

Abstract
Although aberrant mTORC1 signaling has been well established in models of obesity, little is known about its repressor, REDD1. Therefore, the initial goal of this study was to determine the role of REDD1 on mTORC1 in obese skeletal muscle. REDD1 expression (protein and message) and mTORC1 signaling (S6K1, 4E-BP1, raptor-mTOR association, Rheb GTP) were examined in lean vs. ob/ob and REDD1 wild-type (WT) vs. knockout (KO) mice, under conditions of altered nutrient intake [fasted and fed or diet-induced obesity (10% vs. 60% fat diet)]. Despite higher (P < 0.05) S6K1 and 4E-BP1 phosphorylation, two models of obesity (ob/ob and diet-induced) displayed elevated (P < 0.05) skeletal muscle REDD1 expression compared with lean or low-fat-fed mouse muscle under fasted conditions. The ob/ob mice displayed elevated REDD1 expression (P < 0.05) that coincided with aberrant mTORC1 signaling (hyperactive S6K1, low raptor-mTOR binding, elevated Rheb GTP; P < 0.05) under fasted conditions, compared with the lean, which persisted in a dysregulated fashion under fed conditions. REDD1 KO mice gained limited body mass on a high-fat diet, although S6K1 and 4E-BP1 phosphorylation remained elevated (P < 0.05) in both the low-fat and high-fat-fed KO vs. WT mice. Similarly, the REDD1 KO mouse muscle displayed blunted mTORC1 signaling responses (S6K1 and 4E-BP1, raptor-mTOR binding) and circulating insulin under fed conditions vs. the robust responses (P < 0.05) in the WT fed mouse muscle. These studies suggest that REDD1 in skeletal muscle may serve to limit hyperactive mTORC1, which promotes aberrant mTORC1 signaling responses during altered nutrient states.
Keywords: Rheb, raptor, diet-induced obesity, fasted, fed
nearly two-thirds of the U.S. population are overweight, a third of which are obese (55). These conditions are associated with insulin insensitivity, metabolic inflexibility (51), and reduced activities of daily living. Despite nutrient excess, studies of obesity present data showing lower relative muscle mass that point toward aberrant growth signaling, satellite cell activation, and protein synthesis (6, 7, 13, 21, 34, 54, 57, 67). Since skeletal muscle is a major site for insulin action and glucose disposal, and comprises a large portion of fat-free mass, and is positively associated with metabolic homeostasis (2), the maintenance of muscle mass is central to metabolic homeostasis (72, 73). The key mechanism of regulating skeletal muscle protein synthesis is by the translation of messenger RNA (mRNA) (30, 48). mRNA translation is a tightly controlled process, dictating peptide formation and protein synthesis, and is regulated by hormonal and nutrient cues, such as those that occur in a fasted or fed state, to the mammalian target of rapamycin (mTOR) kinase (5, 38, 39). Skeletal muscle from obese rodents has an attenuated response to nutrient and growth stimuli (35, 49, 76), associated with dysregulated mTOR signaling.
mTOR comprises two separate multiprotein complexes, raptor containing mTOR complex 1 (mTORC1) and rictor containing mTOR complex 2 (mTORC2) (36, 64). mTORC1 phosphorylates two downstream substrates, the eukaryotic initiation factor (eIF) 4E binding protein-1 (4E-BP1) and p70 ribosomal protein S6 kinase-1 (S6K1) (12). In its unphosphorylated state, 4E-BP1 binds to eIF4E, inhibiting translation initiation. mTORC1 phosphorylation of 4E-BP1 promotes its release from the cap-binding protein eIF4E. eIF4E is then free to bind with the initiation factor, eIF4G, increasing eIF4F complex formation (27). During the initiation phase of mRNA translation, phosphorylation of eIF3-bound S6K1 by mTORC1 results in its release from the 43S preinitiation complex, allowing it to phosphorylate substrates such as eIF4B and PDCD4 (29, 59). In addition, during the pioneering round of mRNA translation, activated S6K1 also phosphorylates the exon junction complex constituent S6K1 ALY-REF-like target (SKAR), which upregulates the process (61, 65). Moreover, it has been suggested that SKAR may act to recruit S6K1 to the newly synthesized mRNA, allowing it to phosphorylate proteins such as eIF4B to promote mRNA translation (44). The eIF4F complex then binds with the mRNA and 40S ribosomal subunit so that together with a 60S subunit, a functional 80S monosome is formed to begin mRNA translation.
An inhibitor of mTOR, the protein regulated in development and DNA damage responses 1 [REDD1; also known as DNA-damage-inducible transcript 4 (DDIT4), dexamethasone-induced gene 2 (Dig2), and RTP801], is upregulated by various stressors, such as glucocorticoids (78), DNA damage (43), endoplasmic reticulum (ER) stress (58, 79), and hypoxia (11, 69). These same stressors are also observed in the obese (1, 17, 46), and inhibit mTOR (11, 23, 78). However, REDD1 expression is reduced under growth-promoting conditions, including refeeding a fasted animal (46), or in skeletal muscle after a bout of resistance exercise (20). Although REDD1's mechanism of action on mTORC1 signaling remains unclear, findings suggest that it requires release of 14-3-3 proteins from the tuberous sclerosis complex 2 (TSC2) (11, 16, 71). In the absence of REDD1, Akt phosphorylates TSC2 and inhibits its GTPase activator function toward Rheb (71). When Rheb is associated with GTP, but not GDP, Rheb activates mTORC1 (32, 74). However, little is known about mTORC1 and its regulation during obesity, as it relates to REDD1 expression. Therefore, the initial goal of this study was to determine the role of REDD1 on mTORC1 in obese skeletal muscle. To achieve this goal we tested the working hypothesis that REDD1 is a significant regulator of mTORC1 in obese skeletal muscle, contributing to aberrant mTORC1 responses to nutrient stimuli.
EXPERIMENTAL DESIGN AND METHODS
Materials.
The rodent diets were purchased from Research Diets (New Brunswick, NJ). Kits for blood analysis of glucose, insulin, and nonesterified fatty acids were purchased from Sigma-Aldrich (St. Louis, MO; GAHK20), Alpco (Salem, NH; 80-INSMSU-E01), and Wako Chemical [Richmond, VA; HR Series NEFA-HR(2)], respectively. A Coomassie (Bradford) protein assay was performed using Coomassie Plus Reagent from Thermo Scientific (Rockford, IL). Western blotting was performed using a Bio-Rad Mini-PROTEAN Tetra Cell system. Polyvinylidene difluoride (PVDF) membrane was purchased from Bio-Rad Laboratories (Hercules, CA). Active Rheb kit was purchased from New East Biosciences (81201; King of Prussia, PA). Primary antibody for anti-actin (sc-8432) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for anti-phospho S6K1 T389 (9234), anti-phospho-Akt S473 (9202), anti-phospho 4E-BP1 T37/46 (9459), anti-4E-BP1 (9452), anti-mTOR (2972), anti-raptor (2280), and anti-GAPDH (2118) were purchased from Cell Signaling Technology (Beverly, MA). The antibody for REDD1 (10638-1-AP) was purchased from ProteinTech (Chicago, IL) and the HIF1α (NB100-105) was purchased from Novus Biologicals (Littleton, CO). The anti-rabbit IgG (7074) and anti-mouse IgG (7076) HRP-linked secondary antibodies were obtained from Cell Signaling Technology. Chemiluminescence imaging was performed on a Bio-Rad ChemiDoc MP Imager. Enhanced chemiluminescence (ECL) reagent was purchased from Thermo Scientific (Pierce ECL, West Pico, and West Femto) and Bio-Rad Laboratories (Clarity Western ECL).
Animal protocol.
