

Abstract
Forebrain γ-aminobutyric acid (GABA) interneurons have crucial roles in high-order brain function via modulating network activities and plasticity, and they are implicated in many psychiatric disorders. Availability of enriched functional human forebrain GABA interneurons, especially those from people affected by GABA interneuron deficit disease, will be instrumental to the investigation of disease pathogenesis and development of therapeutics. We describe a protocol for directed differentiation of forebrain GABA interneurons from human embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) in a chemically defined system. In this protocol, human PSCs are first induced to primitive neuroepithelial cells over 10 d, and then patterned to NKX2.1-expressing medial ganglionic eminence progenitors by simple treatment with sonic hedgehog or its agonist purmorphamine over the next 2 weeks. These progenitors generate a nearly pure population of forebrain GABA interneurons by the sixth week. This simple and efficient protocol does not require transgenic modification or cell sorting, and it has been replicated with multiple human ESC and iPSC lines.
INTRODUCTION
GABA-secreting neurons form the major inhibitory system in the brain and spinal cord. There are numerous subtypes of GABA interneurons, especially in the human cerebral neocortex, depending on their developmental origins, distribution (localization), synaptic connections, coexpression of molecular and transmitter markers, intrinsic electrophysiological properties and function in adults1. GABA interneurons are synaptically integrated into neural networks during development, as well as in adult life, and they thus have crucial roles in modulating network activities and plasticity. Dysfunction of GABA interneurons in the cerebral cortex is involved in diverse developmental and psychiatric disorders, from Down’s syndrome to autism and schizophrenia1,2.
Developmentally, GABA interneurons in the forebrain originate mostly from the medial ganglionic eminence (MGE) and, to a lesser degree, from the ventral lateral ganglionic eminence (vLGE) and the anterior dorsal ganglionic eminence3,4. These GABA interneuron progenitors migrate throughout the forebrain, especially to the neocortex, by following tangential or radial pathways, and they differentiate into post-mitotic neurons and make a connection with local glutamatergic neurons5,6. In primates, some GABA interneurons in the cortex are thought to originate from the cortical plate7,8. Given their origin in the MGE, the vast majority of forebrain GABA interneuron progenitors express the MGE transcription factor NKX2.1 (ref. 9) in addition to the telencephalic transcription factor FOXG1 (refs. 2,10), and they can be distinguished from other types of GABA neurons in the CNS, including the striatal GABA projection neurons, which originate in the LGE11-13. The MGE progenitors have the potential to differentiate into GABA interneurons and cholinergic neurons14, and the transcription factor LHX6 appears to bias the progenitors toward GABA interneurons15,16, which allows segregation of GABA interneuron progenitors from cholinergic neuron progenitors. Forebrain GABA interneurons can be divided into many subgroups on the basis of molecular markers and their expression of neuropeptides or calcium-binding proteins, including somatostatin (SST), parvalbumin (PV), calretinin (CR), calbindin (CB) and neuropeptide Y. The MGE progenitors give rise mostly to SST and PV interneurons1,4. GABA interneuron subtypes in humans mature slowly, with SST, CR and CB GABA neurons appearing earlier, followed by PV neurons1,17,18.
Functionally, GABA interneurons in the forebrain, especially in the cerebral cortex, remain the most complex neuronal type because of their different subtypes, migration patterns and functional diversities. Given that it is now possible to produce iPSCs from patients with neurological conditions, it is desirable to produce high-purity human GABA interneurons in a large scale from human ESCs or iPSCs that represent forebrain or cortical GABA interneurons to further study.
The protocol we describe here is based on our primary research paper19. Two further groups have also reported the generation of robust forebrain GABA interneurons from hPSCs17,18. These methods require the combination of multiple small molecules and/or growth factors (SB-431542 + LSB + XAV939 + SHH + purmorphamine or SB-431542 + BMPRIA-Fc + DKK1 + purmorphamine + Y27632), which increases the experimental cost and variability. In our protocol, we generate GABA neurons by simply patterning the neuroepithelia to a near-pure population of NKX2.1-expressing MGE progenitors with a single molecule: sonic hedgehog (SHH) or a smoothened activator purmorphamine (Pur)20,21. SHH is derived from the ventral part of the developing embryo, such as the notochord, and from the floor plate of the neural tube. The SHH protein diffuses dorsally, thereby affecting the morphogenesis of the developing neural tissues. MGE develops from the ventral-most part of the forebrain. Hence, MGE cells receive a high concentration of SHH. As is the case in development in vivo, we found that a high concentration of SHH is necessary to induce the expression of NKX2.1 (ref. 19). To reduce the cost of recombinant SHH, we have adapted the protocol to use its agonist Pur (Table 1). Under a commonly used, chemically defined medium (without the presence of nerve growth factor (NGF)), these MGE progenitors differentiate into GABA neurons with a purity of over 90%. The reason for omitting NGF is that MGE progenitors can generate cholinergic neurons besides GABA interneurons19, and the survival of cholinergic neurons is dependent on NGF22. Thus, the chemically defined culture system without the presence of NGF favors GABA neuron generation. This protocol does not require transgenic modification or cell sorting. It is hence useful for dissecting the molecular mechanisms underlying human cortical GABA interneuron development. It can also be applied to the generation of nearly pure cortical GABA interneurons from human iPSCs with specific pathological traits, thus facilitating the development of tools for disease modeling and drug discovery.
TABLE 1.
Comparison of morphogens.
| Pur (StemGent) | C24 (1845-SH-025/CF) | C25 (464-SH-025/CF) | |
|---|---|---|---|
| Patterning concentration | 1.5 μM | 1,000 ng ml−1 | 300 ng ml−1 |
| Maintaining cell proliferation | 0.5 μM | 50 ng ml−1 | 20 ng ml−1 |
| Cost for patterning (US dollars ($) per 10 ml of mediuma) | 0.149 | 119.6 | 38.28 |
Only calculate ventralizing morphogens.
Experimental design
Our GABA interneuron differentiation procedure follows three major developmental stages (Fig. 1). First, human pluripotent stem cells (hPSCs) are induced into primitive neuroepithelia or neural stem cells over the first 10 d (Steps 1–18; Fig. 2). Second, the primitive neuroepithelia is patterned into ventral forebrain progenitors with the MGE feature (Steps 19–26; Fig. 3). Finally, the MGE-like progenitors are differentiated into GABA interneurons (Steps 27–36; Fig. 3). The precursor cells at each of the stages are identified by characteristic morphologies, expression of transcription factors and other markers, as well as functional traits as revealed by electrophysiology (Fig. 4). In long-term cultures, subtypes of GABA interneurons may be distinguished on the basis of their expression of transmitters and other molecular markers (Fig. 5).
Figure 1.
