

Abstract
AIM: To study the expression of endothelial and inducible nitric oxide synthases (eNOS and iNOS) and their role in inflammatory bowel disease (IBD).
METHODS: We examined the effect of sera obtained from patients with active Crohn’s disease (CD) and ulcerative colitis (UC) on the function and viability of human umbilical vein endothelial cells (HUVEC). HUVECs were cultured for 0-48 h in the presence of a medium containing pooled serum of healthy controls, or serum from patients with active CD or UC. Expression of eNOS and iNOS was visualized by immunofluorescence, and quantified by the densitometry of Western blots. Proliferation activity was assessed by computerized image analyses of Ki-67 immunoreactive cells, and also tested in the presence of the NOS inhibitor, 10-4 mol/L L-NAME. Apoptosis and necrosis was examined by the annexin-V-biotin method and by propidium iodide staining, respectively.
RESULTS: In HUVEC immediately after exposure to UC, serum eNOS was markedly induced, reaching a peak at 12 h. In contrast, a decrease in eNOS was observed after incubation with CD sera and the eNOS level was minimal at 20 h compared to control (18% ± 16% vs 23% ± 15% P<0.01). UC or CD serum caused a significant increase in iNOS compared to control (UC: 300% ± 21%; CD: 275% ± 27% vs 108% ± 14%, P<0.01). Apoptosis/necrosis characteristics did not differ significantly in either experiment. Increased proliferation activity was detected in the presence of CD serum or after treatment with L-NAME. Cultures showed tube-like formations after 24 h treatment with CD serum.
CONCLUSION: IBD sera evoked changes in the ratio of eNOS/iNOS, whereas did not influence the viability of HUVEC. These involved down-regulation of eNOS and up-regulation of iNOS simultaneously, leading to increased proliferation activity and possibly a reduced anti-inflammatory protection of endothelial cells.
Keywords: Crohn’s disease, Human umbilical vein endothelial cells, Inflammatory bowel disease, Nitric oxide synthase, Ulcerative colitis
INTRODUCTION
The two major forms of inflammatory bowel disease (IBD), ulcerative colitis (UC) and Crohn’s disease (CD) are characterized by chronic inflammatory ulceration of the intestine. In an early[1], and in some recent studies[2,3], the contribution of mucosal microvascular dysfunction is implicated in the development of CD. Histological analysis of IBD colonic mucosa indicated structural changes of the vascular system. In CD, arterial thickening and the appearance of an increasing number of capillaries were observed[1,3]. Dysfunction in the vasodilatory capacity of microvessels from areas of chronic inflammatory damage was also shown in IBD[2]. Inflammatory cytokines modify and regulate the level of NO, and they are also involved in vascular endothelial response to inflammatory injury[3,4]. Apoptosis of endothelial cells induced by proinflammatory cytokines and reactive oxygen species is counteracted by the endothelial nitric oxide (NO). The suppression of apoptosis may contribute to the anti-inflammatory and pro-angiogenic effect of endothelial NO[5].
The endothelial form of nitric oxide synthase (eNOS) plays a significant protective role against experimental colitis[6]. Decrease of eNOS immunoreactivity in the endothelium is suggested as a putative cause of arterial thickening and the appearance of an increasing number of capillaries in the mucosa of Crohn’s colitis[3]. Moreover, weak eNOS immunoreactivity in the endothelium together with structural alteration of the vascular wall, both found in biopsies of Crohn’s patients suggest that malfunction of local blood supply may also contribute to the development of the disease[3]. Human umbilical vein endothelial cells (HUVEC) are frequently used as a specific model for studying endothelial function. HUVECs are also sensitive to pro-inflammatory stimuli induced by interleukins. NO is produced in HUVEC constitutively by eNOS and by inducible nitric oxide synthase (iNOS) in the case of different stimuli[7,8]. NO also contributes to the regulation of apoptosis, cell differentiation and proliferation (angiogenesis) in this cell type[9-11].
The changes in the activity of eNOS and iNOS as well as their role in the endothelium of IBD patients have not been examined in detail. In the present study, we examined the effect of sera from patients diagnosed with UC and CD on cultured HUVEC. The expression and localization of eNOS and iNOS were determined in highly confluent and in semi-confluent HUVEC cultures, and the apoptotic/necrotic characteristics and proliferation activity upon distinct treatments were also assessed.
MATERIALS AND METHODS
Collection of blood sera
Blood was sampled for examination from patients who had relapsing UC or CD. Among 21 UC patients, 16 were on their 1st relapse (mean time from diagnosis [mtd] was 1 yr), and 5 on 2nd relapse (mtd = 3 yr), whereas all the 18 CD patients were on their 1st relapse (mtd = 4 yr). Control sera were obtained from 16 healthy volunteers. They received no systemic or locally applied corticosteroids, and their medication was stable for the last four weeks before sampling. All the patients were on 5ASA oral therapy. Patients diagnosed as having UC had left side colitis (Table 1). Crohn’s patients had colonic localization and a non-stricturating, non-penetrating form of the disease (Table 1) according to the Vienna classification (1998), and an elevated C-reactive protein level (CRP>10 mg/L), without abscess or sign of infection (all of the patients had negative abdominal ultrasound). In cases which raised the suspicion of abdominal or pelvic abscess, CT or MRI (to exclude the presence of perianal / perirectal abscesses) was performed. Five milliliters of blood were taken from patients of UC, CD and control subjects respectively, placed into collecting tubes, and centrifuged at 2 000 r/min for 10 min. The sera were removed and mixed, and kept frozen at -80°C until use.
Table 1.
Data of patients with ulcerative colitis and Crohn’s disease
| Group | n | Age (yr)(range) | M/F | Endoscopic activity score | Clinical activity score | CRP (mg/L) | Treatment |
| UC | 21 | 27 (18-33) | 3/8 | G1: 4 G2: 9 G3: 8 | 4≤UC-DAI | >10 | 5-ASA |
| CD | 18 | 31 (23-45) | 13/5 | 8≤CDEIS≤14 | 150≤CDAI≤450 | >10 | 5-ASA |
| Control | 16 | 29 (25-39) | 8/8 |
G0-3: Endoscopic grading of UC (Br Med J 1964; 1:89); CDEIS: Crohn’s disease endoscopic index of severity (Gut 1989; 30:983-89); UC-DAI: Ulcerative colitis disease activity index (Br Med J 1955; 2:1041); CDAI: Crohn’s disease activity index (Gastroenterology 1979; 77:843)
Preparation and culturing of HUVEC
Freshly isolated umbilical cords were obtained from the Department of Obstetrics and Gynecology (University of Debrecen, Medical and Health Science Center). Umbilical cords were immersed into a sterile flask containing culture medium (components are described below) and carried to the cell culture room. Both ends of the cords were bound and the ends of the umbilical vein were cannulated. The vein vessel was washed through the cannulas by sterile PBS, followed by Hanks’ Balanced Salt Solution (HBSS), and M199 media (all purchased from Sigma, Budapest, Hungary). Then, the vein vessel was filled with 1 g/L collagenase in M199, and placed in a CO2 incubator for 12 min at 37 °C. Endothelial cells were isolated in a sterile centrifuge tube from the vessel by washing twice with 20 mL M199 medium. Cell suspension was centrifuged at 1000 r/min for 15 min, and the supernatant was discarded and the cells were resuspended in the culture medium.