The Institutional Animal Care and Use Committee of the University at Buffalo, SUNY approved all of the animal protocols and procedures. All mice were housed in an environmentally controlled room with a 12:12-h light-dark cycle on standard chow, unless where specified. First, 3-mo-old lean (Lepob/+ or Lepob/?+) and ob/ob (Lepob/Lepob) from Jackson Laboratories (Bar Harbor, ME; cat. no. 000632) male mice were euthanized following a 12-h fast. Also, 3-mo-old C57Bl/6 male mice (Jackson Laboratories; cat. no. 000664) or 3- to 4-mo-old wild-type and RTP801 (REDD1) knockout C57Bl/6x129SvEv mice (generated by Lexicon; Woodland, TX for Quark Pharmaceuticals; Fremont, CA) were fed a diet containing 10% fat (Research Diets; cat. no. D12492i) or 60% fat (Research Diets; cat. no. D12450Bi) for 8 wk, representing the low-fat (LF) or the high-fat diet (HF), respectively. These mice were fasted for 12 h, then euthanized. For the fasted/fed experiments, another group of 3-mo-old lean (Lepob/+ or Lepob/?+) and ob/ob (Lepob/Lepob) from Jackson Laboratories or 3- to 4-mo-old wild-type and RTP801 (REDD1) knockout C57Bl/6x129SvEv male mice were food deprived (fasted) or allowed to consume food ad libitum (fed) for the 18 h prior to the experiment, then euthanized. Following the respective experiment and/or treatment period, the plantar flexor complex (medial and lateral gastrocnemius, soleus, and plantaris) was removed tendon-to-tendon under 3% isoflurane anesthesia, weighed, and processed for immediate analysis or frozen in liquid nitrogen for subsequent analysis. The mice were then euthanized.
Blood metabolites and hormones.
Blood was collected by cardiac puncture into heparinized tubes, and plasma was separated from whole blood by centrifugation at 1,000 g for 10 min. Glucose was measured using a glucose (HK) kit per manufacturer's specifications (Sigma; St. Louis, MO). Insulin was measured using an ELISA per manufacturer's specifications (ALPCO; Salem, NH). Nonesterified free fatty acids were analyzed by enzymatic kit in accordance with the manufacturer's instructions (Wako Chemical; Richmond, VA).
Tissue homogenization.
Gastrocnemius samples were homogenized in 10 vol of CHAPS-containing buffer [40 mM HEPES (pH 7.5), 120 mM NaCl, 1 mM EDTA, 10 mM pyrophosphate, 10 mM β-glycerophosphate, 40 mM NaF, 1.5 mM sodium vanadate, 0.3% CHAPS, 0.1 mM PMSF, 1 mM benzamidine, 1 mM DTT, and protease inhibitor cocktail]. The resulting homogenate was centrifuged at 1,000 g for 5 min (at 4°C). A small aliquot of the fraction was taken, prior to adding 2X sodium dodecyl sulfate (SDS) sample buffer, for the determination of protein concentration for each sample. An equal volume of 2X SDS sample buffer was then added to an aliquot of this fraction for Western blot analysis. Samples were then boiled for 5 min and stored at −20°C for future analysis.
Protein assay.
A Coomassie/Bradford protein assay was performed in 96-well microplate. Bovine serum albumin (BSA) standard was used with concentrations ranging from 0 to 2,000 μg/ml. Samples were loaded in triplicate at dilutions of 1:10 and read using a Bio-Rad iMark Microplate Absorbance Reader at 595 nm, and then the protein concentrations were calculated.
Western blotting.
Equal quantities of protein were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred onto a PVDF membrane. After blocking in 5% milk in Tris-buffered saline (TBS) plus 0.1% Tween-20 (TBS-T) for 1 h, membranes were incubated with the specified primary antibody in TBS-T overnight at 4°C. Membranes were then washed and incubated with the respective secondary antibody for 1 h in a 5% milk/TBS-T solution at room temperature. The membranes were washed in TBS-T, then visualized via ECL and then quantified by measuring the luminescent signal using a Bio-Rad ChemiDoc MP Imager. The blots were analyzed using Bio-Rad Image Lab software, and the data are expressed as a percentage of the respective control group for each individual blot. As a positive control of REDD1 protein expression (see Fig. 1, inset), REDD1 and HIF1α protein expression in C2C12 myotubes were induced after 3 h of hypoxia exposure (vs. normoxic conditions) using a BBL GasPak System (Becton-Dickinson; Cockeysville, MD) using standard culturing conditions (82).
Fig. 1.
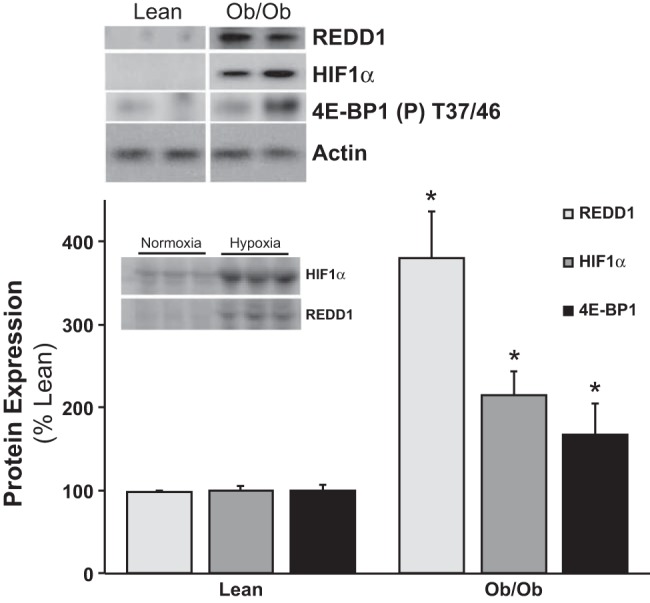
Obese skeletal muscle has elevated REDD1 (regulated in development and DNA damage responses 1) protein expression. Equal protein from fasted lean and ob/ob male mice plantar flexor complex muscle homogenates were analyzed by Western blot analysis for REDD1, hypoxia-inducible factor (HIF)-1α, phospho-4E-BP1 T37/46, and actin, then normalized to actin. Representative Western blots are shown. Means marked with an * are significantly different, P < 0.05 vs. lean (n = 10/group). Inset: C2C12 myotube lysates analyzed for REDD1 and HIF1α protein expression following either 3 h of normoxia or hypoxia.
Analysis of polysome aggregation.
Sucrose density gradient centrifugation was employed to analyze muscle polysome aggregation state (18, 83) following 8 wk of consuming a low- or a high-fat diet. Plantar flexor complex muscles were homogenized in 10 vol of buffer [50 mM HEPES (pH 7.4), 75 mM KCl, 5 mM MgCl2, 250 mM sucrose, 1% Triton X-100, 1.3% deoxycholate, and 100 μg/ml cycloheximide (per 5 ml)] using a drill press homogenizer. Homogenates were incubated on ice for 5 min, and then 150 μl/ml Tween-deoxycholate mix (1.34 ml Tween 20, 0.66 g deoxycholate, 18 ml sterile water) was added, and the samples were thoroughly mixed. Samples were incubated on ice for 15 min and then centrifuged at 1,000 g for 15 min at 4°C. The resulting supernatant (600 μl) was layered on a 20–47% linear sucrose density gradient [50 mM HEPES (pH 7.4), 75 mM KCl, 5 mM MgCl2] and centrifuged in a SW41 rotor at 40,000 rpm for 4 h at 4°C. Following centrifugation, the gradient was displaced upward (2 ml/min) using Fluorinert (Isco, Lincoln, NE) through a spectrophotometer, and the optical density at 254 nm was continuously recorded (chart speed, 150 cm/h).
RNA isolation.
First, a sample of the muscle homogenate was taken prior to gradient fractionation that would represent the total RNA fraction. Also, based upon the subpolysomal/polysomal profile, sample was collected from the polysome-containing fraction (18) in equal volume of TRIzol Reagent (Invitrogen) for RNA isolation. The RNA was extracted using the standard manufacturer's protocol and resuspended in RNA Storage Solution (Ambion). RNA (1 μg) was reverse transcribed and subjected to quantitative real-time PCR with primers corresponding to the mouse REDD1 and GAPDH mRNAs were used for amplification [REDD1 forward primer 5′-TGGTGCCCACCTTTCAGTTG-3′, reverse primer 5′-GTCAGGGACTGGCTGTAACC-3′; GAPDH forward primer 5′-GTTGTCTCCTGCGACTTCA-3′, reverse primer 5′-TGCTGTAGCCGTATTCATTG-3′ per Kimball et al. (37)]. REDD1 mRNA expression levels were normalized to GAPDH mRNA expression.
Analysis of raptor:mTOR association.
