

Summary
Inactivation of the von Hippel-Lindau (VHL) gene is associated with the development of highly vascularized tumors. pVHL targets the α-subunits of Hypoxia Inducible Factor (HIF) for ubiquitin-mediated degradation in an oxygen-dependent manner. Although pVHL-deficient tumor cell lines demonstrate constitutive stabilization and activation of HIF, it has yet to be shown that loss of murine Vhl alone is sufficient to dysregulate HIF. We utilized a genetic approach to demonstrate that loss of Vhl is sufficient not only to stabilize HIF-α subunits under normoxia, but also fully activate HIF-mediated responses. These studies have implications for the hierarchy of signaling events leading to HIF stabilization, nuclear translocation, and target gene expression. We further demonstrate that loss of murine Vhl does not promote teratocarcinoma growth, indicating that other genetic changes must occur to facilitate Vhl-mediated tumorigenesis.
Introduction
von Hippel-Lindau disease is an autosomal dominant hereditary cancer syndrome. Patients heterozygous for one inactivating mutation within the von Hippel-Lindau (VHL) gene are predisposed to the development of pheochromocytomas and a variety of highly vascularized tumors, including renal clear cell carcinomas, retinal angiomas, and hemangioblastomas (Maher and Kaelin 1997). Within these tumors the second VHL allele has been rendered inactive by deletions, mutations, or hypermethylation. VHL is therefore a classic tumor suppressor conforming to the Knudson two hit hypothesis in which the second inactivating mutation occurs within somatic cells. A majority of sporadic renal clear cell carcinomas also exhibit biallelic loss of VHL function, further supporting VHL's role as a tumor suppressor (Foster et al. 1994; Gnarra et al. 1994). Animal models demonstrate that deletion of murine Vhl function results in lethality, as Vhl-/- mice die in utero between E10.5-12 due to placental vascular abnormalities (Gnarra et al. 1997). Hepatocyte-specific loss of Vhl in adult mice results in the formation of hemangiomas within the liver, a phenotype rarely observed in human VHL patients (Haase et al. 2001).
pVHL, the protein product of the VHL gene, interacts through its α-domain with elongins B/C and Cul2 to form the VBC complex, an E3 ubiquitin ligase. This multi-protein complex targets proteins for ubiquitin-mediated degradation, analogous to the S.cerevisiae SCF (Skpl/Cdc53/F-box) ubiquitination machinery (Pause et al. 1997; Lonergan et al. 1998; Lisztwan et al. 1999). Although pVHL has no sequence homology to any known proteins, it is functionally similar to the F-box protein of SCF by binding specific substrates through its β-domain (Ohh et al. 2000).
Major targets of the VBC complex include the regulatory α-subunits of the heterodimeric transcription factor Hypoxia Inducible Factor (HIF). HIF modulates both cellular and systemic responses to changes in oxygen (O2) concentrations by stimulating the expression of genes involved in energy metabolism, angiogenesis, hematopoiesis and O2 delivery. Proteasome inhibitor studies show that at high levels of O2 (21%), the VBC complex binds HIF-α subunits (HIF-1α and HIF-2α), targeting them for ubiquitination and degradation via the 26S proteasome (Maxwell et al. 1999). Low O2 (hypoxia) disrupts the association of pVHL with HIF-α subunits, which then dimerize with constitutively expressed ARNT/HIF-β to form transcriptionally active HIF. Mutations within the β-domain of pVHL disrupt its interaction with HIF-α subunits leading to constitutive HIF-α stabilization and HIF activity (Cockman et al. 2000; Ohh et al. 2000). Furthermore, VHL-/- renal clear cell carcinoma (RCC) cell lines fail to degrade HIF-α subunits under normoxic conditions. These cells display constitutive expression of HIF targets including genes encoding erythropoietin (EPO), vascular endothelial growth factor (VEGF), glucose transporter (GLUT-1) and phosphoglycerate kinase (PGK) (Iliopoulos et al. 1996; Maxwell et al. 1999; Krieg et al. 2000). In addition to its role as part of an E3 ubiquitin ligase, pVHL is required for proper fibronectin deposition, cytoskeletal reorganization and growth arrest upon serum withdrawal and contact inhibition, although the underlying mechanisms for these properties remain unclear. (Ohh et al. 1998; Pause et al. 1998; Hoffman et al. 2001; Kamada et al. 2001; Bindra et al. 2002). Defects within all of these pathways can be reversed by reintroduction of wild-type pVHL into VHL-/- RCC cells.
The interaction of pVHL with HIF-α is one of several O2-dependent events thought to regulate HIF activity. First, pVHL binding to HIF-α subunits is dependent upon hydroxylation of key proline residues within the O2-dependent degradation (ODD) domain of HIF-α (Ivan et al. 2001; Jaakkola et al. 2001). A recently defined family of HIF prolyl-hydroxylases (HTF-PHs) catalyzes this iron- and O2-dependent reaction. All three elements of this pathway are conserved in C. elegans, Drosophila and mammals (Bruick and McKnight 2001; Epstein et al. 2001). Second, hypoxia is also believed to induce the nuclear accumulation of stabilized HIF-α subunits, protecting them from pVHL-mediated degradation (Kallio et al. 1999; Tanimoto et al. 2000). Third, hypoxia-mediated HIF transactivation of target genes requires interactions with coactivators p300 and CBP (Arany et al. 1996; Jiang et al. 1997; Pugh et al. 1997; Ebert and Bunn 1998; Carrero et al. 2000). Recent studies suggest that hydroxylation of a C-terminal activation domain (CAD) asparagine residue by an iron- and O2- dependent asparagyl hydroxylase disrupts HIF-α binding to coactivators p300 and CBP, thereby preventing transactivation during normoxia (Lando et al. 2002). Furthermore, hydroxylation within the CAD is thought to occur in a pVHL-independent manner (Sang et al. 2002). Therefore, under normal O2 concentrations, hydroxylation of HIF-α on proline and asparagine residues regulates HIF's transcriptional activity by targeting the protein for proteolysis and inhibiting binding to coactivators, respectively.
Thus far, most studies correlating loss of VHL function with HIF dysregulation have been conducted in renal tumors and fully transformed cell lines. As cancer development requires multiple genetic lesions, these cells undoubtedly contain a highly abnormal genetic background. Although recent results have shown that VHL inactivation is correlated with stimulation of the HIF pathway in early kidney lesions (Mandriota et al. 2002). other genetic abnormalities were not examined. Indeed, cytogenetic analysis of renal clear cell carcinomas shows that a majority of these tumors have lost the short arm of chromosome 3 and contain a myriad of structural aberrations and unbalanced translocations (Gunawan et al. 2001). Although reintroduction of VHL into RCC lines eliminates their tumorigenic phenotype (Gnarra et al. 1996; Iliopoulos et al. 1996), this occurs within a background of uncharacterized mutations, making it difficult to identify the effects of VHL loss alone. Until now, a formal genetic study to assess the tumorigenic effects of VHL loss in euploid primary cells has not been performed. Furthermore, although the regulation of HIF activity is believed to be a multi-step process responsive to O2 levels, it is unclear to what degree these events can occur in normoxic conditions. Here, we utilize gene targeting of Vhl in mouse embryonic stem (ES) cells to demonstrate that loss of Vhl is sufficient to dysregulate HIF degradation, nuclear accumulation, DNA binding activity and downstream target gene activation, conferring a hypoxic phenotype to normoxic Vhl-/- ES cells. Unexpectedly, Vhl-/- teratocarcinomas are smaller by volume and mass compared to Vhl+/- tumors, suggesting that stimulation of the HIF pathway is not sufficient to induce tumor growth and additional genetic changes must occur to facilitate tumorigenesis in the absence of Vhl.
Results
Generation of Vhl-/- ES cells
Although renal cancer cell lines deficient for pVHL exhibit constitutive expression of HIF target genes under normoxic conditions, loss of VHL alone has not been shown to result in complete disruption of HIF regulation and promotion of tumor formation. To examine the precise role of Vhl in these processes, we utilized a genetic strategy based on Vhl+/- mouse J1 embryonic stem (ES) cells (Gnarra et al. 1997). The recombined locus maintains intact the Vhl promoter region, the first 23 codons, and 3′ untranslated region (Fig. 1A). Homozygous Vhl-/- clones were generated from Vhl+/- ES cells by selecting for loss of the remaining wild-type allele with increasing concentrations of G418 (Mortensen et al. 1992). Approximately 200 clones were screened for loss of heterozygosity by Southern blot and/or PCR assays (Figs. 1B and C). Three independently derived Vhl-/- clones were identified, none of which expressed detectable amounts of pVHL when compared to Vhl+/+ and Vhl+/- ES cells (Fig. 1D), and these were further assayed for HIF function.
Figure 1. Generation of Vhl-/- ES cells.