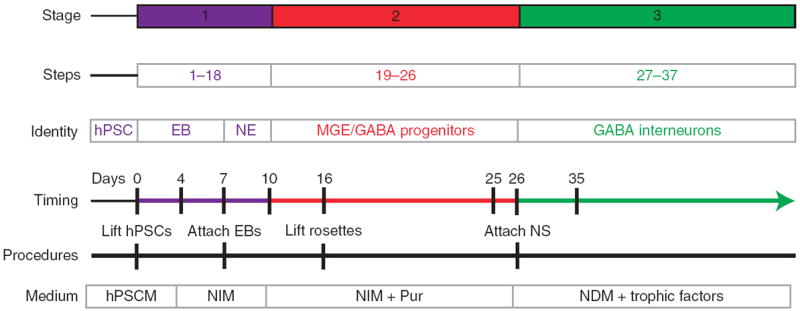
Timeline of forebrain GABA interneuron generation. Cells are differentiated under a chemically defined system. Differentiation of human PSC to forebrain GABA interneurons involves 37 steps and three stages, including induction of neuroepithelial cells, patterning of MGE progenitors and differentiation to GABA neurons. Pur is the major factor for MGE progenitor patterning. NE, neuroepithelium; NS, neuroepithelial spheres.
Figure 2.
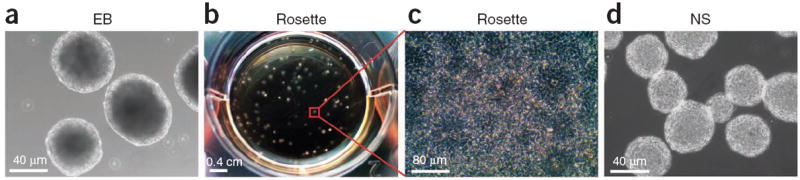
Step-by-step illustration of neuroepithelial differentiation. (a) Under a chemically defined system, hPSCs form EBs in suspension culture for the first 7 d. (b) The EBs are then plated on day 7 at the density as shown. (c) By day 10, each EB will develop into a colony that contains neuroepithelial cells in the form of rosettes. (d) At day 16, the rosette-containing colonies are detached and grown in suspension to form neuroepithelial spheres (NS).
Figure 3.
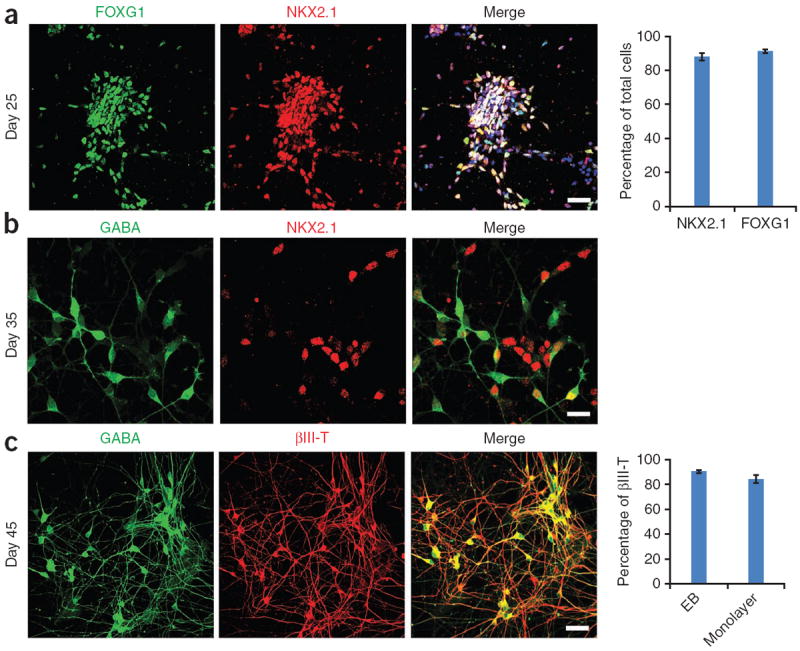
Characteristics of MGE progenitors and forebrain GABA interneurons. (a) At day 25, nearly all the cells express the forebrain marker FOXG1 (green), and the MGE marker NKX2.1 (red). Hoechst is shown in blue. Quantification is on the right; n = 5 different differentiation cultures. We counted 800–1,200 cells per replicate. Data are presented as means ± s.e.m. (b) At day 35, MGE cells (NKX2.1+; red) begin to express GABA (green), indicating that the GABA neurons are coming from the ventral forebrain area. (c) At day 45, nearly all the neurons express GABA (green). Quantification of the EB method and monolayer method is on the right; n = 5 different differentiation cultures. We counted 800–1,200 cells per replicate. Data are presented as means ± s.e.m. Scale bars, 50 μm. βIII-T, βIII-tubulin.
Figure 4.
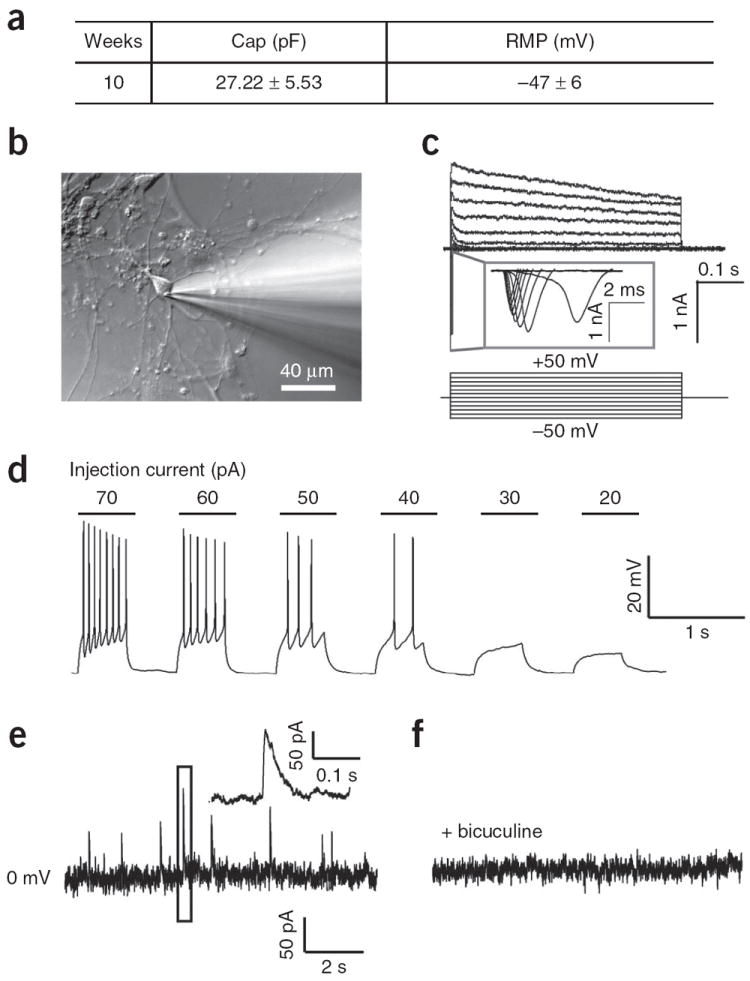
Human PSC-derived forebrain GABAergic interneurons are functional in vitro. (a) Electrophysiological characteristics of hPSC-derived GABAergic neurons, which were differentiated for 10 weeks in vitro. n = 12; data are means ± s.e.m. (b) Experimental setup showing the whole-cell recording on a neuron. (c) Inward and outward currents were triggered upon −50 to 50-mV voltage steps. (d) Action potentials were induced by injection of current as low as 40 pA into neurons. (e,f) Spontaneous synaptic currents were recorded (e), and the GABA receptor activity was blocked by application of bicuculine (f).
Figure 5.