Similar to our previous work (83), muscle was homogenized in CHAPS buffer [40 mM HEPES (pH 7.5), 120 mM NaCl, 1 mM EDTA, 10 mM pyrophosphate, 10 mM β-glycerolphosphate, 40 mM NaF, 1.5 mM sodium vanadate, 0.3% CHAPS, 0.1 mM PMSF, 1 mM benzamidine, and 1 mM DTT], and the homogenate was mixed on a platform rocker for 20 min at 4°C and then clarified by centrifugation at 1,000 g for 3 min (4°C). An aliquot of supernatant containing 250 μg of protein was combined with 1.4 μl of anti-mTOR antibody, and mixed on a platform rocker overnight at 4°C. Immune complexes were isolated with a goat, anti-rabbit BioMag IgG (PerSeptive Diagnostics) bead slurry in 0.1% nonfat dry milk in CHAPS buffer. After a 1-h incubation at 4°C, the beads were collected using a magnetic stand, washed twice with CHAPS buffer, and once in CHAPS buffer containing 200 mM instead of 120 mM NaCl and 60 mM instead of 40 mM HEPES. The immunoprecipitates were solubilized in 1X SDS sample buffer, and then boiled for 5 min. The beads were removed by centrifugation and the supernatant was collected and subjected to SDS-PAGE. The proteins in the gel were transferred to nitrocellulose membranes, which were then incubated in anti-raptor or anti-mTOR antibody overnight at 4°C. The blots were visualized by ECL, and then the ratios of raptor to total mTOR were calculated.
Active Rheb.
Per the kit manufacturer's specifications (New East Biosciences; cat. no. 81201), configuration-specific anti-Rheb-GTP monoclonal antibody was incubated with 250 μg of muscle cytosolic lysates (CHAPS buffer described previously; 1,000 g). As a negative and a positive control in vitro, GDP and GTPγS loading of Rheb, respectively, was performed on fasted muscle lysates and included in the immunoprecipitation. The bound active Rheb was immunoprecipitated by protein A/G agarose (4°C, 1 h), then pelleted and washed three times in CHAPS buffer. After the final wash, the pellet was resuspended in 2X SDS-sample buffer and boiled. The precipitated active Rheb was detected by Western immunoblot analysis (described above) using anti-Rheb rabbit polyclonal antibody provided in the kit to measure the active Rheb-GTP levels.
Statistical analysis.
Statistics were performed using IBM SPSS v. 22.0.0 software. The results are expressed as means ± SE. Comparisons were made for each variable using a t-test (2-tailed) or an ANOVA (1-way or 2-way) with a Tukey HSD post hoc test, to establish significant differences between groups, only after the F statistic indicated an overall significance in the data. The significance levels was set a priori at P < 0.05.
RESULTS
Our first observation of increased REDD1 expression in obese skeletal muscle stemmed from a proteomics screen, which was verified with Western blot analysis as shown in Fig. 1. As a positive control for REDD1 and HIF1α protein expression, myotube cultures were subjected to normoxic and hypoxic conditions (see Fig. 1, inset). Compared with fasted lean skeletal muscle that had low REDD1 and HIF1α protein expression levels, fasted leptin-resistant, ob/ob mouse muscle expression was significantly higher (P < 0.05 vs. lean control). In these same mice, we have previously reported a dysregulation of mTORC1 signaling (hyperactive S6K1) and translational capacity in fasted ob/ob skeletal muscle (18), as corroborated by the high 4E-BP1 phosphorylation (Fig. 1; P < 0.05) in muscle from fasted obese mice. While the aforementioned results were conducted in fasted conditions, McGhee et al. (46) have shown that REDD1 expression was differentially regulated by food deprivation and feeding in both nondiabetic and type I diabetic rats. Thus we followed-up the aforementioned observations by examining skeletal muscle REDD1 expression in fasted and fed obese mice. Fasted blood fatty acids, glucose, and insulin concentrations were higher (P < 0.05 vs. fasted lean) in the ob/ob mice (Table 1). Expectedly, nutrient (fasted vs. fed) status contributed to significant increases (P < 0.05) in circulating glucose and insulin, and a reciprocal decrease in fatty acids regardless of group (Table 1), although more so in the ob/ob fed group (P < 0.05 vs. fed lean). Fasted obese mouse muscle S6K1 and Akt phosphorylation were higher (Fig. 2, A and B; P < 0.05) than lean fasted controls, and fed mice (lean and obese) had significantly higher (Fig. 2, A and B; P < 0.05) S6K1 and Akt phosphorylation vs. the fasted groups. This resulted in a fasted-to-fed percent change of 107.3 ± 6.5 vs. 29.9 ± 4.4 (P = 0.001) for S6K1 and 247.4 ± 64.8 vs. 168.2 ± 26.6 (P = 0.37) for Akt phosphorylation in the lean and ob/ob groups, respectively. Fasted obese mouse muscle had higher REDD1 expression (Fig. 2C; P < 0.05) compared with the lean fasted group. Despite an overall reduction (P < 0.05) of REDD1 expression in the fed mice (vs. the fasted groups), REDD1 remained higher (Fig. 2C; P < 0.05) in the ob/ob mice vs. the lean, which translated into a trending fasted-to-fed percent change of −68.3.3 ± 8.3 vs. −42.3 ± 4.8 (P = 0.06) in the lean and ob/ob groups, respectively. The direct regulator of mTOR, active Rheb (Rheb-GTP), was trending higher (P = 0.12) in the fasted obese mouse muscle vs. the lean fasted (Fig. 2D), while the lean fed muscle had significantly higher (P < 0.05) Rheb-GTP vs. the lean fasted group. Rheb-GTP was minimally higher in the obese fed group vs. fasted lean, with no difference from the ob/ob fasted and lower (P < 0.05) than the lean fed, resulting in an increased fasted-to-fed percent change of 98.6 ± 22.9 vs. 17.15 ± 17.8 (P = 0.02) in the lean and ob/ob groups, respectively. Similar to our previous observations (18, 19), raptor associated with mTOR and AMPK phosphorylation were lower (Fig. 2, E and F; P < 0.05) in fasted ob/ob mouse muscle compared with the lean. The raptor association with mTOR (P < 0.05) and AMPK phosphorylation (trending P = 0.13) were lower (Fig. 2, E and F) in the lean fed mice vs. the lean fasted mice, despite a lack of change in the fed ob/ob mice (vs. the lean fasted). These findings exhibited a fasted-to-fed percent change of −25.4 ± 7.4 vs. 10.0 ± 15.6 (P = 0.03) for raptor:mTOR association and −15.1 ± 11.3 vs. 67.2 ± 22.7 (P = 0.04) for AMPK phosphorylation in the lean and ob/ob groups, respectively.
Table 1.
Characteristics of fasted and fed, lean and ob/ob mice
| Lean Fasted | ob/ob Fasted | Lean Fed | ob/ob Fed | |
|---|---|---|---|---|
| Body weight, g | 26.2 ± 1.4 | 46.2 ± 2.0* | 28.9 ± 0.8 | 50.2 ± 2.2* |
| Plantar flexor complex, g | 0.15 ± 0.01 | 0.12 ± 0.01* | 0.16 ± 0.0.01 | 0.11 ± 0.01* |
| Liver, g | 0.9 ± 0.1 | 2.3 ± 0.1* | 1.2 ± 0.1*† | 2.8 ± 0.3*† |
| eWAT, g | 0.5 ± 0.1 | 4.2 ± 0.2* | 0.6 ± 0.1 | 3.9 ± 0.1* |
| Glucose, mg/dl | 72.5 ± 1.4 | 225.2 ± 16.2* | 223.1 ± 21.2* | 414.1 ± 34.5*† |
| Insulin, μIU/ml | 2.69 ± 0.03 | 3.37 ± 0.25* | 3.67 ± 0.50* | 13.31 ± 1.94*† |
| HOMA-IR | 0.47 ± 0.01 | 2.04 ± 0.14* | 1.84 ± 0.39* | 12.97 ± 2.39*† |
| NEFA, mmol/l | 0.66 ± 0.05 | 1.07 ± 0.24* | 0.40 ± 0.03*† | 0.65 ± 0.07 |
Values are means ± SE. eWAT, epididymal white adipose tissue; HOMA-IR, homeostasis model assessment of insulin resistance; NEFA, nonesterified fatty acid.
Statistically different from lean fasted (P < 0.05).
Significantly different from ob/ob fasted (P < 0.05).
Fig. 2.

