

Abstract
Most homogenous gold catalyses demand ≥0.5 mol % catalyst loading. Due to the high cost of gold, these reactions are unlikely to be applicable in medium or large scale applications. Here we disclose a novel ligand design based on the privileged biphenyl-2-phosphine framework that offers a potentially general approach to dramatically lowering catalyst loading. In this design, an amide group at the 3’ position of the ligand framework directs and promotes nucleophilic attack at the ligand gold complex-activated alkyne, which is unprecedented in homogeneous gold catalysis considering the spatial challenge of using ligand to reach antiapproaching nucleophile in a linear P-Au-alkyne centroid structure. With such a ligand, the gold(I) complex becomes highly efficient in catalyzing acid addition to alkynes, with a turnover number up to 99,000. Density functional theory calculations support the role of the amide moiety in directing the attack of carboxylic acid via hydrogen bonding.
Introduction
One of the exciting thrusts in organic synthesis in the past dozen years is homogeneous gold catalysis.(1–7) Various versatile synthetic methods, offering efficient access to a broad array of functional structures, have been developed based on an initial coordination of a C-C triple bond to a LAu+, a cationic gold(I) species with one coordinating ligand (i.e., L), and a subsequent anti attack by a nucleophile at the alkyne (Fig. 1A). Due to the linear nature of the L-Au-alkyne complex and, making the matter worse, to the anti nucleophilic approach, it is highly challenging to introduce bonding interaction between the ligand and the nucleophile due to apparent spatial constrain. To date, there is no reported reaction of this nature. Even though in enantioselective gold catalysis,(8, 9) ligands/counter anions to the metal center could impose a chiral environment in the approaching path of nucleophiles (often with allene substrates), the controls are invariably steric and offer little, if not to the opposite effect, rate acceleration or efficiency improvement. On the other hand, with a few exceptions,(10–15) most homogenous gold catalyses demand ≥0.5 mol % catalyst loading. Due to the high cost of gold, these reactions are unlikely applicable in medium or large scale applications, which in turn has continuously reinforced the prevailing perception that gold chemistry would be too expensive to be useful for industrial scale processes. This general shortcoming in gold catalysis could be overcome by substantially improving the turnover numbers (TONs) of gold catalysts and hence dramatically lowering their loadings. Ligand tuning (16) is a well-practiced strategy in transition metal catalysis to achieve high catalyst TONs, but little success has been reported in this regard in homogeneous gold catalysis..
Fig. 1.

A novel ligand design for highly efficient gold catalysis. (A) anti attack at gold(I)-activated alkyne by a nucleophile. (B) A general concept to achieve quasi-intramolecular, ligand directed nucleophilic attack. (C) A design based on 2-biphenylphosphine framework.
Herein we disclose a ligand design that offers a conceptually new and potentially general approach to achieving highly efficient gold catalysis with ultra-low catalyst loadings. With an optimized ligand based on the privileged biphenyl-2-phosphine framework yet possessing an unprecedented and strategical 3’-amide group, additions of a diverse range of carboxylic acids to alkynes are achieved with typically ppm-level catalyst loadings and catalyst TON up to 99,000.
Results
Design and Synthesis of Novel Biphenyl-2-Phosphine Ligands
It is envisioned that a ligand with a rigid and extending framework could project a functional group farther enough to reach to an approach nucleophile (Fig. 1B).(17) The functional group could be designed such that it could have attractive interaction with the nucleophile and hence be capable of directing its attack at the gold-activated triple bond. The rigidity of the backbone would minimize the entropy cost for organizing transition states, and the directing group should be flexible enough to accommodate a broad substrate scope. This strategy is expected to facilitate the initial nucleophilic attack drastically by converting it from an intermolecular process to a quasi-intramolecular event and may also accelerate subsequent transformations. As a result, higher catalyst TONs and hence more efficient catalysis could be expected. Herein we report an implementation of this design in a gold-catalyzed addition of carboxylic acids to alkynes with catalyst TONs up to 99,000, a broad reaction scope and high to excellent efficiencies; moreover, the general applicability of the design in other gold catalyses is demonstrated by highly efficient hydration and hydroamination of alkynes.
Bulky and electron-rich 2-biphenylphosphines belong to a privileged class of phosphorus-based ligands that are developed by Buchwald (18–20) for various versatile Pd catalysis.(21, 22) Lately, these ligands have been borrowed in gold catalysis with much success,(23–30) but modification of them for gold catalysis is rare.(31) Perhaps due to the nature of Pd catalysis, the lower half of the bottom phenyl ring of these ligands (i.e., C3’, C4’ and C5’) has seldom been substituted with functional groups. The only exceptions are a 3’-sulfonate derived from SPhos and a 4’-sulfonate from XPhos for the purpose of increasing catalyst aqueous solubility.(32) In contrast to square planar Pd(II) complexes, bis-coordinated gold(I) complexes exhibit linear structures, which should invite a ligand design philosophy different from that for Pd catalysis. In the context of biphenylphosphine ligands (Fig. 1C), by fixing the P-C2 rotation using bulky substituents on the phosphorus, the linear P-Au-alkyne axis should be parallel to and bisect the pendant phenyl ring, thus putting the coordinated alkyne on the top of the lower half of that ring. It is envisioned if these remote positions on that part of the phenyl ring, with the activated alkyne lying above, were to be functionalized with H-bond acceptors, these functional groups could extend further enough to direct neutral nucleophiles via H-bonding interactions and/or proton stabilization to attack the activated alkyne in an ‘intramolecular’ anti attack. In this manner the design concept shown in Fig. 1B could be implemented.