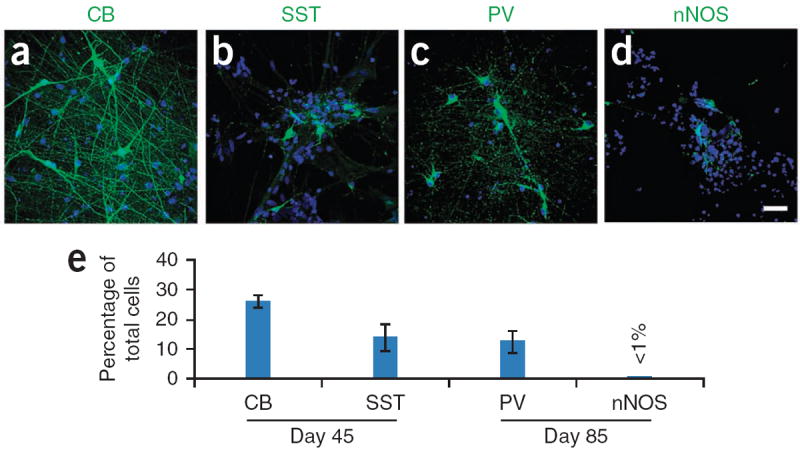
Maturation of GABA interneuron subtypes. (a) CB+ and (b) SST+ GABA neurons are seen from day 35, and their populations increase over time. (c) PV+ GABA neurons appear later. They are observed at around day 65, and their proportion increases over time. (d) nNOS+ neurons can be also observed after day 65. (e) Quantification of GABA interneuron subtypes. n = 5 different differentiation cultures. Nuclei are stained with Hoechst (blue). We counted 800–1,200 cells per culture. Data are presented as means ± s.e.m. Scale bar, 50 μm.
In the first stage, hPSCs maintained on mouse embryonic fibroblasts (MEFs) are differentiated into neuroepithelia under a chemically defined culture system for 10 d. The procedure below is essentially the same as that described in ref. 21, with minimal modifications. The neuroepithelia show columnar morphology, often organize into neural tube–like rosettes23 (Fig. 2b,c) and express the neuroepithelial markers PAX6, SOX1, N-cadherin, SOX2 and nestin, as well as the forebrain markers FOXG1 (refs. 24,25). Because of the characteristic rosette morphology in the adherent culture, non-neural colonies (without rosettes) can be removed easily. The neuroepithelial differentiation in the first stage may be enhanced by using inhibitors of bone morphogenetic proteins (BMPs) and transforming growth factor (TGF)-β (ref. 26) (Table 2); this leads to the culture being ready for patterning the neuroepithelia (stage 2) at an earlier time point (day 1).
TABLE 2.
Comparison of the chemically defined and monolayer dual-inhibitor protocols.
| Days | Chemically defined (EB method) | Monolayer dual inhibitors |
|---|---|---|
| 0–4 | Suspension; hESC medium | Adherent; NIM; Pur; dual inhibitors |
| 4–6 | Suspension; NIM | Adherent; NIM; Pur; dual inhibitors |
| 7–9 | Attach; NIM | Attach NIM; Pur |
| 10–12 | Attach; NIM; Pur | Suspension; NIM; Pur |
| 12–16 | Attach; NIM; Pur | Suspension; NIM; low Pura |
| 17–25 | Suspension; NIM; Pur | Suspension; NIM; low Pura |
| 26– | Suspension; NIM or Adherent; NDM medium with neuron factors |
Application of a high concentration of Pur for a long time period decreases cell proliferation. After patterning, low concentrations of Pur (0.5 μM) maintain the ventral identity during specification.
The second stage is the most crucial part of the protocol. In this stage, early or primitive neuroepithelial cells are patterned to MGE progenitors by a high concentration of SHH or Pur, which activates the hedgehog pathway. Here the timing and concentration of SHH/Pur are crucial. In general, early application of SHH (i.e., at days 8–14) is required for efficient ventral patterning19. Even earlier SHH application is necessary when BMP or TGF inhibitors are used to enhance neuroepithelial differentiation (Table 2). Because the MGE is in the ventral part of the fore-brain, a relatively high concentration of SHH or Pur is needed, as opposed to a medium concentration for striatal GABA neurons13,19. Another important issue in this stage is to maintain the forebrain identity of the specified neural progenitors, for which the commonly used neural inducers such as retinoic acid (RA)27 or mitogens such as FGF2 should be avoided, as they caudalize the neuroepithelia to various degrees28. By the end of the stage (day 25), more than 90% of the cells become NKX2.1+ and FOXG1+ MGE progenitors (Fig. 3a). These progenitors can then be expanded, frozen or differentiated to mature GABA neurons.
The final stage is the differentiation of the MGE-like progenitors to GABA interneurons. As described above, MGE progenitors possess the potential to give rise to cholinergic (projection) neurons and GABA interneurons19. For this reason, we built in two simple steps in the protocol to bias the differentiation toward GABA interneurons. One is to omit the NGF in our differentiation culture medium, which is essential for the survival of cholinergic neurons22,29. The other is to dissociate MGE progenitor clusters into single cells for differentiation, which is based on the consideration that the cholinergic projection neurons are generated from MGE progenitors earlier and are more vulnerable to cell dissociation–induced cell death. In addition, we apply inhibitors for cell division in order to synchronize the population of postmitotic neurons. After 2 weeks of differentiation, more than 90% of the neurons are GABA interneurons, as confirmed by immunostaining and electrophysiological analysis.
MATERIALS
REAGENTS
Antibodies; refer to Table 3 for details
2-Mercaptoethanol (β-mercaptoethanol, Sigma-Aldrich, cat. no. M3148) ! CAUTION Avoid direct contact or inhalation.
Ascorbic acid (Sigma-Aldrich, cat. no. A4403)
B27 supplement, without vitamin A, 50× (Gibco, cat. no. 12587-010)
Brain-derived neurotrophic factor (BDNF; PeproTech, cat. no. 450-02)
BSA (Sigma-Aldrich, cat. no. A-7906)
Cyclic AMP (cAMP; Sigma-Aldrich, cat. no. D-0260)
Compound E (EMD Biosciences, cat. no. 565790-500UG)
Dispase (Gibco, cat. no. 17105-041)
DMSO (Sigma-Aldrich, cat. no. D8418) ! CAUTION Avoid direct contact or inhalation.
DMEM/F-12, 1×, liquid, 1:1 (Gibco, cat. no. 11330)
Dulbecco’s PBS, 1×, liquid (DPBS; Gibco, cat. no. 14190)
FBS (Gibco, cat. no. 10437)
Fibroblast growth factor 2 (FGF2; R&D, cat. no. 233-FB)
Glial cell line–derived neurotrophic factor (GDNF; PeproTech, cat. no. 450-10)
Glass coverslips (Bellco, cat. no. 1943-10012)
GlutaMAX-I, 100×, liquid (Gibco, cat. no. 35050)
Heparin sodium salt from porcine intestinal mucosa (Sigma-Aldrich, cat. no. H3149)
hESC line H9 (Wisconsin International Stem Cell Bank, cat. no. WA09) ! CAUTION Manipulate human cells in a biosafety level 2 area.
