

Abstract
Recent reports underscore the unparalleled potential of antisense-oligonucleotide (ASO)-based approaches to ameliorate various pathological conditions. However, in vivo studies validating the effectiveness of a short ASO (<10-mer) in the context of a human disease have not been performed. One disease with proven amenability to ASO-based therapy is spinal muscular atrophy (SMA). SMA is a neuromuscular disease caused by loss-of-function mutations in the survival motor neuron 1 (SMN1) gene. Correction of aberrant splicing of the remaining paralog, SMN2, can rescue mouse models of SMA. Here, we report the therapeutic efficacy of an 8-mer ASO (3UP8i) in two severe models of SMA. While 3UP8i modestly improved survival and function in the more severe Taiwanese SMA model, it dramatically increased survival, improved neuromuscular junction pathology, and tempered cardiac deficits in a new, less severe model of SMA. Our results expand the repertoire of ASO-based compounds for SMA therapy, and for the first time, demonstrate the in vivo efficacy of a short ASO in the context of a human disease.
Keywords: antisense oligonucleotide, cardiac, short ASO, skeletal muscle, SMA mouse model, SMN2, spinal muscular atrophy, splicing
Introduction
The survival motor neuron (SMN) gene is ubiquitously expressed and produces an essential protein.1 Low SMN levels cause spinal muscular atrophy (SMA) (OMIM 253300; 253550; 253400; 271150), an autosomal recessive neuromuscular disease that is the leading hereditary cause of infant death.2,3 SMA is characterized by the loss of motor neurons within the spinal cord and resultant muscle weakness and atrophy.4 Clinically, SMA is heterogeneous, and patients are stratified into clinical types based on the age at disease-onset and maximum motor function achievements.5,6,7,8,9 Genetically, SMA is homogeneous in that all forms of the disease are caused by the loss of function mutations within the SMN1 gene, with concurrent retention of at least one copy of a highly similar gene, SMN2.2 SMN2 differs from SMN1 functionally by a single C to T nucleotide transition at position 6 (C6U transition in transcript) in exon 7 (ref. 10). The C6U transition creates a silencer and/or strengthens an inhibitory motif, which leads to a predominant amount of the transcripts skipping SMN2 exon 7 (refs. 11,12), and reviewed in Singh et al.13 SMNΔ7, the translated product from exon 7-skipped transcripts, is less functional and rapidly degraded.14
A recent update on potential therapeutic options for SMA treatment identified antisense-oligonucleotide (ASO)-based therapy as one of the most promising strategies.15 Blocking of intronic splicing silencer N1 (ISS-N1) by an ASO fully corrects SMN2 exon 7 splicing and restores a high level of SMN from endogenous SMN2.16 The original study performed with the ISS-N1 target employed a 20-mer ASO encompassing uniform phosphorothioate backbone and 2′-O-methyl modifications (2′OMeP).16 Subsequent preclinical studies have revealed efficacy differences between ASO chemistries and the route of ASO administrations.17 ASO-10–27, an 18-mer ASO encompassing phosphorothioate backbone and 2′-O-methoxyethyl modifications (abbreviated as “MOE”), has shown better efficacy when delivered peripherally but not when delivered through an intracerebroventricular (ICV) route.18 Twenty-mer or longer phosphorodiamidate morpholino oligomers (abbreviated as “PMOs”) have shown unprecedented efficacies when delivered through the ICV route.19,20 These findings underscore the challenge in selecting an optimal ASO with the right size and chemistry. Short ASOs offer several advantages over longer ASOs, including low to zero tolerance for mismatch base pairing, reduced cost of synthesis, and ease of modifications.21 Despite these advantages, the lack of a validated short target coupled with the possibility of rapid clearance from the bloodstream, have limited exploring the in vivo efficacy of very short ASOs.
The lessons learnt from ISS-N1 as an antisense target underscore certain basic principles that could be exploited for the validation of additional targets for SMA therapy.16 For instance, any strong inhibitory element within an intronic sequence, sequestration of which does not interfere with downstream processes, could serve as an ideal antisense target. Incidentally, an eight-nucleotide GC-rich sequence (GCRS) partially overlaps with ISS-N1, and simultaneously participates in the formation of an internal stem formed by long-distance interaction (LDI), termed internal stem through LDI-1 (ISTL1), a unique inhibitory RNA structure.21,22 Sequestration of GCRS by an 8-mer ASO encompassing 2′OMeP (3UP8) fully corrects aberrant SMN2 exon 7 splicing and restores high levels of SMN in SMA patient cells.21 Encouraged by these findings, here we examine the therapeutic efficacy of 3UP8i, an improved version of 3UP8 in two severe mouse models of SMA. Our results provide the first evidence of therapeutic efficacy of a very short ASO in the context of a preclinical study of a human disease.