To put this concept into practice, we designed a series of 2-biphenyldi-(1-adamentyl)phosphine ligands (i.e., L1 – L8) with either C3’ or C4’ functionalized (Table 1). They can be readily synthesized via consecutive cross-couplings of 1,2-dihalobenzene with arylboronic acids and di(1-adamentyl)phosphine (Ad2PH). Ad2P was chosen because of its ease of installation(33) and, more importantly, the steric bulk of the Ad groups. The structure of the ligand L8 is confirmed by the X-ray diffraction study of its gold complex L8AuCl (Fig. 2).
Table 1.
New ligands prepared in study and C=O stretching wavelengths of carbonyls in ligands and catalysts.
 |
Fig. 2.

The ORTEP drawing of L8AuCl at 50% ellipsoid probability
Screening Ligands to Maximize Catalyst Turnover Numbers
The addition of a carboxylic acid to an alkyne is chosen as the model reaction. This transformation has been previously realized using 5 mol % Ph3PAuCl/AgPF6 with catalyst TONs up to 18(34), and the best catalyst TON (i.e., 124) of this type of reaction was achieved via Ru catalysis.(35) Importantly, the alkenyl esters products possess mildly activated acyl groups and are versatile substrates of broad synthetic utilities.
As shown in Table 2, with IPrAuNTf2, a catalyst only different by the counter anion from the previously reported ppm-level IPrAuCl/AgSbF6 for alkyne hydration in 1,4-dioxane/H2O,(12) there was only trace amount of the desired 2-dodecenyl benzoate (i.e., 1a) detected under the indicated conditions (entry 1). While a basic dimethylamino group at the 4’ position in the case of the ligand L2 did not offer any improvement (entry 3), its regioisomer L1 with the substituent at the 3’ position led to some notably improvement (entry 2), hinting that substitution at C3’ might be better than at C4’ for the reaction. With piperidin-1-ylcarbonyl as the functional group (FG) at either the 4’ or the 3’ position in the case of ligand L3 or L7, respectively, a much more efficient reaction with an observed catalyst TON of 80 was realized, suggesting that an amide is a suitable FG for promoting the reaction (entries 4 and 5). By lowering down the gold loading to 0.11%, the TONs reached 609 for L3 (entry 6) and 827 for L7 (entry 7), which are much better than the Ru case, (35) and was further increased to 1681 (entry 8) by further decreasing the loading of the better gold complex L7AuCl. With little success with other FGs at the 3’ position, the reaction optimization was then focused on modifying the amide N-substituents of L7, and the results are shown in entries 9–12. Whereas L8 with a 3’-(pyrrolidin-1-ylcarbonyl) group turned out to be the most efficacious ligand, a qualitative inverse correlation between the reaction yields and the wavenumbers of the amide carbonyl stretch band (νC=O) of both the ligands and the corresponding gold complexes (see Fig. 1D) is revealed. As the higher νC=O is, the less basic the carbonyl oxygen is and hence the weaker H-bond acceptor it is. Therefore, the reaction yield positively correlates to the H-bond acceptor capacity of the amide carbonyl oxygen. This qualitative trend is consistent with the design that necessitates the remote FG to recruit the nucleophile via H-bonding. After further optimization, the catalyst TON using the best ligand L8 could reach 34400 in 12 h with 25 ppm of its cationic gold complex in fluorobenzene when NaBARF (sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate, 0.12%) was added (entry 14), while a 40 ppm catalyst loading allowed full conversion under the same conditions (entry 13). The role of NaBARF is likely to prevent catalyst deactivation by scavenging coordinating anionic impurities. In comparison, under the same conditions a catalyst TON of 40 was reached for JohnPhosAuNTf2 (entry 15). By considering that t-butyl and 1-adamantyl have similar steric bulk, the rate acceleration of 860 fold by L8AuNTf2 over JohnPhosAuNTf2 can be attributed to the mere incorporation of the remote 3’-amide group.
Table 2.