Insulin-like growth factor (IGF1; PeproTech, cat. no. 100-11)
Induced pluripotent stem cell lines of interest. We have used IMR90-4 (Wisconsin International Stem Cell Bank, cat. no. iPS(IMR90)-4), DS1 (derived from a Down’s syndrome patient), DS2U (derived from the same Down’s syndrome patient with DS1, but with a normal chromosome 21) ! CAUTION Manipulate human cells in a biosafety level 2 area.
DMH1 (Tocris Bioscience, cat. no. 4126)
Knockout serum replacement (KOSR; Gibco, cat. no. 10828)
MEM non-essential amino acids solution, 10 mM, 100×, liquid (NEAA; Gibco, cat. no. 11140)
N-2 supplement, 100×, liquid (Gibco, cat. no. 17502-048)
Natural mouse laminin, 1 mg ml−1 (laminin; Invitrogen, cat. no. 23017-015)
Neurobasal medium, 1×, liquid (Gibco, cat. no. 21103)
Paraformaldehyde (PFA; Sigma-Aldrich, cat. no. P6148) ! CAUTION PFA is toxic; manipulate it in a fume hood. Avoid direct contact or inhalation.
Poly-l-ornithine hydrobromide (PLO; Sigma-Aldrich, cat. no. P3655)
Pur (StemGent, cat. no. 04-0009)
Recombinant human ciliary neurotrophic factor (CNTF; R&D Systems, cat. no. 257-NT)
Recombinant human sonic hedgehog (C24II) N-terminus, CF (SHH; R&D Systems, cat. no. 1845-SH-025/CF)
Recombinant human sonic hedgehog (C25II) N-terminus, CF (SHH; R&D Systems, cat. no. 464-SH-025/CF)
Sodium hydroxide (Sigma-Aldrich, cat. no. S8045)
SB-431542 (StemGent, cat. no. 04-0010)
Trypan blue solution (Sigma-Aldrich, cat. no. T8154)
TrypLE Express stable trypsin replacement enzyme without phenol (Invitrogen, cat. no. 12604-021)
Sodium phosphate, monobasic (Sigma-Aldrich, cat. no. S5011-500G)
Sodium phosphate, dibasic (Sigma-Aldrich, cat. no. S0876-1KG)
Hydrochloric acid (Fisher Scientific, cat. no. A144-500)
TABLE 3.
Antibodies.
| Antibody | Isotype | Dilution | Source (cat. no.) |
|---|---|---|---|
| CB | Rabbit IgG | 1:1,000 | Chemicon and Millipore (AB1778) |
| FOXG1 | Rabbit IgG | 1:100 | Abcam (ab18259) |
| GABA | Rabbit IgG | 1:10,000 | Sigma-Aldrich (A2052) |
| GABA | Mouse IgG | 1:100 | Sigma-Aldrich (A0310) |
| nNOS | Rabbit IgG | 1:200 | Chemicon and Millipore (07-517) |
| NKX2.1 | Mouse IgG | 1:500 | Chemicon and Millipore (MAB5460) |
| PV | Rabbit IgG | 1:2,000 | Abcam (ab11427) |
| PV | Mouse IgG | 1:1,000 | Sigma-Aldrich (P3088) |
| SST and receptor | Rat IgG | 1:500 | Chemicon and Millipore (MAB354) |
| βIII-tubulin | Rabbit IgG | 1:10,000 | Covance Research Products (PRB-435P) |
| βIII-tubulin | Mouse IgG | 1:10,000 | Sigma-Aldrich (T8660) |
EQUIPMENT
Plates, six well and 24 well (Nunc, cat. nos. 140675 and 142475)
Disposable borosilicate glass Pasteur pipettes, 9 inch (Fisher Scientific, cat. no. 13-678-20D)
Centrifuge tubes, 15 ml and 50 ml (RPI, cat. no. 163224)
CL2 benchtop centrifuge (Thermo Scientific, cat. no. 004260F)
Hausser Bright-Line counting chamber (Fisher Scientific, cat. no. 02-671-10)
HERAcell 150 CO2 incubator (Thermo Scientific, cat. no. 50077952)
Phase-contrast inverted microscope (Eclipse TS100; Nikon)
Polystyrene disposable serological pipettes, 5, 10 and 25 ml (Fisher Scientific, cat. nos. 13-678-11D, 13-678-11E and 13-678-11)
Steriflip, 50 ml (Millipore, cat. no. SCGP00525)
Steriflip, 500 ml (Millipore, cat. no. SCGPU05RE)
T25 flasks (TPP, cat. no. 90026)
T75 flasks (Nunc, cat. no. 157400)
REAGENT SETUP
Dispase (1 U ml−1)
Dissolve 50 units of dispase into 50 ml of DMEM/F-12 medium. Warm the solution at 37 °C for 10–15 min to dissolve the dispase completely, and then filter the solution with a 50-ml Steriflip. Store this reagent at 4 °C for up to 2 weeks.
Human PSC medium (hPSCM, 500 ml)
Combine 390 ml of sterile DMEM/F-12, 5 ml of NEAA, 5 ml of 100× GlutaMAX, 3.6 μl of 2-mercaptoethanol and 100 ml of KOSR in a sterile hood. Store the medium at 4 °C for up to 2 weeks. ! CAUTION 2-Mercaptoethanol is toxic and an irritant; avoid direct contact or inhalation.
Heparin (20 mg ml−1)
Dissolve 20 mg of heparin into 1 ml of DMEM/F-12 medium. Prepare aliquots and store them at −80 °C for up to 6 months.
DMH1 (10 mM)
Dissolve 10 mg of DMH1 into 1.3 ml of DMSO and 1.3 ml of ethanol; prepare 50-μl aliquots in sterilized dark tubes and store them at −80 °C for up to 6 months.
SB-431542 (10 mM)
Dissolve 5 mg into 0.65 ml of DMSO and 0.65 ml of ethanol; prepare 50-μl aliquots in sterilized dark tubes and store them at 80 °C for up to 6 months.
Compound E (2 mM)
Dissolve 500 μg of compound E into 255 μl of DMSO and 255 μl of ethanol; prepare 50-μl aliquots in sterilized dark tubes and store them at 80 °C for up to 6 months.
Neural induction medium (NIM, 500 ml)
Combine 490 ml of DMEM/F-12, 5 ml of NEAA, 5 ml of N-2 supplement and 50 μl of heparin in a sterile hood. Store the medium at 4 °C for up to 2 weeks.
Neuronal differentiation medium (NDM, 50 ml)
Combine 49 ml of Neurobasal medium, 0.5 ml of NEAA and 0.5 ml of N-2 supplement in a sterile hood. Store the medium at 4 °C for up to 2 weeks.
FGF2 (bFGF, 100 μg ml−1)
Dissolve 25 μg of FGF2 into 250 μl of sterile DPBS with 0.1% (wt/vol) human serum albumin or BSA. Divide the solution into aliquots and store them at −80 °C for up to 6 months.
BDNF, GDNF, IGFI (100 μg ml−1)
Dissolve 100 μg into 1 ml of sterile DPBS with 0.1% (wt/vol) human serum albumin or BSA. Divide the solution into aliquots and store them at −80 °C for up to 6 months.