Results
Optimization of an 8-mer ASO for in vivo application
We previously reported that an 8-mer 2′OMeP ASO (3UP8) that targets a GCRS that partially overlaps with ISS-N1 corrects SMN2 exon 7 splicing in SMA patient cells (Figure 1c, lane 3).21 Unlike large ASOs, 3UP8 showed zero tolerance for single-nucleotide mismatch base pairing.21 Another 8-mer ASO (F8) that targeted the first eight residues of ISS-N1 had no effect on SMN2 exon 7 splicing, suggesting the effect of 3UP8 is independent of ISS-N1 and specific to the GCRS (Figure 1c, lane 2).21 To test the in vivo efficacy of 3UP8, we performed preliminary experiments in healthy, adult Smn heterozygous mice containing human SMN2.23 Intraperitoneal (IP) administration of a moderate dose (20 μg/g body weight) of 3UP8 did not produce any noticeable effect on SMN2 exon 7 splicing in the liver. Among several factors, the lack of the stimulatory effect of 3UP8 in vivo could be attributed to the fast clearance of a short ASO from the blood. To circumvent this problem, we generated 3UP8i, an improved version of 3UP8 by incorporating PEG-282 and propyl modifications at the 5′ and 3′ ends, respectively (Figure 1b). These modifications confer enhanced stability and resistance to enzymatic degradation or modification.24,25 As expected, 3UP8i retained the GCRS-specific stimulatory effect on SMN2 exon 7 splicing in SMA patient cells (Figure 1c, lane 5). A control 8-mer ASO with similar modifications and a single-nucleotide substitution (CON8i) produced no appreciable effect on SMN2 exon 7 splicing (Figure 1c, lane 4). In vivo, 3UP8i (20 μg/g body weight) showed noticeable stimulation of SMN2 exon 7 splicing in liver (Figure 1d, lane 4). We next examined the effect of high dosages (80 μg/g body weight) of 3UP8i and CON8i delivered through IP and subcutaneous (SC) routes. 3UP8i showed substantial stimulation of SMN2 exon 7 inclusion in the liver, whereas CON8i had no noticeable effect (Figure 1d, lanes 5–10). Upon ICV administration of a moderate dose of 3UP8i, we observed substantial inclusion of SMN2 exon 7 in neuronal tissues, with some concomitant peripheral increases as well (Supplementary Figure S1c). These initial experiments demonstrated that 3UP8i shows a wide range of in vivo function.
Figure 1.
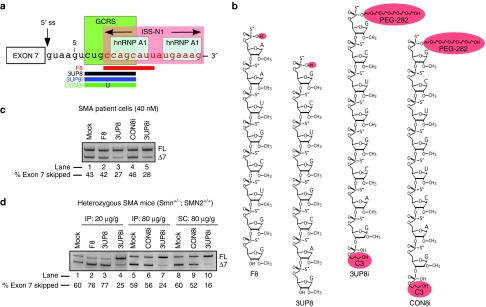
Effect of GC-rich sequence (GCRS)-targeting ASOs on splicing of SMN2 in vitro and in vivo. (a) Diagrammatic representation of annealing positions of ASOs targeting GCRS within SMN2 intron 7. Negative regulators of exon 7 splicing, GCRS, ISS-N1 and hnRNP A1 binding sites are highlighted with boxes. Annealing positions of ASOs (3UP8, 3UP8i, CON8i, and F8) are shown as horizontal bars. Numbering of nucleotides starts from the first position of intron 7. (b) Sequences of ASOs used in this study. All ASOs employed for mouse studies had PEG-282 and propyl modifications at the 5′ and 3′ ends, respectively. (c) SMN2 exon 7 inclusion in SMA patient fibroblasts after transfection with various ASOs. (d) SMN2 exon 7 inclusion in the liver of heterozygous mice (Smn+/−; SMN2+/+) after treatment with various ASOs. Each ASO was administered by intraperitoneal (IP) or subcutaneous (SC) route at the indicated dose. Exon 7-included and exon 7-skipped products are indicated as FL and Δ7, respectively.
In vivo efficacy of 3UP8i in severe 5058-Hemi SMA mice
To examine the in vivo efficacy of 3UP8i, we employed the “Taiwanese” severe SMA mouse model, which we refer to as 5058-Hemi SMA mice.26 This model harbors two copies of the SMN2 transgene and a targeted mutation of murine Smn exon 7 (ref. 27). Previous studies established that early, peripheral ASO delivery was more effective than an ICV delivery strategy for long-term rescue of the SMA phenotype in this SMA model.18 Thus, in blinded studies, we administered 3UP8i via IP injection starting at postnatal day 0 (P0), with two subsequent doses at P5 and P10. Both low (20 µg/g body weight) and high (80 µg/g body weight) doses were tested to assess the dose-dependent effect (Figure 2). The low dose of 3UP8i produced no apparent benefit in terms of survival or weight gain compared to CON8i, although a significant improvement in righting reflex was seen by P9 (Figure 2a–c). At the higher dose, 3UP8i resulted in a modest, yet statistically significant increase in weight gain starting at P8 compared to the CON8i-treated 5058-Hemi SMA mice (Figure 2a). Compared to heterozygous littermates, the 3UP8i-treated SMA mice were still noticeably underdeveloped (Figure 2d). The higher 3UP8i dose also resulted in a significant increase in median survival compared to CON8i, from 10 to 13 days (Figure 2b). Although general mobility was diminished, motor function of 3UP8i-treated SMA mice, as assayed by the time-to-right, was indistinguishable from heterozygous controls (Figure 2c). Overall, although 3UP8i showed modest efficacy, it did not produce the level of therapeutic efficacy seen with the longer ASOs in the same model.18
Figure 2.

3UP8i modestly improves weight, survival, and righting reflex in severe SMA mice. 5058-Hemi FVB/N SMA mice (SMN2tg/o;Smntm1Hung/tm1Hung) were administered three doses at P0, P5, P10 of 3UP8i or mismatch control (CON8i) by IP injection (HET, n = 5; SMA-3UP8i (20 µg/g body weight), n = 5; SMA-3UP8i (80 µg/g body weight), n = 7; SMA-CON8i, n = 5). (a) SMA mice treated with 3UP8i show improved weight over CON8i-treated SMA mice. Values show as mean ± SEM. *P = 0.04825, **P = 0.0231, Student's t-test. (b) Kaplan–Meier survival curve. The median survival of pairwise comparison between 3UP8i (80 µg/g body weight) and CON8i-treated SMA mice was significantly different, by log-rank test, P = 0.00260. Median survival, HET = undefined (n = 5); 3UP8i (80 µg/g body weight) = 13 d (n = 7); 3UP8i (20 µg/g) = 8 d (n = 5); CON8i = 10 d (n = 5). (c) Righting reflex. Righting time ≥60 seconds considered a failed test. Data presented as a mean ± SEM. *P = 0.02761, Student's t-test. (d) Representative image of a P11 SMA mouse treated with 3UP8i at 80 µg/g body weight shown alongside a littermate control (HET).
