

Abstract
The divergent synthesis of syn-1, 2-aminoalcohol or syn-1,2-diamine precursors from a common terminal olefin has been accomplished using a combination of palladium(II) catalysis with Lewis acid co-catalysis. Palladium(II)/bis-sulfoxide catalysis with a silver triflate co-catalyst leads for the first time to anti-2-aminooxazolines (C—O) in good to excellent yields. Simple removal of the bis-sulfoxide ligand from this reaction results in a complete switch in reactivity to afford anti-imidazolidinone products (C—N) in good yields and excellent diastereoselectivities. Mechanistic studies suggest the divergent C—O versus C—N reactivity from a common ambident nucleophile arises due to a switch in mechanism from allylic C—H cleavage/functionalization to olefin isomerization/oxidative amination.
Keywords: Amination, Oxidation, Ambident, Aminoalcohol, Diamine, Heterobimetallic catalysis, C—H oxidation, Palladium, Sulfoxide
Introduction
The advent of powerful C—H oxidation reactions1 enables reactive oxygen and nitrogen functionality to be carried through synthetic sequences “masked” as inert C—H bonds and strategically unveiled at late stages to improve efficiency and increase diversity.2 A powerful approach to increase diversity would be to take a common hydrocarbon starting material with an appended ambident nucleophile and, by fully exploiting its distinguishable reactive centers, selectively transform a C—H bond into multiple functionalized products. In this regard, homoallylic ureas could produce either imidazolidinones (cyclic ureas, C—N functionalization) or 2-aminooxazolines (isoureas, C—O functionalization), two medicinally important heterocycles3 and precursors to 1,2-diamines or 1,2-aminoalcohols (Scheme 1).4 While C— O olefin functionalizations with ureas have been demonstrated using stoichiometric hypervalent iodine reagents with limited scope,5 in palladium-catalyzed reactions proceeding through either π-allylPd or amino-palladated intermediates,6–11 ambident O/N nucleophiles have reportedly provided primarily8e C—N functionalization products (Scheme 1).
Scheme 1. Ambident Nucleophile Reactivity.

Design Principles
Palladium(II)/sulfoxide catalysis has proven itself to be a general platform or allylic C—H to C—O12 and C—N13 reactions of terminal olefins under mildly acidic, oxidative conditions. For example, 1,2- and 1,3-aminoalcohol precursors are readily accessible from homoallylic and bishomoallylic carbamates (respectively, Scheme 1). Under similar conditions, homoallylic ureas could furnish either valuable 1,2-diamine (C—N) precursors or 1,2-aminoalcohol (C— O) precursors. Under the reductive, basic conditions of Pd(o)-catalyzed allylic substitutions, ambident urea nucleophiles have provided exclusively C—N products in the form of imidazoidinones.6 This is thought to be due to both a kinetic (base promoted deprotonation at nitrogen) and thermodynamic preference for C—N alkylation [Pd(0)-catalyzed equilibration].14 Given that Pd(ll)/suloxide catalysis proceeds under acidic, oxidative conditions that do not favor nitrogen deprotonation or promote reversible functionalization, O-alkylation may compete with N-functionalization using urea terminal olefin substrates. Additionally, O-alkylation may be further promoted by the addition of an azaphilic Lewis acid additive that binds to the nitrogen and retards nucleophilic attack. Significantly, the 1,2-aminoalcohol products furnished by this method would have the opposite regiochemistry to those previously obtained from carbamate nucleophiles (Scheme 1). With the goal of increasing diversity by tuning the reactivity of an ambident urea nucleophile, herein we show that readily accessible homoallylic N-nosyl urea substrates can be transformed into 2-aminooxazolines (C— O) or imidazolidinones (C—N) by a simple switch in the Pd(II) catalyst in the presence of azaphilic Lewis acid co-catalysts.