Pur (10 mM)
Dissolve 5 mg of Pur in 480 μl of ethanol and 480 μl of DMSO; prepare 50-μl aliquots in sterilized dark tubes and store them at −80 °C for up to 6 months. ▲ CRITICAL The working concentration of Pur is narrowly defined. Prepare the stock solution as accurately as possible.
cAMP (1 mM)
Dissolve 4.914 mg of cAMP in 10 ml of sterilized water. Divide the solution into aliquots and store them at −80 °C for up to 6 months.
PFA, 4% (wt/vol)
In a fume hood, warm 50 ml of water up to 60 °C in a beaker. Add 4 g of PFA in 50 ml of 60 °C water while stirring. Add 3–5 drops of 1 N NaOH to clear the PFA solution. Dissolve 220 mg of sodium phosphate monobasic and 1.22 g of sodium phosphate dibasic into 50 ml of water. Combine the two solutions and cool the mixture to room temperature (20–25 °C). Adjust the pH to 7.3–7.4 with 1 N NaOH and HCl, filter it with a Steriflip and store the solution at 4 °C for up to 2 weeks. ! CAUTION PFA is toxic; manipulate it in a fume hood. Avoid direct contact or inhalation. ▲ CRITICAL PFA solution should be cold when it is applied for the fixation of cells.
PLO, 10× (1 mg ml−1)
Dissolve 100 mg of PLO into 100 ml of sterile water. Filter the solution with a Steriflip and store it at −20 °C for up to 6 months.
SHH, 500 μg ml−1
Dissolve 500 μg of SHH into 1 ml of sterile DPBS with 0.1% (wt/vol) human serum albumin or BSA for a 500-μg ml−1 stock. Divide the solution into aliquots and store them at −80 °C for up to 6 months.
Initial culture plate setup
Irradiate MEFs by λ-ray and plate them in a six-well plate at a density of 1.87 × 105 cells per well. ▲ CRITICAL MEFs vary from lot to lot, and each lot should be tested for its ability to support hPSC growth. Please visit http://www.wicell.org for specifics.
EQUIPMENT SETUP
PLO and laminin-coated coverslips
Dilute the PLO stock solution by 1:10 in sterile water (1 ml of PLO stock in 10 ml of water). Insert one acid-etched coverslip into each well of a 24-well plate in sterile conditions. Add 75 μl of diluted PLO onto each coverslip. Be careful not to allow the solution to flow out of the coverslip, and store the plate in a 37 °C incubator overnight. On the second day, aspirate all of the PLO solution and dry the plate for ~30 min. Wash the coverslips with sterile water three times. Leave the plate in a hood until it is completely dry, and then store it at −20 °C for up to 1 month. One hour before plating neurons, dilute 25 μl of laminin solution into 1.2 ml of NDM, and place 50 μl of the diluted laminin onto each of the PLO-pretreated coverslips. Avoid spreading the solution out of the coverslip onto the edge of the well. Incubate the coverslips at 37 °C for at least 1 h before plating the cells.
PROCEDURE
Induction of forebrain neuroepithelial cells from human PSCs ● TIMING 10 d (days 0–10)
-
On day 0, maintain human pluripotent cells on a six-well plate containing irradiated MEFs with 2.5 ml of hPSCM containing 4 ng ml−1 of FGF2. When this hPSC culture becomes ~80% confluent, remove the old medium and add 1 ml of prewarmed dispase (1 U ml−1) to each well. Incubate the culture at 37 °C, 5% CO2 for 2–5 min.
▲ CRITICAL STEP A uniform population of undifferentiated hPSCs is needed. Partially differentiated PSCs or heterogeneous PSC cultures will decrease the synchronized neuroepithelial differentiation.
When the edges of hPSC colonies begin to curl, aspirate off the dispase.
Rinse the hPSCs with 2 ml of DMEM/F12 medium gently; do not disturb the loosely attached hPSC colonies.
Add 2 ml of fresh hPSCM to each well.
-
Gently swirl the plate, and then blow off the colonies with a 5-ml pipette or a 1,000-μl pipette tip. Pipette the hPSC colonies to break the detached colonies to the size of 50–100 μm.
▲ CRITICAL STEP Breaking down the hPSC colonies into too-small fragments will result in a lower neural differentiation rate. Avoid pipetting hPSC colonies more than five times.
Collect hPSC colonies into a 15-ml tube, and centrifuge at 50g for 1 min. Alternatively, let the hPSC colonies settle by letting the tube rest for 3–5 min.
Aspirate off the supernatant without disturbing the cells.
Rinse the cells with hPSCM.
-
Resuspend the cells with 5 ml of hPSCM without FGF2.
▲ CRITICAL STEP Exogenous FGF2 is not necessary for promoting neural induction, as FGFs are produced by differentiating cells28. FGF2 also caudalizes cells and interferes with dorsal-ventral patterning20, thus decreasing the efficiency of MGE progenitor induction.
-
Transfer the cells into a T75 flask, and then add hPSCM up to 40 ml. Incubate the cells at 37 °C, 5% CO2 overnight.
▲ CRITICAL STEP The exact amount of hESCM to use ranges from 25 to 40 ml, depending on the number of embryoid bodies (EBs) in one T75 flask: usually one six-well plate of hPSCs requires 40 ml of hPSCM.
On day 1, hPSCs should have formed EBs (Fig. 2a). Lay the flask at a 45-degree angle to let cell aggregates (EBs) sink to one corner. Gently aspirate off two-thirds of the medium without disturbing the spheres. Add 25–30 ml of hPSCM without FGF2. Incubate the cells for a further 24 h.
On days 2 and 3, repeat Step 11, thus changing the medium every day.
On day 4, use a 10-ml pipette to pipette EBs gently to remove cell debris from the surface of the EBs, and then let them settle as in Step 11. Remove the hPSCM and feed the culture with the NIM. Continue to incubate the cells for a further 3 d, feeding the culture every other day.
On day 7, collect the EBs into a 50-ml tube, and then let them sink by letting the tube stand for 3–5 min.
Aspirate off the supernatant and add 0.5 ml of fresh NIM to prevent the EBs from drying.
Add 1.5 ml of NIM containing 5% (vol/vol) FBS into each well of a six-well plate.
-
By using a 200-μl pipette tip, transfer the EBs from the 50-ml tube to the six-well plate. Plate 35–50 EBs per well of the six-well plate (Fig. 2b). Disperse the EBs evenly. Avoid swirling the plate, as it will aggregate the EBs in the center (Fig. 2b). Incubate the culture at 37 °C, 5% CO2 for 5–8 h until the EBs attach, but do not incubate the culture for longer than 10 h.
▲ CRITICAL STEP Too many EBs in one well (too high a density) will decrease the neural induction efficiency; too few EBs in one well would waste the medium, plate and morphogens, as well as increase labor cost. See the density in Figure 2b.
▲ CRITICAL STEP The 5% (vol/vol) FBS is intended to promote attachment of EBs. As FBS inhibits neural differentiation, do not incubate the EBs with FBS for too long. Alternatively, simply use laminin-coated plates for EB attachment.
-
After the EBs have attached, gently remove the plate from the incubator. Remove the old medium and add fresh NIM. Be careful to add the medium from the edge of the well gently so as to avoid dislodging the attached EBs. Feed the culture with the same medium every other day until day 10, when neural rosettes should appear.
