

Abstract
Anti-cancer topoisomerase I (Top1) inhibitors (camptothecin and its clinical derivatives irinotecan and topotecan, and the indenoisoquinolines) induce lethal DNA lesions by stabilizing Top1-DNA cleavage complex (Top1cc). These lesions are repaired by parallel repair pathways including the tyrosyl-DNA phosphodiesterase 1 (TDP1)-related pathway and homologous recombination. As TDP1-deficient cells in vertebrates are hypersensitive to Top1 inhibitors, small molecules inhibiting TDP1 should augment the cytotoxicity of Top1 inhibitors. We developed a cell-based high-throughput screening assay for the discovery of inhibitors for human TDP1 using a TDP1-deficient chicken DT40 cell line (TDP1-/-) complemented with human TDP1 (hTDP1). Any compounds showing a synergistic effect with the Top1 inhibitor camptothecin (CPT) in hTDP1 cells should either be a TDP1 inhibitor or an inhibitor of alternate repair pathways for Top1cc. We screened the 400,000-compound Small Molecule Library Repository (SMLR, NIH Molecular Libraries) against hTDP1 cells in the absence or presence of CPT. After confirmation in a secondary screen using both hTDP1 and TDP1-/- cells in the absence or presence of CPT, five compounds were confirmed as potential TDP1 pathway inhibitors. All five compounds showed synergistic effect with CPT in hTDP1 cells, but not in TDP1-/- cells, indicating that the compounds inhibited a TDP1-related repair pathway. Yet, in vitro gel-based assay revealed that the five compounds did not inhibit TDP1 catalytic activity directly. We tested the compounds for their ability to inhibit poly(ADP-ribose)polymerase (PARP) because PARP inhibitors are known to potentiate the cytotoxicity of CPT by inhibiting the recruitment of TDP1 to Top1cc. Accordingly, we found that the five compounds inhibit PARP activity by ELISA and Western blotting. We identified the most potent compound (Cpd1) that offers characteristic close to veliparib, a leading clinical PARP inhibitor. Cpd1 may represent a new scaffold for the development of PARP inhibitors.
Keywords: TDP1, PARP, topoisomerases, drug discovery, combination therapy
1. Introduction
Tyrosyl-DNA phosphodiesterase 1 (TDP1) is a DNA repair enzyme (for review see [1, 2]) that removes topoisomerase I (Top1) cleavage complexes (Top1 cc) resulting from the trapping of Top1 on DNA. Reversible Top1 cc are generated during replication and transcription to relax DNA supercoiling. However, they can also be trapped by DNA lesions including abasic sites, oxidized bases and carcinogenic adducts [3-5]. Anticancer Top1 inhibitors such as topotecan, irinotecan and non-camptothecin indenoisoquinolines stabilize Top1cc and cause lethal DNA double-strand ends when Top1cc are collided by replication forks [6-10] and transcription complexes [11-14]. TDP1 repairs Top1cc by excising the covalent bond between the catalytic tyrosine residue of Top1 linked and the DNA 3’-phosphate group [15-17]. TDP1 can also remove a wide variety of 3’-DNA blocking lesions including 3’-phosphoglycolates [18, 19], 3’-deoxyribose phosphate [20, 21] and chain-terminating nucleotides [22]. In addition, TDP1 plays a role in the backup of the topoisomerase II (Top2) cleavage complexes repair pathway [23, 24]. TDP1 is part of the XRCC1 complex that interacts with poly(ADP-ribose)polymerase 1 (PARP1) in the repair of Top1cc [25-27]. PARP1 poly-ADP-ribosylates TDP1, which enhances TDP1 stability and its recruitment to DNA damage sites [25]. Hence, PARP1 is emerging as a key component driving the repair of Top1cc by TDP1, which, at least in part [28, 29], accounts for the synergistic effect of PARP inhibitors with Top1 inhibitors.
The rationale for developing TDP1 inhibitors is rooted in the hypersensitivity of TDP1-deficient cells to Top1 inhibitors [12, 13, 30], to monofunctional alkylating agents including temozolomide [31, 32] and to chain terminating nucleosides [22]. Therefore, TDP1 inhibitors should exhibit synergistic activity when administered in combination with Top1 inhibitors, temozolomide, and chain terminators. Furthermore, cancer cells are very often deficient in alternative DNA repair and/or checkpoint pathways [33]. For example, deficiency in XPF-ERCC1, which belong to the exonuclease pathway for Top1cc repair, selectively sensitizes cancer cells to the combination of PARP and Top1 inhibitors [27]. It is also possible that deficiency in BRCA1, a key player of homologous recombination (HR) might sensitize familial breast and ovarian cancers to combination therapy with Top1 inhibitors and TDP1 inhibitors, in addition to the established activity of PARP inhibitors in BRCA-deficient tumors [34, 35]. This is because both the HR and TDP1 pathways act in parallel (synthetic lethality paradigm) for the repair of Top1-induced lesions. Still, there is currently no TDP1 inhibitor with desirable properties for drug development [for review see [36]]. In this study, we developed a cell-based high throughput-screening method for TDP1 pathway inhibitors, screened 400,000-compounds, and report small molecules that potentiate the cytotoxicity of CPT, and which all turn out to be PARP inhibitors.