Results and Discussion
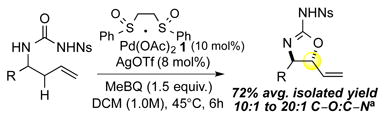
Reaction Discovery: C—O Functionalization
We began our study by subjecting N-nosyl urea 3 to our standard Pd(II)/sulfoxide catalysis. Interestingly, we found that although C-N (5) product was formed in slight excess, an appreciable amount of C–O (4) product was present (Table 1, entry 1). As expected, inclusion of a Brønsted base co-catalyst (Table 1, entry 2) completely shut down the C—O functionalization pathway and resulted in a strong preference for C—N functionalization. However, we found that catalytic B(C6F5)315 (entry 3) or silver triflate16,17 (entry 4), previously shown to have azaphilic properties with amides, increased the overall selectivity favoring the formation of the desired 2-aminooxazoline (4.9:1 and 7:1, respectively) furnishing 4 in 50% and 76% yield, respectively. In contrast, inclusion of an oxophilic Lewis acid co-catalyst [Cr(salen)Cl]13a, previously shown to activate intermediate π-allylPd(BQ) electrophiles towards functionalization, led to diminished yields and did not change the C–O to C–N selectivity (entry 5). As had been previously observed with carbamate nucleophiles, N-tosyl ureas are less reactive (entry 6). Catalytic triflic acid (TfOH) additive promotes reactivity but with lower selectivity for anti-aminooxazoline 4 (entry 7). This result is consistent with experimental observations that water, which may form triflic acid in situ, erodes C—O:C—N ratios. Deletion experiments showed that the Pd/bissulfoxide catalyst 1 was critical for reactivity (entries 8 and 9). To the best of our knowledge, this represents the first example of successfully tuning the reactivity of an ambident O/N nucleophile under palladium catalysis to favor C–O bond formation.
Table 1. Reaction Development.
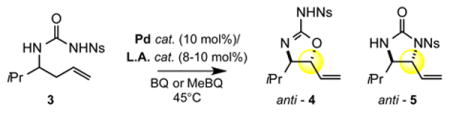
| |||||||
|---|---|---|---|---|---|---|---|
| entrya,b | R |
Pd catalyst (10 mol%) |
L.A. co-catalyst (8 - 10 mol%) |
Yield of 4 (%)c,d |
Yield of 5 (%)c,d |
4:5 | |
| 1e | Ns | 1 | none | 19% | 28% | 1:1.6 |
|
| 2f | Ns | 1 | DIPEA | 0% | 38% | 1:>20 | |
| 3e | Ns | 1 | B(C6F6)3 | 50% | 7% | 4.9:1 | |
| 4g | Ns | 1 | AgOTf | 76% | 9% | 7:1 | |
| 5h | Ns | 1 | Cr(salen)Cl | 8% | 8% | 1:1 | |
| 6g | Ts | 1 | AgOTf | 56% | trace | >20:1 | |
| 7g | Ns | 1 | TfOH | 64% | 28% | 2.3:1 | |
| 8i | Ns | none | TfOH | 0% | 0% | - | |
| 9g,i | Ns | none | AgOTf | 0% | 0% | - | |
| 10e | Ns | 2 | AgOTf | 0% | 46% | 1:>20 | Pd(OAc)2 cat. 2 |
| 11 j | Ns | 2 | AgOTf | 0% | 59% | 1:>20 | |
| 12j | Ns | 2 | B(C6F6)3 | 0% | 62% | 1:>20 | |
| 13j | Ns | 2 | none | 0% | 32% | 1:>20 | |
| 14i,j | Ns | none | B(C6F6)3 | 0% | 0% | - | |
All reactions were run using THF (1.0M) as solvent, MeBQ (methyl-p-benzoquinone, 1.5 equiv.) as terminal oxidant and 8 mol% L.A. co-catalyst for 6 hours unless otherwise noted.
Reactions with cat. 1 were run with an additional 5 mol% BisSO ligand [1,2-bis(phenylsulfinyl)ethane].
Average of 2 runs.
Observed diastereoselectivities in crude reaction mixtures: >20:1 dr for 4 for all entries; dr for 5: entry 1, 1.3:1 dr anti:syn; entry 2, 4.4:1 dr; entry 4, 1:1 dr; entries 3, 5, 10, 11, 12, 13 >20:1 dr; dr determined by 1H NMR analysis of the crude reaction mixture.
Reactions run to complete conversion: entry 1, 24h; entry 3, 2h; entry 10, 72h.
Reaction stopped at 18h (37% rsm); no improved conversion was observed at 48h or 72h.
DCM (dichloromethane, 1.0M); For entry 4, 1 equiv. of DMA was added.
Reaction stopped before complete conversion at 2h. Increased reaction times resulted in product decomposition.
>99% rsm, 0% E-internal olefin.
Reaction run using THF (1.66M) as solvent, BQ (p-benzoquinone, 1.05 equiv.) as terminal oxidant and 10 mol% L.A. co-catalyst for 72 hours.