? TROUBLESHOOTING
Patterning neuroepithelia to NKX2.1-expressing MGE progenitors ● TIMING 15 d (days 11–25)
-
19
When neural rosettes become obvious at around day 10 during stage I differentiation (Fig. 2c), the primitive neuroepithelia has formed. The neuroepithelia is now ready to be patterned toward regional progenitors. First, remove non-rosette colonies: if a colony is dark, contains a cyst or contains only large flat cells30, mark the colony with an objective marker that is mounted in the microscope. Next, manually scrape the marked colonies using a pipette tip in the hood.
▲ CRITICAL STEP The presence of non-neural cells interferes with neuroepithelial induction and subsequent patterning.
-
20
Aspirate off the old medium, and then add 2–3 ml (it depends on the density of rosette colonies) of NIM containing 1,000 ng ml−1 of SHH (C24II, 1:500 dilution from the stock solution) or 300 ng ml−1 of SHH (C25II, 1:1,667 dilution from the stock solution/add 6 μl of 500 μg ml−1 stock solution into 10 ml NIM). Alternatively, 1.5 μM Pur (1:6,666 dilution of the stock solution/add 1.5 μl of 10 mM stock solution into 10 ml NIM) can replace SHH from this step onward. Incubate the culture at 37 °C, 5% CO2 for 48 h. We use a concentration of SHH or Pur selected on the basis of our previous studies13,19.
▲ CRITICAL STEP Pur should be diluted completely in NIM first before feeding the cells.
▲ CRITICAL STEP There are different types of SHH (C24II, C25II) and SHH agonists (Pur). We have titrated the concentrations for each (Table 1).
-
21
On days 12–16, feed the culture every other day with the same medium and procedures as in Step 20.
? TROUBLESHOOTING
-
22
On day 16, the differentiating colonies should contain neural tube–like rosettes formed by multiple layers of columnar epithelia surrounded by a ring of flat cells (Fig. 2c). Aspirate off the old medium and add 1.5 ml of fresh NIM to each well. Gently blow off the neural tube–like rosettes with a 1-ml pipette until most of the colonies are detached.
▲ CRITICAL STEP Avoid pipetting too hard and repeatedly. The rosettes attach to the well loosely, whereas the surrounding flat cells adhere more tightly. Pointing the pipette tip toward the colonies will effectively blow off the colonies without much force and leave the surrounding cells attached.
-
23
Gently transfer the lifted cell clusters into a 15-ml conical tube and centrifuge the cells for 1 min at 50g at room temperature. Alternatively, simply let the cell clusters settle down for 2–5 min without centrifugation.
-
24
Aspirate off the supernatant. Add 10 ml of NIM containing B27 (1:50 from stock solution) and SHH (C24, 1:500 dilution from the stock solution) or 300 ng ml−1 of SHH (C24, 1:1,666.67 dilution from the stock solution) or Pur (1.5 μM). Transfer cell clusters into a T75 flask (cells from a six-well plate may be transferred to one T75 flask). Add an additional 15 ml of the same medium to a total volume of 25 ml. Culture the cells at 37 °C, 5% CO2 for 48 h.
▲ CRITICAL STEP Two to three wells of rosettes can be collected into a T25 flask with 10 ml of medium; four to six wells of rosettes can be collected into a T75 flask with 25 ml of medium. (Around 50 neuroepithelial colonies are collected from each well.)
-
25
On day 18, neuroepithelial cells form neurospheres (Fig. 2d). Use a 10-ml pipette to gently pipette the spheres two or three times to remove dead cells on the surface of the spheres, and then lay the flask at a 45-degree angle to let the spheres sink to one corner. Gently aspirate off two-thirds of the medium without disturbing the spheres. Add 15 ml of the same fresh NIM at Step 24.
-
26
On days 20–25, change two-thirds of the medium every other day with the same medium used in Step 24. By day 20, the differentiating cells should become MGE progenitors. This can be confirmed by plating the cells onto coverslips and carrying out immunostaining for the MGE marker NKX2.1 (Box 1). After day 20, there should be ~90% NKX2.1+ and FOXG1+ cells13,19 (Fig. 3a).
? TROUBLESHOOTING
Box 1. Plating and staining cells.
Dissociate the neural progenitor spheres into single cells by TrypLE and plate them onto PLO/laminin-coated coverslips at a density of 10,000 cells per coverslip.
Incubate the cells overnight in NDM.
Fix the coverslips in 4% (wt/vol) PFA and carry out immunostaining for NKX2.1, FOXG1 and other markers (Table 3).
■ PAUSE POINT The MGE progenitors can be cryopreserved at days 25–30. Two days before cell freezing, triturate MGE progenitor clusters into small spheres (20–50 μm). Spin down the spheres and resuspend about 200 spheres in 1 ml of cryopreservation medium that contains 10% (vol/vol) DMSO, 30% (vol/vol) FBS and 60% (vol/vol) NIM. Place the cryopreservation vial in a cryopreservation container; condition the container at −80 °C overnight before transferring the vials to a liquid nitrogen tank.
Differentiation of MGE progenitors to GABA interneurons ● TIMING 20 d (days 26–45)
-
27
On day 26, collect MGE progenitor spheres into a 15-ml tube. Spin the tube down for 1 min at 50g at room temperature. Aspirate off the supernatant, add 1 ml of TrypLE to resuspend the spheres, and then incubate the mixture for 2–4 min at 37 °C until the spheres become loose.
-
28
Spin the tube at 50g for 1 min at room temperature. Aspirate off the supernatant, and then add 5 ml of NIM to wash the spheres.
-
29
Repeat Step 18 once.
-
30
Aspirate off the supernatant, add 1 ml of NDM containing 1 μM cAMP (1:1,000 dilution of stock solution), 10 ng ml−1 BDNF, 10 ng ml−1 GDNF, 10 ng ml−1 IGF1 (all are 1:10,000 dilution of stock solution), and then gently pipette the spheres three or four times. The addition of the neural trophic factors will support neuronal survival and maturation. See Figure 6 for information on pipetting MGE-like neurospheres.
▲ CRITICAL STEP Avoid pipetting the spheres too harshly.
-
31
Take 10 μl of the cell suspension, mix it with the Trypan Blue solution at 1:1 and count the cell numbers.
-
32
Plate 10,000 live trypan blue–negative cells in 50 μl of the NDM onto a PLO- and laminin-pretreated coverslip. Move the plate(s) gently to the incubator, and then culture the cells at 37 °C, 5% CO2 for 3–4 h. Do not disturb or touch the plate until the cells have attached.
▲ CRITICAL STEP Do not plate progenitors at a density higher than 10,000 cells per coverslip. High density inhibits progenitors from differentiation and maturation.
▲ CRITICAL STEP Alternatively, neural spheres can be directly plated on the PLO- and laminin-coated coverslips. In this case, the GABA interneuron differentiation rate (~60–70%) will be lower than the plated single cells (~90%) because of the persistence of progenitors in the cluster, but less cell death will occur in the culture.
-
33
MGE cells usually attach onto the coverslip after 3–4 h. Add 500 μl of NDM containing 1 μM cAMP (1:1,000 dilution of stock solution), 10 ng ml−1 BDNF, 10 ng ml−1 GDNF, 10 ng ml−1 IGF1 (all are 1:10,000 dilution of stock solution) and compound E (1:10,000 dilution from stock solution) to each well.