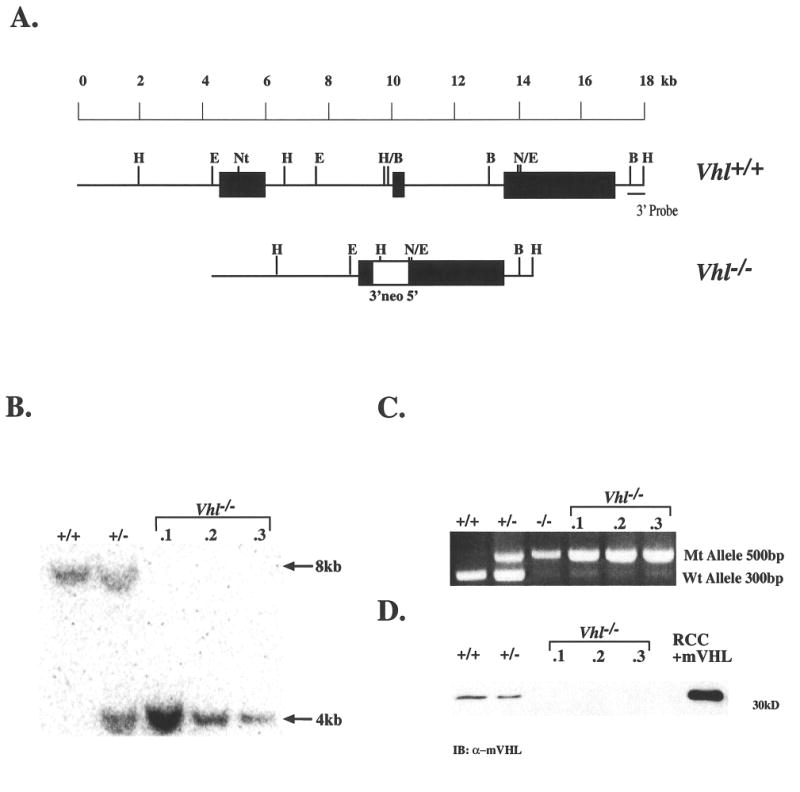
A: Filled boxes represent the three Vhl exons while the open box depicts the pgk-neo selectable marker within the targeting vector. The 3′ flanking probe used in Southern blots is indicated. B: Detection of wild-type and targeted alleles in ES cells by HindIII digestion and hybridization to the 3′ flanking probe revealed an 8-kb wild-type fragment and 4-kb mutant fragment in Vhl+/- and ES clones 1, 2, and 3. C: PCR genotyping of ES clones demonstrated a 300-bp wild-type and a 500-bp mutant product. The lower band visible within lanes 3-6 is a background signal thought not to represent the wild-type allele because of its greater size and decreased intensity. D: Immunoblot for pVHL in ES clones detected a 30kd protein in wild-type and Vhl+/- ES cell whole cell extracts but not in clones 1, 2, and 3. A pVHL-deficient RCC cell line stably transfected with HA-tagged murine pVHL was used as a control.
HIF is constitutively stabilized and maintains DNA binding activity in the absence of Vhl
We first investigated the stabilization of HIF-α proteins by western blot and electrophoretic mobility shift assay (EMSA). Immunoblots of nuclear extracts demonstrated that Vhl+/+ and Vhl+/- ES cells express low levels of HIF-1α and HIF-2α at 21% O2 (normoxia) (Fig. 2A), which increased dramatically at 1.5% O2 (hypoxia). In contrast, Vhl-/- cells accumulated high levels of HIF-1α and HIF-2α under normoxia. Moreover, HIF-1α and HIF-2α protein levels remained largely unchanged within normoxic and hypoxic extracts when normalized to the Rad50 loading control. The constitutive stabilization of HIF-α subunits in Vhl-/- cells is consistent with data reported for RCC lines (Maxwell et al. 1999; Krieg et al. 2000).
Figure 2. HTF stabilization and DNA binding activity in the absence of Vhl.
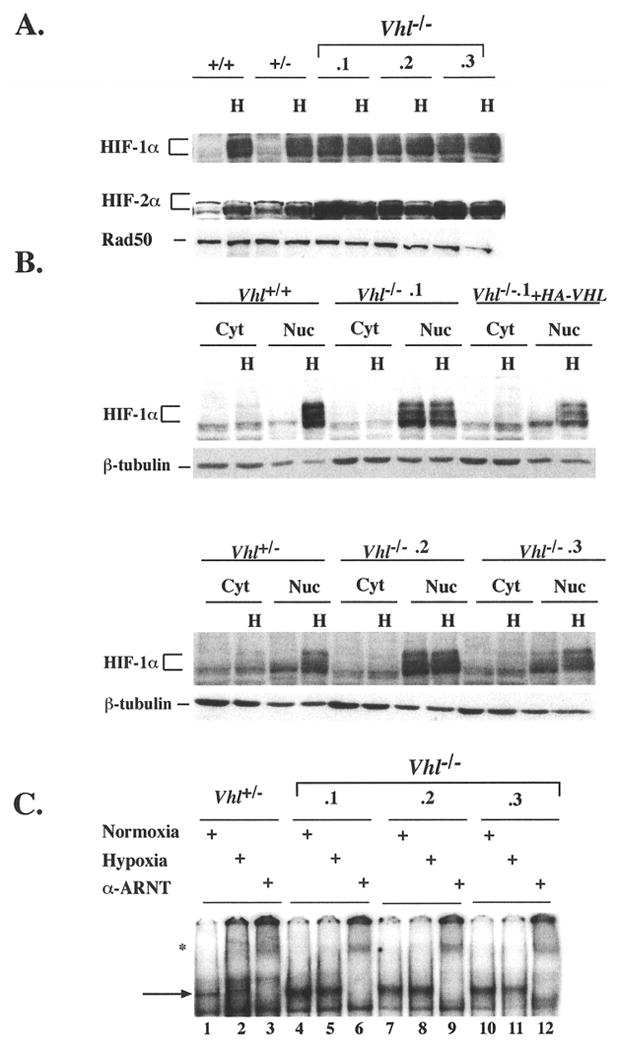
A: Nuclear extracts were prepared from ES cells cultured under normoxia (21% O2) or hypoxia (1.5% O2) for 4hrs. Low levels of HTF-lα and HIF-2α proteins were present under normoxia and become markedly induced by hypoxia in Vhl+/+ and Vhl+/- ES cells. In the absence of Vhl, HIF-α proteins became stabilized under normoxia at higher levels than Vhl+/+ and Vhl+/- ES cells and were not substantially induced by hypoxia. Nuclear protein Rad50 was utilized as a loading control. B: Immonoblot for HTF-1α in cytoplasmic and nuclear fraction revealed that stabilized HTF-1α accumulated exclusively in the nucleus of Vhl-/- ES cells. HIF-1α regulation was restored by stable transfection of HA-VHL into clone Vhl-/-.1. Loading control β-tubulin, although more abundant in cytoplasmic fractions, demonstrated equivalent protein amounts were loaded per cell sample. C: EMSA of 5.0μg nuclear extracts prepared from cells grown under normoxic or hypoxic conditions for 4hrs where indicated. HIF DNA binding activity was maintained in Vhl-/- cells under normoxia. The arrow represents the HIF/ARNT complex, which was shown to be specific by supershifting with an α-ARNT antibody, marked by an asterisks.
The nuclear translocation of HTF-α subunits has been reported to be an O2-regulated step, independent of pVHL function (Tanimoto et al. 2000). To detect the localization of constitutively stabilized HIF-1α in Vhl-/- cells, western blots were performed using cytoplasmic and nuclear fractions of extracted proteins (Fig. 2B). In Vhl+/+ and Vhl+/- cells, stabilized HIF-lα was exclusively detected in the nuclear fraction of hypoxic extracts (Fig. 2B). All Vhl-/- clones exhibited exclusively nuclear accumulation of HIF-1 α under normoxia and hypoxia. Introduction of hemagglutin-tagged (HA)-VHL into clone Vhl-/-.1 restored the hypoxic regulation of HIF-lα, consistent with previously reported data (Maxwell et al. 1999; Krieg et al. 2000). This “rescued” Vhl-/- clone expressed levels of pVHL comparable to RCC lines rescued with HA-VHL (data not shown) (Iliopoulos et al. 1996).
HIF DNA binding activity in normoxic and hypoxic Vhl-/-ES cells was assessed by EMSA. Vhl+/- ES cells exhibited hypoxic induction of a DNA complex confirmed to be HIF by “supershifting” with an α-ARNT antibody (Fig. 2C, lanes 2 and 3). In contrast, high levels of HIF were detected in normoxic Vhl-/- ES cells (see lanes 4, 7, and 10), which was also by supershifted with the ARNT antibody (lanes 6,9, and 12). We conclude from these results that HIF-α stabilization, subcellular localization, and DNA binding activity are dependent upon the presence of functional pVHL regardless of O2 concentration.
Loss of Vhl is sufficient to activate HIF-mediated transcription under normoxia
To assess the transcriptional activity of HIF in Vhl-/- ES cells, reporter assays using hypoxia response element (HRE)-driven luciferase constructs were performed (Fig. 3A). Normoxic Vhl+/+ and Vhl+/- ES cells demonstrated low levels of HIF transcriptional activity induced more than 10-fold by hypoxia. In contrast, although the levels of HIF activity varied between Vhl-/- clones within these assays, the trend consistently showed high levels of HIF activity under normoxic conditions, which were only slightly elevated by hypoxia. Use of a mutant HRE construct, which contains three base pair substitutions to disrupt HIF binding, ablated the hypoxic induction of transcriptional activity in all cell lines demonstrating its HIF-dependence. Furthermore, Hif-1α-/- ES cells exhibited reduced levels of luciferase activity in both normoxic and hypoxic conditions, comparable to levels detected using the mutant HRE construct. Importantly, cotransfection of a plasmid encoding HA-VHL with HRE-reporter constructs repressed normoxic HIF activity and restored hypoxic induction of HRE-dependent transcription in Vhl-/- cells (Fig. 3B).