Effect of 3UP8i on SMN2 exon 7 splicing is age-dependent
Based on the modest improvements in survival and function of 5058-Hemi SMA mice treated with 3UP8i, we postulated that 3UP8i might possess developmentally variable efficacy. To test this, we examined the ability of 3UP8i to increase full-length SMN2 (FL-SMN2) transcripts over time. We used homozygous 5058-SMA mice, which harbor four copies of SMN2 on a Smn mutant background; other than mild peripheral necrosis, they are phenotypically akin to unaffected wild-type mice.27,28,29 Quantitative RT-PCR (qRT-PCR) of liver samples from 5058-homozygous mice administered 3UP8i or CON8i revealed that 3UP8i had limited SMN2 exon 7 splice-correcting activity at early postnatal time points (P2: 3UP8i, 1.55 ± 0.180; CON8i, 0.876 ± 0.126) (Figure 3a, left panel). At later time points, 3UP8i displayed a substantial increase in FL-SMN2 transcript relative to CON8i (P10: 3UP8i, 2.12 ± 0.153; CON8i, 0.759 ± 0.437. P15: 3UP8i, 3.88 ± 0.128; CON8i, 2.73 ± 0.115). End-point PCR of the same samples showed a similar pattern (Figure 3b). The qRT-PCR analysis at P20 showed only a modest increase of FL-SMN2 transcripts (P20: 3UP8i, 1.38 ± 0.124; CON8i, 0.974 ± 0.085). However, the end point PCR analysis at P20 showed the highest ratio of FL:Δ7 transcripts of any time-point (Figure 3b, panel 4). The Δ7 transcripts of the CON8i-treated samples were significantly higher for all time-points except P2 (Figure 3a, right panel). These results confirmed that SMN2 exon 7 inclusion by 3UP8i is age-dependent.
Figure 3.

3UP8i splicing efficacy is age-dependent. 5058-4copy mice (SMN2tg/o; SMN2tg/o) were administered 3UP8i (80 µg/g body weight, once daily) of or mismatch control (CON8i) starting at days P0, P8, P13, and P18. Liver was harvested at P2, P10, P15, and P20 was analyzed via qRT-PCR for full-length (FL) SMN2 (Left Panel) and ▵7-SMN2 (Right Panel). (a) SMN2 exon-7 inclusion. SMN2 levels were normalized to Gapdh. Data presented as mean fold change (log2−▵▵Ct) of 3UP8i relative to untreated ± S.D, n = 3 at each time point. Student's t-test, **P < 0.01, N.S., not significant. (b) FL- and ▵7-SMN2 end point RT-PCR of samples analyzed in (a). Exon 7-included and exon 7-skipped products are indicated as FL and Δ7, respectively.
Effect of 3UP8i in a less severe mouse model of SMA
5058-Hemi SMA mice are one of the more severe models of SMA, with a limited therapeutic window.18,26,27 Knowing that the current formulation of 3UP8i is not entirely suitable for such early interventions, we developed an SMA mouse with a slightly larger therapeutic window. This new SMA model, termed 5058-Hemi hybrid, was generated using the same breeding strategy for 5058-Hemi SMA mice, except instead of intercrossing 5058 homozygous FVB/N mice with the outbred Δ7Smn allele from this line (Smntm1Hung), we used a different, but equivalent, Δ7Smn allele (Smntm1.1Jme) in a C57/BL6 background (Smntm1.1Jme).27,30 By generating an F1 SMA hybrid, we more than doubled the median survival (P8 versus P22), and the age at which weight differentiated from controls (P4 versus P8) (Figure 4).
Figure 4.

3UP8i improves pathology and survival in 5058-Hemi hybrid SMA mice. Hemi-hybrid SMA mice (SMN2tg/o;Smntm1Hung/tm1.1Jme) were administered three doses (80 µg/g body weight at P0, P5, P10) of 3UP8i or mismatch control (CON8i) by IP injection. (a) Representative images of SMA mice treated with 3UP8i (P12, middle panel; P26, right panel) or CON8i (left panel). Red arrow highlights severe hind limb edema in the CON8i-treated SMA mice. Note necrosed tail (right panel). (b) Kaplan–Meier survival curve. The median survival of pairwise comparison between 3UP8i- and CON8i-treated SMA mice was significantly different, by log-rank test, P < 0.0001. Median survival, HET = undefined (n > 20); SMA-3UP8i = undefined at P50 (n = 12); SMA- CON8i = 16 days (n = 11); SMA-untreated (Unt.) = 22 days (n = 19). (c) Weight of 5058 Hemi-hybrid SMA mice treated with 3UP8i increases compared to CON8i-treated mice (HET, n > 20; SMA-3UP8i, n = 12; SMA-CON8i, n = 11, SMA-Unt., n = 19). Data presented as mean ± SEM. *P = 0.028456, Student's t-test. (d) SMN2 exon-7 inclusion. Liver was harvested at P12 or P50, and analyzed via qRT-PCR for full-length FL-SMN2 (left Panel) and ▵7-SMN2 (right panel). SMN2 levels were normalized to Gapdh. Data presented as fold change (log2−▵▵Ct) ± S.D. (n = 4) relative to either untreated (P12) or HET (P50). Student's t-test, **P = 0.00000003 (FL); P = 0.00004661 (▵7). (e) FL- and ▵7-SMN2 end point RT-PCR of samples analyzed in (d). (f) Liver was harvested at P12 or P50, and subjected to immunoblot with α-SMN and α-tubulin antibodies. (g) SMN levels were normalized to tubulin. Data presented as mean ± S.D. (n = 3). Student's t-test, *P = 0.0018, **P = 0.0001; N.S., not significant. Unt., untreated.