Screening various ligands and the optimization of reaction conditions.
 | ||||||
|---|---|---|---|---|---|---|
| entry | [Au] | Solvent, Temperature, Conc. |
time | NMR yielda | ||
| 1a | TON | 2a | ||||
| 1 | IPrAuNTf2 (1%) | DCE, 40 °C, 0.5 M | 8 h | 0.5% | 0.5 | 0 |
| 2 | L1AuCl/AgNTf2 (1.1%/1%) | DCE, 40 °C, 0.5 M | 8 h | 6% | 5 | 0 |
| 3 | L2AuCl/AgNTf2 (1.1%/1%) | DCE, 40 °C, 0.5 M | 8 h | trace | - | 0 |
| 4 | L3AuCl/AgNTf2 (1.1%/1%) | DCE, 40 °C, 0.5 M | 8 h | 89% | 80 | 7% |
| 5 | L7AuCl/AgNTf2 (1.1%/1%) | DCE, 40 °C, 0.5 M | 8 h | 89% | 80 | 7% |
| 6 | L3AuCl/AgNTf2 (0.11%/0.1%) | DCE, 40 °C, 0.5 M | 8 h | 67% | 609 | 0 |
| 7 | L7AuCl/ AgNTf2 (0.11%/0.1%) | DCE, 40 °C, 0.5 M | 8 h | 91% | 827 | 2% |
| 8 | L7AuCl/AgNTf2 (220/200 ppm) | DCE, 40 °C, 1 M | 12 h | 37% | 1681 | 0 |
| 9 | L4AuCl/AgNTf2 (220/200 ppm) | DCE, 40 °C, 1 M | 12 h | 27% | 1227 | 0 |
| 10 | L5AuCl/AgNTf2 (220/200 ppm) | DCE, 40 °C, 1 M | 12 h | 35% | 1590 | 0 |
| 11 | L6AuCl/AgNTf2 (220/200 ppm) | DCE, 40 °C, 1 M | 12 h | 36% | 1636 | 0 |
| 12 | L8AuCl/AgNTf2 (220/200 ppm) | DCE, 40 °C, 1 M | 12 h | 43% | 1954 | 0 |
| 13 | L8AuNTf2 (40 ppm)/ NaBArF (0.12 %) | PhF, 80 °C, 2 M | 12 h | 97% | 24250 | 0 |
| 14 | L8AuNTf2 (25 ppm)/ NaBArF (0.12 %) | PhF, 80 °C, 2 M | 12 h | 86% | 34400 | 0 |
| 15 | JohnPhosAuNTf2 (500 ppm)/ NaBArF (0.12 %) | PhF, 80 °C, 2 M | 12 h | 2% | 40 | 0 |
The NMR yield calculated by assuming that the triplet at around 0.9 ppm corresponds to the terminal methyl groups of all compounds derived from 2-dodecyne. NaBARF, sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate; DCE, 1,2-dichloroethane.
The Reaction Scope with Ultra-Low Catalyst Loadings
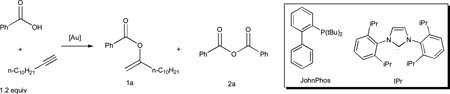
With the low sub-micromolar catalysis realized, its applicability to other carboxylic acids was first examined using 1-hexyne or 1-dodecyne as the alkyne partner. As shown in Fig. 3A, various acids possessing a diverse range of functional groups were allowed, mostly resulting in near quantitative yields while with a catalyst loading of 100 ppm or less. Some notable functional groups include Bpin (e.g., 1h), thiophene (e.g., 1i), diene (e.g., 1l), different halogens (e.g., 1e–1g, 1p, and 1q), and protected α-amino groups (e.g., 1t and 1u). Whereas the reaction of N-protected proline leading to 1t require higher catalyst loading, the reaction of much more acidic trifluoroacetic acid was realized with 10 ppm catalyst, giving a catalyst TON of at least 99,000. In addition, hindered acids such as 1-adamentanecarboxylic acid (in the case of 1n) and 2,6-dichlorobenzoic acid (in the case of 1g) reacted smoothly. The scope with different alkynes was then probed using benzoic acid. As shown in Fig. 3B, terminal alkynes with different functionalities and internal alkynes were all suitable substrates, and the reaction yields were mostly nearly quantitative. These reactions, however, generally required higher catalyst loadings. In the cases of an enyne (i.e., 1z) and non-activated internal alkynes (i.e., 1aa and 1ab), the catalyst loadings were relatively high, likely due to lower reactivities of these substrates in comparison to aliphatic terminal alkynes.
Fig. 3.

The reaction scope with isolated yields reported. (A) Reactions of various acids with 1-hexyne or 1-dodecyne. (B) Reactions of various alkynes with benzoic acid. Reactions run under N2 in vial. NaBARF, sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate.
Discussion
This dramatic increase of catalyst TON in this gold catalysis using the remotely amide-functionalized L8 as ligand can be ascribed to the H-bonding between the ligand amide group and the approaching carboxylic acid, which makes the reaction quasi-intramolecular and hence decrease of the entropy loss in the transition state and moreover, materializes a general base catalysis by the basic amide moiety, via which the nucleophilicity of the acid is enhanced. In addition, the protonated amide after the initial step should serve as intramolecular proton source for rapid protodeauration and hence accelerate catalyst turnover.