Figure 3. HIF transcriptional activity is dysregulated in Vhl-/- ES cells.
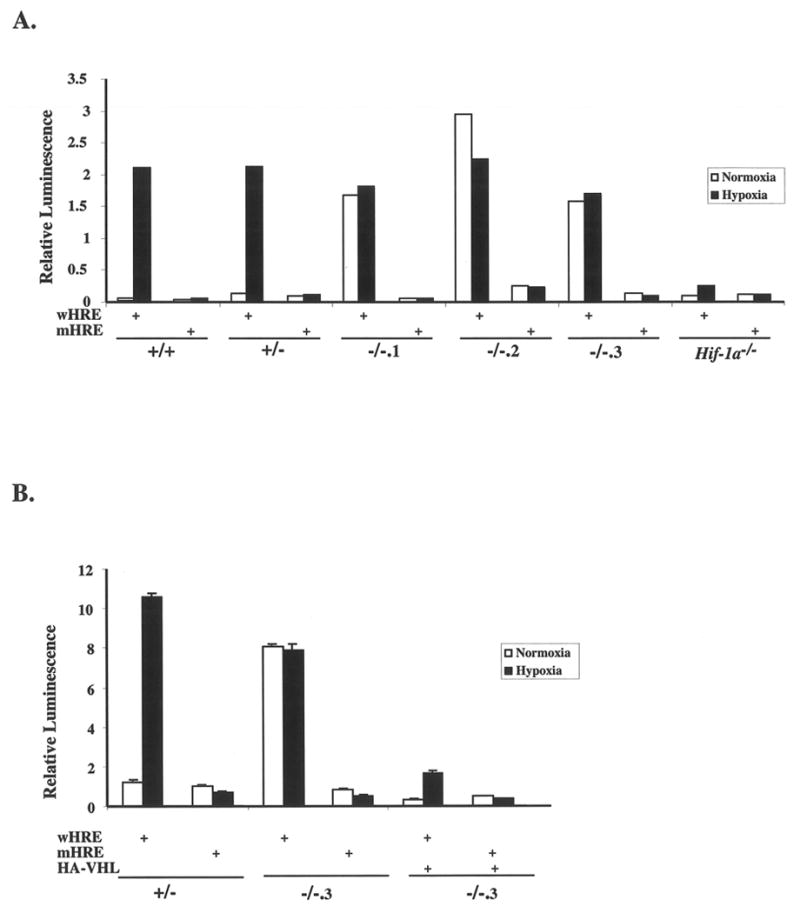
A: ES cells were transiently transfected with wild-type (wHRE) or mutant (mHRE) reporter constructs and transcriptional activity was measured in normoxic (21% O2) or hypoxic (1.5% O2) cells at 16hrs. Luciferase activity detected in normoxic Vhl-/- clones was not further induced by hypoxia. Data represents one of three independent assays in which similar results were produced. B: ES cells were cotransfected with wHRE or mHRE plus or minus a plasmid encoding HA-VHL. The presence of HA-VHL decreased the normoxic levels of HIF activity and restored hypoxic induction. Assays were performed in triplicate and bars represent standard error.
To further investigate the function of stabilized HIF in Vhl-/- cells, the activation of endogenous HIF target genes was examined. mRNA expression analysis of several known hypoxia responsive genes (Glut-1, Vegf, Pgk, Alda and Bnip3) revealed low levels of expression in normoxic Vhl+/+ and Vhl+/- ES cells, which were markedly induced by hypoxia (Fig. 4A). In direct contrast, the expression of HIF target genes in multiple normoxic Vhl-/- ES clones was comparable to, if not greater than, their expression in hypoxic in Vhl+/+ and Vhl+/- ES cells. Consistent with reporter gene data, HIF target gene expression remained high and was not greatly affected by hypoxia in two Vhl-/- ES clones. Clone Vhl-/-. 3 exhibited high levels of HIF target gene expression under normoxia that was moderately induced by hypoxia, albeit to a lesser degree than wild-type controls. This could be simply a result of clonal variation, as extensive genotyping indicated the absence of contaminating Vhl+/- cells in this clone. In all other assays that analyzed HIF target gene activation, Vhl-/-. 3 behaved similarly to the other clones. The expression of all target genes was primarily regulated by HIF, as shown by their greatly reduced expression in Hif-1α-/- ES cells. However, Glut-1 and Vegf transcripts can be stabilized by hypoxia independently of HIF (Ikeda et al. 1995; Levy et al. 1996; Levy et al. 1996), which may account for the residual hypoxic induction of these genes in Hif-1α-/- ES cells.
Figure 4. Induction of HIF target genes in normoxic Vhl-/- ES cells.
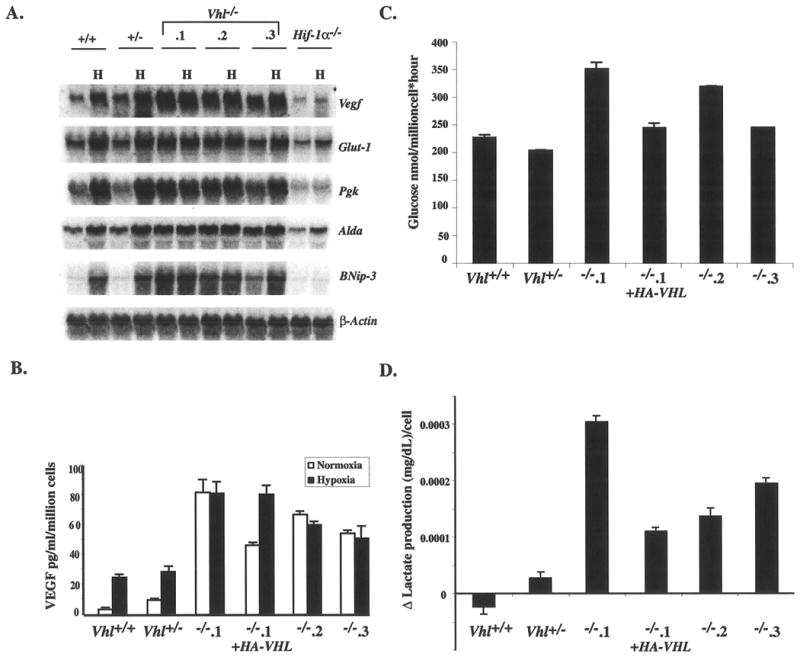
A: Total RNA isolated from Vhl +/+, Vhl +/-, Vhl-/- clones and Hif-1α-/- ES cells was probed for Vegf, Glut-1, Pgk, Alda, Bnip-3, and β-actin , which served as a loading control. Expression of HIF target genes was abundant under normoxia and failed to be further induced by hypoxia in Vhl-/- ES clones. B: In vitro secretion of VEGF was measured by ELISA. VEGF secretion was greatly induced by hypoxia in Vhl+/+ and Vhl+/- ES cells but not in Vhl-/- clones. The high level of VEGF secretion by Vhl-/-.1 was suppressed by reintroduction of HA-VHL. C: A representative measurement of the glycolytic rate assessed as the rate of conversion of 3H-Glucose to 3H20 (Liang et al. 1997). Vhl-/- ES cells consumed glucose at a higher rate than controls, which was suppressed by reintroduction of HA-VHL. D: Change in lactate secretion measured over the course of 24 hrs is depicted. Vhl-/- ES cells secreted greater amounts of lactate when compared to controls, which was rescued by reintroduction of HA-VHL. All assays were performed in triplicate and bars represent standard error.
To further assess the activation of HIF targets, VEGF secretion was measured by ELISA (Fig. 4B). Although the absolute amounts of VEGF secretion varied between all clones, the pattern of Vegf regulation observed by Northern blot was recapitulated. VEGF secretion by Vhl+/+ and Vhl+/- cells was substantially induced by hypoxia, while secretion by Vhl-/- cells under the same conditions remained similar to their normoxic levels. The induction of VEGF secretion by hypoxia could be restored by reintroduction of HA-VHL into clone Vhl-/-.1.
When O2 is limiting, cells switch from oxidative phosphorylation to glycolysis as the primary generator of ATP (Pasteur effect). Since the constitutive stabilization of HIF in Vhl-/- cells mimicked a hypoxic state, the glycolytic rate of and lactic acid production by these cells was measured. Consistent with their upregulation of glycolytic genes (Glut-1, Pgk, and Aldd), Vhl-/- cells consumed more glucose than Vhl+/+and Vhl+/- cells over the course of a one-hour normoxic incubation (Fig. 4C). The higher glycolytic rate of Vhl-/-ES cells resulted in a substantially increased rate of lactate production over the course of 24 hours, when compared to Vhl+/+ and Vhl+/- cells (Fig. 4D). The increased glycolytic rate in and lactate production by Vhl-/-.1 was suppressed by reintroduction of HA-VHL, demonstrating that these effects on glycolysis are pVHL-dependent. Therefore, by the criteria of stabilization, nuclear localization, DNA binding activity, target gene activation, and cellular metabolism, loss of Vhl alone is sufficient to constitutively activate HIF to hypoxic levels, independent of O2 signaling.
