

Abstract
We previously reported that the halogenase RebH catalyzes selective halogenation of several heterocycles and carbocycles, but product yields were limited by enzyme instability. Here, we use directed evolution to engineer a RebH variant, 3-LR, with a Topt over 5 °C higher than that of wild type, and 3-LSR, with a Tm 18 °C higher than that of wild type. These enzymes provided significantly (up to 4-fold) improved conversions for halogenation of tryptophan and several non-natural substrates. This initial demonstration of RebH evolution not only provides improved enzymes for immediate synthetic applications, but also establishes a robust protocol for further halogenase evolution.
Keywords: RebH, halogenase, thermostability, biocatalysis, directed evolution
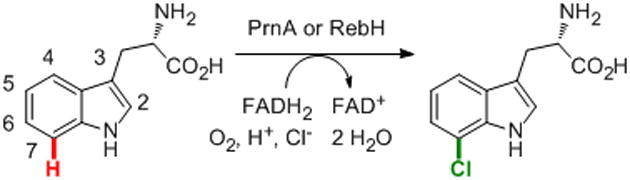
Halogenated organic compounds pervade chemistry, playing important roles as industrial, agrochemical, pharmaceutical, and materials products, as well as functioning as essential building blocks and intermediates in organic synthesis.[1-3] Halogenated arenes comprise a particularly important class of compounds, but conventional approaches to arene halogenation via electrophilic aromatic substitution require harsh chemical oxidants and of ten suffer from poor regioselectivity.[4,5]. In nature, selective arene halogenation is catalyzed by flavin-dependent halogenases,[6] which employ halide salts and air as the halogen source and terminal oxidant respectively (Scheme 1). Several groups have used halogenases, including RebH,[7] PrnA,[8] and point mutants of these enzymes,[9,10] to halogenate tryptophan or related small molecules on an analytical scale. We recently explored the substrate scope and selectivity of RebH and showed that this enzyme can halogenate a range of substituted indoles and naphthalenes on a preparative scale.[11] While the scope, selectivity, and mild reaction conditions we employed highlight the synthetic utility of enzymatic halogenation, the high enzyme loadings required to achieve synthetically useful product yields hinder the practicality of RebH. During the preparative-scale bioconversions conducted in our laboratory, extensive RebH precipitation was observed after several hours of reaction (well after the window in which kinetic data were acquired), which suggests that significant improvements in product yield might be possible by increasing the stability of this enzyme.
Scheme 1. RebH- or PrnA-catalyzed 7-chlorination of tryptophan.
Stability is an important property of all enzymes, particularly those exposed to the reaction conditions encountered in industrial processes or subjected to laboratory evolution experiments.[12,13] Improving enzyme thermostability has multiple benefits, including prolonging catalyst lifetime, increasing enzyme tolerance to stresses such as proteolysis or organic solvents, and enabling reactions to be conducted at higher temperatures, which can increase reaction rates and thus decrease reaction times.[14-16] Stable enzymes can also better tolerate mutations introduced to alter other properties, such as substrate scope and specific activity, since random mutations are generally destabilizing.[17] To our knowledge, no halogenases from thermophilic organisms have been characterized. Therefore, in this work, we describe the first use of directed evolution to increase both the thermostability and the optimal operating temperature of RebH.
To improve thermostability without losing catalytic activity, we employed a screen that involved incubating libraries of RebH mutants at elevated temperatures and examining reaction conversions at room temperature.[18] Error-prone PCR was used to generate a library of RebH variants with an average of two residue mutations per sequence. The library was expressed in E. coli in 96-well expression plates, the cells were lysed, and the supernatants were transferred to microtiter plates for heat treatment. Tryptophan halogenation reactions were conducted overnight, and reaction conversions determined by HPLC analysis.
The first-generation mutant library was constructed using wild-type (WT) RebH as the parent, and 1,365 colonies were screened following incubation at 42 °C for 2 h. Mutants providing twice the conversion of WT were identified and these improved conversions were confirmed following purification and incubation at 49 °C for 2 h. In addition, the melting temperature (Tm), defined as the midpoint of the thermal unfolding transition curve, of an improved mutant with a single amino acid mutation, S2P, was analyzed by circular dichroism (CD) spectroscopy. The S2P mutant has a Tm 2 °C higher than that of WT RebH, indicating increased stability. The beneficial mutations identified in improved variants from the first round were recombined using overlap extension PCR, and the best variant (designated 1-PVM, with the mutations S2P, M71V, and K145M) from this library showed an almost 20-fold improvement in conversion compared to WT (Figure 1A).
