects. All lines were backcrossed to w1118 for at least six generations allowing the use of w1118 as controls (Figs 5–8, unless otherwise stated) (39).
Electrophysiology
TEVC recordings using sharp-electrodes were made from ventral longitudinal Muscle 6 in abdominal segments 2 and 3 of third instar larvae using pClamp 10, an Axoclamp 900A amplifier and Digidata 1440A (Molecular Devices, USA) in hemolymph-like solution 3 (HL-3) (60). Recording electrodes (10–30 MΩ) were filled with 3 m KCl. mEJCs were recorded in the presence of 0.5 μm tetrodotoxin (Tocris, UK). All synaptic responses were recorded from muscles with input resistances ≥4 MΩ and resting potentials more negative than −60 mV at 25°C as differences in recording temperature cause changes in glutamate receptor kinetics and amplitudes (93). Holding potentials were −60 mV. The extracellular HL-3 contained (in mm): 70 NaCl, 5 KCl, 20 MgCl2, 10 NaHCO3, 115 sucrose, 5 trehalose, 5 HEPES and 0.5–3 CaCl2 (as specified). Average single eEJC amplitudes (stimulus: 0.1 ms, 1–5 V) are based on the mean peak eEJC amplitude in response to ten presynaptic stimuli (recorded at 0.2 Hz). Nerve stimulation was performed with an isolated stimulator (DS2A, Digitimer). Paired-pulse experiments were performed by applying five repetitive stimuli (0.2 Hz) at different intervals (20, 40, 100 and 200 ms) for each cell at each inter-spike-interval. Materials were purchased from Sigma-Aldrich (UK) unless otherwise stated.
All data were digitized at 10 kHz and for miniature recordings, 200 s recordings we analyzed to obtain mean mEJC amplitudes, decay and frequency values. QC was estimated for each recording by calculating the ratio of eEJC amplitude/average mEJC amplitude followed by averaging recordings across all NMJs for a given genotype. mEJC and eEJC recordings were off-line low-pass filtered at 500 Hz and 1 kHz, respectively.
Ca2+co-operativity
Ca2+ co-operativity was analyzed from synaptic current amplitudes recorded for each [Ca2+]e from several muscles of different larvae. Co-operativity coefficients were derived by fitting linear regression lines to log-transformed individual data points for Ca2+ concentrations ≤1 mm and the slopes of the regression lines were statistically compared. Co-operativity coefficients estimated this way closely match coefficients derived by recording from several Ca2+ concentrations in single cells giving coefficients of ∼3–4 in wild-type larvae.
Variance–mean analysis of eEJCs
Approximately 40 eEJCs were elicited at different [Ca2+]e, ranging from 0.5 to 3 mm to give mean eEJC amplitudes (I). The mean eEJC is given by I = Npvrq (52,94) with N being the number of independent release-ready vesicles, pvr the vesicular release probability and q the quantal size at each given [Ca2+]e. The eEJC variance was calculated as previously described (52,95). The plots of the variance–mean were obtained for each cell and fitted with the parabolic function Var(I) = I2/N + qI that was constraint to pass through the origin. Upon fitting the parabola, pvr and q were calculated using the equations: q = A/(1 + CV2) and pvr = I(B/A)(1 + CV2) where CV2 is the coefficient of variation of the eEJC amplitudes at a given [Ca2+]e concentration calculated as: CV2 = (eEJCs standard deviation/mean amplitude)2, A and B were obtained from the fitting parameters (53,96). Estimated values were not corrected for variability in mEJC amplitude distributions or latency fluctuations (51,97).
Cumulative postsynaptic current analysis
The apparent size of the RRP was probed by the method of cumulative eEJC amplitudes (50), which was applied to the Drosophila NMJ previously (42). Muscles were clamped to −60 mV and eEJC amplitudes during a stimulus train (50 Hz, 500 ms) were calculated as the difference between peak and baseline before stimulus onset of a given eEJC. Receptor desensitization was not blocked as it did not affect eEJC amplitudes since a comparison of the decay of the first and the last eEJC within a train did not reveal any significant difference in decay kinetics. The number of release-ready vesicles was obtained by back-extrapolating a line fit to the linear phase of the cumulative eEJC plot (the last 200 ms of the train) to time zero. The number of release-ready vesicles was then obtained by dividing the cumulative eEJC amplitude at time zero by the mean mEJC amplitude recorded in the same cell. To calculate the QC in the train, we used mean mEJC amplitudes measured before the train.