To substantiate this rationale and, hence, to offer theoretical support to our original design, density functional theory (DFT) (36, 37) studies were performed with GAUSSIAN09 (38) program and PBE1PBE (39, 40) method. In the calculation, the ligand L8, one molecule of benzoic acid and one molecule of propyne were used; moreover, 6-31+G** basis set was used for the reactants, the NTf2− anion and the 3’-(pyrrolidin-l-ylcarbonyl) group, 6-311+G** basis set was used for P, 6-31G* basis set was used for other atoms in the ligand, and the SDD basis set with an Effective Core Potential (ECP) (41) was used for Au. Geometry optimization was performed in dichloroethane using the SMD (42) method. Harmonic vibration frequency calculations were carried out and each of the optimized transition states has one imaginary frequency.
We proposed that, in the catalytic reaction with JohnPhos as the ligand, the NTf2− counter anion will activate the benzoic acid and assist the proton transfer process.(43–45) However, in the catalytic reaction with L8 the amide group is a better alternative to the NTf2− anion. To facilitate the comparison of these two scenarios, a reaction pathway using L8 as ligand but without involving its 3’-amide group was used to mimic the case of JohnPhos. As shown in Fig. 4, in the transition state TS-NTf2, the NTf2− anion stabilize the proton of the benzoic acid while its carbonyl oxygen attacks the C2 of propyne; notably, the amide group is a bystander. In TS-Amide, the amide group stabilizes the proton, and the NTf2− anion is far away from the reaction center, binding to the system via weak hydrogen bonds. The TS-Amide is 2.1 kcal/mol more favorable in free energy than TS-NTf2, consistent with the experimental observation that L8 is a better ligand than JohnPhos. In addition, in TS-Amide NTf2− does not participate in the reaction and hence needs not to be bound.(45) Such a binding in TS-Amide causes rigid body translational and rotational entropy loss, which was estimated to be 3.6~4.8 kcal/mol for the binding of small molecules to protein (46). Based on this estimation, TS-Amide should be more than 5 kcal/mol more favorable than TS-NTf2, which is in line with the observed 860 fold increase of the TONs as the estimated energy barrier difference is around 4.7 kcal/mol if the TON difference is treated as the rate difference. These theoretical studies corroborate that the basic amide group and the intramolecular nature of the reaction are critical for the much enhanced TONs.
Fig. 4.

Optimized transition states. The optimized NTf2− anion activated transition state TS-NTf2 and amide group activated transition state TS-Amide. The selected bond lengths are in angstroms, the relative energies ΔEsol and free energies ΔGsol (in italic, 298K) in dichloroethane are in kcal/mol.
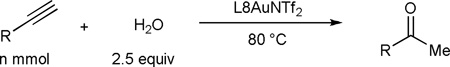
The remote amide group in L8 can also facilitate nucleophilic attack by H2O, resulting in substantially accelerated hydration of alkynes. As shown in Table 3, when the reaction was run in non-polar toluene with 2 equivalents of H2O, L8 was a much more effective ligand than the remotely non-functionalized 2-biphenyphosphine ligand JohnPhos and the NHC ligand IPr; the same trend was observed with MeOH as solvent, and notably without optimization the hydration was easily accomplished with 100 ppm of L8AuNTf2 and a catalyst TON of at least 10,000. These results again highlight the essential role of the remote amide group, and more importantly suggest that the design principle can be generally applicable to other reactions. To further substantiate the point, we performed hydroamination of alkynes with aniline. As shown in Fig. 5A, using L8AuNTf2 as the catalyst, its TONs with phenylacetylene as the substrate reached 8500, a nevertheless pleasing number albeit lower than that (i.e., 22000) achieved by the best catalyst developed by Lavallo (14); however, with 1-dodecyne as the substrate, L8AuNTf2 was a much better catalyst than that by Lavallo as its TON reached 3900 while a TON of 435 is reported in the latter case.
Table 3.
Gold-catalyzed alkyne hydration.
 | ||
|---|---|---|
| Catalyst | Yield/TON | |
| R = n-decyl, [Au] (500 ppm), toluene (0.25 × n mL), 5 h |
R = n-butyl, [Au] (100 ppm), MeOH (0.3 × n mL), 6 h |
|
| JohnPhosAuNTf2 | 8%/160 | 44%/4400 |
| IPrAuNTf2 | 9%/180 | 69%/6900 |
| L8AuNTf2 | 38%/760 | 100%/10000 |
Fig. 5.

Synthesis of amides. (A) Gold-catalyzed hydroamination of alkynes. (B) Application in amide synthesis.a NMR yields using diethyl phthalate as the internal reference.b Isolated yields. NaBARF, sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate; DCE, 1,2-dichloroethane.
The utility of this chemistry is exemplified by a two-step, high yielding synthesis of amides (Fig. 5B), where the gold catalysis offered mild activation of carboxylic acids in a highly efficient manner. A notable advantage of this amide formation is that the byproduct is 2- hexanone, which is volatile and can be readily removed under vacuum.