Loss of Vhl does not promote tumor growth
To investigate whether loss of Vhl can enhance the tumorigenic potential of ES cells, subcutaneous tumors were generated by injecting Vhl+/- and Vhl-/- ES cells into the dorsal region of NIH-III immunodeficient mice. Clones Vhl-/-.1 and Vhl-/-.2 were used because of their high levels of VEGF secretion relative to Vhl+/- cells (Fig. 4B). Surprisingly, Vhl-/- tumors were considerably smaller than Vhl+/- tumors after 21 or 28 days. Table 1 shows the results of four separate experiments in which Vhl-/- tumors were on average 42-68% smaller than Vhl+/- tumors. Vhl-/-.1 tumors grew more slowly than Vhl+/-controls as measured by tumor volume, contrary to the findings of pVHL-deficient RCC-derived tumors (Fig. 6B) (Iliopoulos et al. 1995). Figure 5C shows pooled data from three separate teratocarcinoma experiments at 21 days, in which the masses of tumors formed by clones Vhl-/-.1 and Vhl-/-.2 were combined for an n-value of 20. The difference between the masses of Vhl+/- and Vhl-/- tumors was statistically significant (p= 0.003 by Student t-test). Most importantly, the growth of genetically rescued Vhl-/-.1 + HA-VHL tumors was similar to Vhl+/- controls, demonstrating that this deficiency is functionally dependent upon Vhl (Fig 5C).
Table 1. Vhl-/- tumors are smaller than controls.
| Genotype | T/Sa | Days | Mean Mass (g) | |
|---|---|---|---|---|
| Exp. 1 | Vhl+/- | 6/6 | 21 | 3.052 ± 0.670 |
| Vhl-/-.1 | 5/6 | 21 | 1.036 ± 0.257 | |
| Exp. 2 | Vhl+/- | 10/10 | 21 | 1.223 ± 0.238 |
| Vhl-/-.2 | 10/10 | 21 | 0.709 ± 0.092 | |
| Exp. 3 | Vhl+/- | 5/5 | 21 | 1.62 ± 0.371 |
| Vhl-/-.1 | 4/4 | 21 | 0.53 ± 0.125 | |
| Vhl-/-.1 + HA-VHL | 5/5 | 21 | 1.76 ± 0.296 | |
| Exp. 4 | Vhl+/- | 5/5 | 28 | 4.96 ± 1.19 |
| Vhl-/-.1 | 3/4 | 28 | 0.466 ± 0.318 |
T/S, number of tumors formed/ number of sites injected.
Figure 6. Increased hemorrhagic regions and vasculature of Vhl-/- tumors.
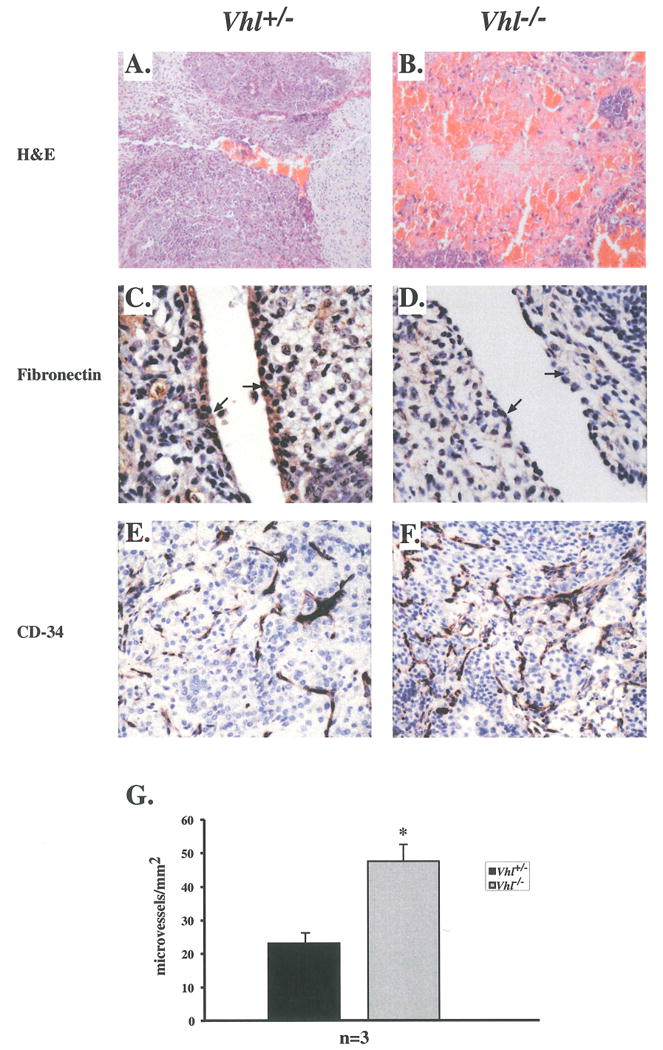
A & B: Hematoxylin and eosin staining of Vhl+/- and Vhl-/- tumors. C & D: Basement membrane expression of fibronectin in Vhl+/- endothelial cells (arrows) and reduced fibronectin deposition in endothelial cells of Vhl-/- tumors (arrows). E: CD-34 staining of Vhl+/- microvessels. F: Microvessel density in Vhl-/- tumors appeared to be increased as demonstrated by α-CD-34 staining. G: Quantitation of tumor microvessel density. The microvessel density of Vhl-/- tumors was greater than controls, n=3. Student t-test revealed statistically significant differences between tumor types, p<0.03. Bars represent standard error. Final magnifications are 100X (A and B), 400X (C and D) and 200X (E and F)
Figure 5. Decrease in growth of Vhl-/- tumors.
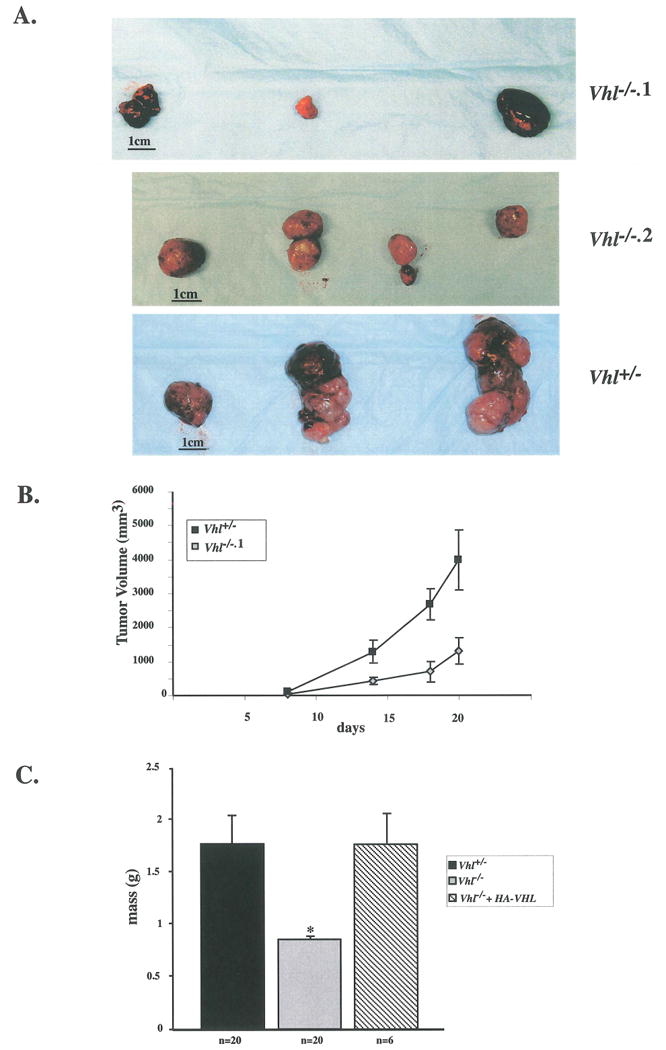
A: Teratocarcinomas were created by injecting VhV-/-, Vhl-/-.1, and Vhl-/-.2 ES cells subcutaneously into nude mice. Vhl-/- tumors ranged in size but were considerably smaller than Vhl+/- derived tumors. B: Rate of tumor growth was measured by tumor volume for 21 days. C: Graph represents the combined results of three separate experiments in which the masses of Vhl+/+, Vhl-/-, and Vhl-/-.1 + HA-VHL tumors after 21 days of growth were measured. N values are as indicated and bars represent standard error. Vhl-/- tumors were significantly smaller than Vhl+/- derived tumors. Genetically rescued Vhl-/-.1 + HA-VHL tumors grew similarly to Vhl+/- tumors. Student t-test revealed a statistically significant difference between tumor masses, p=0.003.