Lean and obese mouse muscle mTOR signaling responses to fasting and feeding. Equal protein from fasted and fed, lean (L) and ob/ob (Ob) male mice plantar flexor complex muscle homogenates were analyzed by Western blot analysis. A: phospho-S6K1 T389 and actin, then normalized to actin. B: phospho-Akt S473 and actin, then normalized to actin. C: REDD1 and actin, then normalized to actin. D: Rheb-GTP from active-Rheb immunoprecipitates. Inset: negative and positive controls from Rheb-GTP. E: raptor and mTOR from mTOR coimmunoprecipitations. F: phospho-AMPK T172. Representative Western blots are shown. Means marked with an * are significantly different, P < 0.05 vs. lean fasted; † are significantly different, P < 0.05 vs. ob/ob fasted; and †† are significantly different, P < 0.05 vs. lean fed (n = 5–8/group).
In an attempt to limit possible genetic influence of the ob/ob model on REDD1, we employed a high-fat diet approach to induce obesity. Similar to our previous report (80), increases in body weight [+26.3% (+10.0 g) vs. low fat] confirmed that consumption of the high-fat diet induced obesity in the mice after 8 wk. Under fasted conditions, diet-induced obesity promoted higher phosphorylation of S6K1 (Fig. 3A; P < 0.05), compared with the low-fat-fed mice. Similar to our findings in ob/ob mice, REDD1 protein expression was higher (Fig. 3B; P < 0.05) in the fasted mouse muscle from the high-fat-fed group vs. the low-fat group. In line with the protein data, high-fat-fed mouse muscle had higher total REDD1 mRNA expression (Fig. 3C; P < 0.05) vs. the low-fat-fed group. Muscle REDD1 mRNA expression was also higher in the polysomal fraction from fasted mice fed a high-fat diet (Fig. 3D; P < 0.05) compared with the low-fat-fed mice.
Fig. 3.

Skeletal muscle from high-fat-fed mice has elevated REDD1 protein and mRNA expression. A and B: equal protein from fasted low (10%)-fat- and high (60%)-fat-fed male mice plantar flexor complex muscle homogenates was analyzed by Western blot analysis for phospho-S6K1 T389, REDD1, and actin, then normalized to actin. Representative Western blots are shown. C and D: skeletal muscle REDD1 mRNA expression from total and polysome fractions. A sample of the muscle homogenate was taken prior to gradient fractionation that would represent the total RNA fraction, and a sample was collected from the polysome-containing fraction for RNA isolation and subjected to quantitative real-time PCR. Polysome aggregation from sucrose density gradient of plantar flexor complex muscle from fasted low-fat and high-fat-fed male mice. A representative profile from each condition is presented; dashed line is low-fat-fed group, and solid line is high-fat-fed group. Peaks corresponding to 40S and 60S ribosomal subunits, 80S monomers, and polysomes are shown. Means marked with an * are significantly different, P < 0.05 vs. low fat (n = 8/group).
To directly address the role of REDD1 in obese skeletal muscle, wild-type (WT) and knockout (KO) RTP801 (REDD1) mice were fed either a low- or a high-fat diet for 8 wk. Similar to the abovementioned diet-induced obesity findings, WT-HF mice gained significantly more weight [+30.8% (+10.4 g); P < 0.05] than WT-LF group (Table 2 and Fig. 4; P < 0.05), whereas the KO-HF group only gained +11.2% (4.7 g) vs. the KO-LF group (Table 2 and Fig. 4). Further support for the diet model of obesity is provided by the observation that the high-fat-fed groups had larger livers and more epididymal white adipose tissue (eWAT) (P < 0.05), with no differences in muscle mass. When the eWAT were normalized to body weight, there was a diet effect (P < 0.05), regardless of group, and normalized muscle mass was lower (P < 0.05) in the WT-HF, KO-LF and -HF vs. the WT-LF. Normalized liver-to-body weight mass showed no differences between groups. Interestingly, although the quantity of food consumed by the KO-HF mice was less (P < 0.05) than the other groups, when expressed as kilocalories per day (LF = 3.8 kcal/g food and HF = 5.2 kcal/g food) or total kilocalories consumed (59 days), both the WT and KO groups fed a high-fat diet consumed more kilocalories (Table 2; P < 0.05) vs. the low-fat-fed groups. Blood glucose, insulin, HOMA index, and fatty acids were significantly higher (P < 0.05) in the high-fat-fed groups, independent of genotype (Table 2). There was a significantly higher fasting blood glucose concentration noted in the WT-HF compared with the KO-HF. Fasting S6K1 and 4E-BP1 activation was significantly higher (Fig. 5, A and B; P < 0.05) in muscle from the WT-HF group vs. the WT-LF group. Although fasted S6K1 activation was higher (P < 0.05) in the KO-LF mice vs. the WT-LF mice, the high-fat diet had no further effect on S6K1 (Fig. 5A) and 4E-BP1 (Fig. 5B) phosphorylation in the KO mice. Similar to the above findings, muscle REDD1 protein expression was elevated in the WT-HF fed group compared with the WT-LF mice (Fig. 5C; P < 0.05), and the KO-LF/-HF mice showed extremely low REDD1 protein expression.
Table 2.
Characteristics of REDD1 WT and KO mice fed a low- or high-fat diet
| WT-LF | WT-HF | KO-LF | KO-HF | |
|---|---|---|---|---|
| Body weight, g | 33.7 ± 1.4 | 44.1 ± 1.8* | 41.7 ± 1.6* | 46.4 ± 1.9* |
| Plantar flexor complex, g | 0.16 ± 0.01 | 0.17 ± 0.01 | 0.16 ± 0.0.01 | 0.17 ± 0.01 |
| Liver, g | 1.3 ± 0.1 | 1.5 ± 0.1* | 1.3 ± 0.1 | 1.7 ± 0.1* |
| eWAT, g | 1.1 ± 0.1 | 2.3 ± 0.1* | 1.7 ± 0.1* | 2.0 ± 0.2* |
| Food intake | ||||
| g/day | 3.5 ± 0.1 | 3.2 ± 0.1 | 3.3 ± 0.1 | 2.9 ± 0.1* |
| kcal/day | 13.4 ± 0.4 | 16.8 ± 0.5‡ | 12.5 ± 0.4 | 15.3 ± 0.5‡ |
| Total kcal | 790.8 ± 20.9 | 989.2 ± 29.7‡ | 740.3 ± 25.7 | 902.7 ± 29.4‡ |
| Glucose, mg/dl | 82.8 ± 33.0 | 266.5 ± 16.9* | 112.8 ± 32.0 | 229.2 ± 13.1*† |
| Insulin, μIU/ml | 2.71 ± 0.02 | 4.28 ± 0.69* | 2.98 ± 0.10 | 5.27 ± 1.47* |
| HOMA-IR | 0.55 ± 0.22 | 2.85 ± 0.63* | 0.84 ± 0.25 | 3.59 ± 0.66* |
| NEFA, mmol/l | 0.85 ± 0.05 | 1.07 ± 0.16* | 0.73 ± 0.04 | 0.58 ± 0.08* |
Values are means ± SE. WT and KO are REDD1 wild type and knockout, respectively; LF, low-fat diet; HF, high-fat diet.
Statistically different from WT-LF (P < 0.05).
Statistically different from the LF group (P < 0.05).
Significantly different from WT-HF (P < 0.05).
Fig. 4.
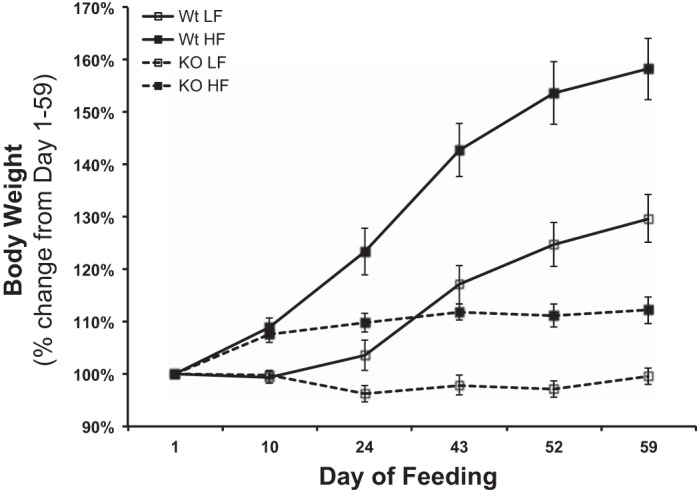
Consumption of a high-fat diet promotes more relative body weight gain in wild-type but not REDD1 knockout mice. The percent change in body weight from the initiation of the study (day 1) to the end of the study (day 59). Body weights were taken every 7–10 days and expressed in g. REDD1 wild-type (WT) and knockout (KO), low (10%)-fat (LF)-, and high (60%)-fat (HF)-fed male mice; (n = 8/group).