Using the same dosing strategy as with 5058-Hemi SMA mice, the median survival of 3UP8i-treated Hemi-hybrid SMA mice was undefined at study end point (P50), whereas the median survival of CON8i-treated Hemi-hybrid SMA was 16 days. This represented at least a threefold increase in median survival of the 3UP8i-treated SMA mice (log rank, P < 0.0001) (Figure 4b). Untreated SMA mice had a median survival of 22 days, which was significantly better than CON8i-treated SMA mice (log rank, P = 0.0016), and likely due to a larger sampling size in conjunction with the stress of dosing the CON8i-treated SMA mice. Alternatively, the CON8i ASO may not have been well tolerated, as there was also a small decrease in overall weight compared to the untreated Hemi-hybrid SMA mice (Figure 4c). The study end point at P50 was chosen since the frequency of the 3UP8i-treated deaths had slowed substantially, and no further decline in health or motor function was evident. Moreover, we found that 3UP8i treatment resulted in a significant improvement in weight gain of 5058-Hemi hybrids, starting at P13 (3UP8i, 6.19g ± 0.457; CON8i, 5.03 ± 0.322) (Figure 4c). This trend continued, and the slope of weight gain was similar to control mice.
Since heart defects and bradyarrythmia had been noted in other SMA mouse models,31,32,33,34 we studied the ECG waveforms of 5058-Hemi hybrid mice. Recording intervals were limited to preweaning ages due to post-weaning variability between the sexes.35 By P10, heterozygote littermates diverged from all SMA mice for heart rate (HR) and PR interval (Supplementary Figure S2a and c, respectively). The 3UP8i-treated SMA mice displayed significant improvement in HR by P18 (bpm: HET, 776 ± 11.2; 3UP8i, 701 ± 31.9; CON8i, 608 ± 39.3; Supplemenatry Figure S2a). The lower HR and elongated PR interval (ms: HET, 23.31 ± 0.459; 3UP8i, 26.4 ± 0.930; CON8i, 30.43 ± 2.49) confirmed that this model presents with bradyarrythmia, although to a lesser extent than in the more severe, 5025-▵7 model and severe inducible SMA mice.32,33 The QRS interval of the SMA pups was slightly elevated compared to control (HET, 9.69 ± 0.211; 3UP8i, 10.7 ± 0.594; CON8i, 10.75 ± 0.543) (Supplementary Figure S2d). This elevation was indicative of delayed ventricular depolarization times, and suggested a bundle branch block.36 Treatment with 3UP8i resulted in a corrective, downward trend toward heterozygote levels, but it did not reach statistical significance compared to CON8i-treated SMA mice by P18. No significant difference was observed in heart rate variability (HRV) between SMA mice and control littermates (Supplementary Figure S2b). Together, the bradyarrythmia and heart block suggested that the 5058-Hemi hybrid SMA mice suffer from deficits in sympathetic innervation, and to some extent, 3UP8i was able to correct this.
One property of this particular model is the appearance of hind limb edema and tail necrosis, starting at ~P8 in untreated mice (Figure 4a, left panel). Treatment with 3UP8i prevented hind limb edema, and delayed tail necrosis (Figure 4a, middle panel). Although the tail eventually necrosed in treated mice, they were generally spared from any additional peripheral necrosis (Figure 4a, right panel). Further, 3UP8i-treated SMA mice did not show any noticeable deficits in motor function over the course of the study.
The primary mode-of-action of 3UP8i is to promote the inclusion of exon 7 in SMN2 transcripts. Thus, we expected the levels of FL-SMN2 transcripts to substantially increase in the surviving 5058-Hemi hybrid SMA population that received 3UP8i. We examined the relative SMN2 expression levels in the liver of 5058-Hemi hybrid mice by TaqMan qRT-PCR, and found that 2 days postfinal dose, 3UP8i-treated liver samples showed a fourfold increase in FL-SMN2 transcripts compared to CON8i-treatment (3UP8i, 5.95 ± 0.220; CON8i, 1.40 ± 0.113) (Figure 4d, left panel). Correspondingly, the exon 7-skipped isoform (Δ7-SMN) significantly decreased threefold (3UP8i, 0.591 ± 0.117; CON8i, 1.78 ± 0.190) (Figure 4d, right panel). By P50, there was no significant difference in amount of FL-SMN2 and Δ7-SMN2 transcripts between 3UP8i-treated SMA mice and untreated heterozygotes (Figure 4d). This suggested that the pharmacodynamic range of 3UP8i is less than 40 days in vivo. End-point PCR of the same samples confirmed the trend seen via qRT-PCR, with a noticeable increase in the FL-versus Δ7-SMN2 transcript ratio following 3UP8i treatment at P12 (Figure 4e). Previous studies have demonstrated that peripheral administration of ASOs produces a splice-correction that is generally most pronounced in the liver, with other peripheral tissues displaying a reduced response.18 Examination of FL-SMN2 levels in the quadriceps of 3UP8i-treated mice revealed consistent findings. FL-SMN2 levels were elevated compared to CON8i-treated mice, although less than the change seen in the liver (Supplementary Figure S3a, b).
Ultimately, SMA is caused by a deficiency in SMN protein levels. Therefore, we determined whether the increases seen at the transcript level in the liver of 3UP8i-treated 5058-Hemi hybrid SMA mice translated to increased levels of SMN. Compared to CON8i-treated mice, 3UP8i treatment resulted in nearly a threefold increase in SMN levels (HET, 1.821 ± 0.133; CON8i, 0.441 ± 0.096; 3UP8i, 1.317 ± 0.183) (Figure 4g). These increased levels persisted through P50, although they were substantially lower compared to heterozygous littermates (HET, 3.205 ± 0.177; 3UP8i, 1.683 ± 0.311). As expected, the decreased levels of FL-SMN2 transcripts from P12 to P50 translated to lower SMN levels (Figure 4f,g). However, the SMN levels from 3UP8i-treated mice were still much higher at P50 than the levels of CON8i-treated mice at P12. Moreover, SMN levels were also elevated in muscle, albeit to a smaller extent than in the liver (Supplementary Figure S3c,d). Taken together, the SMN protein levels and SMN2 exon 7 splicing data verified the molecular basis for the improved survival and function in the 5058-Hemi hybrid SMA mice.