Figure 1.
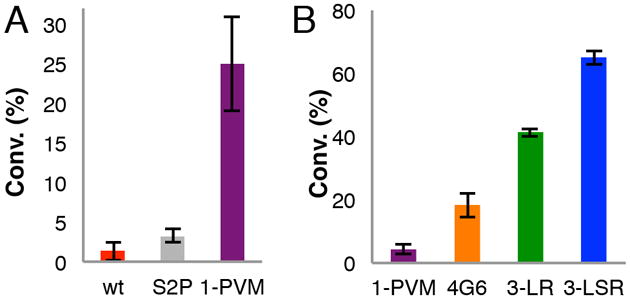
Halogenation conversions (conv.) following incubation at 49 °C for 2 h. Reactions were performed on tryptophan with 2 % (A) and 0. 5 % (B) enzyme loading.
The 1-PVM mutant was used as the parent for a second-generation random mutagenesis library. Of the 1,008 colonies screened following incubation at 51 °C for 2 h, variant 4G6 provided a 2.5-fold increase in conversion relative to the parent as a result of amino acid mutations E423D and E461G. The third-generation random mutagenesis library used 4G6 as the template and contained another 1,008 colonies. The three best-performing variants from the third round of screening following incubation at 54 °C for 3 h each contained single amino acid mutations. Following recombination, the two best variants were identified as 3-LR (S130L, Q494R) and 3-LSR (S130L, N166S, Q494R) (Figure 1B).
The melting temperatures of the best mutants identified throughout the rounds of genetic diversification, screening, and recombination were analyzed to probe the relationship between halogenase conversion and thermostability (Figure 2A). WT RebH has a melting temperature of 52.4 °C, and that of the most thermostable variant, 3-LSR, is 70.0 °C. The 18 °C increase in Tm indicates significant improvement in enzyme stability. To determine if improved thermostability enables the use of higher reaction temperatures, conversion-temperature profiles of RebH variants were constructed (Figure 2B). With the accumulation of beneficial mutations, the optimum temperature for halogenation (Topt) of tryptophan based on total conversion to halogenated product (not initial rate) increased by at least 5 °C, from between 30 and 35 °C for WT RebH to 40 °C for 3-LR. Mutant 3-LR produced 100% more 7-chlorotryptophan than WT RebH when each acted at their respective Topt on an analytical scale.
Figure 2.
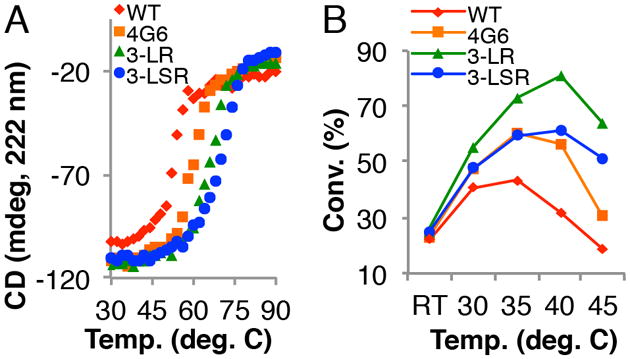
A) Thermal denaturation curves obtained using CD at 222 nm. B) Conversion (conv.)-temperature profiles of RebH enzymes (0.4 mol% RebH).
To establish the relevance of these thermostability improvements to preparative-scale biocatalysis, halogenation of several substrates was examined using 3-LR and 3-LSR (Scheme 2, Table 1). Reaction of tryptophan with 3-LR at 40 °C afforded a 2.8-fold increase in the yield of 1 relative to the reaction of tryptophan with WT RebH at 35 °C, under optimal reaction conditions for both enzymes,[19] based on HPLC analysis. Furthermore, a 69% isolated yield of 1 was obtained using only a 0.4 mol% 3-LR loading compared to a 37% yield using the same loading of WT RebH.
Scheme 2.