The cells (2 ×109 cells/L) were inoculated into a plastic culture dish. The culture medium contained the following components: 500 mL/L normal human serum, 20 g/L HEPES, 10 g/L L-glutamine, 10 g/L amphotericin, 10 g/L penicillin, 450 mL/L sterile filtered M199. One day after inoculation, dishes were washed with 37 °C HBSS, and put into the culture media again. After 4-5 d of culturing, during which time cells grew extensively over the entire space of the dish, cells were washed with HBSS, then digested with 1 mL tripsin-EDTA (tripsin : EDTA = 1 : 25). The cells were suspended in 1 mL culture medium and collected into a sterile flask, then mixed well. The cells were inoculated into the culture dish and the cell propagation and collection procedure were repeated again as described above. Then, cells were passed onto culturing plates at about 2 ×109 cell/L concentration. Cell monolayers that reached 20%-30% or 70%-80% confluency were used for the experiments. The cells were kept in 50% normal, 50% UC or 50% CD sera dissolved in culturing medium for 2, 4, 8, 12, 20, 24, 36, and 48 h, then processed for visualization of eNOS or iNOS.
Western blot
After removal of the culture media, cells were washed with ice-cold PBS, then harvested by a cell scraper into 50 µL RIPA lysis buffer (0.01 mol/L NaH2PO4, 10 g/L Nonident P-40, 10 g/L Na-deoxycholate, 1 g/L Na-dodecyl sulfate (SDS), 0.15 mol/L NaCl, 2 mmol/L EDTA), containing 100 kU/L protease inhibitor mix (Sigma-Aldrich, Budapest, Hungary). Cell lysis was accelerated by continuous shaking. Five microliters of this mixture were added to 45 µL RIPA buffer for protein measurement. All protein solutions were frozen and kept at -80 °C until use.
The protein concentration of the lysates was determined by the BCA method using BSA as standard in an ELISA reader at 540 nm. Data were analyzed by GraphPad Prism software using linear regression to determine the protein concentration (g/L). Proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using the conditions of Laemmli (1970) on 100 g/L acrylamide gels in a Bio-Rad MiniProtean apparatus[3]. Samples for immunoblot analysis were diluted in the RIPA buffer, dissolved in an equal volume of 2 ×SDS sample buffer (100 mmol/L Tris pH 6.8, 2 g/L bromophenol blue, 200 mL/L glycerol, 200 mL/L β-mercaptoethanol) and boiled for 5 min. From each solution, aliquots (30 µg protein) were applied to SDS-PAGE and electrophoresed at 120 V for 1 h. Separated proteins were blotted onto a nitrocellulose membrane (Pharmacia, Vienna, Austria) at 100 V for 90 min during the continuous mixing and cooling of the transfer buffer (192 mmol/L glycine, 25 mmol/L Tris-base, 200 mL/L methanol). Membranes were rinsed twice in 0.1 mol/L PBS (pH 7.6) containing 1 g/L Tween 20 (PBS-Tween), then blocked for 1 h with 50 mL/L non-fat dried milk in PBS-Tween. After 3 × 5 min of washing with PBS-Tween, the membranes were incubated overnight at 4 °C with the anti-eNOS (diluted into 1∶5000, Cat. No.: 482726, Merck-Calbiochem, Darmstadt, Germany) or anti-iNOS (1∶2000, Cat. No.: AB5384, Chemicon, Temecula, CA) polyclonal antibodies raised in rabbits. After washing with PBS-Tween for 3 ×5 min, membranes were incubated with horseradish peroxidase conjugate of anti-rabbit IgG (diluted into 1∶5000 in PBS-Tween, Vector, Bulingame, CA) for 30 min. After washing the membranes subsequently with PBS-Tween for 2 ×5 min, and once with PBS for 5 min. The blots were developed with the enhanced chemiluminescent (ECL) reagent kit (Pierce, Rockford, IL) and the immunoreactions were visualized on X-ray films.
Densitometry of the blots was performed and the scans were analyzed by the Volume Analyse feature of the Molecular Analyst Software (Bio-Rad, Hercules, CA). The densities for the NOS bands were normalized in each blot to the NOS-IR band obtained from the sample of 2 h treatment with normal serum and expressed as percentage of this control. Means of the percentages obtained from three independent experiments were presented in figures as mean ± SEM. The significance of the data between groups (normal, UC, CD) was tested by analysis of variance (ANOVA). Differences were considered significant for values of P < 0.01.
Immunofluorescence
Cells were cultured on coverglasses coated with sterile filtered 100 g/L gelatine in PBS. Cultures showing 20%-30% confluence were used for detection of proliferation, cell distribution and apoptosis/necrosis characteristics, whereas those with 70%-80% confluency were used for visualization of NOS isoforms. After treatments, cells were fixed in an ascendant ethanol gradient (500 ml/L, 700 mL/L, 960 mL/L), then rehydrated with PBS. For specific detection of eNOS and iNOS proteins, non-specific binding sites of primary antibodies were blocked by incubation of the fixed cells for 1 h with 50 mL/L normal goat serum in PBS also containing 1 g/L bovine serum albumin (BSA), 1 g/L Triton-X 100 and 0.1 g/L Na-azide (referred to as solution-A below). NOS primary antibodies used for immunocytochemistry were the same as those used for immunoblotting. Both eNOS and iNOS antibodies were applied at 1∶200 dilution in solution-A overnight at 4 °C. After extensive washing with PBS (3×5 min), cells were incubated with FITC conjugated anti-rabbit IgG diluted to 1∶40 in solution-A for 30 min at room temperature. Cells were washed for 3 ×5 min with PBS and cover glasses were mounted on microscope slides (with one drop of PBS-glycerol (1∶1) solution on them).