Larval locomotor assay
Age-matched third instar larvae (∼100–120 h) were selected, washed and placed onto a moist, food-free surface (with constant temperature of 20°C (63)). Crawling activities were imaged over 30 min using AnyMaze software v4.98 (Stoelting Co., USA) and data were analyzed off-line.
Immunohistochemistry
Third instar larvae were dissected in ice-cold phosphate buffered saline (PBS) then fixed in 4% paraformaldehyde. After permeabilization with PBS-0.1% Triton (PBS-T) and blocking with PBS-T containing 0.2% bovine serum albumin and 2% normal goat serum, larval fillets were incubated at 4°C overnight in solutions of primary antibody. The following antibody dilutions were used: AH6 anti-PrP (TSE Reagent Resource Centre, Compton, UK) 1:1000 dilution; anti-vGlut C-terminus (kind gift from Hermann Aberle, University of Düsseldorf) 1:2000 dilution; NC82 (supernatant) anti-Brp (Bruchpilot; Developmental Studies Hybridoma Bank) 1:200 dilution and α-tubulin 1:4000 dilution. After 3 × 10 min washes in PBS-T, larvae were incubated with AlexaFluor 488 goat anti-rabbit and/or AlexaFluor 546 goat anti-mouse 1:1000 dilution for 90 min at room temperature. Larvae were mounted using Vectashield mounting medium (Vector Labs) and NMJ 6/7 (segments A2 and A3) images were acquired with a Zeiss laser-scanning confocal microscope (LSM 510, Carl Zeiss International). Image analysis was performed with ZEN (Carl Zeiss International) and Volocity software. Mean number of active zones per NMJ was calculated by dividing the number of NC82 (anti-Brp) puncta per NMJ by the total NMJ area detected by the vGlut antibody from maximum projections of z-stack images.
PK digestion assay/immunoblotting
Five Third instar larvae heads were homogenized in 30 µl radioimmunoprecipitation assay buffer (150 mm NaCl, 10 mm Tris–HCl, 0.1% sodium dodecyl sulphate (SDS), 1% Triton X-100,1% sodium deoxycholate, 5 mm ethylenediaminetetraacetic acid) with Protease Cocktail Inhibitor (Promega) added. Homogenates were centrifuged at 4°C for 15 min at 16 000g. Ten micrograms of protein was incubated with the appropriate concentration of PK at 37°C for 30 min. The reaction was stopped by the addition of 2 mm phenylmethylsulfonyl fluoride, 4× Laemmli buffer was added and samples were boiled for 5 min before SDS-polyacrylamide gel electrophoresis and western blot analysis was performed. Prion protein aggregation (PrPSC) causes a lack of PK sensitivity resulting in digestion starting at ∼5–7 μg/ml relative to PrPC digestion starting at ∼0.1 μg/ml.
Electron microscopy
Third instar larvae were ‘filleted’ in phosphate-buffered saline at room temperature and then fixed in 2% (wt/vol) glutaraldehyde in 0.1 m sodium cacodylate buffer (pH 7.4) at 4°C overnight. They were post-fixed with 1% (wt/vol) osmium tetroxide/1% (wt/vol) potassium ferrocyanide for 1 h at room temperature and then stained en bloc, overnight, with 5% (wt/vol) aqueous uranyl acetate at room temperature, dehydrated and embedded in Taab epoxy resin (Taab Laboratories Equipment Ltd, Aldermaston, UK). Semi-thin sections, stained with toluidine blue, were used to identify areas containing synaptic regions (muscle 6/7 in regions A2/A3). Ultra-thin sections were cut from these areas, counterstained with lead citrate and examined in a JEOL 100-CXII electron microscope [JEOL (UK) Ltd, Welwyn Garden City, UK]. Images were recorded using a SIS Megaview III digital camera with iTEM software. SV measurements were made using ImageJ software. A total of ∼480–980 SVs were measured in 4–10 boutons from three animals per genotype.
Statistics
Statistical analysis was performed with Prism 6 and InStat 3 (Graphpad, San Diego, USA). Statistical tests were carried out using an ANOVA test where applicable with a posteriori test or unpaired Student's t-test as indicated. Cumulative frequency distributions were compared using the Kolmogorov–Smirnov test (K–S test). Data are expressed as mean ± SEM or ± SD (as indicated) where n is the number of boutons (Fig. 3)/NMJs (Fig. 2 and 4–7)/larvae (Fig. 8) and significance is shown as *P < 0.05, **P < 0.01, ***P < 0.001.
FUNDING
This work was supported by the Medical Research Council and BBSRC, UK. Funding to pay the Open Access publication charges for this article was provided by the Medical Research Council.