In summary, we have advanced a novel ligand design based on the privileged biphenylphosphine framework that could dramatically improve gold catalysis. It employs functional groups at the remote 3’ position to direct and promote nucleophilic attack of gold-activated alkynes, which is unprecedented in homogeneous gold catalysis considering the spatial challenge of using ligand to reach anti-approaching nucleophile in a linear L-Au-alkyne centroid structure. With a basic, H-bond accepting (pyrrolidin-1-yl)carbonyl group at the 3’ position, the gold(I) complex derived from the biphenylphosphine ligand becomes highly efficient in catalyzing acid addition to alkynes, with its TON up to 99,000. DFT calculations support the role of the amide moiety in directing the attack of carboxylic acid via H-bonding. Further applications of this catalyst to hydration and hydroamination of alkynes likewise realized high catalyst TONs, therefore confirming the general applicability of this ligand design. We anticipated that this design and the general principle of engaging bonding interactions between ligand and reactants could be employed to substantially improve an array of other gold catalyses, thereby promoting the application of gold chemistry in industrial scale processes, and might also be useful for improving catalysis by other transition metals.
Methods
General procedure for synthesis of ligand and catalyst
As shown in Fig. 6, to a dispersion of 10 mmol 3-iodobenzoic acid (1 equiv) in 50 mL dry CH2Cl2 was added 25 mmol oxalyl chloride (2.5 equiv) and three drops of DMF, and the mixture was stirred for 2 – 4 h at room temperature. The reaction mixture was evaporated under reduced pressure and dried under vacuum to yield 3-iodobenzoyl chloride, which was dissolved in 50 mL dry CH2Cl2 again and cooled in an ice bath. A 10 mL CH2Cl2 solution containing 15 mmol amine (1.5 equiv) and 20 mmol Et3N (2 equiv) was then added and the reaction mixture was stirred at room temperature under a nitrogen atmosphere. After 1 h, the solution was treated with 50 mL water and 100 mL DCM, and the organic phase was separated, dried, evaporated, and then purified by column chromatography to yield compound A in 90 – 95% yield.
Fig. 6.

Synthesis of ligands and catalysts. DMF, N, N-dimethylformamide; DCM, dichloromethane; Dippf, bis(diisopropylphosphinyl)ferrocene; Tol, toluene; DMS, dimethylsulfide.
A mixture of 8 mmol A (1 equiv), 8.8 mmol 2-bromophenylboronic acid (1.1 equiv) and 24 mmol Et3N (3 equiv) in 40 mL DMF was stirred and bubbled with N2 gas for 15 minutes, and then 0.4 mmol Pd(PPh3)4 (5 mol %) was added; the reaction mixture was heated at 90 °C for 4 – 8 h under nitrogen atmosphere. Once TLC indicated A was completely consumed, the reaction was diluted with 500 mL Et2O and washed with water to remove DMF. Then the organic layer was dried over MgSO4, filtrated, evaporated, and then purified by column chromatography to yield product B in 85 – 92% yield.
Under nitrogen atmosphere 2 mmol B (1 equiv), 0.1 mmol Pd(OAc)2 (5 mol%), 0.12 mmol DiPPF (1,1'-bis(diisopropylphosphino)ferrocene, 6 mol%), 2.4 mmol NaOt-Bu (1.2 equiv) and 5 mL dry Toluene were added to a flamed dried Schlenk flask and the resulting suspension was stirred until apparently homogeneous (around 15 min). Added 2.2 mmol di(1-adamantyl)phosphine (1.1 equiv), the flask was heated at 110 °C in oil bath for 12 h, which then was cooled to room temperature, and purified by column chromatography without work-up to yield the final ligand L in 60 – 80% yield.
To a suspension of 1 mmol ligand L in 5 mL anhydrous DCM was added chloro(dimethylsulfide)gold(I) (294.5 mg, 1 mmol). The mixture was stirred for 30 min at room temperature and the solvent was evaporated off under reduced pressure to give the desired gold complex LAuCl in quantitative yield.
For the detailed procedures and NMR and HRMS data, see Supplementary Methods. For the NMR and HRMS spectra of the ligands and catalysts, see Supplementary Figures 1–68.
General procedure for preparation of enol esters
In a sealed 1 dr reaction vial equipped with a magnetic stirring bar, 3 mmol carboxylic acids (1 equiv), 4.4 mmol 1-hexyne (1.45 equiv) or 3.6 mmol other alkynes (1.2 equiv) and 3.1 mg NaBARF (1200 ppm) were added to 0.6 mL fluorobenzene. L8AuNTf2 (50 µL of a 3.09 mg/mL solution in PhF, 0.150 µmol, 50 ppm) was added to the above vial and then the reaction mixture was heated at 80 °C for 12 – 24 h. Once the reaction finished by TLC, it was concentrated and left on the high vacuum pump for overnight to give the NMR pure product. If crude NMR was not pure, the residue was further purified through silica gel flash chromatography to give the desired product.