For evaluation of the proliferation activity, HUVECs were incubated in a medium containing 500 mL/L normal, 500 mL/L UC or 500 mL/L CD sera, or in a medium containing 500 mL/L normal sera plus 10-4 mol/L nitro-L-arginine methyl-ester (L-NAME, Sigma-Aldrich), an NOS inhibitor. Cells were exposed to a monoclonal antibody (at 1∶100 dilution) specific for the proliferation marker Ki-67 antigen (clone No. MIB-1, Dako, Denmark) for 2 h at room temperature. In this case, FITC-conjugated anti-mouse IgG (1∶40 in solution-A, Dako) was used as a secondary antibody. For testing of the effect of normal and IBD sera on the viability of HUVEC, apoptotic/necrotic cells were assessed by the annexin-V-biotin kit (Cat. No. 1828690, Boehringer-Mannheim, Germany). Briefly, after removal of the incubation media and washing with PBS, cells were labeled with the annexin-V-biotin/propidium iodide solution (1∶50 annexin-V-biotin stock solution diluted in a 0.01 mol/L HEPES buffer, pH 7.4, containing 0.14 mol/L NaCl, 0.005 mol/L CaCl2, and 1 mg/L propidium iodide) for 15 min. After subsequent washing with HEPES, cells were fixed with methanol-ethanol (1∶1) solution for 1 min, and labeled with 5 mg/L avidin-fluorescein for 15 min. After consecutive washing with HEPES and PBS, cells were put on microscope slides as described above. Preparations used for the assay of proliferation activity and assessment of apoptosis/necrosis were stained prior to covering with 10 g/L toluidin blue in PBS. The latter labeled the cell nuclei and helped to determine total cell numbers for calculation of apoptotic/necrotic indices. The integrity of the cytoskeleton was examined by visualization of actin filaments by incubation with 0.2 mg/L phalloidin-FITC (Sigma-Aldrich) for 1 h.
Examination was carried out and digital images were captured with an Olympus AX-70 fluorescence microscope. In each case, the brightness-contrast of the images was set in the same manner by the Jasc software Paintshop Pro 7.0. Images were taken from Ki-67 immunoreactive (IR), annexin-labeled, and propidium iodide reactive cell nuclei, respectively. The same sites of the preparations were also captured in bright visual illumination to count the toluidin blue stained nuclei (total cell number). The images were quantitatively analyzed by the computer image analysis software Olympus analysis 2.11 (SiSoft), developed and validated by Olympus (Tokyo, Japan). At least 103 cells were counted for each data point and the ratios of proliferative, apoptotic and necrotic cells were given as the percentage of total cell numbers.
Statistical analyses
Statistical analyses were performed to compare the data obtained from the different treatments groups of IBD and normal sera using the ANOVA test. P < 0.05 was
considered as significant.
RESULTS
eNOS
Western blots from HUVEC lysates detected eNOS immunoreactive bands at approximately 160 kU, corresponding to the molecular mass of human eNOS (Figure 1A). HUVEC was cultured in a medium containing 500 mL/L normal human serum. The amount of eNOS did not change significantly during the experimental period (0-48 h). However, in the presence of 500 mL/L UC serum, eNOS level increased after 8 h, and peaked at 12 h (relative optical density value was 188% ± 23% and 229 %± 26%, P < 0.01, respectively). At 12 h, eNOS level appeared to have doubled compared to the normal level at 2 h (100% ± 15%). Over the 12 h period in the presence of 500 mL/L UC serum, eNOS level began to decline (36 h, 153% ±23%, P < 0.01), and reached control level at 48 h (100%± 8%). In the presence of 500 ml/L CD serum, the level of eNOS was close to normal until 8 h, and decreased after 12 h (41% ± 12%, P < 0.01), reaching the lowest level at 20-24 h (18% ± 16% and 23% ± 15%, P < 0.01, respectively), and then slowly increased rising close to normal at 48 h (65% ± 8%, Figure 1A, B).
Figure 1.
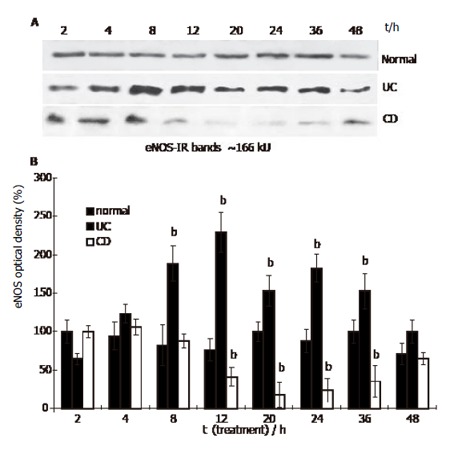
eNOS content in HUVEC treated with IBD sera (Western blot. Mean ± SE, n = 3; bP<0.01 vs normal).
Localization of eNOS in HUVEC by immunofluorescence revealed that the enzyme mostly accumulated in the nuclear area of the cytoplasm and was also detected at the periphery of the cells (Figure 2). Normal human serum had no effect on the distribution and intensity of eNOS immunoreactivity in HUVEC. Prolonged treatment (12 h) with UC serum extended eNOS immunoreactive area and simultaneously enhanced slightly the intensity of eNOS immunofluorescence in HUVEC. In contrast, in the
Figure 2.
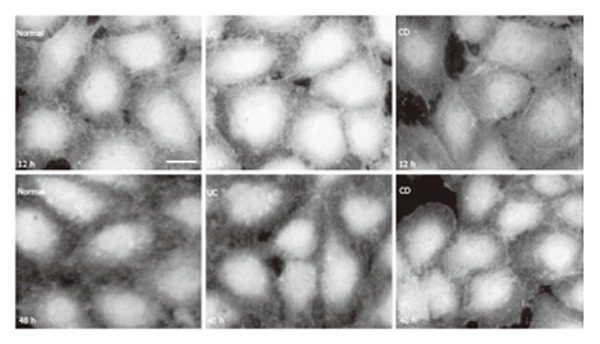
eNOS in HUVEC (immunoreaction was visualized by FITC. bar: 10 µm).
presence of CD serum (after more than 4 h), eNOS immunoreactivity seemed to contract toward the centre of the cells and its immunofluorescence intensity decreased (Figure 2). At the end of the experimental period (48 h treatment), no visible difference could be detected in eNOS immunofluorescence of HUVEC in the presence of normal, UC or CD sera.
iNOS
Western blots in HUVEC lysates detected faint iNOS immunoreactive bands at around 130 kU, corresponding to the molecular mass of human iNOS (Figure 3A). HUVEC was cultured in the presence of 500 mL/L normal human serum, the relatively low level of iNOS expression did not change significantly during the 48 h incubation period. In the presence of 500 mL/L UC serum, iNOS level increased at 4 h (relative optical density value was 225% ± 20%, P < 0.01), peaked at 8 h (300% ± 21%, P < 0.01), and then declined to about normal level at 48 h (117% ± 15%). In the presence of 500 mL/L CD serum the iNOS level in HUVEC increased after 2 h (201% ±19%, P < 0.01) and peaked at 8 h (275% ± 27%, P < 0.01). From 12 h iNOS level declined and was not distinguishable from the control during the 24-48 h period (at 48 h: 92% ± 18%, Figure 3A, B).
Figure 3.