Effect of 3UP8i on neuromuscular junction pathology
SMA is characterized as a disease of the motor unit, and viable therapeutics should improve synaptic and muscular pathologies. To examine possible improvements in motor unit pathology following 3UP8i treatment, we analyzed the neuromuscular junction (NMJ) of intercostal (IC) muscles from P12 5058-Hemi hybrid SMA mice (Figure 5a). IC muscles are critical for maintaining proper respiratory function, and are one of the more vulnerable muscle groups in both SMA mouse models and human SMA patients.37,38,39 The ICs from CON8i-treated SMA mice showed a significant decrease in fully innervated motor endplates (CON8i, 62.00% ± 1.780 versus HET, 86.25% ± 2.18) with a concomitant increase in partial innervations (CON8i, 36.00% ± 3.34 versus HET, 13.75% ± 2.18) (Figure 5b). Treatment with 3UP8i significantly increased the percentage of fully innervated endplates relative to CON8i-treated SMA mice (3UP8i, 77.25% ± 1.93 versus CON8i, 62.00% ± 1.780); and accordingly, brought partial innervations back toward those observed in control mice (HET, 13.75% ± 2.18; 3UP8i, 21.50% ± 2.10; CON8i, 36.00% ± 3.34) (Figure 5b). There was no change in motor end plate size (Figure 5c). These results indicated an inherent defect in either the formation and/or maintenance of the NMJ within the 5058-Hemi hybrid SMA mice, which was responsive to 3UP8i treatment.
Figure 5.

3UP8i improves NMJ pathology. 5058-Hemi hybrid SMA mice (SMN2tg/o;Smntm1Hung/tm1.1Jme) were administered three doses (80 µg/g body weight at P0, P5, P10) of 3UP8i or mismatch control (CON8i) by IP injection (n = 4, n > 50 NMJs/pup). (a) Whole mount NMJ analysis of P12 intercostal (IC) muscles stained with neurofilament H (NF-H)/synaptic vesicle 2 (SV2) (green), and α-Bungarotoxin (BTX) (red). Scale bar = 10 µm. (b) Quantification of IC innervation status. Data presented as mean ± SEM. Two-way analysis of variance repeated measures, with Bonferroni posthoc comparisons (*P = 0.0128, **P = 0.0002, ***P = 0.0001). Asterisks above error bars indicate significance relative to untreated control (HET); asterisks above line indicate significance between 3UP8i and CON8i-treated SMA mice. (c) Quantification of IC motor end plate area. Data are presented as mean ± SEM; N.S., not significant.
Discussion
Recent preclinical studies employing ASO-based strategies aimed at SMN2 exon 7 splicing correction have achieved important milestones for the treatment of SMA, the leading genetic cause of infant mortality. While 18-mer or larger ASOs have been successfully employed in preclinical and clinical studies for the potential treatment of several diseases including SMA, in vivo studies that demonstrate the therapeutic potential of a splice-switching short ASO have yet to be completed. The only in vivo study reported thus far with a short ASO pertains to an 8-mer locked nucleic acid (LNA) against the seed sequence of a microRNA.40 Interestingly, an iso-sequential 8-mer ASO with 2′OMeP modifications targeting the same microRNA turned out to be totally ineffective.40 Although interesting, these findings indicated a limited scope of a short ASO to the LNA-based chemistry. On the other hand, our earlier report that a GCRS-targeting 8-mer ASO (3UP8) could efficiently correct SMN2 exon 7 splicing in SMA patient cells suggested the therapeutic potential of the widely used 2′OMeP chemistry.21 However, despite stabilizing 2′OMeP modifications, in vivo 3UP8 failed to correct SMN2 exon 7 splicing. This was possibly due to terminal degradation and/or fast clearance from the blood. To combat these potential limitations, we explored chemistries that would confer greater in vivo efficacy. 3UP8 with PEG-282 and C3-propyl terminal spacers (3UP8i) induced robust SMN2 exon 7 inclusion in vivo, and a high level of specificity—the introduction of a single mismatch base in the middle of ASO completely eliminated the antisense response.
Bolstered by these initial results, we first tested the in vivo efficacy of 3UP8i in 5058-hemizygous mice, a severe model of SMA. A high dose (80 μg/g body weight) of 3UP8i, we observed an encouraging trend of body weight gain, improved righting reflex, and a modest 3-day extension in survival of these severe mice. The outcome was more convincing in the milder 5058-Hemi hybrid SMA mice, which provided a wider therapeutic window. In fact, treatment of 5058-Hemi hybrid SMA mice with 3UP8i produced improvements in weight and survival that began to mirror the efficacy of larger ISS-N1 targeting ASOs reported recently. Although it is difficult to pinpoint the exact cause of the 5058-Hemi hybrid's lessened severity, we suspect that this was the result of hybrid vigor. The noticeable caveat of this model is the development of hind limb edema. This pathology limits the types of motor tests that can be performed. Central nervous system (CNS) administration of ISIS-SMNRx, an ISS-N1 targeting MOE ASO, into SMA type III mice (5058-4copy) rescued a similar phenotype,29 which may explain why peripheral administration routes were unable to correct this necrosis. The improvements in the Hemi-hybrid SMA mice corresponded to increased FL-SMN2 transcript and total SMN levels at P12. The P50 3UP8i-treated SMA mice showed decreased FL-SMN2 transcript and SMN levels compared to levels at P12, yet they were functionally normal aside from mild necrosis and decreased body size. Previous studies have indicated that the need for increased SMN levels is more critical at early as opposed to later time periods.18
While previous studies have demonstrated the effectiveness of very small ASOs (7-mer-11-mer) in vitro,41,42 our findings with 3UP8i validate, for the first time, the therapeutic potential of a very small splice switching oligonucleotide in an animal model of a human disease. In addition to a better uptake, a small ASO imparts greater efficacy, provided it maintains target specificity.43,44,45 Considering 3UP8i did not tolerate a single mismatch with the target, it fulfilled one of the rigorous requirements of target specificity. Future studies will be aimed at determining global off target effects of 3UP8i, particularly at higher concentrations. This information will be valuable for SMA research and more generally regarding the effects of short ASOs as they are developed for the treatment of other human diseases. The small helix generated by the annealing of 3UP8i to the GCRS is predicted to disrupt ISTL1 that we recently reported to be an inhibitory RNA structure formed by a unique LDI.22 Hence, the mechanism of 3UP8i function is likely to be distinct from the group of ISS-N1 targeting ASOs that displace inhibitory protein(s) hnRNP A1/A2.21,46 To further underscore the promising therapeutic potential of 3UP8i, it did not elicit an inflammatory response, as measured by qRT-PCR of common inflammation markers AIF1, CD68, IL-1A, IFNA, and IL-18 (Supplementary Figure S4). Our results also establish the potential of combining new chemistries to further enhance 3UP8i efficacy. In a broader context, our results validate the ability of a short ASO (8-mer) to correct a human disease in an animal model. This has important implications for the development of other therapeutics directed at small targets, including seed sequences of microRNAs, where the use of large ASOs carries the risk of poor specificity.