ACKNOWLEDGEMENTS
Prion fly stocks were kindly provided by Prof. P.J. Dolph (Dartmouth College Hanover, USA) with the help of Archana Murali. The University of Iowa Developmental Studies Hybridoma Bank (DSHB) and Prof. Hermann Aberle, University of Düsseldorf provided essential reagents for this project. Many thanks to Dr David Read (MRC Toxicology Unit) for support in confocal microscopy and Prof. Ian Forsythe and Thomas Tagoe (University of Leicester) for discussions on the manuscript and data analysis, respectively.
Conflict of Interest statement. The authors declare no competing financial interests.
REFERENCES
- 1.Linden R., Martins V.R., Prado M.A., Cammarota M., Izquierdo I., Brentani R.R. Physiology of the prion protein. Physiol. Rev. 2008;88:673–728. doi: 10.1152/physrev.00007.2007. [DOI] [PubMed] [Google Scholar]
- 2.Caughey B., Baron G.S., Chesebro B., Jeffrey M. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annu. Rev. Biochem. 2009;78:177–204. doi: 10.1146/annurev.biochem.78.082907.145410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prusiner S.B. Prions. Proc. Natl. Acad. Sci. USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Collinge J., Clarke A.R. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 5.Herms J., Tings T., Gall S., Madlung A., Giese A., Siebert H., Schurmann P., Windl O., Brose N., Kretzschmar H. Evidence of presynaptic location and function of the prion protein. J. Neurosci. 1999;19:8866–8875. doi: 10.1523/JNEUROSCI.19-20-08866.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moya K.L., Sales N., Hassig R., Creminon C., Grassi J., Di Giamberardino L. Immunolocalization of the cellular prion protein in normal brain. Microsc. Res. Tech. 2000;50:58–65. doi: 10.1002/1097-0029(20000701)50:1<58::AID-JEMT9>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 7.Um J.W., Nygaard H.B., Heiss J.K., Kostylev M.A., Stagi M., Vortmeyer A., Wisniewski T., Gunther E.C., Strittmatter S.M. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012;15:1227–1235. doi: 10.1038/nn.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Um J.W., Kaufman A.C., Kostylev M., Heiss J.K., Stagi M., Takahashi H., Kerrisk M.E., Vortmeyer A., Wisniewski T., Koleske A.J., et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron. 2013;79:887–902. doi: 10.1016/j.neuron.2013.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu G., Nakajima K., Takeyama N., Yukawa M., Taniuchi Y., Sakudo A., Onodera T. Species-specific anti-apoptotic activity of cellular prion protein in a mouse PrP-deficient neuronal cell line transfected with mouse, hamster, and bovine Prnp. Neurosci Lett. 2008;446:11–15. doi: 10.1016/j.neulet.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 10.Carulla P., Bribian A., Rangel A., Gavin R., Ferrer I., Caelles C., Del Rio J.A., Llorens F. Neuroprotective role of PrPC against kainate-induced epileptic seizures and cell death depends on the modulation of JNK3 activation by GluR6/7-PSD-95 binding. Mol. Biol. Cell. 2011;22:3041–3054. doi: 10.1091/mbc.E11-04-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hetz C., Maundrell K., Soto C. Is loss of function of the prion protein the cause of prion disorders. Trends Mol. Med. 2003;9:237–243. doi: 10.1016/s1471-4914(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 12.Machado C.F., Beraldo F.H., Santos T.G., Bourgeon D., Landemberger M.C., Roffe M., Martins V.R. Disease-associated mutations in the prion protein impair laminin-induced process outgrowth and survival. J. Biol. Chem. 2012;287:43777–43788. doi: 10.1074/jbc.M112.428235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Senatore A., Colleoni S., Verderio C., Restelli E., Morini R., Condliffe S.B., Bertani I., Mantovani S., Canovi M., Micotti E., et al. Mutant PrP suppresses glutamatergic neurotransmission in cerebellar granule neurons by impairing membrane delivery of VGCC alpha(2)delta-1 Subunit. Neuron. 2012;74:300–313. doi: 10.1016/j.neuron.2012.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collinge J., Whittington M.A., Sidle K.C., Smith C.J., Palmer M.S., Clarke A.R., Jefferys J.G. Prion protein is necessary for normal synaptic function. Nature. 1994;370:295–297. doi: 10.1038/370295a0. [DOI] [PubMed] [Google Scholar]