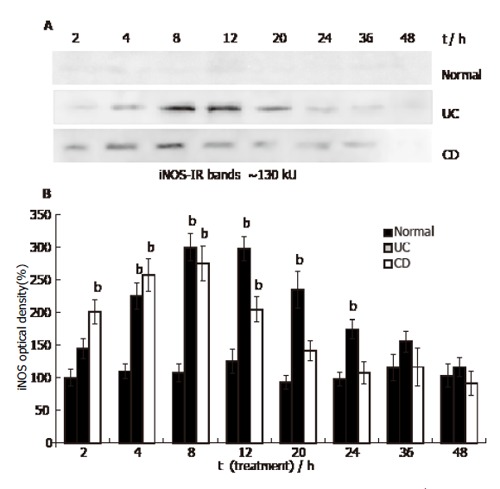
iNOS content in HUVEC (Western blot. Mean ± SE, n=3; bP<0.01 vs normal).
In the presence of 500 mL/L normal serum, HUVEC exhibited faint iNOS immunofluorescence (Figure 4). Expression of iNOS in HUVEC by immunofluorescence could obviously be detected in the presence of IBD sera during the 4-12 h period. Figure 4 shows that iNOS was dispersed in the cytoplasm, accumulated in certain areas showing a high intensity of fluorescent signal. Both UC and CD sera significantly increased iNOS immunofluorescence at the 4-12 h period. Before and after this time interval, iNOS could not be detected unequivocally in HUVEC by immunofluorescence (Figure 4).
Figure 4.
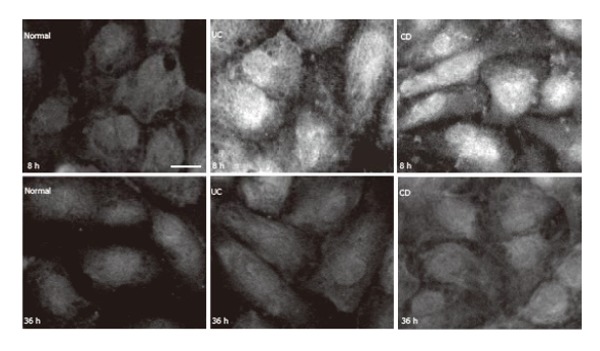
iNOS in HUVEC (immunoreaction was visualized by FITC. bar: 10 µm).
HUVEC proliferation activity
Ten to fifteen percent of HUVEC was observed to be in the state of proliferation in the cultures with 20%-30% confluency at the beginning of the experiments as revealed by Ki-67 immunostaining (Figure 5). During the 2 to 48 h period no difference was detected in the ratio of Ki-67-IR/toluidin blue stained cell nuclei in cultures in the presence of 500 mL/L normal (at 48 h: 19% ± 10%) or 500 mL/L UC sera (at 48 h: 10% ± 3%). In the presence of 500 mL/L CD serum a significant increase in the number of proliferating cells was apparent at 20 h (41% ± 12%, P < 0.01). About 50%-65% of HUVEC nuclei exhibited Ki-67 immunofluorescence in the presence of CD serum for 24-48 h (Figure 5). The presence of 10-4 mol/L L-NAME in the medium containing 500 ml/L normal serum, increased the proliferation activity significantly from 24 h (42% ± 5%, P < 0.01) to 48 h (48% ± 7%, P < 0.01).
Figure 5.
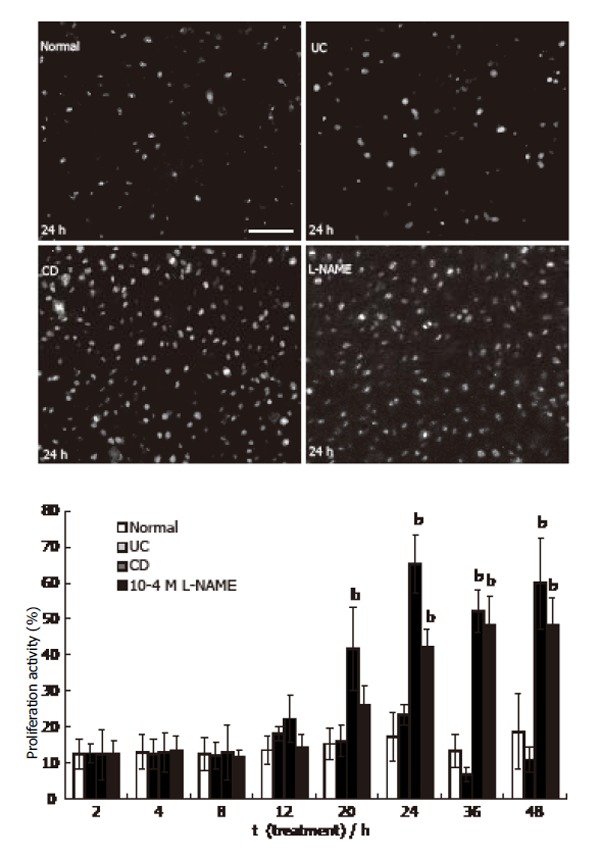
Proliferation activity of HUVEC (Ki-67 immunoreactivity was visualized by FITC. bar: 50 µm. mean ± SE, n = 3; bP<0.01 vs normal).
HUVEC viability
By the annexin-V-biotin method, early apoptotic cells revealed fluorescent signals only on the cell membrane (Figure 6A), whereas necrotic cells showed propidium iodide binding to DNA (nuclear fluorescent signal, Figure 6B). Both signals could be detected in late apoptotic cells (Figure 6C). During the experimental period (2-48 h), the number of apoptotic cells was low (1%-3%) in the presence of 500 mL/L normal serum, but a continuous increase of apoptotic cell numbers was observed in the presence of 500 mL/L CD serum (2%-5%, with a maximum level at 36 h), which proved not to be significant (Figure 7A). In the presence of UC serum the apoptotic index did not differ from that of the control. Incubation of HUVEC with normal or CD sera slightly increased the number of necrotic cells during the examination period (2-48 h), but no significant changes were observed between the necrotic indices (Figure 7B). Actin filaments visualized by phalloidin-FITC seemed to be intact in cells independent of the media used and the time of incubation (Figure 6D).
Figure 6.
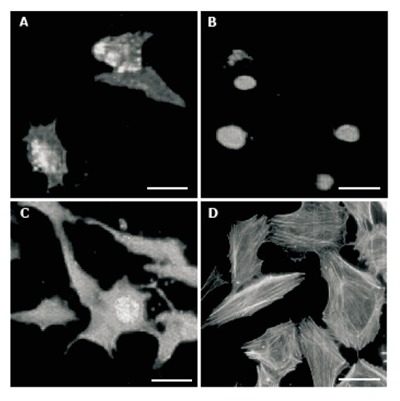
HUVEC viability. A: Early apoptotic cells; B: necrotic cells; C: Late apoptotic cell; D: Phalloidin conjugated with TRITC labeled actin filaments. bars: 10 µm.