Fig. 5.

Skeletal muscle mTORC1 signaling from high-fat-fed REDD1 knockout mice. Equal protein from fasted REDD1 WT and KO, LF- and HF-fed male mice plantar flexor complex muscle homogenates were analyzed by Western blot analysis. A: phospho-S6K1 T389 and GAPDH, then normalized to GAPDH. B: 4E-BP1 activation by examining gamma:total 4E-BP1. C: REDD1 and GAPDH, then normalized to GAPDH. Representative Western blots are shown. Means marked with an * are significantly different, P < 0.05 vs. WT-LF; and † are significantly different, P < 0.05 vs. WT-HF (n = 8/group).
Although indicative that REDD1 contributes to body weight homeostasis, the results of the high-fat feeding study also suggested an altered response to nutrients. Thus WT and KO mice were subjected to the same fasted and fed paradigm performed in the lean and ob/ob mice (see Table 1 and Fig. 2) to examine responses in mTOR signaling. As expected, KO mouse muscle had extremely low REDD1 protein expression vs. the WT mice (Fig. 6A; P < 0.05), and there was a trend (P = 0.07) for REDD1 expression to be lower in fed compared with fasted WT mice (Fig. 6A; P < 0.05 vs. fasted WT), resulting in an increased fasted-to-fed percent difference of −47.3 ± 14.6 for the WT group. Fasted S6K1 and 4E-BP1 phosphorylation was higher (Fig. 6, B and C; P < 0.05) in fasted KO vs. WT mice. In the fed state, the WT mice displayed higher S6K1 and 4E-BP1 phosphorylation vs. the WT fasted (Fig. 6, B and C; P < 0.05), although the effect appeared to be blunted in the KO mice. This resulted in a fasted-to-fed percent difference of 322.1 ± 150.4 vs. −24.7 ± 20.3 (P < 0.05) for S6K1 and 137.0 ± 27.7 vs. 9.76 ± 8.21 (P < 0.05) for 4E-BP1 phosphorylation in the WT and KO groups, respectively. Other than lower (Fig. 6D; P < 0.05) raptor:mTOR association in muscle from the fed vs. WT mouse muscle vs. fasted WT mice, no other differences were observed between groups. This resulted in a fasted-to-fed percent Δ of −20.6 ± 2.9 vs. −11.7 ± 10.6 (P = 0.47) in the WT and KO groups, respectively. Consistent with a recent report (15) in serum-starved REDD1−/− MEFs (mouse embryonic fibroblasts), Akt S473 phosphorylation was significantly lower in the fasted KO mouse muscle vs. the fasted WT (Fig. 6E; P < 0.05). Yet, Akt phosphorylation remained low in the fed KO mouse muscle compared with the WT fed (Fig. 6C; P < 0.05). The resultant fasted-to-fed percent difference was 67.4 ± 35.7 vs. 53.8 ± 25.1 (P = 0.75) for Akt phosphorylation in the WT and KO groups, respectively. Correspondingly, circulating insulin concentrations were significantly lower in the fasted KO mouse muscle vs. the fasted WT (Table 3; P < 0.05). Although higher in the fed state (P < 0.05 vs. KO fasted), insulin remained lower in the fed KO mouse muscle compared with the WT fed (Table 3; P < 0.05). Notably, despite the lower insulin concentrations, no difference in blood glucose levels were observed between WT and KO mice in either the fed or fasted state (Table 3).
Fig. 6.

REDD1 WT and KO mouse muscle mTOR signaling responses to fasting and feeding. Equal protein from fasted and fed, REDD1 WT and KO male mice plantar flexor complex muscle homogenates were analyzed by Western blot analysis. A: REDD1 and actin, then normalized to actin. B: phospho-S6K1 T389 and actin, then normalized to actin. C: phospho-4E-BP1 T37/46 and actin, then normalized to actin. D: raptor and mTOR from mTOR coimmunoprecipitations. E: phospho-Akt S473 and actin, then normalized to actin. Representative Western blots are shown. Means marked with an * are significantly different, P < 0.05 vs. WT fasted; † are significantly different, P < 0.05 vs. KO groups (n = 7/group).
Table 3.
Characteristics of fasted and fed, REDD1 WT and KO mice
| WT Fasted | KO Fasted | WT Fed | KO Fed | |
|---|---|---|---|---|
| Insulin, μIU/ml | 4.93 ± 0.62 | 2.83 ± 0.71* | 8.72 ± 0.71*† | 5.36 ± 0.23‡ |
| Glucose, mg/dl | 87.5 ± 8.3 | 155.5 ± 52.7 | 268.2 ± 11.7* | 281.1 ± 36.1* |
| NEFA, mmol/l | 0.76 ± 0.03 | 0.65 ± 0.05 | 0.64 ± 0.06 | 0.57 ± 0.03* |
Values are means ± SE.
Statistically different from WT fasted (P < 0.05).
Significantly different from KO groups (P < 0.05).
Significantly different from KO fasted (P < 0.05).
DISCUSSION
The inhibitory role of REDD1 in inhibiting mTOR activity in skeletal muscle was first demonstrated in dexamethasone-treated rodents (78). Similar findings in skeletal muscle were reported during models of muscle atrophy (33, 41, 46, 52). Likewise, exogenously increased expression of REDD1 in skeletal muscle promotes smaller soleus fibers vs. control muscle under normoxic conditions (24). In contrast, a handful of studies have reported that anabolic stimuli (20, 46) reduced REDD1 expression in skeletal muscle. Although aberrant mTORC1 signaling has been well established in models of obesity, and REDD1 upregulation has a negative impact on regulators of skeletal muscle growth, little is known about the possible role REDD1 might play in the augmentation of the aberration. The findings from the present study show that in skeletal muscle displaying either abnormally high or a loss of REDD1 protein expression promotes aberrant mTORC1 signaling that has negative consequences during altered nutrient states.
Our laboratory (18, 19) and others (54, 62, 75, 76) have demonstrated the dysregulation of mTOR signaling in obese skeletal muscle. While hyperactive mTOR under fasted conditions is associated with limited responses to insulin (35, 62, 76), S6K1−/− mice are more insulin sensitive and resistant to a high-fat diet (76). Similarly, treating obese mice with AICAR, an inhibitor of mTOR (10), normalizes metabolic and growth processes (18). Thus the prevailing paradigm would suggest that expression of REDD1 would be low in skeletal muscle from the obese. The current findings show the converse in two different models of obesity (ob/ob and diet-induced), despite hyperactive S6K1 and 4E-BP1 phosphorylation in fasted obese skeletal muscle. While fed, lean mice displayed a robust activation of mTORC1 (increases in S6K1 phosphorylation and Rheb-GTP, and less raptor bound to mTOR), fed obese mice displayed modest increases in mTORC1 pathway activation. Rheb-GTP was slightly elevated under fasted conditions in the ob/ob mouse muscle vs. the lean, which corroborates findings in diet-induced obese heart tissue (66). Consistent with our previous findings (18, 19), raptor associated with mTOR and AMPK phosphorylation state were lower in muscle in fasted obese vs. the lean fasted mice that appeared to increase with feeding. In line with this, activated AMPK can promote TSC complex formation (directly) and a higher affinity of raptor for mTOR (28, 81, 83), thus inhibiting its function. AMPK phosphorylation was reduced with feeding in the lean, but actually increased in the obese, which may partially explain the aberrant raptor-mTOR response to feeding, possibly independent of REDD1 (71). The current data coupled with our previous findings suggest that elevated REDD1 expression in fasted muscle from the ob/ob mice is associated with reduced TSC2 complex formation (18) and can promote Rheb GTP loading and mTORC1 signaling (low raptor-mTOR association). Although it is not currently known, the Rag proteins may play a role in obesity. Given the high circulating concentrations of amino acids in the obese (8, 53), the Rag pathway (and mTOR) may be constitutively activated. Although unknown, an upregulation of the Rag pathway could help explain the relative resistance of mTORC1 to REDD1 expression in obese muscle, since REDD1 and growth factors signal to mTORC1 through TSC/Rheb rather than Rag GTPases (14, 15, 63).