-
▲ CRITICAL STEP The fresh medium includes compound E to inhibit cell division. The application of compound E will synchronize cell differentiation31.
▲ CRITICAL STEP Add NDM to the well gently and slowly from the side in order to avoid blowing off the adherent cells.
-
34
Change half of the same medium used in Step 33 every 3 d.
-
35
Continue to incubate the cells, replacing half of the medium twice a week with the NDM containing 1 μM cAMP, 10 ng ml−1 BDNF, 10 ng ml−1 GDNF and 10 ng ml−1 IGF1, but without compound E.
▲ CRITICAL STEP Compound E is intended for stopping cell proliferation; it is needed only once (3 d). If there is obvious cell proliferation, compound E may be extended for 1 week.
-
36
By day 35, more than 90% of cells should be neurons, which are βIII-tubulin+. Half of the neurons should be GABA+, which coexpress the MGE marker NKX2.1 (Fig. 3b). At day 45, 90% of the neurons should be GABA+ (Fig. 3c). If desired, at the 10th week, perform electrophysiological recording using whole-cell voltage clamping to assess the functional maturation of GABA neurons (Fig. 4a–e).
Figure 6.
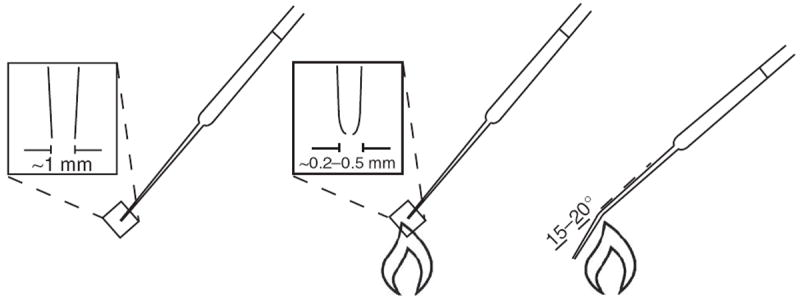
Pasteur pipette technique for triturating MGE-like neurospheres. Flame the end of a cotton-filtered Pasteur pipette to round the edges and narrow the opening to about 0.2–0.5 mm in diameter. Flame the narrow part of the shaft to introduce a 15–20° smooth curve. Rinse the pipette wall with the culture medium three times. Pipette neuroepithelial clusters using the treated Pasteur pipette by pulling the cells in and push them out one or two times. The narrow opening and bend in the pipette will help shear the clusters into smaller pieces of a roughly uniform size. Do not triturate more than three times.
Maturation of GABA interneuron subtypes ● TIMING 40 d (days 46–85)
-
37
Continue culturing cells as described in Steps 36 and 37 for a longer time period to discern GABA interneuron subtypes.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 4. See Figure 6 for an illustration of the Pasteur pipette technique for triturating MGE-like neurospheres.
TABLE 4.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 18 | EBs do not attach well | EBs are too big or too small; they contain too many dead cells. Reduce the time for treatment with dispase | Avoid overtrituration of EBs. Gently pipette the EBs during medium changes to remove dead cells from the surface |
| Low neural induction efficiency of hiPSCs | Variation in neural induction of iPSCs | Treat EBs with SB 431542 (10 μM) and DMH1 (10 μM) for the first 6 d | |
| 21 | The medium becomes orange or yellow too quickly | Too many cells in one flask | Cells should be fed every day or split into two flasks. An acidic environment may increase cell death |
| 26 | Large spheres | Cell proliferation | Triturate the spheres with a glass Pasteur pipette (Fig. 6) |
● TIMING
Steps 1–18, induction of forebrain neuroepithelial cells from human PSCs: 10 d
Steps 19–26, patterning neuroepithelia to NKX2.1-expressing MGE progenitors: 15 d
Steps 27–36, differentiation of MGE progenitors to high proportion of GABA interneurons: 20 d
Step 37, maturation of GABA interneuron subtypes: 40 d
Box 1, plating and staining cells: 1 h plus overnight (plating); 2 d (staining)
ANTICIPATED RESULTS
In the first stage, cells are grown as EBs and then adherent colonies (Fig. 2a–c). At the end of the first stage (Steps 1–18), usually over 90% of the colonies contain neural rosettes and at least 90% of the cells are PAX6-expressing neuroepithelial cells. As no inducers, inhibitors or morphogens are involved in this stage, it is ideal for dissecting the effects of exogenous factors on the earliest phase of neural differentiation. The weakness of this part of the protocol is that it does require undifferentiated hPSCs as a starting material as well as healthy and even-sized EBs (Fig. 2a), which can be difficult for a novice. One alternative is to use inhibitors of BMP and TGF, which will enhance neuroepithelial differentiation and also speed up the process26. The downside of the dual inhibitor protocol is its influence on multiple signaling pathways, thus interfering with subsequent regional patterning by morphogens. Earlier application of SHH or Pur can partly overcome this problem.
The primitive neuroepithelial cells from the first stage of culture are extremely sensitive to morphogens, with an exquisite dose-dependent response to Pur or SHH13. We expect 90% of the cells to be NKX2.1+ MGE (Fig. 3a) progenitors by 3 weeks of differentiation from hPSCs (days 20–25) without any cell sorting. An overwhelmingly common practice in the field is to expand the neural progenitors with mitogens, such as FGF2, during this stage. Expansion of neural progenitors with FGF2 often caudalizes early progenitors, thus decreasing the efficiency of forebrain GABA neurons or other forebrain neurons.
GABA interneurons in the forebrain comprise many different subtypes, and they are born over a long period of time1. Once the MGE progenitors are subjected to differentiation, especially in the presence of inhibitors of cell division, GABA neurons are observed as early as 3 d after plating. We expect that by day 45 almost all the cells are neurons and over 90% of the neurons are GABA+ neurons (Fig. 3c). We found that the cell capacitance was 27.22 ± 5.53 pF (n = 12 patched neurons, data are presented as means ± s.e.m.), and that resting membrane potential was 47 ± 6 mV (Fig. 4a). GABA neurons showed both inward Na and outward K currents (Fig. 4c), indicating the maturation of Na and K channels and resulting in the occurrence of action potentials upon current injection (Fig. 4d). Notably, we observed spontaneous GABA release events in cultures (Fig. 4e), which were eliminated by the application of bicuculine, an antagonist of GABA receptors (Fig. 4e), thus indicating that a functional synaptic network had been formed with surrounding neurons.
GABA interneurons appear early in culture. However, GABA interneuron subtypes appear according to their developmental schedules. CB+ and SST+ GABA neurons are seen from day 35, and their populations increase over time (Fig. 5a,b, day 45). PV+ GABA neurons appear later. They are observed at around day 65 (Fig. 5c, day 85), and their proportion increases over time. Neuronal nitric oxide synthase (NOS)+ neurons can also be observed after day 65 (Fig. 5d). The late generation of PV- or nNOS-expressing neurons is mainly due to the long-term maturation process1.