Figure 7.
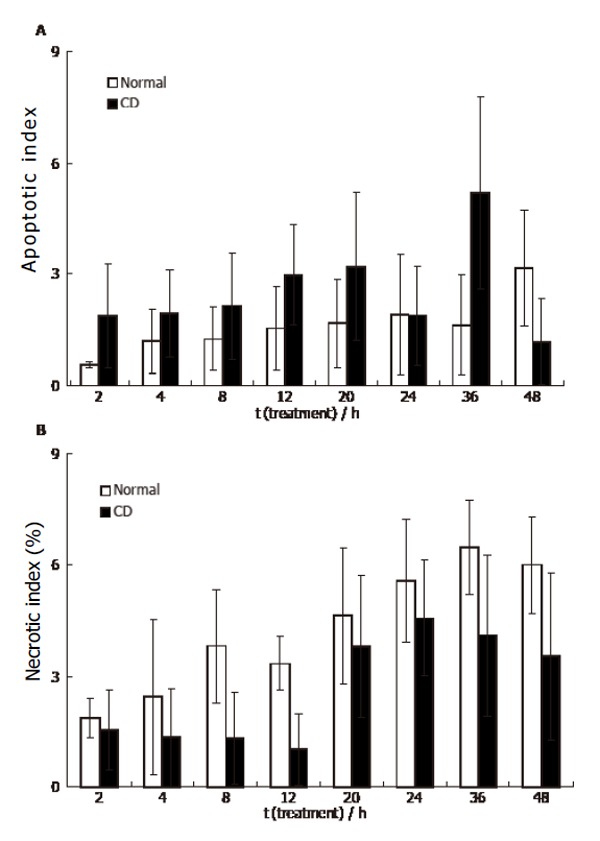
HUVEC 48 h incubation with CD sera (mean ± SE, n = 3).
Shape and structure of HUVEC
HUVEC cultures of 20%-30% confluency were used also to investigate the effect of IBD sera on the distribution and shape of the cells. Cells visualized by eNOS immunofluorescence were partly found to be accumulated in islands exhibiting polygonal shape and attached to each other. In addition, single cells exhibiting long processes were also present. In the presence of 500 ml/L normal or 500 ml/L UC sera cells did not change in distribution and shape over the 48 h experimental period. However, in the presence of 500 mL/L CD serum, after 20 h cells were distributed in a two dimensional reticular network, which changed the shape of the culture reminiscent of the appearance of early vascular vessels in tissues (Figure 8).
Figure 8.
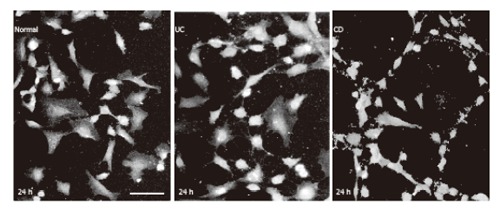
HUVEC visualized by eNOS immunofluorescence in culture. bar: 50 µm.
DISCUSSION
In this study, we investigated the effect of sera from patients with active UC or CD on the expression and localization of eNOS and iNOS in HUVEC cultures. Induction of both iNOS and eNOS was observed in the presence of UC serum. In contrast, upon addition of CD serum to HUVEC media a transient increase in the expression of iNOS was observed in parallel with a marked decrease in the expression of eNOS. The number of apoptotic and necrotic cells was not altered significantly, and the structural integrity of the cytoskeleton (revealed by staining of the actin filaments) remained intact during the exposure of the cells to either CD or UC serum.
It has been shown previously that NO might enhance or reduce the cascade reaction of chronic inflammation in humans and animal models[12-14]. We found opposite changes in the expression of eNOS in HUVEC exposed to UC and to CD sera. The generation of eNOS is modified by elevated levels of cytokines (Th1 in CD) and the cytokine-induced expression and presentation of endothelial cell adhesion molecules[15]. UC and CD sera are known to contain a distinct cytokine and pro-inflammatory molecule profile. It was reported that TNF-α reduced eNOS protein expression in a time dependent manner in human endothelial cells[16,17]. Moreover, TNF-α downregulated eNOS mRNA by decreasing its stability[18]. In our experiment, the low eNOS level in the presence of CD serum, could be due to TNF-α, which is a key cytokine in Th1 mediated inflammation in CD. Inflammation could result in functional and antigenic changes of the endothelium, including eNOS down-regulation and consequently increased expression of leukocyte adhesion molecules (ICAM, VCAM, E-selectin)[19-23]. At the same time, a lag in the fall of enzyme activity was observed which may reflect a greater stability of eNOS protein compared to its mRNA, or may involve posttranslational modification(s) in the increase and stabilization of the enzyme activity[9]. Leukocyte-endothelial binding is exacerbated in an eNOS deficient state and injury during ischemia-reperfusion is more severe[24,25].
A high amount of iNOS is generated during inflammation which can be harmful, worsening the course of disease in the animal colitis model[26]. In Crohn’s colitis, human intestinal microvascular endothelial cells may lack the capacity to express iNOS, which makes them susceptible to leukocyte mediated injury[27,28]. In UC, it has been shown that there is an elevation in iNOS level which is well correlated with disease severity[29]. Elevated iNOS activity was demonstrated in the mesenteric microvascular endothelial cells in active UC, indicating a close relationship between vascular activation and the pathogenesis of UC[30]. Stimulation of HUVEC with pro-inflammatory cytokines (Il-1β, TNF-α) or hormones (eg. relaxin, estrogen) induces the expression of iNOS and the NO produced increases the biosynthesis of prostaglandins and other mediators[31-33]. The maximal level of iNOS detected in primary cell cultures was not related to lipopolysaccharide contamination. It did not affect cell proliferation and was decreased when cells became confluent[34]. Dual modulation of NOS expression was detected upon estradiol treatment in a concentration dependent manner; estradiol in nanomolar range could induce eNOS, whereas in micromolar range it could evoke iNOS expression[35]. In previous studies, INF-γ in conjunction with TNF-α, or together with IL-1β, increased NOS activity, whereas these cytokines alone had different effects on NOS expression[9,36,37]. Up-regulation of iNOS by IBD sera may be an adaptive response of HUVEC to this culture environment.
The effect of UC and CD sera on eNOS expression could be of special interest and may be implicated in the distinct pathophysiological and morphological features of the two diseases. Inhibition of NO production in vivo results in reduced VEGF induced angiogenesis and vascular permeability[38]. Recent studies have provided evidence that basic fibroblast growth factor (bFGF), which is an important component of CD serum, induced angiogenesis by different mechanisms[39]. An impaired VEGF response was suggested in CD with down-regulation of the VEGF receptor (VEGF-Ets-1) and the angiogenic cascade supporting the vascular hypothesis of the disease[40-43]. The deficiency in eNOS expression and concomitant alteration of VEGF-induced angiogenesis and vascular permeability could be an important step in the pathomechanism of CD. However, bFGF, the most potent angiogenic molecule in CD has an NO independent activity[39,44]. In addition, the pathomechanism of UC appears to be VEGF independent[39,44]. The reciprocal reaction of cytokines and angiogenic molecules results in changes in the expression of eNOS and iNOS and causes an imbalance between the two isoforms which may play a role in the dysfunction of angiogenesis in IBD.