Microvessel density is increased in Vhl-/- tumors
Gross morphological analysis revealed a higher incidence of hemorrhagic regions within Vhl-/- tumors (Figures 6A and B). In addition, primitive tissues derived from all three germ layers were present in Vhl-/- tumors, however, more differentiated tissue types such as skeletal muscle, cartilage, neuronal axons, adipocytes and secretory glands were absent or extremely rare, compared to Vhl+/- tumors (data not shown). The presence of a subset of these tissues was restored in Vhl-/-.1 + HA-VHL tumors (data not shown). The ability of Vhl-/- ES cells to deposit fibronectin in the extracellular matrix was assessed by immunohistochemistry. Vhl+/- tumors exhibited abundant fibronectin staining in the basement membrane of blood vessel endothelial cells (Fig. 6C). Fibronectin staining within Vhl-/- tumors was greatly reduced in this cell type (Fig, 6D), consistent with previously reported data for RCCs and Vhl-/- embryos (Ohh et al. 1998; Hoffman et al. 2001). Histological staining for an endothelial cell-specific antigen (CD34) to demarcate tumor vasculature indicated an increase in microvessel density of Vhl-/- tumors (47.5 microvessels/mm2 ± 5.0) when compared to Vhl+/- tumors (23.1 microvessels/mm2± 3.1) (Figures 6E and F). This statistically significant difference correlated with increased VEGF secretion by Vhl-/- cells in vitro and is consistent with data reported for RCC-derived tumors (Kondo et al. 2002), (p=0.0256 by Student t-test) (Fig 6G). When compared to controls, the phenotype of Vhl-/- teratocarcinomas paralleled that of VHL-/-RCCs in terms of fibronectin deposition and microvessel density. However pVHL-deficient teratocarcinomas are smaller than controls, emphasizing a discontinuity between angiogenesis and tumor proliferation.
Vhl-/- tumors are deficient in proliferation but do not exhibit an increase in apoptosis
A possible mechanism for the reduction in tumor volume and mass could be a decrease in cell growth or an increase in cell death. Although undifferentiated Vhl-/- cells do not exhibit these defects in vitro (data not shown), growth in an in vivo tumor microenvironment might reveal such a phenotype. Teratocarcinoma sections were therefore assessed for expression of cell cycle markers. Immunostaining for the proliferation marker Ki-67 (present in the nuclei of all cells not in GO) revealed highly proliferating regions in Vhl+/- tumors, most notably in ectoderm-derived tissue (Fig. 7A). Examination of similar tissue types within Vhl-/- tumors also revealed Ki-67 positive staining, but at lower levels (Fig. 7B). Morphometric analysis of these regions revealed that 41.5% of cells within Vhl-/- tumors were Ki-67+ as compared to 68.1% in Vhl+/- tumors and 60.2% in the genetically rescued tumors Vhl-/-.1 + HA-VHL (Fig 7C), a statistically significant difference (p=0.0028 by Student t-test). This reduction in proliferation rate, although modest, may account for the slower growth rate of Vhl-/- tumors over the course of 21 days.
Figure 7. Vhl-/- tumors exhibited a decrease in proliferation but no increase in apoptosis.
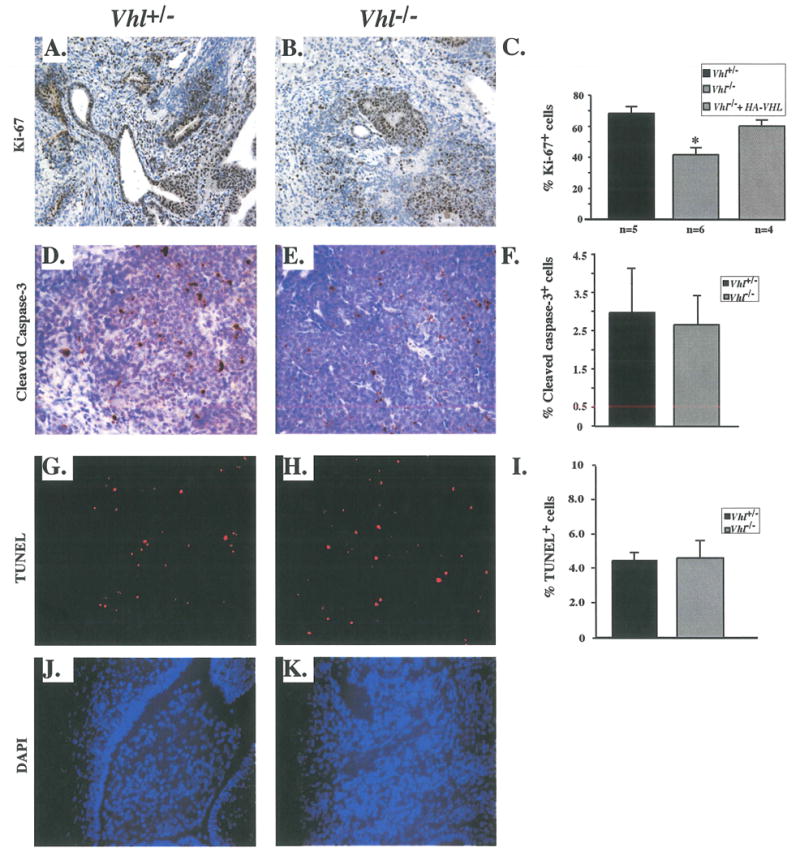
A: Proliferation marker Ki-67 indicated highly proliferating regions of Vhl+/- tumors. B: Expression of Ki-67 in Vhl-/- tumors was reduced compared to morphologically similar Vhl+/- regions. C: Quantitation of Ki-67+ cells in Vhl+/-, Vhl-/- and Vhl-/- + HA-VHL tumors. Vhl-/- tumors were less proliferative than controls. Student t-test revealed statistically significant differences between tumors types, p< 0.003. D& E: Expression of apoptotic marker cleaved caspase-3 in non-necrotic regions of Vhl+/- tumors. Cleaved caspase-3 staining was not increased in Vhl-/- tumors. F: Quantitation of cleaved caspase-3+ cells in Vhl+/- and Vhl-/- tumors. No significant differences were observed between genotypes. G & H: TUNEL staining demarcated apoptotic cells in non-necrotic regions of Vhl+/- and Vhl-/- tumors. I: Quantitation of TUNEL+ cells in Vhl+/- and Vhl-/- tumors. No significant differences were apparent between genotypes. N values are as indicated and bars represent standard error. Magnifications 200X (A-H).
A concomitant increase in the rate of apoptosis could also cause the reduced mass of Vhl-/- teratocarcinomas. To assess this possibility, sections were stained for the apoptotic marker cleaved caspase-3. Although Vhl-/- tumors were significantly smaller than Vhl+/- tumors, there was no corresponding increase in apoptosis outside of necrotic regions (Figures 7D and E). Morphometric analysis did not reveal any differences between the number of cleaved caspase-3+ cells in Vhl-/- and Vhl+/- sections (2.56% and 2.97% respectively) (Fig. 7F). In addition, apoptosis assessed by TUNEL staining (Figures 7G and H), did not reveal any quantitative differences between Vhl-/- and Vhl+/-tumors, (4.67% and 4.47% respectively), when normalized to DAPI stained nuclei (Figures 7I-K). Although the percentage of TUNEL+ cells was greater than that of cleaved caspase-3+ cells, both apoptosis detection methods revealed no significant differences between the tumor types. We conclude that although loss of Vhl is sufficient to dysregulate HIF, this alone does not promote teratocarcinoma growth in a subcutaneous tumor model. To the contrary, loss of Vhl seems to impair the growth of primary cells in vivo.
Discussion
Loss of the tumor suppressor VHL is correlated with the development of highly vascularized tumors within the kidney, central nervous system, and retina (Maher and Kaelin 1997). Insight into VHL's role as a tumor suppressor can be found in its ability to bind and target HIF-α subunits for ubiquitin-mediated degradation (Maxwell et al. 1999; Cockman et al. 2000; Tanimoto et al. 2000). We have shown that whereas loss of Vhl is sufficient to completely dysregulate HIF, it does not enhance the growth of subcutaneous tumors derived from primary cells. In contrast, VHL-/- RCC cells produce rapidly growing xenografts in nude mice, the growth of which is suppressed by reintroduction of functional pVHL. The combined effects of pVHL deficiency and additional mutations within the RCC cells may explain this apparent discrepancy.
The activation of HIF is thought to be an O2-regulated, multi-step pathway requiring α-subunit stabilization, nuclear translocation and recruitment of coactivators (Jiang et al. 1997; Pugh et al. 1997; Kallio et al. 1999; Tanimoto et al. 2000; Lando et al. 2002). Recent evidence also suggests that full activation of HIF is not only dependent upon inhibition of HIF-α prolyl hydroxylation but also asparagyl hydroxylation within the C-terminal activation domain (CAD) (Lando et al. 2002; Sang et al. 2002). In our studies, normoxic Vhl-/- ES cells displayed constitutive HIF-α stability and HIF activity, neither of which were substantially induced by hypoxia. The nuclear accumulation of stabilized HIF-1α seen in Vhl-/- cells is consistent with a recent report suggesting that HIF-1α nuclear export is pVHL-dependent (Groulx and Lee 2002). Constitutively stabilized HIF-1α in Vhl-/- cells maintained DNA binding activity and maximally stimulated the expression of downstream target genes. Proper HIF regulation was restored by genetic rescue with HA-VHL. Hence, loss of Vhl leads to the full activation of HIF in ES cells, even in the absence of additional hypoxic signaling.