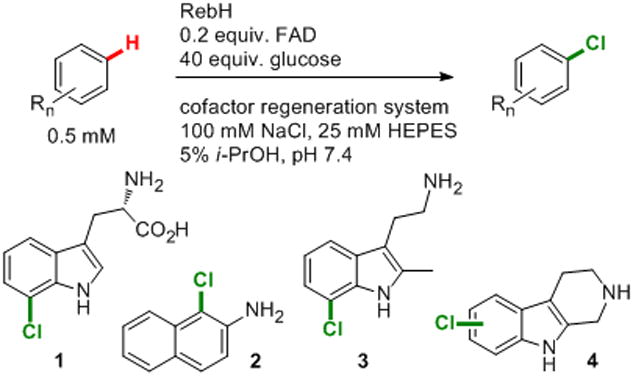
General scheme for RebH-catalyzed arene halogenation, and substrates used to examine enzyme scope.
Table 1.
Representative yields for preparative 3-L(S)R-catalyzed[a] halogenation reactions and comparisons to WT RebH-catalyzed reactions.
| Product | Enzyme (mol%) | Temp. (°C) | Time (h) | Yield (%)[a] | Fold Improvement[b] |
|---|---|---|---|---|---|
| 1 | 3-LR (0.4) | 40 | 16 | 69 | 2.8 |
| 2 | 3-LSR (0.8) | 21 | 30 | 62 | 2.3 |
| 3 | 3-LSR (1.0) | 40 | 36 | 56 | 4.1 |
| 4 | 3-LSR (2.5) | 21 | 48 | 67[c] | 1.7 |
Isolated yield of pure product.
Ratio of product concentrations relative to internal standard from HPLC analysis of crude reaction mixtures using 3-L(S)R or WT RebH under the conditions specified in each entry[19].
88:12 ratio of 5:6 halogenation products.
Improved conversion (1.7-4.1 fold) of the non-natural substrates 2-aminonaphthalene, 2-methyltryptamine, and tryptoline to 2-4, respectively, was also observed with 3-LSR relative to the WT enzyme (Scheme 2, Table 1). Reactions of each substrate with 3-LSR and WT RebH were conducted at 21 °C and 40 °C for identical times and enzyme loadings. In each case, the highest conversion was observed at the same temperature for both 3-LSR and WT RebH: 21 °C for formation of 2 and 4 and 40 °C for formation of 3. Interestingly, the selectivity of tryptoline halogenation (5:6 position) increased from 60% using RebH to 88% using 3-LSR (no other changes in selectivity were observed). Bioconversions with this enzyme continued for 30 hours at up to 40 °C, while WT RebH begins precipitating into inactive aggregates within hours of initiating reactions, clearly illustrating the stability of 3-LSR. [11] Thus, directed evolution can be used to improve halogenase stability, lifetime, and selectivity, and 3-L(S)R should provide a good starting point for evolving RebH variants with activity on a range of additional substrates.[20]
To better illustrate the extended lifetime of these enzymes, the halogenation of 2-methyltryptamine (10 mg) with 3-LSR and WT RebH was monitored using optimal conditions for each enzyme[19] at 40 °C. As noted above, maximum product yields for both enzymes were observed at this temperature; however, the reaction profiles (Fig. 3) show that 3-LSR remains active for significantly longer (ca. 3×) than WT RebH, allowing approximately four-fold improvement in product yield (Table 1, entry 3). Steady state kinetic analysis of these two enzymes shows that WT RebH has a similar kcat/KM relative to 3-LSR at 40 °C (Table 2, entries 1-2), but a significantly higher kcat/KM at 21 °C (Table 2, entries 3-4). These data are consistent with the notion that stabilized enzymes have decreased conformational flexibility relative to non-stabilized enzymes and that while this decrease can be beneficial for stability (prolonging lifetime or enabling reaction at high temp.), it can be detrimental to activity.[21,22] Similarly opposed stability and kinetic data have been reported for other stabilized enzymes that provide increased product yields,[23,24] but stabilized enzymes with essentially unchanged[25,26] or even increased[27,28] kcat/KM have also been reported.
Figure 3. Conversion versus time plots for halogenation of 2-methyltryptoamine by WT RebH and 3-LSR at 40 °C.