- 15.Maglio L.E., Martins V.R., Izquierdo I., Ramirez O.A. Role of cellular prion protein on LTP expression in aged mice. Brain Res. 2006;1097:11–18. doi: 10.1016/j.brainres.2006.04.056. [DOI] [PubMed] [Google Scholar]
- 16.Rangel A., Madronal N., Gruart A., Gavin R., Llorens F., Sumoy L., Torres J.M., Delgado-Garcia J.M., Del Rio J.A. Regulation of GABA(A) and glutamate receptor expression, synaptic facilitation and long-term potentiation in the hippocampus of prion mutant mice. PLoS One. 2009;4:e7592. doi: 10.1371/journal.pone.0007592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tobler I., Gaus S.E., Deboer T., Achermann P., Fischer M., Rulicke T., Moser M., Oesch B., McBride P.A., Manson J.C. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature. 1996;380:639–642. doi: 10.1038/380639a0. [DOI] [PubMed] [Google Scholar]
- 18.Carleton A., Tremblay P., Vincent J.D., Lledo P.M. Dose-dependent, prion protein (PrP)-mediated facilitation of excitatory synaptic transmission in the mouse hippocampus. Pflugers Arch. 2001;442:223–229. doi: 10.1007/s004240100523. [DOI] [PubMed] [Google Scholar]
- 19.Prestori F., Rossi P., Bearzatto B., Laine J., Necchi D., Diwakar S., Schiffmann S.N., Axelrad H., D'Angelo E. Altered neuron excitability and synaptic plasticity in the cerebellar granular layer of juvenile prion protein knock-out mice with impaired motor control. J. Neurosci. 2008;28:7091–7103. doi: 10.1523/JNEUROSCI.0409-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martins V.R., Beraldo F.H., Hajj G.N., Lopes M.H., Lee K.S., Prado M.A., Linden R. Prion protein: orchestrating neurotrophic activities. Curr. Issues Mol. Biol. 2010;12:63–86. [PubMed] [Google Scholar]
- 21.Gama Sosa M.A., De Gasperi R., Elder G.A. Modeling human neurodegenerative diseases in transgenic systems. Hum. Genet. 2012;131:535–563. doi: 10.1007/s00439-011-1119-1. [DOI] [PubMed] [Google Scholar]
- 22.Steinert J.R., Campesan S., Richards P., Kyriacou C.P., Forsythe I.D., Giorgini F. Rab11 rescues synaptic dysfunction and behavioural deficits in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 2012;21:2912–2922. doi: 10.1093/hmg/dds117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giorgini F., Steinert J.R. Rab11 as a modulator of synaptic transmission. Commun. Integr. Biol. 2013;6:e26807. doi: 10.4161/cib.26807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park Y., Kim W., Kim A.Y., Choi H.J., Choi J.K., Park N., Koh E.K., Seo J., Koh Y.H. Normal prion protein in Drosophila enhances the toxicity of pathogenic polyglutamine proteins and alters susceptibility to oxidative and autophagy signaling modulators. Biochem. Biophys. Res. Commun. 2011;404:638–645. doi: 10.1016/j.bbrc.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 25.Gavin B.A., Dolph M.J., Deleault N.R., Geoghegan J.C., Khurana V., Feany M.B., Dolph P.J., Supattapone S. Accelerated accumulation of misfolded prion protein and spongiform degeneration in a Drosophila model of Gerstmann–Straussler–Scheinker syndrome. J. Neurosci. 2006;26:12408–12414. doi: 10.1523/JNEUROSCI.3372-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernandez-Funez P., Zhang Y., Casas-Tinto S., Xiao X., Zou W.Q., Rincon-Limas D.E. Sequence-dependent prion protein misfolding and neurotoxicity. J. Biol. Chem. 2010;285:36897–36908. doi: 10.1074/jbc.M110.174391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rincon-Limas D.E., Casas-Tinto S., Fernandez-Funez P. Exploring prion protein biology in flies: genetics and beyond. Prion. 2010;4:1–8. doi: 10.4161/pri.4.1.10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thackray A.M., Muhammad F., Zhang C., Di Y., Jahn T.R., Landgraf M., Crowther D.C., Evers J.F., Bujdoso R. Ovine PrP transgenic Drosophila show reduced locomotor activity and decreased survival. Biochem. J. 2012;444:487–495. doi: 10.1042/BJ20112141. [DOI] [PubMed] [Google Scholar]