2. Material and Methods
2.1. Cell lines and Drugs
DT40 cells were cultured with RPMI 1640 medium (GIBCO 11875-093) supplemented with 10% fetal calf serum (Gemini Bio-Products 100-106), 1% chicken serum (Invitrogen 16110082), and 50 μM β-mercaptoethanol at 37°C. TDP1-deficient (TDP1-/-) cells, and TDP1-/- cells complemented with human TDP1 (hTDP1) in chicken DT40 B cell line have previously been reported and described here [23]. Wild-type, PARP1-deficient (PARP1-/-), and BRCA2-trancate mutant (BRCA2tr/-) DT40 cells were obtained from Dr. Takeda, Laboratory of Radiation Genetics, Graduate School of Medicine, Kyoto University (Kyoto, Japan), and described before [37, 38]. CPT and veliparib were obtained from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, DCTD, NCI. Tetra-n-octylammonium bromide was obtained from Sigma-Aldrich (St. Louis, MO).
2.2. Primary high-throughput screening assay
Conditions and details for our cell-based high-throughput screening assay can be found on PubChem under the AID# 686978 and 686979 (https://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=686981&loc=ea_ras). Briefly, DT40 hTDP1 cells cultured without or with 20 nM CPT were dispensed at 400 cells in 5 μl per well in 1536-well white wall/solid bottom assay plates (Greiner Bio-One North America, NC) using a Multidrop Combi 8 channel dispenser (Thermo Fisher, Waltham, MA). Compounds at various concentrations were transferred in a volume of 23 nl to the assay plates using a pintool station (Kalypsys, San Diego, CA). Assay plates were then incubated at 37°C for 48 h, followed by addition of 3 μl/well of CellTiter-Glo reagent (cell viability assay, Promega, Madison, WI) for measurement of intracellular ATP level. After 30 min incubation at room temperature in the dark, the luminescence intensity of the plates was measured using a ViewLux plate reader (PerkinElmer, Shelton, CT).
2.3. Secondary screening assay
Drug cellular sensitivity was measured as previously described [39]. Briefly, cells were continuously exposed to various drug concentrations for 72 h in triplicate. DT40 cells were seeded at 200 cells per well into 384-well white plate (PerkinElmer) in 40 μl of medium. Cell viability was determined at 72 h by adding 20 μl of ATPlite solution (ATPlite 1-step kit, PerkinElmer). After 5 min incubation, luminescence was measured on an EnVision Plate Reader (PerkinElmer). The ATP level in untreated cells was defined as 100% percent and viability of treated cells was defined as (ATP level of treated cell/ ATP level of untreated cells) ×100.
2.4. PAR ELISA assay
Poly-ADP-ribosylation was measured by the previously reported PAR enzyme-linked immunosorbent assay (ELISA) [40, 41]. Detailed procedure can be viewed at http://dctd.cancer.gov/ResearchResources/biomarkers/PolyAdenosylRibose.htm.
2.5. PAR Immunoblotting assay
Poly-ADP-ribosylation was also measured by immunoblotting. Briefly, five million DT40 cells in 10 ml medium were treated without or with drug for 30 min Cells were collected and lysed with CelLytic™M lysis reagent (C2978, Sigma-Aldrich, St Louis, MO). After thorough mixing and incubation at 4°C for 30 min, lysates were centrifuged at 20,000 g (~15,000 rpm) at 4°C for 10 min, and supernatants were collected. Immunoblotting was carried out using standard procedures. Rabbit polyclonal anti-PAR polymer antibody (#4336-BPC-100) was from Trevigen (Gaithersburg, MD). Secondary antibodies were horseradish peroxidase (HRP)-conjugated antibodies to rabbit IgG (GE Healthcare, Pittsburgh, PA).
2.6. Gel-based TDP1 inhibition assay
TDP1 gel-based assays were performed as described [31]. Briefly, a 5’-[32P]-labeled single-stranded 14 nt DNA oligonucleotide containing a 3’-phosphotyrosine (N14Y) was incubated with 5 pM recombinant human TDP1 in the absence or presence of inhibitor for 15 min at room temperature in a buffer containing 50 mM Tris HCl, pH 7.5, 80 mM KCl, 2 mM EDTA, 1 mM DTT, 40 μg/ml BSA and 0.01% Tween-20. Reactions were terminated by the addition of 1 volume of gel loading buffer [99.5% (v/v) formamide, 5 mM EDTA, 0.01% (w/v) xylene cyanol, and 0.01% (w/v) bromophenol blue]. Samples were subjected to a 16% denaturing PAGE and gels were exposed after drying to a PhosphorImager screen (GE Healthcare). Gel images were scanned using a Typhoon 8600 (GE Healthcare).