GABA interneurons generated by this protocol are nearly pure, which has been shown by immunostaining (Fig. 3c) and further confirmed by the spontaneous inhibitory, but seldom excitatory, synaptic activities recorded in the cultures (Fig. 4e). Therefore, this highly enriched population of GABA interneurons is ideal for molecular analysis. Technically, the MGE progenitors are amenable to freezing and thawing. Given the involvement of GABA interneurons in so many neurological conditions, particularly psychiatric disorders, the availability of large-scale enriched populations of forebrain GABA interneurons should prove to be useful for understanding disease mechanisms and for potential therapeutic discoveries.
Acknowledgments
This study was supported in part by the US National Institutes of Health (NS045926, MH099587, NS076352), the Busta Family Foundation, the Bleser Family Foundation and the US National Institute of Child Health and Human Development (P30 HD03352).
Footnotes
AUTHOR CONTRIBUTIONS All authors contributed to the design of the experiments, analysis of data and writing of this paper.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Le Magueresse C, Monyer H. GABAergic interneurons shape the functional maturation of the cortex. Neuron. 2013;77:388–405. doi: 10.1016/j.neuron.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 2.Goulburn AL, Stanley EG, Elefanty AG, Anderson SA. Generating GABAergic cerebral cortical interneurons from mouse and human embryonic stem cells. Stem Cell Res. 2012;8:416–426. doi: 10.1016/j.scr.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 3.Nobrega-Pereira S, et al. Postmitotic Nkx2-1 controls the migration of telencephalic interneurons by direct repression of guidance receptors. Neuron. 2008;59:733–745. doi: 10.1016/j.neuron.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fishell G, Rudy B. Mechanisms of inhibition within the telencephalon: “where the wild things are”. Annu Rev Neurosci. 2011;34:535–567. doi: 10.1146/annurev-neuro-061010-113717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kriegstein AR, Noctor SC. Patterns of neuronal migration in the embryonic cortex. Trends Neurosci. 2004;27:392–399. doi: 10.1016/j.tins.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 6.Marin O, Rubenstein JL. Cell migration in the forebrain. Annu Rev Neurosci. 2003;26:441–483. doi: 10.1146/annurev.neuro.26.041002.131058. [DOI] [PubMed] [Google Scholar]
- 7.Letinic K, Zoncu R, Rakic P. Origin of GABAergic neurons in the human neocortex. Nature. 2002;417:645–649. doi: 10.1038/nature00779. [DOI] [PubMed] [Google Scholar]
- 8.Jakovcevski I, Mayer N, Zecevic N. Multiple origins of human neocortical interneurons are supported by distinct expression of transcription factors. Cereb Cortex. 2011;21:1771–1782. doi: 10.1093/cercor/bhq245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sussel L, Marin O, Kimura S, Rubenstein JL. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development. 1999;126:3359–3370. doi: 10.1242/dev.126.15.3359. [DOI] [PubMed] [Google Scholar]
- 10.Danjo T, et al. Subregional specification of embryonic stem cell-derived ventral telencephalic tissues by timed and combinatory treatment with extrinsic signals. J Neurosci. 2011;31:1919–1933. doi: 10.1523/JNEUROSCI.5128-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell K. Dorsal-ventral patterning in the mammalian telencephalon. Curr Opin Neurobiol. 2003;13:50–56. doi: 10.1016/s0959-4388(03)00009-6. [DOI] [PubMed] [Google Scholar]
- 12.Olsson M, Campbell K, Wictorin K, Bjorklund A. Projection neurons in fetal striatal transplants are predominantly derived from the lateral ganglionic eminence. Neuroscience. 1995;69:1169–1182. doi: 10.1016/0306-4522(95)00325-d. [DOI] [PubMed] [Google Scholar]
- 13.Ma L, et al. Human embryonic stem cell–derived GABA neurons correct locomotion deficits in quinolinic acid–lesioned mice. Cell Stem Cell. 2012;10:455–464. doi: 10.1016/j.stem.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Y, et al. The LIM-homeobox gene Lhx8 is required for the development of many cholinergic neurons in the mouse forebrain. Proc Natl Acad Sci USA. 2003;100:9005–9010. doi: 10.1073/pnas.1537759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flandin P, et al. Lhx6 and Lhx8 coordinately induce neuronal expression of Shh that controls the generation of interneuron progenitors. Neuron. 2011;70:939–950. doi: 10.1016/j.neuron.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manabe T, et al. L3/Lhx8 is involved in the determination of cholinergic or GABAergic cell fate. J Neurochem. 2005;94:723–730. doi: 10.1111/j.1471-4159.2005.03261.x. [DOI] [PubMed] [Google Scholar]
- 17.Maroof AM, et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 2013;12:559–572. doi: 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nicholas CR, et al. Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell. 2013;12:573–586. doi: 10.1016/j.stem.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, et al. Medial ganglionic eminence-like cells derived from human embryonic stem cells correct learning and memory deficits. Nat Biotechnol. 2013;31:440–447. doi: 10.1038/nbt.2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li XJ, et al. Directed differentiation of ventral spinal progenitors and motor neurons from human embryonic stem cells by small molecules. Stem Cells. 2008;26:886–893. doi: 10.1634/stemcells.2007-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu BY, Zhang SC. Differentiation of spinal motor neurons from pluripotent human stem cells. Nat Protoc. 2009;4:1295–1304. doi: 10.1038/nprot.2009.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, et al. Regulation of TrkA and ChAT expression in developing rat basal forebrain: evidence that both exogenous and endogenous NGF regulate differentiation of cholinergic neurons. J Neurosci. 1995;15:2888–2905. doi: 10.1523/JNEUROSCI.15-04-02888.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang SC, Wernig M, Duncan ID, Brustle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol. 2001;19:1129–1133. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, et al. Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell. 2010;7:90–100. doi: 10.1016/j.stem.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pankratz MT, et al. Directed neural differentiation of human embryonic stem cells via an obligated primitive anterior stage. Stem Cells. 2007;25:1511–1520. doi: 10.1634/stemcells.2006-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chambers SM, et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cooper O, et al. Differentiation of human ES and Parkinson’s disease iPS cells into ventral midbrain dopaminergic neurons requires a high activity form of SHH, FGF8a and specific regionalization by retinoic acid. Mol Cell Neurosci. 2010;45:258–266. doi: 10.1016/j.mcn.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LaVaute TM, et al. Regulation of neural specification from human embryonic stem cells by BMP and FGF. Stem Cells. 2009;27:1741–1749. doi: 10.1002/stem.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reilly JO, Karavanova ID, Williams KP, Mahanthappa NK, Allendoerfer KL. Cooperative effects of sonic hedgehog and NGF on basal forebrain cholinergic neurons. Mol Cell Neurosci. 2002;19:88–96. doi: 10.1006/mcne.2001.1063. [DOI] [PubMed] [Google Scholar]
- 30.Krencik R, Zhang SC. Directed differentiation of functional astroglial subtypes from human pluripotent stem cells. Nat Protoc. 2011;6:1710–1717. doi: 10.1038/nprot.2011.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogura A, Morizane A, Nakajima Y, Miyamoto S, Takahashi J. Gamma-secretase inhibitors prevent overgrowth of transplanted neural progenitors derived from human-induced pluripotent stem cells. Stem Cells Dev. 2013;22:374–382. doi: 10.1089/scd.2012.0198. [DOI] [PubMed] [Google Scholar]