- 29.Sanchez-Garcia J., Arbelaez D., Jensen K., Rincon-Limas D.E., Fernandez-Funez P. Polar substitutions in helix 3 of the prion protein produce transmembrane isoforms that disturb vesicle trafficking. Hum. Mol. Genet. 2013;22:4253–4266. doi: 10.1093/hmg/ddt276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi J.K., Jeon Y.C., Lee D.W., Oh J.M., Lee H.P., Jeong B.H., Carp R.I., Koh Y.H., Kim Y.S. A Drosophila model of GSS syndrome suggests defects in active zones are responsible for pathogenesis of GSS syndrome. Hum. Mol. Genet. 2010;19:4474–4489. doi: 10.1093/hmg/ddq379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spielhaupter C., Schatzl H.M. PrPC directly interacts with proteins involved in signaling pathways. J. Biol. Chem. 2001;276:44604–44612. doi: 10.1074/jbc.M103289200. [DOI] [PubMed] [Google Scholar]
- 32.Re L., Rossini F., Re F., Bordicchia M., Mercanti A., Fernandez O.S., Barocci S. Prion protein potentiates acetylcholine release at the neuromuscular junction. Pharmacol. Res. 2006;53:62–68. doi: 10.1016/j.phrs.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 33.Cunningham C., Deacon R., Wells H., Boche D., Waters S., Diniz C.P., Scott H., Rawlins J.N., Perry V.H. Synaptic changes characterize early behavioural signs in the ME7 model of murine prion disease. Eur. J. Neurosci. 2003;17:2147–2155. doi: 10.1046/j.1460-9568.2003.02662.x. [DOI] [PubMed] [Google Scholar]
- 34.Jeffrey M., Halliday W.G., Bell J., Johnston A.R., MacLeod N.K., Ingham C., Sayers A.R., Brown D.A., Fraser J.R. Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie-infected murine hippocampus. Neuropathol. Appl. Neurobiol. 2000;26:41–54. doi: 10.1046/j.1365-2990.2000.00216.x. [DOI] [PubMed] [Google Scholar]
- 35.Wadsworth J.D., Joiner S., Linehan J.M., Cooper S., Powell C., Mallinson G., Buckell J., Gowland I., Asante E.A., Budka H., et al. Phenotypic heterogeneity in inherited prion disease (P102L) is associated with differential propagation of protease-resistant wild-type and mutant prion protein. Brain. 2006;129:1557–1569. doi: 10.1093/brain/awl076. [DOI] [PubMed] [Google Scholar]
- 36.Fernandez-Funez P., Casas-Tinto S., Zhang Y., Gomez-Velazquez M., Morales-Garza M.A., Cepeda-Nieto A.C., Castilla J., Soto C., Rincon-Limas D.E. In vivo generation of neurotoxic prion protein: role for hsp70 in accumulation of misfolded isoforms. PLoS Genet. 2009;5:e1000507. doi: 10.1371/journal.pgen.1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DiAntonio A., Petersen S.A., Heckmann M., Goodman C.S. Glutamate receptor expression regulates quantal size and quantal content at the Drosophila neuromuscular junction. J. Neurosci. 1999;19:3023–3032. doi: 10.1523/JNEUROSCI.19-08-03023.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haas K.F., Miller S.L., Friedman D.B., Broadie K. The ubiquitin–proteasome system postsynaptically regulates glutamatergic synaptic function. Mol. Cell Neurosci. 2007;35:64–75. doi: 10.1016/j.mcn.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Müller M., Liu K.S., Sigrist S.J., Davis G.W. RIM controls homeostatic plasticity through modulation of the readily-releasable vesicle pool. J. Neurosci. 2012;32:16574–16585. doi: 10.1523/JNEUROSCI.0981-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mozer B.A., Sandstrom D.J. Drosophila neuroligin 1 regulates synaptic growth and function in response to activity and phosphoinositide-3-kinase. Mol. Cell Neurosci. 2012;51:89–100. doi: 10.1016/j.mcn.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karunanithi S., Marin L., Wong K., Atwood H.L. Quantal size and variation determined by vesicle size in normal and mutant Drosophila glutamatergic synapses. J. Neurosci. 2002;22:10267–10276. doi: 10.1523/JNEUROSCI.22-23-10267.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weyhersmüller A., Hallermann S., Wagner N., Eilers J. Rapid active zone remodeling during synaptic plasticity. J. Neurosci. 2011;31:6041–6052. doi: 10.1523/JNEUROSCI.6698-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanse E., Gustafsson B. Paired-pulse plasticity at the single release site level: an experimental and computational study. J. Neurosci. 2001;21:8362–8369. doi: 10.1523/JNEUROSCI.21-21-08362.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katz B., Miledi R. The role of calcium in neuromuscular facilitation. J. Physiol. 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zucker R.S., Regehr W.G. Short-term synaptic plasticity. Annu. Rev. Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