For the detailed procedures and NMR and HRMS data, see Supplementary Methods. For the NMR and HRMS spectra of the ligands and catalysts, see Supplementary Figures 69–158.
General procedure for one-pot sequence preparation of amides
In a sealed 1 dr reaction vial equipped with a magnetic stirring bar, 3 mmol carboxylic acids (1 equiv), 4.4 mmol 1-hexyne (1.45 equiv) and 3.1 mg NaBARF (1200 ppm) were added to 0.6 mL fluorobenzene. L8AuNTf2 (50 µL of a 3.09 mg/mL solution in PhF, 0.150 µmol, 50 ppm or 100 µL, 100 ppm) was added to the above vial and then the reaction mixture was heated at 80 °C for 12 – 18 h. Once the reaction finished by TLC, it was concentrated and left on the high vacuum pump for overnight to give the crude product, followed by adding 3.6 mmol amine (1.2 equiv) and stirring at 100 °C for 6 – 24 h. Once the reaction completed by TLC, the mixture was purified through silica gel flash chromatography (eluents: ethyl acetate: hexanes = 1: 3) to give the desired product.
For the detailed procedures and NMR and HRMS data, see Supplementary Methods.
DFT calculation
The details of the theoretical calculations are provided in the Supplementary Methods and Supplementary Data 1.
Supplementary Material
Acknowledgements
The authors appreciate the financial support of NIH (NIGMS (R01 GM084254), NSF (CHE-1301343), and the Natural Science Foundation of China (Grant No. 21172248, 21121062).
Footnotes
Author contributions
L.Z. came up with the design and wrote the manuscript. Y.W. planned and conducted the experiments. Z.W. performed some experiments and helped with revising the manuscript. Y.L. performed computational study. G.W. and Z.C. helped collecting some experimental data.
Accession codes: The X-ray crystallographic coordinates for structure L8AuCl reported in this Article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 985188. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Competing financial interest: The authors declare no competing financial interests.
Supplementary Materials: experimental procedures, compound characterization data and NMR spectra, the structures of the optimized transition states and the calculated total energies and geometrical coordinates.
References
- 1.Hashmi ASK. Gold-Catalyzed Organic Reactions. Chem. Rev. 2007;107:3180–3211. doi: 10.1021/cr000436x. [DOI] [PubMed] [Google Scholar]
- 2.Fürstner A, Davies PW. Catalytic Carbophilic Activation: Catalysis by Platinum and Gold π Acids. Angew. Chem., Int. Ed. 2007;46:3410–3449. doi: 10.1002/anie.200604335. [DOI] [PubMed] [Google Scholar]
- 3.Zhang L, Sun J, Kozmin SA. Gold and Platinum Catalysis of Enyne Cycloisomerization. Adv. Synth. Catal. 2006;348:2271–2296. [Google Scholar]
- 4.Gorin DJ, Sherry BD, Toste FD. Ligand Effects in Homogeneous Au Catalysis. Chem. Rev. 2008;108:3351–3378. doi: 10.1021/cr068430g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiménez-Núñez E, Echavarren AM. Gold-Catalyzed Cycloisomerizations of Enynes: A Mechanistic Perspective. Chem. Rev. 2008;108:3326–3350. doi: 10.1021/cr0684319. [DOI] [PubMed] [Google Scholar]
- 6.Abu Sohel SM, Liu R-S. Carbocyclisation of alkynes with external nucleophiles catalysed by gold, platinum and other electrophilic metals. Chem. Soc. Rev. 2009;38:2269–2281. doi: 10.1039/b807499m. [DOI] [PubMed] [Google Scholar]
- 7.Arcadi A. Alternative Synthetic Methods through New Developments in Catalysis by Gold. Chem. Rev. 2008;108:3266–3325. doi: 10.1021/cr068435d. [DOI] [PubMed] [Google Scholar]
- 8.Pradal A, Toullec PY, Michelet V. Recent Developments in Asymmetric Catalysis in the Presence of Chiral Gold Complexes. Synthesis. 2011:1501–1514. [Google Scholar]
- 9.Sengupta S, Shi X. Recent Advances in Asymmetric Gold Catalysis. Chemcatchem. 2010;2:609–619. [Google Scholar]
- 10.Teles JH, Brode S, Chabanas M. Cationic gold(I) complexes: highly efficient catalysts for the addition of alcohols to alkynes. Angew. Chem., Int. Ed. 1998;37:1415–1418. doi: 10.1002/(SICI)1521-3773(19980605)37:10<1415::AID-ANIE1415>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 11.Mizushima E, Hayashi T, Tanaka M. Au(I)-Catalyzed Highly Efficient Intermolecular Hydroamination of Alkynes. Org. Lett. 2003;5:3349–3352. doi: 10.1021/ol0353159. [DOI] [PubMed] [Google Scholar]