Materials and methods
Mice. Animals were cared for and monitored in accordance with the approved Lurie Children's Hospital Institutional Animal Care and Use Committee protocols. Animals were kept in a controlled environment at 25 °C and 50% humidity, with a 12 hours light/dark photoperiod, and were monitored daily for health. Three SMA mouse models were utilized in this study (see Supplementary Table S1): 5058-homozygous mice (SMN2tg/o;SMN2tg/o;Smntm1Hung/tm1Hung) were generated as described;28,29 5058-Hemi FVB/N SMA mice (SMN2tg/o;Smntm1Hung/tm1Hung) were generated as described;18,27 5058-Hemi hybrid SMA mice (SMN2tg/o; Smntm1Hung/tm1.1Jme) were generated by crossing 5058-homozygous FVB/N mice (SMN2tg/o;SMN2tg/o;Smntm1Hung/tm1Hung) with the heterozygous mice harboring the ▵7Smn Melki allele (Smntm1.1Jme/Wt; C57-congenic), which produces litters in which 50% of the pups are SMA. The resulting SMA mice are FVB/N:C57/BL6 F1-hybrids, hemizygous for the SMN2 allele and compound heterozygotes for the Δ7Smn allele (SMN2tg/o;Smntm1Hung/tm1.1Jme). All offspring were genotyped on P0 using multiplex PCR of tail biopsies as previously described.28 Mice were culled or combined to maintain litter sizes between six and nine pups, with at least two heterozygous siblings per cage. When possible, each cage of SMA mice under study (5058-Hemi FVB/N and 5058-Hemi hybrid) was evenly split between mismatch control (CON8i) and 3UP8i, which were delivered as blinded drugs known as oligo A and oligo B. One caveat of note was that by P6 or later, 3UP8i-treated 5058-hemizygous mice could often be unblinded based on weight and righting reflex.
Drug treatments and phenotyping. The 3UP8i and mismatch control (CON8i) 8-mer ASOs incorporated 2′OMeP, with a 5′ PEG-282 modification (Spacer 18) and a 3′ C3-propyl spacer, were synthesized (TriLink Technologies, San Diego, CA) as previously described.16,21 ASOs were resuspended in 0.9% saline and passed through a 0.22 µm filter. Samples were coded as oligo A or oligo B to blind the studies. Mice were dosed on P0, P5, and P10 at either 20 or 80 µg/g of body weight via IP injection. Mice were monitored every morning for survival, weight, righting reflex (5058-Hemi only), and ECG (5058-Hemi hybrid only). For the righting reflex test, mice were placed in a supine position and assayed for the time-to-right on all four paws. Failure to right was set at 60 seconds. Functional death was defined as two consecutive days of greater than 20% weight loss or righting reflex failure. Noninvasive ECG measurements of conscious 5058-Hemi hybrid mice were recorded between P8 and P18, using the ECGenie system (Mouse Specifics, Quincy, MA) as previously described.35,47
RNA and protein analysis. Total RNA was isolated from 50 mg of tissue using the TRIzol reagent (Life Technologies, Carlsbad, CA) as per the manufacturer's instructions. DNase treatment and first-strand cDNA synthesis were performed as previously described.48 FL- and Δ7-SMN2 end point PCR was performed as previously described.29 Cycle conditions: 94 °C/45 seconds, 60 °C/45 seconds, 72 °C/45 seconds, 30 cycles. TaqMan assays of FL- and Δ7-SMN2 were performed on an Applied Biosystems (Carlsbad, CA) 7500 Fast Real-Time PCR System utilizing the following conditions: 1 cycle: 2 minutes 50 °C, 1 cycle: 10 minutes 95 °C, 40 cycles: 15 seconds 95 °C, 1 minutes 60 °C. The data were analyzed using the ΔΔCt method. Primer and probe sets used were previously described.29 Mouse Gapdh (#4352339E, Applied Biosystems) was used to normalize samples. The data were analyzed using the ΔΔCt method.49 Radioactive RT-PCR was performed as previously described.21 5058-Hemi hybrid mouse tissue was collected and homogenized as previously described.50 Lysates were analyzed via immunoblot using the Li-COR Odyssey (Li-COR Technologies, Lincoln, NE) system as previously described.48
Immunostaining and microscopy. NMJ staining and imaging of whole-mount intercostal (IC) muscles from P12 5058-Hemi hybrid mice were performed as previously described.32 Images were blinded and randomized prior to the analysis of NMJ motor endplate area and innervation status. Innervation status was determined as previously described.28 Motor endplate area was defined by measuring the area of the background-subtracted of the α-BTX-594 signal using NIH Image J software (National Institutes of Health, Bethesda, MD). For each pup, n = 50 or more NMJs were analyzed.