- 46.Xu-Friedman M.A., Regehr W.G. Structural contributions to short-term synaptic plasticity. Physiol. Rev. 2004;84:69–85. doi: 10.1152/physrev.00016.2003. [DOI] [PubMed] [Google Scholar]
- 47.Dobrunz L.E., Stevens C.F. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- 48.Atwood H.L., Karunanithi S. Diversification of synaptic strength: presynaptic elements. Nat. Rev. Neurosci. 2002;3:497–516. doi: 10.1038/nrn876. [DOI] [PubMed] [Google Scholar]
- 49.Graf E.R., Valakh V., Wright C.M., Wu C., Liu Z., Zhang Y.Q., Diantonio A. RIM promotes calcium channel accumulation at active zones of the Drosophila neuromuscular junction. J. Neurosci. 2012;32:16586–16596. doi: 10.1523/JNEUROSCI.0965-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schneggenburger R., Meyer A.C., Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- 51.Scheuss V., Neher E. Estimating synaptic parameters from mean, variance, and covariance in trains of synaptic responses. Biophys. J. 2001;81:1970–1989. doi: 10.1016/S0006-3495(01)75848-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Silver R.A. Estimation of nonuniform quantal parameters with multiple-probability fluctuation analysis: theory, application and limitations. J. Neurosci. Methods. 2003;130:127–141. doi: 10.1016/j.jneumeth.2003.09.030. [DOI] [PubMed] [Google Scholar]
- 53.Clements J.D., Silver R.A. Unveiling synaptic plasticity: a new graphical and analytical approach. Trends Neurosci. 2000;23:105–113. doi: 10.1016/s0166-2236(99)01520-9. [DOI] [PubMed] [Google Scholar]
- 54.Stewart B.A., Mohtashami M., Trimble W.S., Boulianne G.L. SNARE proteins contribute to calcium cooperativity of synaptic transmission. Proc. Natl. Acad. Sci. U.S.A. 2000;97:13955–13960. doi: 10.1073/pnas.250491397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fedchyshyn M.J., Wang L.Y. Developmental transformation of the release modality at the calyx of Held synapse. J. Neurosci. 2005;25:4131–4140. doi: 10.1523/JNEUROSCI.0350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kochubey O., Han Y., Schneggenburger R. Developmental regulation of the intracellular Ca2+ sensitivity of vesicle fusion and Ca2+-secretion coupling at the rat calyx of Held. J. Physiol. 2009;587:3009–3023. doi: 10.1113/jphysiol.2009.172387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Atwood H.L., Govind C.K., Wu C.F. Differential ultrastructure of synaptic terminals on ventral longitudinal abdominal muscles in Drosophila larvae. J. Neurobiol. 1993;24:1008–1024. doi: 10.1002/neu.480240803. [DOI] [PubMed] [Google Scholar]
- 58.Dodge F.A., Jr, Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J. Physiol. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jan L.Y., Jan Y.N. Properties of the larval neuromuscular junction in Drosophila melanogaster. J. Physiol. 1976;262:189–214. doi: 10.1113/jphysiol.1976.sp011592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stewart B.A., Atwood H.L., Renger J.J., Wang J., Wu C.F. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J. Comp. Physiol. A. 1994;175:179–191. doi: 10.1007/BF00215114. [DOI] [PubMed] [Google Scholar]
- 61.Staples J., Broadie K. The cell polarity scaffold Lethal Giant Larvae regulates synapse morphology and function. J. Cell Sci. 2013;126:1992–2003. doi: 10.1242/jcs.120139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dani N., Broadie K. Glycosylated synaptomatrix regulation of trans-synaptic signaling. Dev. Neurobiol. 2012;72:2–21. doi: 10.1002/dneu.20891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Steinert J.R., Kuromi H., Hellwig A., Knirr M., Wyatt A.W., Kidokoro Y., Schuster C.M. Experience-dependent formation and recruitment of large vesicles from reserve pool. Neuron. 2006;50:723–733. doi: 10.1016/j.neuron.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 64.Jakubowski B.R., Longoria R.A., Shubeita G.T. A high throughput and sensitive method correlates neuronal disorder genotypes to Drosophila larvae crawling phenotypes. Fly (Austin) 2012;6:303–308. doi: 10.4161/fly.21582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen S., Mange A., Dong L., Lehmann S., Schachner M. Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol. Cell Neurosci. 2003;22:227–233. doi: 10.1016/s1044-7431(02)00014-3. [DOI] [PubMed] [Google Scholar]