Given the limited data on the mechanism involved in the regulation of REDD1 expression in skeletal muscle and/or obesity, we turn to regulators of REDD1, such as HIF1α. A lack of, or reductions in, HIF1α promotes higher oxidative capacity and an increased capillary density (45) and mitigates the negative effects of diet-induced obesity (68), whereas obese skeletal muscle has reduced capillary density, muscle perfusion, and ability to perform work (25). Moreover, mTOR-mediated phosphorylation of 4E-BP1 has been shown to promote translation of HIF1α mRNA, leading to increased expression of the REDD1 mRNA (22). More evidence for support from simultaneous elevation of REDD1 and mTOR signaling comes from findings in clear-cell renal cell carcinoma (ccRCC), where the von Hippel-Lindau (VHL) gene is frequently inactivated, leading to an upregulation of HIF-1 and REDD1 (40), coinciding with hyperactive mTOR signaling. Interestingly, VHL gene expression and protein alterations have been reported in human skeletal muscle (3), and obese individuals present VHL gene defects (47).
In inducing obesity in REDD1 KO mice by feeding a high-fat diet, we unexpectedly observed that they did not gain significant amounts of weight compared with the WT-HF mice, despite a hyperactive mTORC1 pathway under fasted conditions. To follow up on this, REDD1 WT and KO mice that underwent the fasted/fed paradigm displayed comparable responses similar to the lean and ob/ob mice, respectively. Fasted KO mouse muscle displayed higher S6K1 and 4E-BP1 phosphorylation vs. the WT mice, although the response of these kinases was blunted in the fed KO mice vs. a robust increase in the fed WT mice. Similarly, when REDD1−/− MEFs are deprived of serum, mTORC1 signaling was higher vs. REDD1+/+ MEFs (15). Interestingly, the circulating insulin concentrations were lower in the KO mice regardless of nutrient status compared with the WT mice, suggesting that insulin secretion and/or production is low in the REDD1 KO mice, which was supported by lower Akt phosphorylation on S473 in muscle. Unlike the KO mice that are unable to increase REDD1 expression to limit mTORC1, the obese WT mice may induce REDD1 to limit hyperactive mTORC1 signaling, as a defense mechanism under conditions of nutrient insufficiency (i.e., fasting) and/or reduced substrate mobilization. Thus the combination of higher circulating insulin and a large increase in Akt phosphorylation may have provided for a more positive mTORC1 fasted-to-fed response in the ob/ob vs. the REDD KO mice.
There are two models that may describe a limited response in the fed vs. the fasted animals exhibiting aberrant mTORC1 signaling. Consistent with the current data's models displaying hyperactive mTORC1 (e.g., ob/ob, high-fat diet, REDD1 KO), the previous work by Um et al. (76) and others since (56, 62) shows that hyperactive mTORC1, specifically through S6K1, negatively feeds back to IRS-1, downregulating insulin signaling. Elevated insulin concentrations are associated with increased REDD1 expression (26, 60) although the data of McGhee et al. suggest that insulin is not a factor in type 1 diabetes (46). Notwithstanding differences in mTOR signaling under fasted conditions, the obese mice in the current study display elevated REDD1 expression in muscle comparable to those observed in type 1 diabetic mice (31, 46). Despite the larger increase in circulating insulin observed in the ob/ob fed group (vs. lean fed), both models show similar relative reductions in REDD1 protein when comparing the fasted vs. fed state that was still higher than the lean controls (46). Similar to previous observations in high-fat-fed rodents in the fasted state (9, 70, 77), we observed an increase in Akt phosphorylation compared with the lean, although the fed response was similar between both groups. While not measured, glucocorticoids may also be elevated in the obese under both fasted and fed conditions (4, 50, 84), contributing to the irregular and blunted mTORC1 responses by the obese (46). The findings of Frost et al. (26) support the potential for high circulating insulin concentrations regulating REDD1 expression in muscle through a PI3-kinase-dependent mechanism. However, akin to the REDD1 KO model, hyperactive mTOR might play a role in β-cell loss in type 2 diabetes, as suggested during raptor knockdown (i.e., mTOR inactivation) of INS-1 cells increased insulin production and secretion (42). Thus the hyperactivation of mTORC1 due to a loss of REDD1 may reduce insulin production and secretion, negatively affecting insulin-sensitive tissues, although this remains to be determined.
In conclusion, the collective findings from the studies reported herein show that skeletal muscle displaying either significantly high (ob/ob and diet-induced obesity) or a complete loss of REDD1 protein expression (REDD1 KO) promotes aberrant mTORC1 signaling responses to altered nutrient states. The role of REDD1 in skeletal muscle is not as clear as previously thought, and high or extremely low expression of REDD1 exhibits a more complex model of nutrient and hormonal regulation of mTORC1.
GRANTS
This research was supported by University at Buffalo, SUNY (D. L. Williamson) and National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-15658 (S. R. Kimball).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: D.L.W., R.M.T., E.F., and S.R.K. conception and design of research; D.L.W., Z.L., and C.M.D. performed experiments; D.L.W., Z.L., and C.M.D. analyzed data; D.L.W., S.R.K., and C.M.D. interpreted results of experiments; D.L.W. and C.M.D. prepared figures; D.L.W. drafted manuscript; D.L.W., Z.L., R.M.T., E.F., S.R.K., and C.M.D. edited and revised manuscript; D.L.W., Z.L., R.M.T., E.F., S.R.K., and C.M.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank A. Raslawsky for technical support and Dr. J. Wilson for laboratory equipment use; Dr. W. Sigurdson at the Confocal Microscope and Flow Cytometry Facility for technical support; and Dr. D. Pendergast and Dr. D. Wright for critique of the manuscript.
REFERENCES
- 1.Al-Aubaidy HA, Jelinek HF. Oxidative DNA damage and obesity in type 2 diabetes mellitus. Eur J Endocrinol 164: 899–904, 2011 [DOI] [PubMed] [Google Scholar]
- 2.Alley DE, Chang VW. The changing relationship of obesity and disability, 1988–2004. JAMA 298: 2020–2027, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Ameln H, Gustafsson T, Sundberg CJ, Okamoto K, Jansson E, Poellinger L, Makino Y. Physiological activation of hypoxia inducible factor-1 in human skeletal muscle. FASEB J 19: 1009–1011, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Anagnostis P, Athyros VG, Tziomalos K, Karagiannis A, Mikhailidis DP. Clinical review: The pathogenetic role of cortisol in the metabolic syndrome: a hypothesis. J Clin Endocrinol Metab 94: 2692–2701, 2009 [DOI] [PubMed] [Google Scholar]
- 5.Anthony JC, Reiter AK, Anthony TG, Crozier SJ, Lang CH, MacLean DA, Kimball SR, Jefferson LS. Orally administered leucine enhances protein synthesis in skeletal muscle of diabetic rats in the absence of increases in 4E-BP1 and S6K1 phosphorylation. Diabetes 51: 928–936, 2002 [DOI] [PubMed] [Google Scholar]
- 6.Augert G, Monier S, Le Marchand-Brustel Y. Effect of exercise on protein turnover in muscles of lean and obese mice. Diabetologia 29: 248–253, 1986 [DOI] [PubMed] [Google Scholar]
- 7.Augert G, Van de Werve G, Le Marchand-Brustel Y. Effect of work-induced hypertrophy on muscle glucose metabolism in lean and obese mice. Diabetologia 28: 295–301, 1985 [DOI] [PubMed] [Google Scholar]
- 8.Batch BC, Shah SH, Newgard CB, Turer CB, Haynes C, Bain JR, Muehlbauer M, Patel MJ, Stevens RD, Appel LJ, Newby LK, Svetkey LP. Branched chain amino acids are novel biomarkers for discrimination of metabolic wellness. Metabolism 62: 961–969, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bollheimer LC, Buettner R, Pongratz G, Brunner-Ploss R, Hechtl C, Banas M, Singler K, Hamer OW, Stroszczynski C, Sieber CC, Fellner C. Sarcopenia in the aging high-fat fed rat: a pilot study for modeling sarcopenic obesity in rodents. Biogerontology 13: 609–620, 2012 [DOI] [PubMed] [Google Scholar]