Statistical analysis. Statistical analyses were performed using Graph Pad Prism software, V6.0 (GraphPad Software, La Jolla, CA). Kaplan–Meier survival curves were compared using log-rank tests. Statistical significance was determined using either a one- or two-way analysis of variance with a Bonferroni posthoc comparison, or Student's t-test when appropriate.
SUPPLEMENTARY MATERIAL Figure S1. Effect of 3UP8i treatment on splicing of SMN2 in vivo. Figure S2. 3UP8 improves cardiac deficits in Hemi-hybrid SMA mice. Figure S3. 3UP8i increases FL-SMN2 levels in skeletal muscle of Hemi-hybrid SMA mice. Figure S4. 3UP8i ASO does not increase expression of several inflammation-related genes. Table S1. SMA models utilized in this study.
Acknowledgments
This work was supported in part by grants from National Institutes of Health (NIH) (NS072259 and NS055925) and Salsbury Endowment to Ravindra Singh (Iowa State University, Ames, IA), and NIH R01NS060926, the Families of SMA (DID1214) and Muscular Dystrophy Association (255785) to Christine DiDonato (Manne Children's Research Institute, Chicago, IL). GCRS target (Patent# US 20110269820 A1) was discovered at Iowa State University. Therefore, inventors including RNS and Iowa State University could potentially benefit from commercial exploitation of GCRS target.
Supplementary Material
Effect of 3UP8i treatment on splicing of SMN2 in vivo.
3UP8 improves cardiac deficits in Hemi-hybrid SMA mice.
3UP8i increases FL-SMN2 levels in skeletal muscle of Hemi-hybrid SMA mice.
3UP8i ASO does not increase expression of several inflammation-related genes.
SMA models utilized in this study.
References
- Schrank B, Götz R, Gunnersen JM, Ure JM, Toyka KV, Smith AG, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci USA. 1997;94:9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Roberts DF, Chavez J, Court SD. The genetic component in child mortality. Arch Dis Child. 1970;45:33–38. doi: 10.1136/adc.45.239.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- Dubowitz V. Chaos in the classification of SMA: a possible resolution. Neuromuscul Disord. 1995;5:3–5. doi: 10.1016/0960-8966(94)00075-k. [DOI] [PubMed] [Google Scholar]
- Pearn J. Classification of spinal muscular atrophies. Lancet. 1980;1:919–922. doi: 10.1016/s0140-6736(80)90847-8. [DOI] [PubMed] [Google Scholar]
- Wang DF, Helquist P, Wiech NL, Wiest O. Toward selective histone deacetylase inhibitor design: homology modeling, docking studies, and molecular dynamics simulations of human class I histone deacetylases. J Med Chem. 2005;48:6936–6947. doi: 10.1021/jm0505011. [DOI] [PubMed] [Google Scholar]
- Zerres K, Rudnik-Schöneborn S. Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol. 1995;52:518–523. doi: 10.1001/archneur.1995.00540290108025. [DOI] [PubMed] [Google Scholar]
- Zerres K, Rudnik-Schöneborn S, Forrest E, Lusakowska A, Borkowska J, Hausmanowa-Petrusewicz I. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci. 1997;146:67–72. doi: 10.1016/s0022-510x(96)00284-5. [DOI] [PubMed] [Google Scholar]
- Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30:377–384. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003;34:460–463. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- Singh NN, Singh RN. Alternative splicing in spinal muscular atrophy underscores the role of an intron definition model. RNA Biol. 2011;8:600–606. doi: 10.4161/rna.8.4.16224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorson CL, Androphy EJ. An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum Mol Genet. 2000;9:259–265. doi: 10.1093/hmg/9.2.259. [DOI] [PubMed] [Google Scholar]
- Seo J, Howell MD, Singh NN, Singh RN. Spinal muscular atrophy: an update on therapeutic progress. Biochim Biophys Acta. 2013;1832:2180–2190. doi: 10.1016/j.bbadis.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 2006;26:1333–1346. doi: 10.1128/MCB.26.4.1333-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivanesan S, Howell MD, DiDonato CJ, Singh RN. Antisense oligonucleotide mediated therapy of spinal muscular atrophy. Transl Neurosci. 2013;4 doi: 10.2478/s13380-013-0109-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porensky PN, Mitrpant C, McGovern VL, Bevan AK, Foust KD, Kaspar BK, et al. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum Mol Genet. 2012;21:1625–1638. doi: 10.1093/hmg/ddr600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Janghra N, Mitrpant C, Dickinson RL, Anthony K, Price L, et al. A novel morpholino oligomer targeting ISS-N1 improves rescue of severe spinal muscular atrophy transgenic mice. Hum Gene Ther. 2013;24:331–342. doi: 10.1089/hum.2012.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NN, Shishimorova M, Cao LC, Gangwani L, Singh RN. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 2009;6:341–350. doi: 10.4161/rna.6.3.8723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NN, Lawler MN, Ottesen EW, Upreti D, Kaczynski JR, Singh RN. An intronic structure enabled by a long-distance interaction serves as a novel target for splicing correction in spinal muscular atrophy. Nucleic Acids Res. 2013;41:8144–8165. doi: 10.1093/nar/gkt609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- Murakami T, Sumaoka J, Komiyama M. Sensitive RNA detection by combining three-way junction formation and primer generation-rolling circle amplification. Nucleic Acids Res. 2012;40:e22. doi: 10.1093/nar/gkr909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand M, Chevrie K, Chassignol M, Thuong NT, Maurizot JC. Circular dichroism studies of an oligodeoxyribonucleotide containing a hairpin loop made of a hexaethylene glycol chain: conformation and stability. Nucleic Acids Res. 1990;18:6353–6359. doi: 10.1093/nar/18.21.6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogliotti RG, Cardona H, Singh J, Bail S, Emery C, Kuntz N, et al. The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Hum Mol Genet. 