- 66.Santuccione A., Sytnyk V., Leshchyns'ka I., Schachner M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 2005;169:341–354. doi: 10.1083/jcb.200409127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mallucci G.R., Ratte S., Asante E.A., Linehan J., Gowland I., Jefferys J.G., Collinge J. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J. 2002;21:202–210. doi: 10.1093/emboj/21.3.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zanata S.M., Lopes M.H., Mercadante A.F., Hajj G.N., Chiarini L.B., Nomizo R., Freitas A.R., Cabral A.L., Lee K.S., Juliano M.A., et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002;21:3307–3316. doi: 10.1093/emboj/cdf325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beraldo F.H., Arantes C.P., Santos T.G., Machado C.F., Roffe M., Hajj G.N., Lee K.S., Magalhaes A.C., Caetano F.A., Mancini G.L., et al. Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma1 chain. FASEB J. 2011;25:265–279. doi: 10.1096/fj.10-161653. [DOI] [PubMed] [Google Scholar]
- 70.Linden R., Cordeiro Y., Lima L.M. Allosteric function and dysfunction of the prion protein. Cell Mol. Life Sci. 2012;69:1105–1124. doi: 10.1007/s00018-011-0847-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aguzzi A., Weissmann C. Prion research: the next frontiers. Nature. 1997;389:795–798. doi: 10.1038/39758. [DOI] [PubMed] [Google Scholar]
- 72.Samaia H.B., Brentani R.R. Can loss-of-function prion-related diseases exist. Mol. Psychiatry. 1998;3:196–197. doi: 10.1038/sj.mp.4000378. [DOI] [PubMed] [Google Scholar]
- 73.Fournier J.G., Escaig-Haye F., Grigoriev V. Ultrastructural localization of prion proteins: physiological and pathological implications. Microsc. Res. Tech. 2000;50:76–88. doi: 10.1002/1097-0029(20000701)50:1<76::AID-JEMT11>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 74.Gohel C., Grigoriev V., Escaig-Haye F., Lasmezas C.I., Deslys J.P., Langeveld J., Akaaboune M., Hantai D., Fournier J.G. Ultrastructural localization of cellular prion protein (PrPc) at the neuromuscular junction. J. Neurosci. Res. 1999;55:261–267. doi: 10.1002/(SICI)1097-4547(19990115)55:2<261::AID-JNR14>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 75.Shyng S.L., Heuser J.E., Harris D.A. A glycolipid-anchored prion protein is endocytosed via clathrin-coated pits. J. Cell Biol. 1994;125:1239–1250. doi: 10.1083/jcb.125.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Magalhaes A.C., Silva J.A., Lee K.S., Martins V.R., Prado V.F., Ferguson S.S., Gomez M.V., Brentani R.R., Prado M.A. Endocytic intermediates involved with the intracellular trafficking of a fluorescent cellular prion protein. J. Biol. Chem. 2002;277:33311–33318. doi: 10.1074/jbc.M203661200. [DOI] [PubMed] [Google Scholar]
- 77.Graf E.R., Daniels R.W., Burgess R.W., Schwarz T.L., DiAntonio A. Rab3 dynamically controls protein composition at active zones. Neuron. 2009;64:663–677. doi: 10.1016/j.neuron.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu K.S., Siebert M., Mertel S., Knoche E., Wegener S., Wichmann C., Matkovic T., Muhammad K., Depner H., Mettke C., et al. RIM-binding protein, a central part of the active zone, is essential for neurotransmitter release. Science. 2011;334:1565–1569. doi: 10.1126/science.1212991. [DOI] [PubMed] [Google Scholar]
- 79.Matkovic T., Siebert M., Knoche E., Depner H., Mertel S., Owald D., Schmidt M., Thomas U., Sickmann A., Kamin D., et al. The Bruchpilot cytomatrix determines the size of the readily releasable pool of synaptic vesicles. J. Cell Biol. 2013;202:667–683. doi: 10.1083/jcb.201301072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ruiz R., Cano R., Casanas J.J., Gaffield M.A., Betz W.J., Tabares L. Active zones and the readily releasable pool of synaptic vesicles at the neuromuscular junction of the mouse. J. Neurosci. 2011;31:2000–2008. doi: 10.1523/JNEUROSCI.4663-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Marrus S.B., DiAntonio A. Preferential localization of glutamate receptors opposite sites of high presynaptic release. Curr. Biol. 2004;14:924–931. doi: 10.1016/j.cub.2004.05.047. [DOI] [PubMed] [Google Scholar]