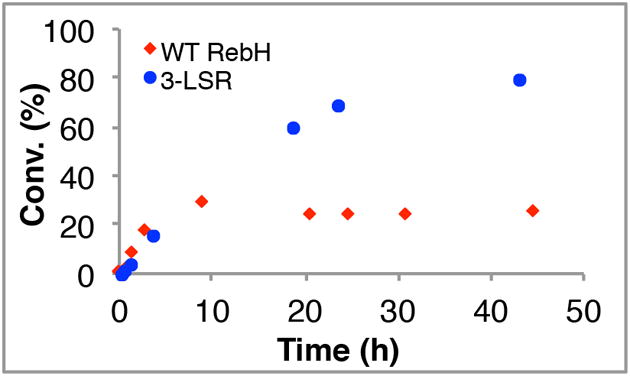
Table 2.
Kinetic data for halogenation of 2-methyltryptamine by RebH and 3-LSR at 40 °C and 21 °C[19].
| Enzyme | Temp. (°C) | KM (μM) | kcat (S-1) | kcat/KM (S-1μM1) |
|---|---|---|---|---|
| RebH | 40 | 280.1 ± 18.4 | 0.25 ± 0.0095 | 8.8 × 10-4 |
| 3-LSR | 40 | 202.5 ± 12.7 | 0.15 ± 0.0093 | 7.9 × 10-4 |
| RebH | 21 | 16.8 ± 3.8 | 0.060 ± 0.0057 | 3.6 × 10-3 |
| 3-LSR | 21 | 40.2 ± 3.7 | 0.013 ± 0.00036 | 3.3 × 10-4 |
A variety of structural features can impart stability to proteins,[29] and in an effort to gain insight into those features in 3-LSR, we solved the crystal structure of this enzyme and compared it with the WT RebH structure. Phases of 3-LSR were obtained by molecular replacement using WT RebH (PDB ID 2OAM) as the search model. The model was refined to 3.05 Å with a final Rwork = 18% and Rfree = 24%. WT RebH and 3-LSR are similar overall with a backbone root mean square deviation (rmsd) of 0.32 Å. The differences in the structures are localized in the eight amino acid changes between the two enzymes.
Investigating the location and nature of the mutations in the structure of 3-LSR may provide a molecular basis for the increase in thermostability and Topt. Mutation Q494R is located on the protein surface and converts the neutral side chain of glutamine into the positively charged side chain of arginine. Increasing the amount of surface charge is a deterrent to protein aggregation.[30] The serine-to-proline mutation of S2P is located at the N-terminus of RebH, and proline residues generally increase protein rigidity by decreasing the flexibility of the polypeptide chain. Indeed, the five other RebH structures in the PDB start their models at amino acid number two or three;[31,32] in 3-LSR, the electron density map extends to amino acid number one, indicating increased order at the N-terminus. The increased rigidity of the N-terminus might also help stabilize the protein by preventing it from acting as a fraying point for thermal unfolding.[33] Mutation K145M is located near the surface of the protein and in the area of two arginine residues (Figure 4). WT RebH increases the density of positive charge in the area with lysine, and 3-LSR might be stabilized by reducing this density by substituting a methionine at this position.[34] Also, the side chain of methionine adopts a conformation that increases its packing with neighboring residues, which might enhance thermostability.[35]
Figure 4.
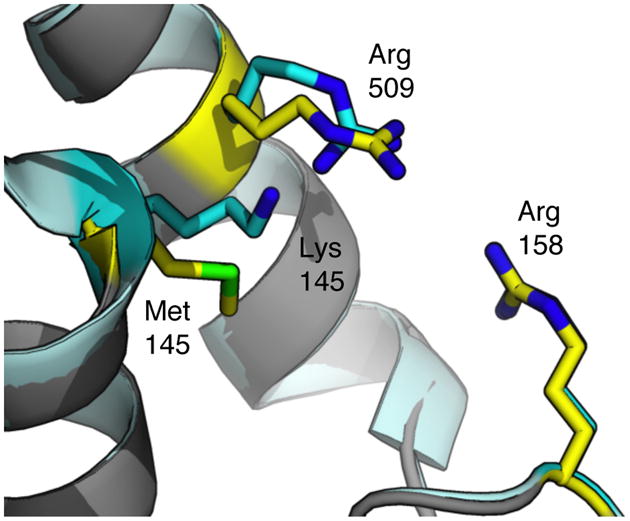
The local environment of the K145M mutation. Overlay of WT RebH (grey backbone and cyan side-chain carbon atoms and blue side-chain nitrogen atoms PDB ID 2OAM) and 3-LSR (light blue backbone and yellow side-chain carbon atoms, blue nitrogen atoms, and green sulfur atom, PDB ID 4LU6).