Reintroduction of wild-type VHL suppresses the growth of pVHL deficient RCC tumors in nude mice (Gnarra et al. 1996; Iliopoulos et al. 1996). In contrast, expression of pVHL containing β-domain mutations that disrupt pVHL/HIF-α interaction do not suppress tumor formation, highlighting the importance of HIF regulation for the tumor suppressor function of VHL (Bonicalzi et al. 2001). pVHL-rescued RCC cells expressing constitutively stabilized HIF-1α produce smaller tumors than those derived from the parental RCC cell line, indicating that loss of VHL contributes to tumor growth through HIF-1α-independent mechanisms (Maranchie et al. 2002). Interestingly, HlF-2α is more frequently upregulated than HIF-1α in renal clear cell carcinomas (Turner et al. 2002) and has been implicated as a critical target for VHL-mediated tumor suppression in renal clear cell carcinomas. Our results suggest that constitutive activation of HIF transcriptional activity in Vhl-/- ES cell-derived teratocarcinomas promotes tumor angiogenesis, but is itself insufficient to increase tumor growth. This may reflect the fact that our targeted Vhl-/- ES cells differ fundamentally from pVHL-deficient RCC cell lines in at least one important way; specifically, they have not undergone selection in vivo for the ability to overcome proliferation checkpoints and apoptotic controls known to suppress tumorigenesis. The chromosomal abnormalities observed in RCC cell lines, (Gunawan et al. 2001), are consistent with the idea that genetic instability underlies the multiple genetic hits that facilitate tumor formation (Hanahan and Weinberg 2000). In contrast, the use of Vhl-/- ES cells permits assessment of pVHL function in an intact genetic background. Taken together, these results suggest that the tumorigenic phenotype of RCC cell lines requires not only pVHL's HIF-dependent and HIF-independent functions, but also other oncogenic mutations.
The precise role of HIF in tumor development is controversial due to the conflicting results of several tumor models. While one group found that genetic disruption of the Hif-1α locus results in the accelerated growth of poorly vascularized Hif-1α-/- teratocarcinomas (Carmeliet et al. 1998), others have shown that the absence of HIF activity in teratocarcinomas and fibrosarcomas impairs tumor growth, but not tumor angiogenesis (Ryan et al. 1998; Ryan et al. 2000; Hopfl et al. 2002). Additional support for HIF as a positive regulator of tumor growth has been obtained from tumor models using non-primary cells. In three different tumor cell lines, loss or disruption of HIF activity also inhibits expression of HIF target genes, tumor vasculature and growth (Maxwell et al. 1997; Kung et al. 2000; Hopfl et al. 2002). Disrupted binding of HIF to coactivators p300 and CBP can also attenuate tumor growth (Kung et al. 2000). Therefore, the full oncogenic consequences of HIF dysregulation are still unresolved and will probably depend considerably on genetic background.
The meager growth of Vhl-/- teratocarcinomas, a rather unexpected phenotype considering VHL's role as a tumor suppressor, did not result from increased apoptosis in Vhl-/- ES cell-derived tumors. This point was of particular interest, as the proapoptotic gene Bnip-3 has recently been described as a novel HIF transcriptional target (Bruick 2000; Sowter et al. 2001). In fact, Bnip-3 expression has been shown to be stimulated in hypoxic regions of human tumors (Sowter et al. 2001). Although the mRNA transcript of Bnip-3 was highly expressed in both normoxic and hypoxic Vhl-/- cell (Fig 4A), this did not equate with an increase in cell death or a growth disadvantage in vitro. In fact, doubling Lime and S phase composition as measured by propidium iodide incorporation of Vhl-/- cells, did not differ significantly from Vhl+/+ or Vhl+/- cells (data not shown). Furthermore, cleaved caspase-3 and TUNEL staining did not reveal an increase in apoptosis in Vhl-/- teratocarcinomas (Figures 7D-I). These results suggest that the expression of Bnip-3 in undifferentiated and differentiated ES cells does not induce apoptosis.
Vhl's effects on early development and tumor growth are contrary to those found for other tumor suppressors such as PTEN. Loss of PTEN also results in embryonic lethality, but still enhances the growth of tumors derived from primary ES cells and embryonic fibroblasts (Di Cristofano et al. 1998; Stiles et al. 2002), The aberrant differentiation of Pten-/- cells, although detrimental to early development, is actually beneficial for tumor proliferation. In contrast, loss of VHL may only be able to induce tumor growth in a background of genetic changes. Cytogenetic abnormalities have been observed in RCC cells, and a minority of RCCs and RCC-derived cell lines have been shown to harbor p53 mutations (Reiter et al. 1993). Amplifications of the oncogenes c-myc and K-ras may also play a role in the generation of renal clear cell carcinomas (Kozma et al. 1997).
It is important to note that our teratocarcinoma model does not recreate the selective pressures that generate pVHL-deficient human cancers. In many ways, the development of heterogeneous tissues within teratocarcinomas reflects the differentiation of ES cells in early embryogenesis. Loss of Vhl in early development results in growth retarded embryos, which die between days E10.5-12.5 (Gnarra et al. 1997). Vhl-/-teratocarcinomas, although less proliferative, appear to contain more primitive tissue types when compared to controls (data not shown). The decreased growth and differentiation of Vhl-/- teratocarcinomas suggests that pVHL may be required for the proliferation and/or initial differentiation of primary cells. For example, recent studies from our lab indicate that Vhl-/- ES cells generate reduced numbers of hematopoietic progenitors in in vitro assays (F.A.M. and M.C.S., unpublished). In conclusion, our data suggest that although inactivation of VHL's tumor suppressor functions can contribute to the malignancy of renal cancer, disruption of these same pathways in pluripotent cells appears to be detrimental to their growth and/or differentiation.
Experimental Procedures
Generation and Genotyping of Vhl+/- ES cells
5.0 × 104 Vhl+/- ES cells were plated on 10cm gelatin-coated tissue culture dishes in media containing 4.0mg/ml of G418, to promote loss of the second wild-type Vhl allele. Resistant clones were isolated and expanded after 6-7 days of selection. Genomic DNA extracted from cultured ES cells was digested with HindIII for Southern blot analysis using a probe generated from an AccI/XbaI fragment from the Vhl 3′ UTR. PCR was also used to genotype ES clones. Oligonucleotide primers included those specific for wild-type Vhl exon 3, 5′-ACT GAA AAC GTC TTC CTC CCT CGG G(reverse) and wild-type exon 1, 5′-GCG GAA GGA CAT ACA GCG ACT GAG CC-3′(forward) and internal neo sequence 5′-TGA CTA GGG GAG GAG TAG AAG GTG GCG-3′ (reverse).
Generation of “rescued” ES cell lines
Wild-type human cDNA encoding pVHL with an N-terminal hemagglutin epitope tag (HA-VHL) was obtained as a generous gift from Dr. W. Kaelin. 2x107 Vhl-/-.1 ES cells were elcctroporated with 20μg pf PvuII linearized expression construct Station II containing HA-VHL with an SV-40 polyadenylation sequence at the C-terminus. Resistant clones were screened by Southern blot using a 900bp probe generated from a HindIII-EcoRI fragment from HA-tagged VHL cDNA. Clones with evidence of an integration event were further analyzed by Western blot.
Western Blot Analysis
For all Western blot and EMSA analyses, 6-7 × 106 cells were plated on 10 cm gelatin coated tissue culture dishes such that the density of the cells at the time of lysis was ∼60-70% confluent. Hypoxia, defined as 1.5% O2, was generated using an In Vivo2 hypoxic workstation (Ruskinn Technologies, Leeds, UK) for Western and EMSA analyses, or an IG750 variable O2 tissue culture incubator (Jouan Inc.) for reporter and Northern analyses.
Whole cell protein lysates were prepared using WCE buffer: 150mM NaCl, 50mM Tris, pH 7.4, 5mM EDTA, 0.1% SDS, and Complete Protease Inhibitor (Roche Molecular Biochemicals). Nuclear and cytoplasmic fractions of protein extracts were prepared using a modified Dignam protocol (Maltepe et al. 2000) with Buffer A further modified to contain 0.1% NP-40 and Buffer C containing 300mM NaCl. For hypoxic extracts, cells were manipulated inside a hypoxic chamber using phosphate-buffered saline and Buffer A that had been equilibrated to the hypoxic environment. Extracts were electrophoresed, transferred, and immunoblotted according to standard protocols using 5% non-fat dry milk (Carnation) in Tris-buffered saline/Tween 20, as a blocking agent. Blots were stained with Ponceau S to ensure equal loading. Antibodies used include: α-mouse pVHL (Santa Cruz), α- human pVHL (Pharmingen), α- mouse HIF-1α and HIF-2α (Novus), α-mouse Rad-50 (Transduction Laboratories) and α-mouse β-Tubulin (InnoGenex). Horseradish peroxidase-conjugated α-rabbit and α-mouse secondary antibodies were purchased from Cell Signaling Technologies and used at dilutions of 1:2000. ECL reagents were purchased from Amersham Biosciences. Blots were stripped in 61.5mM Tris (pH 6.8), 2% SDS and 100mM β-mercaptoethanol at 55°C for 1 h before being blocked and re-probed.