- 82.Melom J.E., Akbergenova Y., Gavornik J.P., Littleton J.T. Spontaneous and evoked release are independently regulated at individual active zones. J. Neurosci. 2013;33:17253–17263. doi: 10.1523/JNEUROSCI.3334-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sheng J., He L., Zheng H., Xue L., Luo F., Shin W., Sun T., Kuner T., Yue D.T., Wu L.G. Calcium-channel number critically influences synaptic strength and plasticity at the active zone. Nat. Neurosci. 2012;15:998–1006. doi: 10.1038/nn.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Awatramani G.B., Price G.D., Trussell L.O. Modulation of transmitter release by presynaptic resting potential and background calcium levels. Neuron. 2005;48:109–121. doi: 10.1016/j.neuron.2005.08.038. [DOI] [PubMed] [Google Scholar]
- 85.Wichmann C., Sigrist S.J. The active zone T-bar—a plasticity module. J. Neurogenet. 2010;24:133–145. doi: 10.3109/01677063.2010.489626. [DOI] [PubMed] [Google Scholar]
- 86.Steinert J.R., Postlethwaite M., Jordan M.D., Chernova T., Robinson S.W., Forsythe I.D. NMDAR-mediated EPSCs are maintained and accelerate in time course during maturation of mouse and rat auditory brainstem in vitro. J. Physiol. 2010;588:447–463. doi: 10.1113/jphysiol.2009.184317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Joshi I., Wang L.Y. Developmental profiles of glutamate receptors and synaptic transmission at a single synapse in the mouse auditory brainstem. J. Physiol. 2002;540:861–873. doi: 10.1113/jphysiol.2001.013506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sigrist S.J., Schmitz D. Structural and functional plasticity of the cytoplasmic active zone. Curr. Opin. Neurobiol. 2011;21:144–150. doi: 10.1016/j.conb.2010.08.012. [DOI] [PubMed] [Google Scholar]
- 89.Gawinecka J., Cardone F., Asif A.R., De Pascalis A., Wemheuer W.M., Schulz-Schaeffer W.J., Pocchiari M., Zerr I. Sporadic Creutzfeldt–Jakob disease subtype-specific alterations of the brain proteome: impact on Rab3a recycling. Proteomics. 2012;12:3610–3620. doi: 10.1002/pmic.201200201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zafar S., von Ahsen N., Oellerich M., Zerr I., Schulz-Schaeffer W.J., Armstrong V.W., Asif A.R. Proteomics approach to identify the interacting partners of cellular prion protein and characterization of Rab7a interaction in neuronal cells. J. Proteome Res. 2011;10:3123–3135. doi: 10.1021/pr2001989. [DOI] [PubMed] [Google Scholar]
- 91.Scott M.R., Supattapone S., Nguyen H.O., DeArmond S.J., Prusiner S.B. Transgenic models of prion disease. Arch. Virol. Suppl. 2000;16:113–124. doi: 10.1007/978-3-7091-6308-5_10. [DOI] [PubMed] [Google Scholar]
- 92.Telling G.C. Transgenic mouse models of prion diseases. Methods Mol. Biol. 2008;459:249–263. doi: 10.1007/978-1-59745-234-2_17. [DOI] [PubMed] [Google Scholar]
- 93.Postlethwaite M., Hennig M.H., Steinert J.R., Graham B.P., Forsythe I.D. Acceleration of AMPA receptor kinetics underlies temperature-dependent changes in synaptic strength at the rat calyx of Held. J. Physiol. 2007;579:69–84. doi: 10.1113/jphysiol.2006.123612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sakaba T., Schneggenburger R., Neher E. Estimation of quantal parameters at the calyx of Held synapse. Neurosci. Res. 2002;44:343–356. doi: 10.1016/s0168-0102(02)00174-8. [DOI] [PubMed] [Google Scholar]
- 95.Scheuss V., Schneggenburger R., Neher E. Separation of presynaptic and postsynaptic contributions to depression by covariance analysis of successive EPSCs at the calyx of held synapse. J. Neurosci. 2002;22:728–739. doi: 10.1523/JNEUROSCI.22-03-00728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Oleskevich S., Walmsley B. Synaptic transmission in the auditory brainstem of normal and congenitally deaf mice. J. Physiol. 2002;540:447–455. doi: 10.1113/jphysiol.2001.013821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Taschenberger H., Scheuss V., Neher E. Release kinetics, quantal parameters and their modulation during short-term depression at a developing synapse in the rat CNS. J. Physiol. 2005;568:513–537. doi: 10.1113/jphysiol.2005.093468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hill A.F., Joiner S., Beck J.A., Campbell T.A., Dickinson A., Poulter M., Wadsworth J.D., Collinge J. Distinct glycoform ratios of protease resistant prion protein associated with PRNP point mutations. Brain. 2006;129:676–685. doi: 10.1093/brain/awl013. [DOI] [PubMed] [Google Scholar]