Reaction Discovery: C—N Functionalization
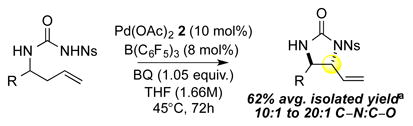
We were intrigued by our discovery of a selective C—O bond-forming reaction using Pd(II)/sulfoxide catalysis, and questioned whether the sulfoxide ligand was critical for the observed selectivity. After removing the ligand from a standard reaction, we made the unexpected observation of the exclusive formation of C—N alkylated product anti-imidazolidinone 5 in 46% yield (entry 10). Upon optimization, synthetically useful yields and excellent diastereoselectivity (>20:1 dr) were obtained using either silver triflate (entry 11, 59% yield) or B(C6F5)3 (entry 12, 62% yield). Notably, omission of the Lewis acid co-catalyst resulted in lower product formation (32% yield) due to an oxidative amination pathway (aminopalladation/β-hydride elimination) of the terminal olefin (entry 13). Additionally, omission of the palladium catalyst gave no reactivity (entry 14).
Collectively, these two processes represent the first examples of transforming readily accessible ambident urea nucleophiles into either C—O or C—N alkylated products by simply switching the Pd(II) catalyst.
Reaction Scope
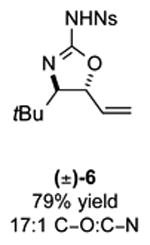
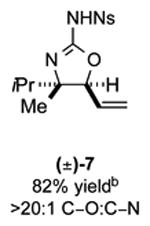
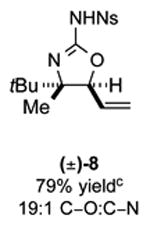
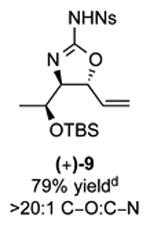
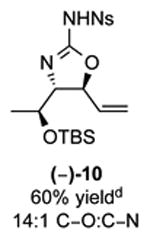
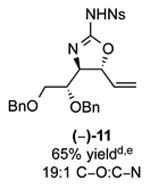
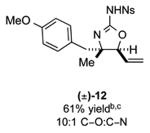
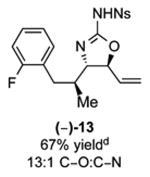
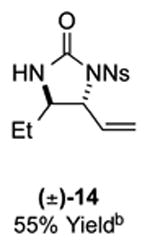
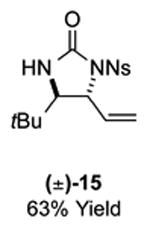
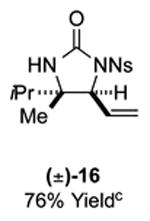
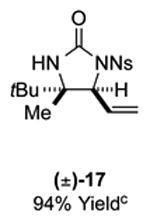
As outlined in Tables 2 and 3, a range of N-nosyl urea olefin substrates were effectively oxidized and aminated using these heterobimetallic catalytic systems to furnish either anti-2-aminooxazolines or anti-imidazolidinones in good yields and selectivities. Substrates containing moderate to high levels of steric bulk adjacent to the urea tether (R = ethyl, isopropyl, t-Bu) furnished the corresponding products in high yields and selectivities with both systems (4, 5, 6, 14, 15). anti-2-Aminooxazolines (7 and 8) and anti-imidazolidinones (16 and 17), derived from common tertiary amines, can also be accessed in excellent yields and selectivities. These represent a class of sterically challenging vicinal diamine and amino-alcohol precursors that are difficult to access otherwise in optically enriched form. In each case, a single starting material was converted selectively into either C—O or C—N functionalized products simply by using the appropriate palladium catalyst. Substrates containing proximal benzylated diols capable of catalyst deactivation via metal chelation are also tolerated under both reaction manifolds [(-)-11, (-)-18, (-)-19]. Aryl moieties were evaluated and the products generated displayed excellent selectivities for either the C—O or the C—N functionalized products [(-)-12, (-)-13, 21]. The diastereoselectivity for both processes appears to be very high; in most cases examined, only the anti-aminooxazolines and anti-imidazolidinones are observed by 1H-NMR of the crude reaction mixtures. For substrates that contain multiple stereogenic centers, the diastereomeric outcome is predictable and controlled entirely by the stereocenter containing the urea ((+)-9, (-)-10, (-)-11, (-)-13, (-)-18, (-)-19). Taken together, these results demonstrate the selective, catalyst-controlled allylic installation of either O or N functionality using a bifunctional urea nucleophile. It is significant to note that alternative methods to generate these products require de novo syntheses of separate starting materials.5-11, 18 Reactions such as these that take a common intermediate and obtain divergent outcomes based on catalyst control have the potential to significantly advance synthetic efficiency and flexibility.