- 12.Marion N, Ramon RS, Nolan SP. [(NHC)Au-I]-Catalyzed Acid-Free Alkyne Hydration at Part-per-Million Catalyst Loadings. J. Am. Chem. Soc. 2009;131:448–449. doi: 10.1021/ja809403e. [DOI] [PubMed] [Google Scholar]
- 13.Tu X-F, Gong L-Z. Highly Enantioselective Transfer Hydrogenation of Quinolines Catalyzed by Gold Phosphates: Achiral Ligand Tuning and Chiral-Anion Control of Stereoselectivity. Angew. Chem., Int. Ed. 2012;51:11346–11349. doi: 10.1002/anie.201204179. [DOI] [PubMed] [Google Scholar]
- 14.Lavallo V, Wright JH, Tham FS, Quinlivan S. Perhalogenated Carba-closo-dodecaborate Anions as Ligand Substituents: Applications in Gold Catalysis. Angew. Chem., Int. Ed. 2013:3172–3176. doi: 10.1002/anie.201209107. [DOI] [PubMed] [Google Scholar]
- 15.Blanco Jaimes MC, Böhling CRN, Serrano-Becerra JM, Hashmi ASK. Highly Active Mononuclear NAC–Gold(I) Catalysts. Angew. Chem., Int. Ed. 2013;52:7963–7966. doi: 10.1002/anie.201210351. [DOI] [PubMed] [Google Scholar]
- 16.Kamer P, Leeuwen PWNMv. Phosphorus(III) ligands in homogeneous catalysis : design and synthesis. Hoboken, N.J.: Wiley; 2012. [Google Scholar]
- 17.Hayashi T, Yamamoto A, Hagihara T, Ito Y. Modification of optically active ferrocenylphosphine ligands for palladium-catalyzed asymmetric allylic alkylation. Tetrahedron Lett. 1986;27:191–194. [Google Scholar]
- 18.Fors BP, Watson DA, Biscoe MR, Buchwald SL. A Highly Active Catalyst for Pd-Catalyzed Amination Reactions: Cross-Coupling Reactions Using Aryl Mesylates and the Highly Selective Monoarylation of Primary Amines Using Aryl Chlorides. J. Am. Chem. Soc. 2008;130:13552–13554. doi: 10.1021/ja8055358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Billingsley K, Buchwald SL. Highly Efficient Monophosphine-Based Catalyst for the Palladium-Catalyzed Suzuki–Miyaura Reaction of Heteroaryl Halides and Heteroaryl Boronic Acids and Esters. J. Am. Chem. Soc. 2007;129:3358–3366. doi: 10.1021/ja068577p. [DOI] [PubMed] [Google Scholar]
- 20.Anderson KW, Tundel RE, Ikawa T, Altman RA, Buchwald SL. Monodentate Phosphines Provide Highly Active Catalysts for Pd-Catalyzed C–N Bond-Forming Reactions of Heteroaromatic Halides/Amines and (H)N-Heterocycles. Angew. Chem., Int. Ed. 2006;45:6523–6527. doi: 10.1002/anie.200601612. [DOI] [PubMed] [Google Scholar]
- 21.Mauger CC, Mignani GA. Synthetic Applications of Buchwald’s Phosphines in Palladium-Catalyzed Aromatic-Bond-Forming Reactions. Aldrichimica Acta. 2006;39:17. [Google Scholar]
- 22.Schlummer B, Scholz U. Palladium-Catalyzed C–N and C–O Coupling–A Practical Guide from an Industrial Vantage Point†. Adv. Synth. Catal. 2004;346:1599–1626. [Google Scholar]
- 23.Ye L, He W, Zhang L. A Flexible and Stereoselective Synthesis of Azetidin-3-ones through Gold-Catalyzed Intermolecular Oxidation of Alkynes. Angew. Chem., Int. Ed. 2011;50:3236–3239. doi: 10.1002/anie.201007624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Ji K, Lan S, Zhang L. Rapid Access to Chroman-3-ones through Gold-Catalyzed Oxidation of Propargyl Aryl Ethers. Angew. Chem., Int. Ed. 2012;51:1915–1918. doi: 10.1002/anie.201107561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nieto-Oberhuber C, Lopez S, Echavarren AM. Intramolecular [4+2] Cycloadditions of 1,3-Enynes or Arylalkynes with Alkenes with Highly Reactive Cationic Phosphine Au(I) Complexes. J. Am. Chem. Soc. 2005;127:6178–6179. doi: 10.1021/ja042257t. [DOI] [PubMed] [Google Scholar]
- 26.Leyva A, Corma A. Isolable Gold(I) Complexes Having One Low-Coordinating Ligand as Catalysts for the Selective Hydration of Substituted Alkynes at Room Temperature without Acidic Promoters. J. Org. Chem. 2009;74:2067–2074. doi: 10.1021/jo802558e. [DOI] [PubMed] [Google Scholar]