Electrophoretic mobility shift assay (EMSA)
EMSA analyses have been previously described (Maltepe et al. 2000). In brief, 5 μg of nuclear extracts were incubated in binding buffer consisting of 10mM Tris-HCl (pH 7.5), 50mM NaCl, 50mM KC1, 1mM MgC12, 5mM dithiothreitol, 1mM EDTA, and 5% glycerol to which 0.05 mg/ml bovine serum albumin, 0.025μg/ml of CREB oligo, and 106 dpm of labeled probe and where indicated 0.5μ1 of polyclonal α-ARNT antiserum (Novus) were added. The 24-bp oligonucleotide probe was derived from the erythropoietin HRE, 5′-GCC CTA CGT GCT GTC CTC A3′.
Reporter Assays
The HRE-luciferase reporter constructs were previously described (Arsham et al. 2002). Transfection efficiency was assessed by cotransfection of a renilla luciferase gene under the control of a minimal tyrosine kinase promoter. Where indicated, cells were also cotransfected with plasmid encoding HA- tagged full-length human pVHL. For transient tansfections, 2x106 ES cells were plated on gelatin treated 6-well plates and allowed to recover overnight. Cells were transfected the next morning using Lipofectamine Plus reagent (Gibco BRL) according to manufacturers protocol. Cells were transfected for 4hrs and one half of each sample was exposed to hypoxia for 18hrs. Proteins were extracted from samples using reporter lysis buffer and dual luciferase assays were performed according to manufacturer's guidelines (Promega).
Northern Analysis
For Northern blots, 2-3 × 106 cells/ 10cm tissue culture dish were plated and allowed to recover overnight. Where indicated, cells were incubated in hypoxia for 18hrs. All cells were lysed in Trizol (InVitrogen) according to manufacturer's instructions in ambient air. 20μg of total RNA were electrophoresed in 1.5% denaturing (formaldehyde) agarose gels and transferred to Hybond N+ membranes (Amersham). Murine Vegf, Pgk, Alda, and Glut-1 probes have been previously described (Maltepe et al. 1997). Murine Bnip3 probe was generated by RT-PCR from cDNA synthesized from murine FL5.12 cell RNA. A 738bp fragment was amplified between bp11 and 749 using primers 5′TGC CCC TGC TAC CTC TCG (forward) and 5′ CAT AGT GCA AAC ACC CAA GG (reverse). PCR products were sequenced and confirmed to be identical to GenBank sequence NM_009760 for murine Bnip3.
VEGF ELISA
VEGF quantitation was performed using the Quantikine M Murine Immunoassay kit, (R&D Systems) according to manufacturers protocol. 7.5x105 cells were seeded on gelatin coated 12-well plates and incubated in hypoxia for 18hrs where indicated. Conditioned medium was incubated with a mouse specific VEGF polyclonal antibody bound to a microtiter plate. After several washes, a second enzyme-linked polyclonal antibody specific for mouse VEGF was added. Sample values were obtained according to manufacturers protocol. Recorded values were normalized for cell number.
Glycolytic Rate Assay
Glycolysis was assayed as the rate of conversion of 3H-Glucose to 3H20 as previously described (Liang et al. 1997). 1x106 cells were washed once in PBS and media was replaced with 0.5ml of Krebs buffer and incubated for 30min at 37°C. Krebs buffer was then replaced with 0.5 ml of Krebs buffer containing 10mM glucose and spiked with 10μCi of 5-3H-glucose. Following incubation for 1h at 37°C, triplicate 50μl aliquots were transferred to uncapped PCR tubes containing 50μ1 of 0.2N HC1, and these tubes were transferred to scintillation vials containing 0.5ml of H2O such that the water in the vial and the contents of the PCR tubes were not allowed to mix. The vials were sealed, and diffusion was allowed to occur for 48hrs. The amounts of diffused and undiffused 3H were determined by scintillation counting. Appropriate 3H-glucose-only and 3H2O-only controls were included, enabling the calculation of 3H2O in each sample and thus the rate of glycolysis.
Lactate Secretion Assay
Lactate concentrations were indirectly determined by a colorimetric assay, which converts the lactic acid present in the media into H2O2, resulting in the oxidative condensation of a chromogen precursor (Sigma). Approximately 5x104 cells were plated on gelatin coated 24-well plates and allowed to recover overnight. Samples were collected and individual cell counts were obtained every 24hrs for two days thereafter
Mouse teratocarcinoma assay
5x106 cells were suspended in 100μl of Phosphate-Buffered Saline (Gibco) and injected subcutaneously into the dorsal area of 4-6 week old female NIH-III immunodeficient mice (Taconic). After 7 days tumors were measured every 2-3 days with calipers in the two greatest dimensions to calculate tumor volume. After 21 or 28 days, tumors were excised, photographed, weighed, frozen for protein assays and fixed in 4% paraformaldehyde.
Immunohistochemistry
Tumor samples were fixed in 4% paraformaldehyde and paraffin embedded by standard techniques. 6μm sections of each sample were incubated overnight with antibodies generated against fibronectin (BD Scientific), cleaved caspase-3 (Calbiochem), Ki-67 (Novacastra), and CD-34 (BD Scientific). A TUNEL assay, In Situ Cell Death Detection Kit, was utilized according to the manufacturers protocol (Roche). α-mouse and α-rat secondary antibodies were biotinylated and staining achieved by a strepavidin-biotin system conjugated with horseradish peroxidase (Vector Lab). Fibronectin, CD-34, and active caspase-3 photographs were taken with a Nikon N6006 35mm camera. Ki-67 photographs were taken with a Spot RT slider digital camera (Diagnostic Instruments Inc.) with a magnification of 0.76X. Morphometric analysis was performed on three to five randomly chosen sections of each tumor using the analytical program Image Pro (Phase 3 Imaging). The number of positive cells was calculated as the total area of positive staining divided by the area of an individual positive cell. The percentage positive cells were calculated as the number of positive cells divided by the total number of cells.
Significance.
Loss of the pVHL tumor suppressor protein is associated with tumors within the CNS, retina and kidney. pVHL targets the α-subunits of Hypoxia Inducible Factor (HIF) for ubiquitin-mediated degradation in an O2-regulated manner. The degree to which constitutive HIF stabilization contributes to VHL disease has been studied in tumor-derived cell lines that contain other genetic lesions. We generated primary Vhl-/- ES cells which display maximal HIF activity under normoxic conditions. These results contrast published reports indicating that full HIF activity requires multiple pVHL-independent, O2-regulated steps, including nuclear transport and transactivation. Surprisingly, Vhl-/- ES cells generate smaller teratocarcinomas than controls, suggesting a growth disadvantage in pVHL-deficient primary cells. Therefore, VHL's tumor suppressor activity may be manifested only in a background of other mutations.
Acknowledgments
We thank Min-min Lu, Q.C. Yu, Cynthia Clendenin, Clint Culpepper, Frank Winslow, Marian Harris, J Thompson, and Nathalie Innocent for reagents and technical assistance. This research was supported by National Institute of Health Grants HL63310 (M.C.S.) and 1F31HD (F.A.M.) and the Abramson Family Cancer Research Institute. M.C.S. is an investigator at the Howard Hughes Institute.