In our previous work with the tryptophan halogenase RebH, product yield was limited by enzyme instability.[11] Even though RebH has a melting temperature >50 °C, improvements in stability under the reaction conditions were desired, so a protein engineering approach was pursued. Three rounds of error-prone PCR, recombination, and screening resulted in variants 3-LR (7 mutations) with a Topt over 5 °C higher than WT and 3-LSR (8 mutations) with a Tm 18 °C higher than that of WT. That different mutants had the highest Tm and Topt values indicates that thermostability and halogenase conversion were not strictly coupled. A common belief accounting for this divergence is that increased rigidity helps stability but hinders activity.[21,22] Ultimately, however, mutant 3-LSR did provide 100% improvement in halogenase conversion at its Topt, and this improvement translated to several non-natural substrates. This initial demonstration of RebH evolution not only provides improved enzymes for immediate synthetic applications, but also establishes a robust protocol for further halogenase optimization.
Supplementary Material
Acknowledgments
This work was supported by an NIH Pathways to Independence Award (5R00GM087551-03) and a Searle Scholar Award (11-SSP-202) to JCL and an NSF predoctoral fellowship to MCA. We thank Prof. Phoebe Rice f or the use of crystallography software, Dr. Elena Solomaha for assistance with CD spectroscopy, and James Payne for optimization of protein expression and lysis conditions in 96-well plate format. This work is based on research conducted at the APS on the NE-CAT beamlines, which are supported by a grant from the NIGMS (P41 GM103403) from the NIH. Use of the APS, an Office of Science User Facility operated for the U.S. DOE Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. We thank all beamline staff at NE-CAT for their assistance.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.20xxxxxxx.
References
- 1.Fauvarque J. Pure Appl Chem. 1996;68:1713–1720. [Google Scholar]
- 2.Jeschke P. Pest Manag Sci. 2010;66:10–27. doi: 10.1002/ps.1829. [DOI] [PubMed] [Google Scholar]
- 3.Hernandes MZ, Cavalcanti SMT, Moreira DRM, de Azevedo WF, Jr, Leite ACL. Curr Drug Targets. 2010;11:303–314. doi: 10.2174/138945010790711996. [DOI] [PubMed] [Google Scholar]
- 4.Smith K, El-Hiti GA. Curr Org Synth. 2004;1:253–274. [Google Scholar]
- 5.Podgoršek A, Zupan M, Iskra J. Angew Chem Int Ed. 2009;48:8424–8450. doi: 10.1002/anie.200901223. [DOI] [PubMed] [Google Scholar]
- 6.van Pée KH, Patallo EP. Appl Microbiol Biotechnol. 2006;70:631–641. doi: 10.1007/s00253-005-0232-2. [DOI] [PubMed] [Google Scholar]
- 7.Yeh E, Garneau S, Walsh CT. Proc Nat Acad Sci USA. 2005;11:3960–3965. doi: 10.1073/pnas.0500755102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holzer M, Burd W, Rebig HU, van Peé KH. Adv Synth Catal. 2001;343:591–595. [Google Scholar]
- 9.Glenn WS, Nims E, O'Connor SE. J Am Chem Soc. 2011;133:19346–19349. doi: 10.1021/ja2089348. [DOI] [PubMed] [Google Scholar]
- 10.Lang A, Polnick S, Nicke T, William P, Patallo EP, Naismith JH, van Pée KH. Angew Int Ed. 2011;50:2951–2953. doi: 10.1002/anie.201007896. [DOI] [PubMed] [Google Scholar]
- 11.Payne JT, Andorfer MC, Lewis JC. Angew Chem Int Ed. 2013;52:5271–52. doi: 10.1002/anie.201300762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adamczak M, Krishna SH. Food Technol Biotechnol. 2004;42:251–264. [Google Scholar]
- 13.Bloom JD, Labthavikul ST, Otey CR, Arnold FH. Proc Natl Acad Sci USA. 2006;103:5869–5874. doi: 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu I, Arnold FH. Biotechnol Bioeng. 2013;110:1874–1883. doi: 10.1002/bit.24864. [DOI] [PubMed] [Google Scholar]
- 15.Liao HH. Enzyme Microb Technol. 1993;15:286–292. doi: 10.1016/0141-0229(93)90151-q. [DOI] [PubMed] [Google Scholar]
- 16.Zhao H, Arnold FH. Protein Eng. 1999;12:47–53. doi: 10.1093/protein/12.1.47. [DOI] [PubMed] [Google Scholar]
- 17.Pakula AA, Sauer RT. Annu Rev Genet. 1989;23:289–310. doi: 10.1146/annurev.ge.23.120189.001445. [DOI] [PubMed] [Google Scholar]
- 18.Cirino PC, Georgescu R. In: Methods in Molecular Biology, vol 230: Directed Enzyme Evolution: Screening and Selection Methods. Arnold FH, Georgiou G, editors. Humana Press; Totowa: 2003. pp. 117–125. [DOI] [PubMed] [Google Scholar]
- 19.In preparative scale reactions, we found that 3-LSR provided the highest conversions without agitation, while WT RebH provided the highest conversions with agitation, so these conditions were used for each enzyme, respectively, in preparative scale reactions. Steady state kinetics for both enzymes were measured for reactions that were shaken to achieve reproducible results.