Table 2. PdII/bis-sulfoxide/Ag catalyzed allylic C–O bond formation.
| |||
|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
||
C-O:C-N = 2-aminooxazoline : anti-imidazolidinone; All yields are reported for the major diastereomer.
7: 5.6:1 dr anti:syn 2-aminooxazoline; 12: 4:1 dr.
Reaction run at 45°C with a 0.12:1 DMA:DCM solvent mixture.
Reaction run at 40°C with a 0.12:1 DMA:DCM solvent mixture.
Unable to determine crude selectivity due to peak overlap; isolated selectivity reported.
Table 3.
PdII/B catalyzed allylic C–N bond formation
| |||
|---|---|---|---|
|
|
|
|
|
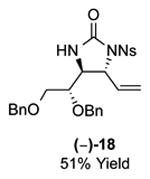
|
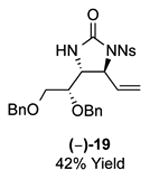
|
|
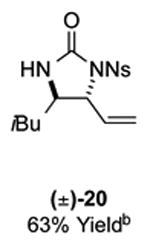
|
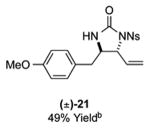
|
||
Diastereoselectivities and C–N:C–O ratios were measured by 1H NMR of the crude reaction mixtures; >20:1 dr and >20:1 C–N:C–O unless otherwise noted.
14: 9.8:1 dr; 20: 7:1 dr; 21: 12:1 dr.
16: 10:1 C–N:C–O; 17: 10:1 C–N:C–O
α,β-Diamino Acids and α-Hydroxy-β-Amino Acids
The vinyl imidazolidinones (ureas) and aminooxazolines (isoureas) generated via these methods are precursors to valuable 1,2-diamines and 1,2-aminoalcohols. For example, the α-olefin moiety can be easily elaborated to furnish α,β-diamino acids and α-hydroxy-β-amino acids, nonproteinogenic amino acids found in numerous biologically active compounds and synthetic peptides.19
Beginning with readily accessible homoallylic N-nosyl urea precursors (4 steps from commercial starting materials, see Supporting Information), application of Pd(II)/bis-sulfoxide/LA-catalyzed oxidation or Pd(II)/LA catalyzed amination methods furnished either the corresponding aminooxazoline (±)-8 or imidazolidinone (±)-20 in excellent yields and selectivities (Tables 2, 3). Elaboration of the vinyl moiety of aminooxazoline (±)-8 via ruthenium tetraoxide-catalyzed oxidative cleavage affords carboxylic acid (±)-22 in 86% yield. Hydrolysis of the/V-nosyl-2-aminooxazoline (±)-22 under acidic conditions afforded the α-hydroxy-β-amino acid (±)-23 in 80% yield. Imidazolidinone (±)-20 could also be readily elaborated to α,β-diamino acid (±)-25 via a three-step sequence involving oxidative cleavage of the olefin (78%), mild PhSH/K2CO3 removal of the N-nosyl group, and acidic hydrolysis (90% over two steps). These methods enable the selective and efficient generation of heterocyclic and acyclic polyoxidized pharmacophores and may expedite discovery of new medicinal agents.
Mechanistic Considerations
Data from the reaction discovery studies suggested that the C—O versus C—N selectivities obtained with the ambident urea nucleophile are dictated by the choice of PdII catalyst (Table 1). In order to confirm that it is a switch in palladium catalyst alone that is causing the selectivity for C—O vs. C—N bond formation in these two reactions, we subjected a common starting material 26 to either 10 mol% Pd(ll)/bis-sulfoxide catalyst 1 or Pd(OAc)2 2 under otherwise identical reaction conditions of azaphilic Lewis acid co-catalyst, solvent, oxidant, and temperature (Scheme 3). Remarkably, both Lewis acid co-catalysts B(C6F5)3 and Ag(OTf) furnished imidazolidinone product anti-15 and 2-aminooxazoline product anti-6 in comparable yields with excellent C—O vs. C—N selectivities of >20:1 that are dependent solely on the nature of the palladium catalyst.
Scheme 3. Ligand Effects on Selectivity.

a Isolated yield, average of 2 runs at 0.3 mmol; b The ratios were determined by 1H NMR analysis of crude reaction mixture.