Angiogenesis is an important pathophysiological process in chronic inflammation and it includes proliferation, migration, and tube formation of endothelial cells. This process is tightly regulated by different angiogenic factors, and one of them is NO which has a pro-angiogenic effect[45,46]. HUVEC, in the presence of CD serum, but not in the presence of UC serum, showed morphological changes reminiscent of vascular like structures, forming loop like rearrangement of the cells. It was shown recently that reactive oxygen species (ROS) have a putative pro-angiogenic activity in vivo possibly via iNOS up-regulation[47]. ROS also play a significant role in proliferation through regulation of eNOS activity, whereas antioxidants stimulate NO production and proliferation of HUVEC[48]. However, it was suggested also that antioxidants may enhance the activity of NOS[49,50]. This dual effect could have a role in the pro-angiogenic and proliferation activity of HUVEC.
Recent data revealed that NO, by regulating mitochondrial respiration and cytochrome c release, triggers a defensive mechanism against cell death induced by pro-apoptotic stimuli[51-53]. In the present study, HUVEC incubated in the presence of CD serum exhibited a low expression level of eNOS, which could explain the slightly elevated apoptotic index. It also suggests a defective anti-apoptotic mechanism. The TNF-α level is known to be high also in CD serum and this could alter cell viability by inducing apoptosis in an NO independent pathway[54]. In contrast, in control and UC sera treated cells, the higher eNOS level may result in protection against apoptosis.
Our present results correlate well with our previous immunohistochemical findings through biopsy studies, and suggest the presence of a cytokine composition, which is able to evoke changes in the eNOS/iNOS ratio in endothelial cells in the blood of IBD patients. Simultaneous down-regulation of eNOS and up-regulation of iNOS, together with the increase of cell proliferation activity may reduce the anti-inflammatory capacity of endothelial cells. The latter may contribute to the initiation of pro-inflammatory cascade reactions leading to endothelial barrier dysfunction and causing structural changes in the microvessels in Crohn’s mucosa.
ACKNOWLEDGEMENTS
The authors thank Mrs. Éva Galáth for her excellent technical work in cell culturing.
Footnotes
Supported by the ˝Mecenatura˝ grant of Debrecen University 3/1999 to K. P., and grants from the Hungarian Ministry of Health (ETT 41/2000 to I. A., and ETT 026/2003 to F. E.), and from the Hungarian Science Research Fund (OTKA 043296 to F. E.).
S- Editor Pan BR L- Editor Zhu LH E- Editor Zhang Y
References
- 1.Wakefield AJ, Sawyerr AM, Dhillon AP, Pittilo RM, Rowles PM, Lewis AA, Pounder RE. Pathogenesis of Crohn's disease: multifocal gastrointestinal infarction. Lancet. 1989;2:1057–1062. doi: 10.1016/S0140-6736(89)91078-7. [DOI] [PubMed] [Google Scholar]
- 2.Hatoum OA, Binion DG, Otterson MF, Gutterman DD. Acquired microvascular dysfunction in inflammatory bowel disease: Loss of nitric oxide-mediated vasodilation. Gastroenterology. 2003;125:58–69. doi: 10.1016/S0016-5085(03)00699-1. [DOI] [PubMed] [Google Scholar]
- 3.Palatka K, Serfozo Z, Veréb Z, Hargitay Z, Lontay B, Erdodi F, Bánfalvi G, Nemes Z, Udvardy M, Altorjay I. Changes in the expression and distribution of the inducible and endothelial nitric oxide synthase in mucosal biopsy specimens of inflammatory bowel disease. Scand J Gastroenterol. 2005;40:670–680. doi: 10.1080/00365520510015539. [DOI] [PubMed] [Google Scholar]
- 4.Reimund JM, Wittersheim C, Dumont S, Muller CD, Kenney JS, Baumann R, Poindron P, Duclos B. Increased production of tumour necrosis factor-α, interleukin-1β, and interleukin-6 by morphologically normal intestinal biopsies from patients with Crohn's disease. Gut. 1996;39:684–689. doi: 10.1136/gut.39.5.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stoclet JC, Martinez MC, Ohlmann P, Chasserot S, Schott C, Kleschyov AL, Schneider F, Andriantsitohaina R. Induction of nitric oxide synthase and dual effects of nitric oxide and cyclooxygenase products in regulation of arterial contraction in human septic shock. Circulation. 1999;100:107–112. doi: 10.1161/01.CIR.100.2.107. [DOI] [PubMed] [Google Scholar]
- 6.Dimmeler S, Zeiher AM. Nitric oxide-an endothelial cell survival factor. Cell Death Differ. 1999;6:964–968. doi: 10.1038/sj.cdd.4400581. [DOI] [PubMed] [Google Scholar]
- 7.Sasaki M, Bharwani S, Jordan P Elrod JW, Grisham MB, Jackson TH, Lefer DJ, Alexander JS. Increased disease activity in eNOS-deficient mice in experimental colitis. Free Radical Biol Med. 2003;35:1679–1687. doi: 10.1016/j.freeradbiomed.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 8.Marsden PA, Schappert KT, Chen HS, Flowers M, Sundell CL, Wilcox JN, Lamas S, Michel T. Molecular cloning and characterization of human endothelial nitric oxide synthase. FEBS Lett. 1992;307:287–293. doi: 10.1016/0014-5793(92)80697-F. [DOI] [PubMed] [Google Scholar]
- 9.Rosenkranz-Weiss P, Sessa WC, Milstien S, Kaufman S, Watson CA, Pober JS. Regulation of nitric oxide synthesis by proinflammatory cytokines in human umbilical vein endothelial cells. Elevations in tetrahydrobiopterin levels enhance endothelial nitric oxide synthase specific activity. J Clin Invest. 1994;93:2236–2243. doi: 10.1172/JCI117221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimmeler S, Haendeler J, Nehls M, Zeiher AM. Suppression of apoptosis by nitric oxide via inhibition of interleukin-1 beta-converting enzyme (ICE)-like and cysteine protease protein (CPP)-32-like proteases. J Exp Med. 1997;185:601–607. doi: 10.1084/jem.185.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Papapetropoulos A, García-Cardeña G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest. 1997;100:3131–3139. doi: 10.1172/JCI119868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heller R, Polack T, Grabner R, Till U. Nitric oxide inhibits proliferation of human endothelial cells via a mechanism independent of cGMP. Atherosclerosis. 1999;144:49–54. doi: 10.1016/S0021-9150(99)00041-6. [DOI] [PubMed] [Google Scholar]