- 20.Lewis JC, Mantovani SM, Fu Y, Snow CD, Komor RS, Wong CH, Arnold FH. ChemBioChem. 2010;11:2502–2505. doi: 10.1002/cbic.201000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Závodszky P, Kardos J, Svingor Á, Petsko GA. Proc Natl Acad Sci USA. 1998;95:7406–7411. doi: 10.1073/pnas.95.13.7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolf-Watz M, Thai V, Henzler-Wildman K, Hadjipavlou G, Eisenmesser EZ, Kern D. Nat Struct Biol. 2004;11:945–949. doi: 10.1038/nsmb821. [DOI] [PubMed] [Google Scholar]
- 23.Pei XQ, Yi ZL, Tang CG, Wu ZL. Bioresour Technol. 2011;102:3337–3342. doi: 10.1016/j.biortech.2010.11.025. [DOI] [PubMed] [Google Scholar]
- 24.McDaniel A, Fuchs E, Liu Y, Ford C. Microbial Biotechnol. 2008;1:523–531. doi: 10.1111/j.1751-7915.2008.00055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang D, Zhu F, Fan W, Tao R, Yu H, Yang Y, Jiang W, Yang S. Appl Microbiol Biotechnol. 2011;90:1361–1371. doi: 10.1007/s00253-011-3114-9. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Fuchs E, da Silva R, McDaniel A, Seibel J, Ford C. Starch. 2006;58:501–508. [Google Scholar]
- 27.Kumar R, Sharma M, Singh R, Kaur J. Mol Cell Biochem. 2013;373:149–159. doi: 10.1007/s11010-012-1483-8. [DOI] [PubMed] [Google Scholar]
- 28.Maté D, García-Burgos C, García-Ruiz E, Ballesteros AO, Camarero S, Alcalde M. Chem Biol. 2010;17:1030–1041. doi: 10.1016/j.chembiol.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 29.Petsko GA. Methods Enzymol. 2001;334:469–478. doi: 10.1016/s0076-6879(01)34486-5. [DOI] [PubMed] [Google Scholar]
- 30.Lawrence MS, Phillips KJ, Liu DR. J Am Chem Soc. 2007;129:10110–10112. doi: 10.1021/ja071641y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeh E, Blasiak LC, Koglin A, Drennan CL, Walsh CT. Biochemistry. 2007;46:1284–1292. doi: 10.1021/bi0621213. [DOI] [PubMed] [Google Scholar]
- 32.Bitto E, Huang Y, Bingman CA, Singh S, Thorson JS, Phillips GN., Jr Proteins. 2008;70:289–293. doi: 10.1002/prot.21627. [DOI] [PubMed] [Google Scholar]
- 33.Blake PR, Park JB, Bryant FO, Aono S, Magnuson JK, Eccleston E, Howard JB, Summers MF, Adams MWW. Biochemstry. 1991;30:10885–10895. doi: 10.1021/bi00109a012. [DOI] [PubMed] [Google Scholar]
- 34.Martin A, Sieber V, Schmid FX. J Mol Biol. 2001;309:717–726. doi: 10.1006/jmbi.2001.4698. [DOI] [PubMed] [Google Scholar]
- 35.Querol E, Perez-Pons JA, Mozo-Villarias A. Protein Eng. 1996;9:265–271. doi: 10.1093/protein/9.3.265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.