- 27.Wang C, Han Z-Y, Luo H-W, Gong L-Z. Highly Enantioselective Relay Catalysis in the Three-Component Reaction for Direct Construction of Structurally Complex Heterocycles. Org. Lett. 2010;12:2266–2269. doi: 10.1021/ol1006086. [DOI] [PubMed] [Google Scholar]
- 28.López-Carrillo Vn, Echavarren AM. Gold(I)-Catalyzed Intermolecular [2+2] Cycloaddition of Alkynes with Alkenes. J. Am. Chem. Soc. 2010;132:9292–9294. doi: 10.1021/ja104177w. [DOI] [PubMed] [Google Scholar]
- 29.Wang T, Zhang J. Chemoselective C-C Bond Cleavage of Epoxide Motifs: Gold(I)-Catalyzed Diastereoselective [4+3] Cycloadditions of 1-(1-Alkynyl)oxiranyl Ketones and Nitrones. Chem. Eur. J. 2011;17:86–90. doi: 10.1002/chem.201002395. [DOI] [PubMed] [Google Scholar]
- 30.Barabé F, Levesque P, Korobkov I, Barriault L. Synthesis of Fused Carbocycles via a Selective 6-Endo Dig Gold(I)-Catalyzed Carbocyclization. Org. Lett. 2011;13:5580–5583. doi: 10.1021/ol202314q. [DOI] [PubMed] [Google Scholar]
- 31.Henrion G, Chavas TEJ, Le Goff X, Gagosz F. Biarylphosphonite Gold(I) Complexes as Superior Catalysts for Oxidative Cyclization of Propynyl Arenes into Indan-2-ones. Angew. Chem., Int. Ed. 2013;52:6277–6282. doi: 10.1002/anie.201301015. [DOI] [PubMed] [Google Scholar]
- 32.Anderson KW, Buchwald SL. General Catalysts for the Suzuki–Miyaura and Sonogashira Coupling Reactions of Aryl Chlorides and for the Coupling of Challenging Substrate Combinations in Water. Angew. Chem., Int. Ed. 2005;44:6173–6177. doi: 10.1002/anie.200502017. [DOI] [PubMed] [Google Scholar]
- 33.Lundgren RJ, Peters BD, Alsabeh PG, Stradiotto M. A P,N-Ligand for Palladium-Catalyzed Ammonia Arylation: Coupling of Deactivated Aryl Chlorides, Chemoselective Arylations, and Room Temperature Reactions. Angew. Chem., Int. Ed. 2010;49:4071–4074. doi: 10.1002/anie.201000526. [DOI] [PubMed] [Google Scholar]
- 34.Chary BC, Kim S. Gold(I)-Catalyzed Addition of Carboxylic Acids to Alkynes. J. Org. Chem. 2010;75:7928–7931. doi: 10.1021/jo101543q. [DOI] [PubMed] [Google Scholar]
- 35.Goossen LJ, Paetzold J, Koley D. Regiocontrolled Ru-catalyzed addition of carboxylic acids to alkynes: practical protocols for the synthesis of vinyl esters. Chem. Commun. 2003:706–707. [PubMed] [Google Scholar]
- 36.Hohenberg P, Kohn W. Inhomogeneous Electron Gas. Phys. Rev. 1964;136:B864–B871. [Google Scholar]
- 37.Kohn W, Sham LJ. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965;140:A1133–A1138. [Google Scholar]
- 38.Frisch MJT, G W, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery A, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision A.02. Wallingford, CT: Gaussian, Inc.; 2009. [Google Scholar]
- 39.Perdew JP, Burke K, Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 40.Perdew JP, Burke K, Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997;78:1396–1396. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 41.Fuentealba P, Preuss H, Stoll H, Von Szentpály L. A proper account of core-polarization with pseudopotentials: single valence-electron alkali compounds. Chem. Phys. Lett. 1982;89:418–422. [Google Scholar]
- 42.Marenich AV, Cramer CJ, Truhlar DG. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. 2009;113:6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 43.Jiang M, Liu L-P, Shi M, Li Y. Gold(I)-Catalyzed Tandem C–H and C–C Activation (Cleavage) Org. Lett. 2010;12:116–119. doi: 10.1021/ol902593f. [DOI] [PubMed] [Google Scholar]
- 44.Xia YZ, Dudnik AS, Gevorgyan V, Li YH. Mechanistic Insights into the Gold-Catalyzed Cycloisomerization of Bromoallenyl Ketones: Ligand-Controlled Regioselectivity. J. Am. Chem. Soc. 2008;130:6940–6941. doi: 10.1021/ja802144t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zuccaccia D, Belpassi L, Tarantelli F, Macchioni A. Ion Pairing in Cationic Olefin–Gold(I) Complexes. J. Am. Chem. Soc. 2009;131:3170–3171. doi: 10.1021/ja809998y. [DOI] [PubMed] [Google Scholar]
- 46.Murray CW, Verdonk ML. The consequences of translational and rotational entropy lost by small molecules on binding to proteins. J. Comput. Aid. Mol. Des. 2002;16:741–753. doi: 10.1023/a:1022446720849. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.