2013;22:4084–4101. doi: 10.1093/hmg/ddt258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, et al. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- Gogliotti RG, Hammond SM, Lutz C, DiDonato CJ. Molecular and phenotypic reassessment of an infrequently used mouse model for spinal muscular atrophy. Biochem Biophys Res Commun. 2010;391:517–522. doi: 10.1016/j.bbrc.2009.11.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Sahashi K, Hung G, Rigo F, Passini MA, Bennett CF, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24:1634–1644. doi: 10.1101/gad.1941310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Diaz C, Frugier T, Tiziano FD, Lacène E, Roblot N, Joshi V, et al. Deletion of murine SMN exon 7 directed to skeletal muscle leads to severe muscular dystrophy. J Cell Biol. 2001;152:1107–1114. doi: 10.1083/jcb.152.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan AK, Hutchinson KR, Foust KD, Braun L, McGovern VL, Schmelzer L, et al. Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum Mol Genet. 2010;19:3895–3905. doi: 10.1093/hmg/ddq300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogliotti RG, Quinlan KA, Barlow CB, Heier CR, Heckman CJ, DiDonato CJ. Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not a cause, of motor neuron dysfunction. J Neurosci. 2012;32:3818–3829. doi: 10.1523/JNEUROSCI.5775-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heier CR, Satta R, Lutz C, DiDonato CJ. Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum Mol Genet. 2010;19:3906–3918. doi: 10.1093/hmg/ddq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shababi M, Habibi J, Yang HT, Vale SM, Sewell WA, Lorson CL. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum Mol Genet. 2010;19:4059–4071. doi: 10.1093/hmg/ddq329. [DOI] [PubMed] [Google Scholar]
- Heier CR, Hampton TG, Wang D, DiDonato CJ. Development of electrocardiogram intervals during growth of FVB/N neonate mice. BMC Physiol. 2010;10:16. doi: 10.1186/1472-6793-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murkofsky RL, Dangas G, Diamond JA, Mehta D, Schaffer A, Ambrose JA. A prolonged QRS duration on surface electrocardiogram is a specific indicator of left ventricular dysfunction [see comment] J Am Coll Cardiol. 1998;32:476–482. doi: 10.1016/s0735-1097(98)00242-3. [DOI] [PubMed] [Google Scholar]
- Murray LM, Comley LH, Thomson D, Parkinson N, Talbot K, Gillingwater TH. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:949–962. doi: 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- Montes J, McDermott MP, Martens WB, Dunaway S, Glanzman AM, Riley S, et al. Six-Minute Walk Test demonstrates motor fatigue in spinal muscular atrophy. Neurology. 2010;74:833–838. doi: 10.1212/WNL.0b013e3181d3e308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling KK, Lin MY, Zingg B, Feng Z, Ko CP. Synaptic defects in the spinal and neuromuscular circuitry in a mouse model of spinal muscular atrophy. PLoS One. 2010;5:e15457. doi: 10.1371/journal.pone.0015457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet. 2011;43:371–378. doi: 10.1038/ng.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan WM, Kothavale A, Wagner RW. Effects of oligonucleotide length, mismatches and mRNA levels on C-5 propyne-modified antisense potency. Nucleic Acids Res. 1996;24:2936–2941. doi: 10.1093/nar/24.15.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner RW, Matteucci MD, Grant D, Huang T, Froehler BC. Potent and selective inhibition of gene expression by an antisense heptanucleotide. Nat Biotechnol. 1996;14:840–844. doi: 10.1038/nbt0796-840. [DOI] [PubMed] [Google Scholar]
- Loke SL, Stein CA, Zhang XH, Mori K, Nakanishi M, Subasinghe C, et al. Characterization of oligonucleotide transport into living cells. Proc Natl Acad Sci USA. 1989;86:3474–3478. doi: 10.1073/pnas.86.10.3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf TM, Melton DA, Jennings CG. Specificity of antisense oligonucleotides in vivo. Proc Natl Acad Sci USA. 1992;89:7305–7309. doi: 10.1073/pnas.89.16.7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straarup EM, Fisker N, Hedtjärn M, Lindholm MW, Rosenbohm C, Aarup V, et al. Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates. Nucleic Acids Res. 2010;38:7100–7111. doi: 10.1093/nar/gkq457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NN, Seo J, Rahn SJ, Singh RN. A multi-exon-skipping detection assay reveals surprising diversity of splice isoforms of spinal muscular atrophy genes. PLoS One. 2012;7:e49595. doi: 10.1371/journal.pone.0049595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu V, Otero JM, Lopez O, Morgan JP, Amende I, Hampton TG. Method for non-invasively recording electrocardiograms in conscious mice. BMC Physiol. 2001;1:6. doi: 10.1186/1472-6793-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond SM, Gogliotti RG, Rao V, Beauvais A, Kothary R, DiDonato CJ. Mouse survival motor neuron alleles that mimic SMN2 splicing and are inducible rescue embryonic lethality early in development but not late. PLoS One. 2010;5:e15887. doi: 10.1371/journal.pone.0015887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Heier CR, DiDonato CJ. Translational readthrough by the aminoglycoside geneticin (G418) modulates SMN stability in vitro and improves motor function in SMA mice in vivo. Hum Mol Genet. 2009;18:1310–1322. doi: 10.1093/hmg/ddp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of 3UP8i treatment on splicing of SMN2 in vivo.
3UP8 improves cardiac deficits in Hemi-hybrid SMA mice.
3UP8i increases FL-SMN2 levels in skeletal muscle of Hemi-hybrid SMA mice.
3UP8i ASO does not increase expression of several inflammation-related genes.
SMA models utilized in this study.