- 10.Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem 277: 23977–23980, 2002 [DOI] [PubMed] [Google Scholar]
- 11.Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG., Jr Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 18: 2893–2904, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci USA 95: 1432–1437, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campion DR, Purchas RW, Merkel RA, Romsos DR. Genetic obesity and the muscle satellite cell. Proc Soc Exp Biol Med 176: 143–147, 1984 [DOI] [PubMed] [Google Scholar]
- 14.Dennis MD, Baum JI, Kimball SR, Jefferson LS. Mechanisms involved in the coordinate regulation of mTORC1 by insulin and amino acids. J Biol Chem 286: 8287–8296, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dennis MD, McGhee NK, Jefferson LS, Kimball SR. Regulated in DNA damage and development 1 (REDD1) promotes cell survival during serum deprivation by sustaining repression of signaling through the mechanistic target of rapamycin in complex 1 (mTORC1). Cell Signal 25: 2709–2716, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev 22: 239–251, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drager LF, Li J, Reinke C, Bevans-Fonti S, Jun JC, Polotsky VY. Intermittent hypoxia exacerbates metabolic effects of diet-induced obesity. Obesity (Silver Spring) 19: 2167–2174, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drake JC, Alway SE, Hollander JM, Williamson DL. AICAR treatment for 14 days normalizes obesity-induced dysregulation of TORC1 signaling and translational capacity in fasted skeletal muscle. Am J Physiol Regul Integr Comp Physiol 299: R1546–R1554, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drake JC, Benninger L, Williamson DL. 8-Weeks of β-GPA treatment reduces body mass while positively altering translation initiation in obese skeletal muscle. J Obes Weight Loss Ther 01/2011; 10.4172/2165-7904.1000101 [DOI] [Google Scholar]
- 20.Drummond MJ, Fujita S, Abe T, Dreyer HC, Volpi E, Rasmussen BB. Human muscle gene expression following resistance exercise and blood flow restriction. Med Sci Sports Exerc 40: 691–698, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Durschlag RP, Layman DK. Skeletal muscle growth in lean and obese Zucker rats. Growth 47: 282–291, 1983 [PubMed] [Google Scholar]
- 22.Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, Clish CB, Murphy LO, Manning BD. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39: 171–183, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ellisen LW, Ramsayer KD, Johannessen CM, Yang A, Beppu H, Minda K, Oliner JD, McKeon F, Haber DA. REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell 10: 995–1005, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Favier FB, Costes F, Defour A, Bonnefoy R, Lefai E, Bauge S, Peinnequin A, Benoit H, Freyssenet D. Downregulation of Akt/mammalian target of rapamycin pathway in skeletal muscle is associated with increased REDD1 expression in response to chronic hypoxia. Am J Physiol Regul Integr Comp Physiol 298: R1659–R1666, 2010 [DOI] [PubMed] [Google Scholar]
- 25.Frisbee JC. Impaired skeletal muscle perfusion in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol 285: R1124–R1134, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Frost RA, Huber D, Pruznak A, Lang CH. Regulation of REDD1 by insulin-like growth factor-I in skeletal muscle and myotubes. J Cell Biochem 108: 1192–1202, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, Sonenberg N. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev 13: 1422–1437, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30: 214–226, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123: 569–580, 2005 [DOI] [PubMed] [Google Scholar]
- 30.Howard G, Steffen JM, Geoghegan TE. Transcriptional regulation of decreased protein synthesis during skeletal muscle unloading. J Appl Physiol 66: 1093–1098, 1989 [DOI] [PubMed] [Google Scholar]
- 31.Hulmi JJ, Silvennoinen M, Lehti M, Kivela R, Kainulainen H. Altered REDD1, myostatin, and Akt/mTOR/FoxO/MAPK signaling in streptozotocin-induced diabetic muscle atrophy. Am J Physiol Endocrinol Metab 302: E307–E315, 2012 [DOI] [PubMed] [Google Scholar]
- 32.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 17: 1829–1834, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kelleher AR, Kimball SR, Dennis MD, Schilder RJ, Jefferson LS. The mTORC1 signaling repressors REDD1/2 are rapidly induced and activation of p70S6K1 by leucine is defective in skeletal muscle of an immobilized rat hindlimb. Am J Physiol Endocrinol Metab 304: E229–E236, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kemp JG, Blazev R, Stephenson DG, Stephenson GM. Morphological and biochemical alterations of skeletal muscles from the genetically obese (ob/ob) mouse. Int J Obes (Lond) 33: 831–841, 2009 [DOI] [PubMed] [Google Scholar]
- 35.Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology 146: 1473–1481, 2005 [DOI] [PubMed] [Google Scholar]
- 36.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110: 163–175, 2002 [DOI] [PubMed] [Google Scholar]
- 37.Kimball SR, Do AN, Kutzler L, Cavener DR, Jefferson LS. Rapid turnover of the mTOR complex 1 (mTORC1) repressor REDD1 and activation of mTORC1 signaling following inhibition of protein synthesis. J Biol Chem 283: 3465–3475, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kimball SR, Jefferson LS, Fadden P, Haystead TAJ, Lawrence JC. Insulin and diabetes cause reciprocal changes in the association of eIF4E AND PHAS-I rat skeletal muscle. Am J Physiol Cell Physiol 270: C705–C709, 1996 [DOI] [PubMed] [Google Scholar]
- 39.Kimball SR, Jurasinki CV, Lawrence JC, Jr, Jefferson LS. Insulin stimulates protein synthesis in skeletal muscle by enhancing the association of eIF-4E and eIF-4G. Am J Physiol Cell Physiol 272: C754–C759, 1997 [DOI] [PubMed] [Google Scholar]
- 40.Kucejova B, Pena-Llopis S, Yamasaki T, Sivanand S, Tran TA, Alexander S, Wolff NC, Lotan Y, Xie XJ, Kabbani W, Kapur P, Brugarolas J. Interplay between pVHL and mTORC1 pathways in clear-cell renal cell carcinoma. Mol Cancer Res 9: 1255–1265, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lang CH, Frost RA, Vary TC. Acute alcohol intoxication increases REDD1 in skeletal muscle. Alcohol Clin Exp Res 32: 796–805, 2008 [DOI] [PubMed] [Google Scholar]
- 42.Le Bacquer O, Queniat G, Gmyr V, Kerr-Conte J, Lefebvre B, Pattou F. mTORC1 and mTORC2 regulate insulin secretion through Akt in INS-1 cells. J Endocrinol 216: 21–29, 2013 [DOI] [PubMed] [Google Scholar]
- 43.Lin L, Qian Y, Shi X, Chen Y. Induction of a cell stress response gene RTP801 by DNA damaging agent methyl methanesulfonate through CCAAT/enhancer binding protein. Biochemistry 44: 3909–3914, 2005 [DOI] [PubMed] [Google Scholar]
- 44.Ma XM, Yoon SO, Richardson CJ, Julich K, Blenis J. SKAR links pre-mRNA splicing to mTOR/S6K1-mediated enhanced translation efficiency of spliced mRNAs. Cell 133: 303–313, 2008 [DOI] [PubMed] [Google Scholar]
- 45.Mason SD, Rundqvist H, Papandreou I, Duh R, McNulty WJ, Howlett RA, Olfert IM, Sundberg CJ, Denko NC, Poellinger L, Johnson RS. HIF-1alpha in endurance training: suppression of oxidative metabolism. Am J Physiol Regul Integr Comp Physiol 293: R2059–R2069, 2007 [DOI] [PubMed] [Google Scholar]
- 46.McGhee NK, Jefferson LS, Kimball SR. Elevated corticosterone associated with food deprivation upregulates expression in rat skeletal muscle of the mTORC1 repressor, REDD1. J Nutr 139: 828–834, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGuire BB, Fitzpatrick JM. BMI and the risk of renal cell carcinoma. Curr Opin Urol 21: 356–361, 2011 [DOI] [PubMed] [Google Scholar]
- 48.Merrick WC, Hershey JWB. The pathway and mechanism of eukaryotic protein synthesis. In: Translational Control, edited by Hershey JWB, Mathews MB, Sonenberg N. Cold Spring Harbor: Cold Spring Harbor Laboratory Press, 1996, p. 31–69 [Google Scholar]