References
- Arany Z, Huang LE, et al. An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci U S A. 1996;93:12969–73. doi: 10.1073/pnas.93.23.12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsham AM, Plas DR, et al. Phosphatidylinositol 3-kinase/Akt signaling is neither required for hypoxic stabilization of HIF-1 alpha nor sufficient for HIF-1-dependent target gene transcription. J Biol Chem. 2002;277:15162–70. doi: 10.1074/jbc.M111162200. [DOI] [PubMed] [Google Scholar]
- Bindra RS, Vasselli JR, et al. VHL-mediated hypoxia regulation of cyclin D1 in renal carcinoma cells. Cancer Res. 2002;62:3014–9. [PubMed] [Google Scholar]
- Bonicalzi ME, Groulx I. Role of exon 2-encoded beta -domain of the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2001;276:1407–16. doi: 10.1074/jbc.M008295200. [DOI] [PubMed] [Google Scholar]
- Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci U S A. 2000;97:9082–7. doi: 10.1073/pnas.97.16.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruick RK, McKnight SL. A Conserved Family of Prolyl-4-Hydroxylases That Modify HIF. Science. 2001;11:11. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Dor Y, et al. Role of HEF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–90. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- Carrero P, Okamoto K, et al. Redox-regulated recruitment of the transcriptional coactivators CREB-binding protein and SRC-1 to hypoxia-inducible factor 1alpha. Mol Cell Biol. 2000;20:402–15. doi: 10.1128/mcb.20.1.402-415.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockman ME, Masson N, et al. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2000;275:25733–41. doi: 10.1074/jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, et al. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–55. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- Ebert BL, Bunn HF. Regulation of transcription by hypoxia requires a multiprotein complex that includes hypoxia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Mol Cell Biol. 1998;18:4089–96. doi: 10.1128/mcb.18.7.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein AC, Gleadle JM, et al. C. elegans EGL-9 and Mammalian Homologs Define a Family of Dioxygenases that Regulate HIF by Prolyl Hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Foster K, Prowse A, et al. Somatic mutations of the von Hippel-Lindau disease tumour suppressor gene in non-familial clear cell renal carcinoma. Hum Mol Genet. 1994;3:2169–73. doi: 10.1093/hmg/3.12.2169. [DOI] [PubMed] [Google Scholar]
- Gnarra JR, Duan DR, et al. Molecular cloning of the von Hippel-Lindau tumor suppressor gene and its role in renal carcinoma. Biochim Biophys Acta. 1996;1242:201–10. doi: 10.1016/0304-419x(95)00012-5. [DOI] [PubMed] [Google Scholar]
- Gnarra JR, Tory K, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet. 1994;7:85–90. doi: 10.1038/ng0594-85. [DOI] [PubMed] [Google Scholar]
- Gnarra JR, Ward JM, et al. Defective placental vasculogenesis causes embryonic lethality in VHL- deficient mice. Proc Natl Acad Sci U S A. 1997;94:9102–7. doi: 10.1073/pnas.94.17.9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnarra JR, Zhou S, et al. Post-transcriptional regulation of vascular endothelial growth factor mRNA by the product of the VHL tumor suppressor gene. Proc Natl Acad Sci U S A. 1996;93:10589–94. doi: 10.1073/pnas.93.20.10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groulx I, Lee S. Oxygen-dependent ubiquitination and degradation of hypoxia-inducible factor requires nuclear-cytoplasmic trafficking of the von Hippel-Lindau tumor suppressor protein. Mol Cell Biol. 2002;22:5319–36. doi: 10.1128/MCB.22.15.5319-5336.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawan B, Huber W, et al. Prognostic impacts of cytogenetic findings in clear cell renal cell carcinoma: gain of 5q31-qter predicts a distinct clinical phenotype with favorable prognosis. Cancer Res. 2001;61:7731–8. [PubMed] [Google Scholar]
- Haase VH, Glickman JN, et al. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc Natl Acad Sci U S A. 2001;98:1583–8. doi: 10.1073/pnas.98.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hoffman MA, Ohh M, et al. von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet. 2001;10:1019–27. doi: 10.1093/hmg/10.10.1019. [DOI] [PubMed] [Google Scholar]
- Hopfl G, Wenger RH, et al. Rescue of hypoxia-inducible factor-1 alpha-deficient tumor growth by wild-type cells is independent of vascular endothelial growth factor. Cancer Res. 2002;62:2962–70. [PubMed] [Google Scholar]
- Ikeda E, Achen MG, et al. Hypoxia-induced transcriptional activation and increased mRNA stability of vascular endothelial growth factor in C6 glioma cells. J Biol Chem. 1995;270:19761–6. doi: 10.1074/jbc.270.34.19761. [DOI] [PubMed] [Google Scholar]
- Iliopoulos O, Kibel A, et al. Tumour suppression by the human von Hippel-Lindau gene product. Nat Med. 1995;1:822–6. doi: 10.1038/nm0895-822. [DOI] [PubMed] [Google Scholar]
- Iliopoulos O, Levy AP, et al. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci U S A. 1996;93:10595–9. doi: 10.1073/pnas.93.20.10595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan M, Kondo K, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for 02 sensing. Science. 2001;292:464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Jiang BH, Zheng JZ, et al. Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J Biol Chem. 1997;272:19253–60. doi: 10.1074/jbc.272.31.19253. [DOI] [PubMed] [Google Scholar]
- Kallio PJ, Wilson WJ, et al. Regulation of the hypoxia-inducible transcription factor 1alpha by the ubiquitin-proteasome pathway. J Biol Chem. 1999;274:6519–25. doi: 10.1074/jbc.274.10.6519. [DOI] [PubMed] [Google Scholar]
- Kamada M, Suzuki K, et al. von Hippel-Lindau protein promotes the assembly of actin and vinculin and inhibits cell motility. Cancer Res. 2001;61:4184–9. [PubMed] [Google Scholar]
- Kondo K, Klco J, et al. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002;1:237–46. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- Kozma L, Kiss I. Investigation of c-myc and K-ras amplification in renal clear cell adenocarcinoma. Cancer Lett. 1997;111:127–31. doi: 10.1016/s0304-3835(96)04527-2. [DOI] [PubMed] [Google Scholar]
- Krieg M, Haas R, et al. Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene. 2000;19:5435–43. doi: 10.1038/sj.onc.1203938. [DOI] [PubMed] [Google Scholar]
- Kung AL, Wang S, et al. Suppression of tumor growth through disruption of hypoxia-inducible transcription. Nat Med. 2000;6:1335–40. doi: 10.1038/82146. [DOI] [PubMed] [Google Scholar]
- Lando D, Peet DJ, et al. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295:858–61. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- Levy AP, Levy NS, et al. Hypoxia-inducible protein binding to vascular endothelial growth factor mRNA and its modulation by the von Hippel-Lindau protein. J Biol Chem. 1996;271:25492–7. doi: 10.1074/jbc.271.41.25492. [DOI] [PubMed] [Google Scholar]
- Levy AP, Levy NS, et al. Post-transcriptional regulation of vascular endothelial growth factor by hypoxia. J Biol Chem. 1996;271:2746–53. doi: 10.1074/jbc.271.5.2746. [DOI] [PubMed] [Google Scholar]
- Liang Y, Buettger C, et al. Chronic effect of fatty acids on insulin release is not through the alteration of glucose metabolism in a pancreatic beta-cell line (beta HC9) Diabetologia. 1997;40:1018–27. doi: 10.1007/s001250050783. [DOI] [PubMed] [Google Scholar]
- Lisztwan J, Imbert G, et al. The von Hippel-Lindau tumor suppressor protein is a component of an E3 ubiquitin-protein ligase activity. Genes Dev. 1999;13:1822–33. doi: 10.1101/gad.13.14.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonergan KM, Iliopoulos O, et al. Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol Cell Biol. 1998;18:732–41. doi: 10.1128/mcb.18.2.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher ER, Kaelin WG., Jr von Hippel-Lindau disease. Medicine (Baltimore) 1997;76:381–91. doi: 10.1097/00005792-199711000-00001. [DOI] [PubMed] [Google Scholar]
- Maltepe E, Keith B, et al. The role of ARNT2 in tumor angiogenesis and the neural response to hypoxia. Biochem Biophys Res Commun. 2000;273:231–8. doi: 10.1006/bbrc.2000.2928. [DOI] [PubMed] [Google Scholar]
- Maltepe E, Schmidt JV, et al. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386:403–7. doi: 10.1038/386403a0. [DOI] [PubMed] [Google Scholar]
- Mandriota SJ, Turner KJ, et al. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell. 2002;1:459–68. doi: 10.1016/s1535-6108(02)00071-5. [DOI] [PubMed] [Google Scholar]
- Maranchie JK, Vasselli JR, et al. The contribution of VHL substrate binding and HEF1-alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell. 2002;1:247–55. doi: 10.1016/s1535-6108(02)00044-2. [DOI] [PubMed] [Google Scholar]
- Maxwell PH, Dachs GU, et al. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci U S A. 1997;94:8104–9. doi: 10.1073/pnas.94.15.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- Mortensen RM, Conner DA, et al. Production of homozygous mutant ES cells with a single targeting construct. Mol Cell Biol. 1992;12:2391–5. doi: 10.1128/mcb.12.5.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohh M, Park CW, et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–7. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- Ohh M, Yauch RL, et al. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell. 1998;1:959–68. doi: 10.1016/s1097-2765(00)80096-9. [DOI] [PubMed] [Google Scholar]
- Pause A, Lee S, et al. The von Hippel-Lindau tumor suppressor gene is required for cell cycle exit upon serum withdrawal. Proc Natl Acad Sci U S A. 1998;95:993–8. doi: 10.1073/pnas.95.3.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pause A, Lee S, et al. The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc Natl Acad Sci U S A. 1997;94:2156–61. doi: 10.1073/pnas.94.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh CW, O'Rourke JF, et al. Activation of hypoxia-inducible factor-1; definition of regulatory domains within the alpha subunit. J Biol Chem. 1997;272:11205–14. doi: 10.1074/jbc.272.17.11205. [DOI] [PubMed] [Google Scholar]
- Reiter RE, Anglard P, et al. Chromosome 17p deletions and p53 mutations in renal cell carcinoma. Cancer Res. 1993;53:3092–7. [PubMed] [Google Scholar]
- Ryan HE, Lo J, et al. HIF-1a is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan HE, Poloni M, et al. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res. 2000;60:4010–5. [PubMed] [Google Scholar]
- Sang N, Fang J, et al. Carboxyl-terminal transactivation activity of hypoxia-inducible factor 1 alpha is governed by a von Hippel-Lindau protein-independent, hydroxylation-regulated association with p300/CBP. Mol Cell Biol. 2002;22:2984–92. doi: 10.1128/MCB.22.9.2984-2992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowter HM, Ratcliffe PJ, et al. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001;61:6669–73. [PubMed] [Google Scholar]
- Stiles B, Gilman V. Essential role of AKT-1/protein kinase B alpha in PTEN-controlled tumorigenesis. Mol Cell Biol. 2002;22:3842–51. doi: 10.1128/MCB.22.11.3842-3851.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimoto K, Makino Y, et al. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. Embo J. 2000;19:4298–309. doi: 10.1093/emboj/19.16.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner KJ, Moore JW, et al. Expression of hypoxia-inducible factors in human renal cancer: relationship to angiogenesis and to the von Hippel-Lindau gene mutation. Cancer Res. 2002;62:2957–61. [PubMed] [Google Scholar]