- 13.García-González MA, Peña AS. [Nitric oxide and inflammatory bowel disease] Rev Esp Enferm Dig. 1998;90:870–876. [PubMed] [Google Scholar]
- 14.Guslandi M. Nitric oxide and inflammatory bowel diseases. Eur J Clin Invest. 1998;28:904–907. doi: 10.1046/j.1365-2362.1998.00377.x. [DOI] [PubMed] [Google Scholar]
- 15.Guihot G, Guimbaud R, Bertrand V, Narcy-Lambare B, Couturier D, Duee P, Chaussade S, Blachier F. Inducible nitric oxide synthase activity in colon biopsies from inflammatory areas: correlation with inflammation intensity in patients with ulcerative colitis but not with Crohn's disease. Amino Acids. 2000;18:229–237. doi: 10.1007/s007260050020. [DOI] [PubMed] [Google Scholar]
- 16.Agnoletti L, Curello S, Bachetti T, Malacarne F, Gaia G, Comini L, Volterrani M, Bonetti P, Parrinello G, Cadei M, et al. Serum from patients with severe heart failure downregulates eNOS and is proapoptotic: role of tumor necrosis factor-alpha. Circulation. 1999;100:1983–1991. doi: 10.1161/01.CIR.100.19.1983. [DOI] [PubMed] [Google Scholar]
- 17.Bachetti T, Comini L, Curello S, Bastianon D, Palmieri M, Bresciani G, Callea F, Ferrari R. Co-expression and modulation of neuronal and endothelial nitric oxide synthase in human endothelial cells. J Mol Cell Cardiol. 2004;37:939–945. doi: 10.1016/j.yjmcc.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 18.Yoshizumi M, Perrella MA, Burnett JC Jr, Lee ME. Tumor necrosis factor downregulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circ Res. 1993;73:205–209. doi: 10.1161/01.RES.73.1.205. [DOI] [PubMed] [Google Scholar]
- 19.Grisham MB, Johnson GG, Lancaster IR Jr. Quantitation of nitrate and nitrite in extracellular fluids. Methods Enzymol. 1996;268:237–246. doi: 10.1016/S0076-6879(96)68026-4. [DOI] [PubMed] [Google Scholar]
- 20.Matsushita H, Chang E, Glassford AJ, Cooke JP, Chiu CP, Tsao PS. eNOS activity is reduced in senescent human endothelial cells: Preservation by hTERT immortalization. Circ Res. 2001;89:793–798. doi: 10.1161/hh2101.098443. [DOI] [PubMed] [Google Scholar]
- 21.Sharp BR, Jones SP, Rimmer DM, Lefer DJ. Differential response to myocardial reperfusion injury in eNOS-deficient mice. Am J Physiol Heart Circ Physiol. 2002;282:H2422–H2426. doi: 10.1152/ajpheart.00855.2001. [DOI] [PubMed] [Google Scholar]
- 22.Krieglstein CF, Cerwinka WH, Laroux FS, Salter JW, Russell JM, Schuermann G, Grisham MB, Ross CR, Granger DN. Regulation of murine intestinal inflammation by reactive metabolites of oxygen and nitrogen: divergent roles of superoxide and nitric oxide. J Exp Med. 2001;194:1207–1218. doi: 10.1084/jem.194.9.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Binion DG, Fu S, Ramanujam KS, Chai YC, Dweik RA, Drazba JA, Wade JG, Ziats NP, Erzurum SC, Wilson KT. iNOS expression in human intestinal microvascular endothelial cells inhibits leukocyte adhesion. Am J Physiol. 1998;275:G592–G603. doi: 10.1152/ajpgi.1998.275.3.G592. [DOI] [PubMed] [Google Scholar]
- 24.Binion DG, Rafiee P, Ramanujam KS, Fu S, Fisher PJ, Rivera MT, Johnson CP, Otterson MF, Telford GL, Wilson KT. Deficient iNOS in inflammatory bowel disease intestinal microvascular endothelial cells results in increased leukocyte adhesion. Free Radic Biol Med. 2000;29:881–888. doi: 10.1016/S0891-5849(00)00391-9. [DOI] [PubMed] [Google Scholar]
- 25.Vento P, Kiviluoto T, Jarvinen HJ, Soinila S. Changes in distribution of three isoforms of nitric oxide synthase in ulcerative colitis. Scand J Gastroenterol. 2001;36:180–189. doi: 10.1080/003655201750065942. [DOI] [PubMed] [Google Scholar]
- 26.Iwashita E, Iwai A, Sawazaki Y, Matsuda K, Miyahara T, Itoh K. Activation of microvascular endothelial cells in active ulcerative colitis and detection of inducible nitric oxide synthase. J Clin Gastroenterol. 1998;27 Suppl 1:S74–S79. doi: 10.1097/00004836-199800001-00012. [DOI] [PubMed] [Google Scholar]
- 27.Tsukahara H, Gordienko DV, Goligorsky MS. Continuous monitoring of nitric oxide release from human umbilical vein endothelial cells. Biochem Biophys Res Commun. 1993;193:722–729. doi: 10.1006/bbrc.1993.1685. [DOI] [PubMed] [Google Scholar]
- 28.Jugdutt BI. Nitric oxide and cardioprotection during ischemia-reperfusion. Heart Fail Rev. 2002;7:391–405. doi: 10.1023/A:1020718619155. [DOI] [PubMed] [Google Scholar]
- 29.Williams IL, Wheatcroft SB, Shah AM, Kerney MT. Obesity, atherosclerosis and the vascular endothelium mechanisms of reduced nitric oxide bioavailability in obese humans. Int J Obes. 2002;26:754–764. doi: 10.1038/sj.ijo.0801995. [DOI] [PubMed] [Google Scholar]
- 30.Price DT, Loscalzo J. Cellular adhesion molecules and atherogenesis. Am J Med. 1999;107:85–89. doi: 10.1016/S0002-9343(99)00153-9. [DOI] [PubMed] [Google Scholar]
- 31.Zadeh MS, Kolb JP, Geromin D, D'Anna R, Boulmerka A, Marconi A, Dugas B, Marsac C, D'Alessio P. Regulation of ICAM-1/CD54 expression on human endothelial cells by hydrogen peroxide involves inducible NO synthase. J Leukoc Biol. 2000;67:327–334. doi: 10.1002/jlb.67.3.327. [DOI] [PubMed] [Google Scholar]
- 32.Miceli F, Tringali G, Tropea A, Minici F, Orlando MT, Lanzone A, Navarra P, Apa R. The effects of nitric oxide on prostanoid production and release by human umbilical vein endothelial cells. Life Sci. 2003;73:2533–2542. doi: 10.1016/S0024-3205(03)00659-3. [DOI] [PubMed] [Google Scholar]