A critical question raised by these studies relates to the means by which a switch in palladium catalyst results in divergent product outcomes from a common urea starting material. We hypothesized that the observed difference in the reactivity of the urea nucleophile when switching from Pd(ll)/bis-sulfoxide 1 to Pd(OAc)2 2 arises from a change in mechanism from allylic C—H oxidation that furnishes anti-2-aminooxazoline products to olefin isomerization/oxidative olefin amination that generates anti-imidazolidinones. Pd(ll)/sulfoxide catalysis with terminal olefins has been well established to proceed via an allylic C—H cleavage/π-allylPd functionalization mechanism with a wide range of oxygen and nitrogen nucleophiles under varying reaction conditions.12,13 Consistent with C—O alkylation proceeding via such a mechanism, stoichiometric π-allyl intermediate 28 affords anti-aminooxazoline 29 as the major product in the presence of AgOTf (Scheme 4a).
Scheme 4. Mechanistic Experiments for C-O Functionalization.

(a) Stoichiometric formation and functionalization of a π-allyl Pd intermediate (b) Functionalization of a preoxidized N-nosyl urea under Pd(0) conditions.
Given that the sulfoxide ligand is known to retard olefin isomerization12b, we envisioned that its exclusion allows Pd(OAc)2/B(C6F5)3 to effect isomerization of the terminal olefin starting material to the internal E-olefin; subsequently, the internal olefin would be capable of undergoing an oxidative amination pathway (aminopalladation/β-hydride elimination). Isomerizations of terminal olefins catalyzed by Pd(ll)-salts20 are well documented and the addition of Lewis acid additives21 has been shown to promote these processes. To test our hypothesis, we evaluated the rate of functionalization for terminal olefin substrate 3 and the corresponding E-internal olefin substrate 33, which would lead to the observed anti-stereochemistry via an aminopalladation mechanism. Consistent with our hypothesis, both substrates formed the anti-imidazolidinone product with comparable overall kinetic profiles, yields and selectivities (Scheme 5).
Scheme 5. Mechanistic Experiments for C–N Functionalization.

(a) Terminal vs internal olefin functionalization under PdII/B catalysis (b) Overall kinetic profile of the terminal olefin and the internal olefin under the standard C–N functionalization conditions
Additionally, 1H-NMR analysis of the reaction with terminal olefin substrate 3 revealed complete conversion to the corresponding E-internal olefin 33 after only 1 hour22. Our studies demonstrate that a change in mechanism from allylic C—H oxidation to isomerization/aminopalladation led to catalyst-controlled, divergent C—O and C—N allylic functionalization reactions using tethered urea nucleophiles.
Interestingly, while inclusion of a Lewis acid results in O-functionalization from a π-allylPd intermediate, exclusive N-functionalization is observed via an aminopalladation pathway. This outcome may be explained by considering that π-allylPd N-functionalization proceeds via external attack that requires an anionic, nucleophilic nitrogen (Scheme 6). In accord with this, we have observed that Pd(0)-catalyzed allylic substitution of an analogous allylic acetate substrate, known to proceed via a π-allylPd intermediate, does not proceed without inclusion of stoichiometric base (Scheme 4b). The Lewis-acid co-catalysts used in this study may coordinate to the urea nosyl nitrogen and block its attack on the Pd/π-allyl complex and/or generate highly electrophilic π-allylPd intermediates that are preferentially attacked by the hard oxygen urea nucleophile (Scheme 6). In contrast, aminopalladation is a process that has been noted to proceed under both acidic and basic reaction conditions.23 Future studies will seek to better understand the divergent reactivity of ambident nucleophiles with π-allylPd and olefin intermediates.
Scheme 6. Proposed mechanisms.

Conclusion
In conclusion, we have developed a novel process that uses palladium catalysis in combination with Lewis acid co-catalysis for the formation of either anti-2-aminooxazolines or anti-imidazolidinone products from common ambident urea precursors. The inclusion of Lewis acid promotes O-alkylation in reactions proceeding with ambident O/N nucleophiles via π-allylPd intermediates, thereby offering a strategy for expanding the scope of nucleophiles subject to palladium catalysis. Moreover, the discovery of facile isomerization/olefin palladation under LA/Pd(II) conditions may broaden the scope of allylic functionalization reactions available from terminal olefins. We anticipate that the general strategy of taking a common hydrocarbon starting material and selectively transforming it into multiple useful products will find immediate use in streamlining and diversifying the synthesis of medicinal agents and natural products.