- 49.Miller AM, Brestoff JR, Phelps CB, Berk EZ, Reynolds TH. Rapamycin does not improve insulin sensitivity despite elevated mammalian target of rapamycin complex 1 activity in muscles of ob/ob mice. Am J Physiol Regul Integr Comp Physiol 295: R1431–R1438, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Misra M, Bredella MA, Tsai P, Mendes N, Miller KK, Klibanski A. Lower growth hormone and higher cortisol are associated with greater visceral adiposity, intramyocellular lipids, and insulin resistance in overweight girls. Am J Physiol Endocrinol Metab 295: E385–E392, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muoio DM, Noland RC, Kovalik JP, Seiler SE, Davies MN, DeBalsi KL, Ilkayeva OR, Stevens RD, Kheterpal I, Zhang J, Covington JD, Bajpeyi S, Ravussin E, Kraus W, Koves TR, Mynatt RL. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab 15: 764–777, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murakami T, Hasegawa K, Yoshinaga M. Rapid induction of REDD1 expression by endurance exercise in rat skeletal muscle. Biochem Biophys Res Commun 405: 615–619, 2011 [DOI] [PubMed] [Google Scholar]
- 53.Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, Rochon J, Gallup D, Ilkayeva O, Wenner BR, Yancy WS, Jr, Eisenson H, Musante G, Surwit RS, Millington DS, Butler MD, Svetkey LP. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 9: 311–326, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nilsson MI, Dobson JP, Greene NP, Wiggs MP, Shimkus KL, Wudeck EV, Davis AR, Laureano ML, Fluckey JD. Abnormal protein turnover and anabolic resistance to exercise in sarcopenic obesity. FASEB J 27: 3905–3916, 2013 [DOI] [PubMed] [Google Scholar]
- 55.Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of Obesity in the United States, 2009–2010. Hyattsville, MD: National Center for Health Statistics; Centers for Disease Control and Prevention's (CDC) and National Center for Health Statistics (NCHS). NCHS data brief, no 82, 2012 [Google Scholar]
- 56.Parra V, Verdejo HE, Iglewski M, Del Campo A, Troncoso R, Jones D, Zhu Y, Kuzmicic J, Pennanen C, Lopez-Crisosto C, Jana F, Ferreira J, Noguera E, Chiong M, Bernlohr DA, Klip A, Hill JA, Rothermel BA, Abel ED, Zorzano A, Lavandero S. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFkappaB-Opa-1 signaling pathway. Diabetes 63: 75–88, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peterson JM, Bryner RW, Alway SE. Satellite cell proliferation is reduced in muscles of obese Zucker rats but restored with loading. Am J Physiol Cell Physiol 295: C521–C528, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Protiva P, Hopkins ME, Baggett S, Yang H, Lipkin M, Holt PR, Kennelly EJ, Bernard WI. Growth inhibition of colon cancer cells by polyisoprenylated benzophenones is associated with induction of the endoplasmic reticulum response. Int J Cancer 123: 687–694, 2008 [DOI] [PubMed] [Google Scholar]
- 59.Raught B, Peiretti F, Gingras AC, Livingstone M, Shahbazian D, Mayeur GL, Polakiewicz RD, Sonenberg N, Hershey JW. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J 23: 1761–1769, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Regazzetti C, Bost F, Le Marchand-Brustel Y, Tanti JF, Giorgetti-Peraldi S. Insulin induces REDD1 expression through hypoxia-inducible factor 1 activation in adipocytes. J Biol Chem 285: 5157–5164, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Richardson CJ, Broenstrup M, Fingar DC, Julich K, Ballif BA, Gygi S, Blenis J. SKAR is a specific target of S6 kinase 1 in cell growth control. Curr Biol 14: 1540–1549, 2004 [DOI] [PubMed] [Google Scholar]
- 62.Rivas DA, Yaspelkis BB, 3rd, Hawley JA, Lessard SJ. Lipid-induced mTOR activation in rat skeletal muscle reversed by exercise and 5′-aminoimidazole-4-carboxamide-1-beta-d-ribofuranoside. J Endocrinol 202: 441–451, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320: 1496–1501, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22: 159–168, 2006 [DOI] [PubMed] [Google Scholar]
- 65.Schoenberg DR, Maquat LE. Regulation of cytoplasmic mRNA decay. Nat Rev Genet 13: 246–259, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, Condorelli G, Sadoshima J. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation 125: 1134–1146, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shargill NS, Ohshima K, Bray GA, Chan TM. Muscle protein turnover in the perfused hindquarters of lean and genetically obese-diabetic (db/db) mice. Diabetes 33: 1160–1164, 1984 [DOI] [PubMed] [Google Scholar]
- 68.Shin MK, Drager LF, Yao Q, Bevans-Fonti S, Yoo DY, Jun JC, Aja S, Bhanot S, Polotsky VY. Metabolic consequences of high-fat diet are attenuated by suppression of HIF-1alpha. PLos One 7: e46562, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shoshani T, Faerman A, Mett I, Zelin E, Tenne T, Gorodin S, Moshel Y, Elbaz S, Budanov A, Chajut A, Kalinski H, Kamer I, Rozen A, Mor O, Keshet E, Leshkowitz D, Einat P, Skaliter R, Feinstein E. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol Cell Biol 22: 2283–2293, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sitnick M, Bodine SC, Rutledge JC. Chronic high fat feeding attenuates load-induced hypertrophy in mice. J Physiol 587: 5753–5765, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sofer A, Lei K, Johannessen CM, Ellisen LW. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol 25: 5834–5845, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Srikanthan P, Hevener AL, Karlamangla AS. Sarcopenia exacerbates obesity-associated insulin resistance and dysglycemia: findings from the National Health and Nutrition Examination Survey III. PLos One 5: e10805, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Srikanthan P, Karlamangla AS. Relative muscle mass is inversely associated with insulin resistance and prediabetes. Findings from the third National Health and Nutrition Examination Survey. J Clin Endocrinol Metab 96: 2898–2903, 2011 [DOI] [PubMed] [Google Scholar]
- 74.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol 13: 1259–1268, 2003 [DOI] [PubMed] [Google Scholar]
- 75.Tremblay F, Gagnon A, Veilleux A, Sorisky A, Marette A. Activation of the mammalian target of rapamycin pathway acutely inhibits insulin signaling to Akt and glucose transport in 3T3–L1 and human adipocytes. Endocrinology 146: 1328–1337, 2005 [DOI] [PubMed] [Google Scholar]
- 76.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431: 200–205, 2004 [DOI] [PubMed] [Google Scholar]
- 77.Wang CY, Kim HH, Hiroi Y, Sawada N, Salomone S, Benjamin LE, Walsh K, Moskowitz MA, Liao JK. Obesity increases vascular senescence and susceptibility to ischemic injury through chronic activation of Akt and mTOR. Science Signaling 2: ra11, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang H, Kubica N, Ellisen LW, Jefferson LS, Kimball SR. Dexamethasone represses signaling through the mammalian target of rapamycin in muscle cells by enhancing expression of REDD1. J Biol Chem 281: 39128–39134, 2006 [DOI] [PubMed] [Google Scholar]
- 79.Wang Z, Malone MH, Thomenius MJ, Zhong F, Xu F, Distelhorst CW. Dexamethasone-induced gene 2 (dig2) is a novel pro-survival stress gene induced rapidly by diverse apoptotic signals. J Biol Chem 278: 27053–27058, 2003 [DOI] [PubMed] [Google Scholar]
- 80.Wen S, Jadhav KS, Williamson DL, Rideout TC. Treadmill exercise training modulates hepatic cholesterol metabolism and circulating PCSK9 concentration in high-fat-fed mice. J Lipids 2013: 908048, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Williamson DL, Bolster DR, Kimball SR, Jefferson LS. Time course changes in signaling pathways and protein synthesis in C2C12 myotubes following AMPK activation by AICAR. Am J Physiol Endocrinol Metab 291: E80–E89, 2006 [DOI] [PubMed] [Google Scholar]
- 82.Williamson DL, Butler DC, Alway SE. AMPK inhibits myoblast differentiation through a PGC-1alpha-dependent mechanism. Am J Physiol Endocrinol Metab 297: E304–E314, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Williamson DL, Kubica N, Kimball SR, Jefferson LS. Exercise-induced alterations in extracellular signal-regulated kinases 1/2 and mammalian target of rapamycin (mTOR) to regulatory mechanisms of mRNA translation in mouse muscle. J Physiol 573: 497–510, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yuen KC, Chong LE, Riddle MC. Influence of glucocorticoids and growth hormone on insulin sensitivity in humans. Diabet Med 30: 651–663, 2013 [DOI] [PubMed] [Google Scholar]