- 33.Kroll J, Waltenberger J. VEGF-A induces expression of eNOS and iNOS in endothelial cells via VEGF receptor-2 (KDR) Biochem Biophys Res Commun. 1998;252:743–746. doi: 10.1006/bbrc.1998.9719. [DOI] [PubMed] [Google Scholar]
- 34.Cristina de Assis M, Cristina Plotkowski M, Fierro IM, Barja-Fidalgo C, de Freitas MS. Expression of inducible nitric oxide synthase in human umbilical vein endothelial cells during primary culture. Nitric Oxide. 2002;7:254–261. doi: 10.1016/S1089-8603(02)00123-4. [DOI] [PubMed] [Google Scholar]
- 35.Quattrone S, Chiappini L, Scapagnini G, Bigazzi B, Bani D. Relaxin potentiates the expression of inducible nitric oxide synthase by endothelial cells from human umbilical vein in in vitro culture. Mol Hum Reprod. 2004;10:325–330. doi: 10.1093/molehr/gah040. [DOI] [PubMed] [Google Scholar]
- 36.Johnson LR, McCormack SA, Yang CH, Pfeffer SR, Pfeffer LM. EGF induces nuclear translocation of STAT2 without tyrosine phosphorylation in intestinal epithelial cells. Am J Physiol. 1999;276:C419–C425. doi: 10.1152/ajpcell.1999.276.2.C419. [DOI] [PubMed] [Google Scholar]
- 37.Lamas S, Michel T, Collins T, Brenner BM, Marsden PA. Effects of interferon-gamma on nitric oxide synthase activity and endothelin-1 production by vascular endothelial cells. J Clin Invest. 1992;90:879–887. doi: 10.1172/JCI115963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Radomski MW, Vallance P, Whitley G, Foxwell N, Moncada S. Platelet adhesion to human vascular endothelium is modulated by constitutive and cytokine induced nitric oxide. Cardiovasc Res. 1993;27:1380–1382. doi: 10.1093/cvr/27.7.1380. [DOI] [PubMed] [Google Scholar]
- 39.Murohara T, Horowitz JR, Silver M, Tsurumi Y, Chen D, Sullivan A, Isner JM. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation. 1998;97:99–107. doi: 10.1161/01.CIR.97.1.99. [DOI] [PubMed] [Google Scholar]
- 40.Ziche M, Morbidelli L, Choudhuri R, Zhang HT, Donnini S, Granger HJ, Bicknell R. Nitric oxide synthase lies downstream from vascular endothelial growth factor-induced but not basic fibroblast growth factor-induced angiogenesis. J Clin Invest. 1997;99:2625–2634. doi: 10.1172/JCI119451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Konno S, Iizuka M, Yukawa M, Sasaki K, Sato A, Horie Y, Nanjo H, Fukushima T, Watanabe S. Altered expression of angiogenic factors in the VEGF-Ets-1 cascades in inflammatory bowel disease. J Gastroenterol. 2004;39:931–939. doi: 10.1007/s00535-004-1423-9. [DOI] [PubMed] [Google Scholar]
- 42.Kapsoritakis A, Sfiridaki A, Maltezos E, Simopoulos K, Giatromanolaki A, Sivridis E, Koukourakis MI. Vascular endothelial growth factor in inflammatory bowel disease. Int J Colorectal Dis. 2003;18:418–422. doi: 10.1007/s00384-003-0495-y. [DOI] [PubMed] [Google Scholar]
- 43.Giatromanolaki A, Sivridis E, Maltezos E, Papazoglou D, Simopoulos C, Gatter KC, Harris AL, Koukourakis MI. Hypoxia inducible factor 1alpha and 2alpha overexpression in inflammatory bowel disease. J Clin Pathol. 2003;56:209–213. doi: 10.1136/jcp.56.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, Buerk DG, Huang PL, Jain RK. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proc Natl Acad Sci U S A. 2001;98:2604–2609. doi: 10.1073/pnas.041359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cook JP. NO and angiogenesis. Atheroscler. 2003;(Suppl. 4):53–60. doi: 10.1016/S1567-5688(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 46.Morbidelli L, Donnini S, Ziche M. Role of nitric oxide in the modulation of angiogenesis. Curr Pharm Des. 2003;9:521–530. doi: 10.2174/1381612033391405. [DOI] [PubMed] [Google Scholar]
- 47.Polytarchou C, Papadimitriou E. Antioxidants inhibit angiogenesis in vivo through downregulation of nitric oxide synthase expression and activity. Free Radic Res. 2004;38:501–508. doi: 10.1080/10715760410001684621. [DOI] [PubMed] [Google Scholar]
- 48.Polytarchou C, Papadimitriou E. Antioxidants inhibit human endothelial cell functions through down-regulation of endothelial nitric oxide synthase activity. Eur J Pharmacol. 2005;510:31–38. doi: 10.1016/j.ejphar.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 49.Tomasian D, Keaney Jr JF, Vita AJ. Antioxidants and the bioactivity of endothelium-derived nitric oxide. Cardiovascular Res. 2000;47:426–435. doi: 10.1016/S0008-6363(00)00103-6. [DOI] [PubMed] [Google Scholar]
- 50.Huang PL. Endothelial nitric oxide synthase and endothelial dysfunction. Curr Hypertens Rep. 2003;6:473–480. doi: 10.1007/s11906-003-0055-4. [DOI] [PubMed] [Google Scholar]
- 51.Beltrán B, Mathur A, Duchen MR, Erusalimsky JD, Moncada S. The effect of nitric oxide on cell respiration: A key to understanding its role in cell survival or death. Proc Natl Acad Sci U S A. 2000;97:14602–14607. doi: 10.1073/pnas.97.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brookes PS, Salinas EP, Darley-Usmar K, Eiserich JP, Freeman BA, Darley-Usmar VM, Anderson PG. Concentration-dependent effects of nitric oxide on mitochondrial permeability transition and cytochrome c release. J Biol Chem. 2000;275:20474–20479. doi: 10.1074/jbc.M001077200. [DOI] [PubMed] [Google Scholar]
- 53.Shiva S, Brookes PS, Patel RP, Anderson PG, Darley-Usmar VM. Nitric oxide partitioning into mitochondrial membranes and the control of respiration at cytochrome c oxidase. Proc Natl Acad Sc. USA. 2001;98:7212–7217. doi: 10.1073/pnas.131128898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim YM, Chung HT, Simmons RL, Billiar TR. Cellular non-heme iron content is a determinant of nitric oxide-mediated apoptosis, necrosis, and caspase inhibition. J Biol Chem. 2000;275:10954–10961. doi: 10.1074/jbc.275.15.10954. [DOI] [PubMed] [Google Scholar]