Experimantal Procedures
General procedure for the allylic oxidation (Table 2)
A ½ dram borosilicate vial containing a Teflon stir bar was flame dried, sealed with a Teflon lined cap and taken inside a glove box. The homoallylic N-nosyl urea starting material (1 equiv., dried over P2O5), freshly sublimed methyl-p-benzoquinone (1.5 equiv.), catalyst 1 (0.10 equiv.), 1,2-bis(phenylsulfinyl)ethane (0.05 equiv.) and freshly recrystallized AgOTf (0.08 equiv.) were added to the ½ dram vial in the glove box, in the order specified. The vial was securely sealed with the Teflon lined cap and taken out of the glove box. Dry dichloromethane (1.0M) (and DMA, if specified in Table 2) was quickly added to the ½ dram vial outside of the glove box, under a flow of Argon gas. The vial was then sealed and placed in an aluminum block to stir at 45°C for exactly 6 hours. If the reaction is allowed to run longer than 6 hours an erosion of the C–O:C–N selectivity is observed. The solution was allowed to cool to room temperature and then transferred using dichloromethane to a 250 mL separatory funnel. The solution was diluted with 15 mL of dichloromethane and rinsed with 1×15 mL aqueous NH4Cl (sat.). The organic layer was collected and dried over MgSO4, filtered and concentrated in vacuo. The crude reaction mixture was purified using flash column chromatography (in general, gradient 25-30% EtOAc/hexanes was used).
General procedure for the allylic amination (Table 3)
A ½ dram borosilicate vial containing a Teflon stir bar was flame dried, sealed with a Teflon lined cap and taken inside a glove box. The homoallylic N-nosyl urea starting material (1 equiv., dried over P2O5), sublimed benzoquinone (1.05 equiv.), Pd(OAc)2 (0.1 equiv.) and B(C6F5)3 (0.1 equiv.) were added to the ½ dram vial in the glove box, in the order specified. The vial was securely sealed with the Teflon lined cap and taken out of the glove box. Dry tetrahydrofuran (1.66M) was quickly added to the ½ dram vial outside of the glove box, under a flow of Argon gas. The vial was then sealed and stirred in an aluminum block at 45°C for 72 hours, unless otherwise noted in Table 3. The solution was allowed to cool to room temperature and the crude reaction mixture was directly purified using flash column chromatography (in general, gradient 20-30% EtOAc/hexanes was used).
Supplementary Material
Scheme 2. Diversification to biologically relevant motifs.

Acknowledgments
We thank Mr. C. Taylor and Dr. G. Rice for preliminary studies on these systems. We thank Dr. M. A. Bigi for assistance with spectral data analysis and Dr. D. Covell for checking the experimental procedure in Table 1, entry 10. We thank Johnson Matthey for a generous gift of Pd(OAc)2.
Funding Sources: Financial support was provided by the NIH/NIGMS (2R01GM 076153B) and ARRA supplemental funding.
Footnotes
Author Contributions: All authors have given approval to the final version of the manuscript.
Supporting Information. Experimental procedures, characterization data, and copies of 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Crabtree RH. J Chem Soc, Dalton Trans. 2001;2437 [Google Scholar]; (b) Labinger JA, Bercaw JE. Nature. 2002;417:507. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]; (c) Dick AR, Sanford MS. Tetrahedron. 2006;62:2439. [Google Scholar]; (d) Giri R, Shi BF, Engle KM, Maugel N, Yu JQ. Chem Soc Rev. 2009;38:3242. doi: 10.1039/b816707a. [DOI] [PubMed] [Google Scholar]; (e) White MC. Science. 2012;335:807. doi: 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]; (f) White MC. Synlett. 2012;23:2746. [Google Scholar]
- 2.(a) Fraunhoffer KJ, Bachovchin DA, White MC. Org Lett. 2005;7:223. doi: 10.1021/ol047800p. [DOI] [PubMed] [Google Scholar]; (b) Covell DJ, Vermeulen NA, Labenz NA, White MC. Angew Chem Int Ed. 2006;45:8217. doi: 10.1002/anie.200603321. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Stang EM, White MC. Nat Chem. 2009;1:547. doi: 10.1038/nchem.351. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Newhouse T, Baran PS, Hoffmann RW. Chem Soc Rev. 2009;38:3010. doi: 10.1039/b821200g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) McMurray L, O'Hara F, Gaunt MJ. Chem Soc Rev. 2011;40:1885. doi: 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]; (f) Vermeulen NA, Delcamp JH, White MC. J Am Chem Soc. 2010;132:11323. doi: 10.1021/ja104826g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Chen MS, White MC. Science. 2007;318:783. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]; (h) Wender PA, Hilinski MK, Mayweg AVW. Org Lett. 2005;7:79. doi: 10.1021/ol047859w. [DOI] [PubMed] [Google Scholar]
- 3.(a) Bakibaev AA, Shtrykova VV. Russ Chem Rev. 1995;64:929. [Google Scholar]; (b) Lopez O, Maya I, Ulgar V, Robina I, Fuentes J, Fernandez-Bolanos JG. Tetrahedron Lett. 2002;43:4313. [Google Scholar]
- 4.(a) Storch de Gracia I, Bobo S, Martin-Ortega MD, Chiara JL. Org Lett. 1999;1:1705. [Google Scholar]; (b) Anastasi C, Crowe MA, Powner MW, Sutherland JD. Angew Chem Int Ed. 2006;45:6176. doi: 10.1002/anie.200601267. [DOI] [PubMed] [Google Scholar]; (c) Tewari N, Tiwari VK, Mishra RC, Tripathi RP, Srivastava AK, Ahmad R, Srivastava R, Srivastava BS. Bioorg Med Chem. 2003;11:2911. doi: 10.1016/s0968-0896(03)00214-1. [DOI] [PubMed] [Google Scholar]; (d) Saibabu Kotti SRS, Timmons C, Li G. Chem Biol Drug Des. 2006;67:101. doi: 10.1111/j.1747-0285.2006.00347.x. [DOI] [PubMed] [Google Scholar]; (e) Bergmeier SC. Tetrahedron. 2000;56:2561. [Google Scholar]
- 5.Urea C-O functionalization of alkenes to furnish 5-monosubstituted oxazolines has been demonstrated with iodine reagents and platinum catalysts; however 4,5-disubstituted 2-aminooxazolines are not accessible via these methods: Farid U, Wirth T. Angew Chem Int Ed. 2012;51:3462. doi: 10.1002/anie.201107703.Cochran BM, Michael FE. Org Lett. 2008;10:5079. doi: 10.1021/ol8022165.Li H, Widenhoefer RA. Tetrahedron. 2010;66:4827. doi: 10.1016/j.tet.2010.03.082.Muniz K, Igelsias A, Fang Y. Chem Commun. 2009:5591. doi: 10.1039/b912139k.
- 6.Oshitari T, Akagi R, Mandai T. Synthesis. 2004;9:1325. [Google Scholar]
- 7.For seminal reports on olefin diamination with stoichiometric palladium or osmium see: Backvall JE. Tetrahedron Lett. 1978;19:163.Chong AO, Oshima K, Sharpless BK. J Am Chem Soc. 1977;99:3420.
- 8.Palladium catalyzed olefin diamination to furnish mono-substituted ureas: Streuff J, Hovelmann CH, Nieger M, Muniz K. J Am Chem Soc. 2005;127:14586. doi: 10.1021/ja055190y. For a dehydrogenative diamination of terminal olefins Wang B, Du H, Shi Y. Angew Chem Int Ed. 2008;47:8224. doi: 10.1002/anie.200803184. Copper catalyzed Zabawa TP, Kasi D, Chemler SR. J Am Chem Soc. 2005;127:11250. doi: 10.1021/ja053335v.Zhao B, Yuan W, Du H, Shi Y. Org Lett. 2007;9:4943. doi: 10.1021/ol702061s. Ami-nooxazolines were observed as side products: Muñiz K, Hövelmann CH, Campos-Gómez E, Barluenga J, González JM, Streuff J, Nieger M. Chem Asian J. 2008;3:776. doi: 10.1002/asia.200700373.
- 9.Palladium catalyzed olefin diamination of stereochemically defined internal olefins to furnish di-substituted sulfamides: McDonald RI, Stahl SS. Angew Chem Int Ed. 2010;49:5529. doi: 10.1002/anie.200906342.van Benthem RATM, Hiemstra H, Longarela R, Speckamp WN. Tetrahedron Lett. 1994;35:9281.
- 10.For a dehydrogenative diamination of terminal olefins to generate disubstituted ureas see: Du H, Zhao B, Shi Y. J Am Chem Soc. 2008;130:8590. doi: 10.1021/ja8027394.
- 11.For palladium-catalyzed diamination of dienes see Bar GLJ, Lloyd-Jones GC, Booker-Milburn KI. J Am Chem Soc. 2005;127:7308. doi: 10.1021/ja051181d.Du H, Zhao B, Shi Y. J Am Chem Soc. 2007;129:762. doi: 10.1021/ja0680562.Du H, Yuan W, Zhao B, Shi Y. J Am Chem Soc. 2007;129:11688. doi: 10.1021/ja074698t.
- 12.Pd-catalyzed allylic C–H esterifications: Chen MS, White MC. J Am Chem Soc. 2004;126:1346. doi: 10.1021/ja039107n.Chen MS, Prabagaran N, Labenz NA, White MC. J Am Chem Soc. 2005;127:6970. doi: 10.1021/ja0500198.Delcamp JH, White MC. J Am Chem Soc. 2006;128:15076. doi: 10.1021/ja066563d.Fraunhoffer KJ, Prabagaran N, Sirois LE, White MC. J Am Chem Soc. 2006;128:9032. doi: 10.1021/ja063096r. (e) Refs. 2a, 2b, 2c, 2g. Gormisky PE, White MC. J Am Chem Soc. 2011;133:12584. doi: 10.1021/ja206013j.Campbell AN, White PB, Guzei IA, Stahl SS. J Am Chem Soc. 2010;132:15116. doi: 10.1021/ja105829t.Thiery E, Aouf C, Belloy J, Harakat D, Le Bras J, Muzart J. J Org Chem. 2010;75:1771. doi: 10.1021/jo902587u.Mitsudome T, Umetani T, Nosaka N, Mori K, Mizugaki T, Ebitani K, Kaneda K. Angew Chem Int Ed. 2006;45:481. doi: 10.1002/anie.200502886.Check CT, Henderson WH, Wray BC, Eynden MJV, Stambuli JP. J Am Chem Soc. 2011;133:18583. doi: 10.1021/ja2089102.Alam R, Pilarski LT, Pershagen E, Szabo KJ. J Am Chem Soc. 2012;134:8778. doi: 10.1021/ja302457p.
- Pd-catalyzed allylic C–H aminations: Reed SA, White MC. J Am Chem Soc. 2008;130:3316. doi: 10.1021/ja710206u.Reed SA, Mazzotti AR, White MC. J Am Chem Soc. 2009;131:11701. doi: 10.1021/ja903939k.Liu G, Yin G, Wu L. Angew Chem Int Ed. 2008;47:4733. doi: 10.1002/anie.200801009.Wu L, Qiu S, Liu G. Org Lett. 2009;11:2707. doi: 10.1021/ol900941t.Fraunhoffer KJ, White MC. J Am Chem Soc. 2007;129:7274. doi: 10.1021/ja071905g.Rice GT, White MC. J Am Chem Soc. 2009;131:11707. doi: 10.1021/ja9054959.Qi X, Rice GT, Lall MS, Plummer MS, White MC. Tetrahedron. 2010;66:4816. doi: 10.1016/j.tet.2010.04.064.Nahra F, Liron F, Prestat G, Mealli C, Messaoudi A, Poli G. Chem Eur J. 2009;15:11078. doi: 10.1002/chem.200901946.
- 14.Moreno-Manas M, Pleixats R. Advances in Heterocyclic Chemistry. 1996;66:73. [Google Scholar]
- 15.For the complexation of B(C6F5)3 with a wide range of nitrogen-containing compounds see: Focante F, Mercandelli P, Sironi A, Resconi L. Coordination Chemistry Reviews. 2006;250:170.
- 16.Ag+ was shown to lower rotational barrier in amides, see: Waghorne WE, Ward AJI, Clune TG, Cox BG. J Chem Soc, Faraday Trans 1. 1980;76:1131. For complexation of electrophilic Cu(II) salts and urea nitrogens see: Maslak P, Sczepanski JJ, Parvez M. J Am Chem Soc. 1991;113:1062. For other metal triflates binding to amide nitrogens see: Ferraris D, Drury WJ, III, Cox C, Lectka T. J Org Chem. 1998;63:4568. doi: 10.1021/jo981079h.
- 17.For a general review on Ag-catalyzed reactions see: Naodovic M, Yamamoto H. Chem Rev. 2008;108:3132. doi: 10.1021/cr068413r.
- 18.There are no direct, catalytic methods to make 4,5-disubstituted 2-aminooxazolines: syntheses require lengthy sequences from amino-alcohols (See Supporting Information).
- 19.(a) Viso A, Fernandez de la Pradilla R, Garcia A, Flores A. Chem Rev. 2005;105:3167. doi: 10.1021/cr0406561. [DOI] [PubMed] [Google Scholar]; (b) Cardillo G, Tomasini C. Chem Soc Rev. 1996:117. [Google Scholar]
- 20.Davies NR. Aust J Chem. 1964;17:212. [Google Scholar]
- 21.Lim HJ, Smith CR, RajanBabu TV. J Org Chem. 2009;74:4565. doi: 10.1021/jo900180p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.It is significant to note that while it takes just four steps to furnish the terminal olefin starting material 3, it requires 5 steps including a challenging metal/ammonia reduction to generate the stereochemically defined E-olefin 33.
- 23.Liu G, Stahl SS. J Am Chem Soc. 2007;129:6328. doi: 10.1021/ja070424u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.