3. Results and discussion
3.1. Screening strategy and experimental design
A cell-based assay was developed in a quantitative high throughput-screening (qHTS) format to discover novel TDP1 pathway inhibitors capable of synergizing with CPT. We used chicken DT40 B lymphoma cells that are widely used for reverse genetic studies [42]. DT40 cells have several advantages for drug screening, including efficient gene targeting, a stable phenotype, rapid proliferation, and easy handling [23, 39]. These advantages are suitable for qHTS implementation, which requires fast inhibition of growth within 48 h. Our overall screening strategy is summarized in Figure 1. The first assay (primary qHTS screen, Fig. 1 upper panel) consisted in a 1536-well plate format robotic cell-based high throughput-screening (HTS) assay measuring viability of hTDP1 cells (TDP1-/- cells complemented with human TDP1) exposed to a range of concentrations for each compound of the library in the absence or presence of CPT. Since hTDP1 cells are much more tolerant to CPT compared to TDP1-/- cells [23], TDP1 inhibitors were therefore expected to show a synergistic effect in the presence of CPT and to reduce cell viability to levels similar to TDP1-/- cells (Fig. 1A). This hypersensitivity should not be observed in the absence of CPT. Compounds identified in the primary qHTS screen for their synergistic effect in the presence of CPT were then characterized in a cell-based assay secondary screen (Fig. 1B). In this secondary cell viability assay, both hTDP and TDP1-/- cells were exposed to the compound of interest in the absence or presence of CPT. Inhibitors of the TDP1 pathway are supposed to maintain their synergistic effect with CPT in hTDP1 cells but not in TDP1-/- cells (Fig. 1B).
Figure 1.
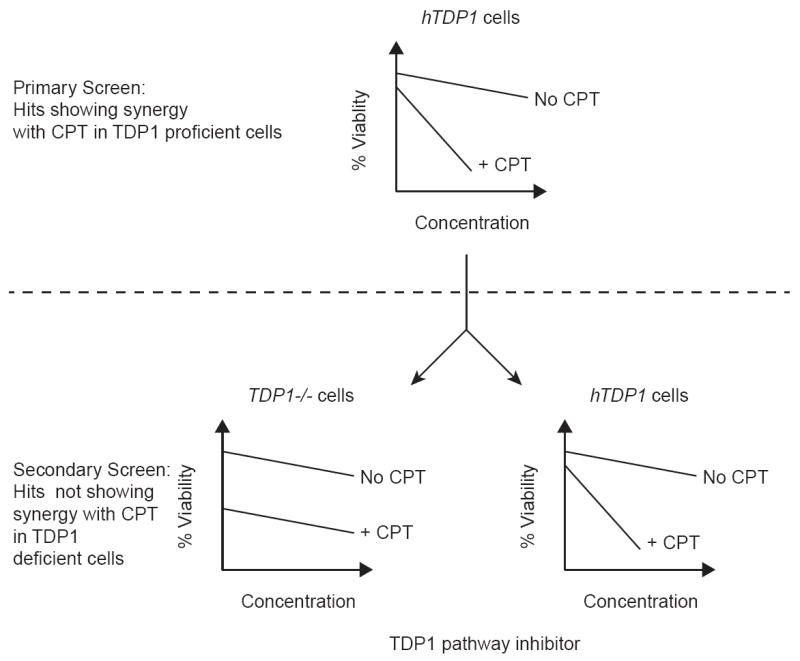
Screening strategy. A: A quantitative robotic high throughput screening (qHTS) assay was run as a primary screen using DT40 chicken B lymphoma cells genetically modified to express human TDP1 (hTDP1) in a knockout background for the chicken TDP1 gene (TDP1-/-) [23]. The compound library was the NIH Roadmap Molecular Libraries 400,000 compound repository. Positive hits were selected based on cellular hypersensitivity in the presence of the Top1 inhibitor camptothecin (CPT). B: Positive hits identified during the HTS assay were confirmed in a secondary screen against both hTDP1 and TDP1-/- cells. Inhibitors of the TDP1 pathway were selected for further characterization based on supra-additive cytotoxicity in the presence of CPT in hTDP1 cells but not in TDP1-/- cells.
Because we recently demonstrated that PARP1 appears to drive the TDP1-related repair pathway [25, 27], we used veliparib (ABT-888) as a positive control in the screening assay. Tetra-n-octylammonium bromide, a highly cytotoxic compound, was used as a non-specific control (Supplemental Figure S1). Veliparib showed average IC50 values (Inhibitory concentration 50%) of 20.4 μM for untreated cells (No CPT) and 0.064 μM for the cells treated with 20 nM CPT, resulting in a 438-fold increase in potency, which recapitulates our recent data [25]. On the other hand, tetra-n-octylammonium bromide as a non-specific control showed average IC50 values of 1.3 and 2.4 μM for untreated cells and cells treated with 20 nM CPT, respectively.
3.3. Primary Screen
The 400,000-compound Small Molecule Library Repository (NIH Molecular Libraries) was screened on the robotic platform of the NIH Chemical Genomics Center (NCGC, now is part of the National Center for Advancing Translational Sciences, NCATS). The entire results were deposited into PubChem (https://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=686981&loc=ea_ras) under AID# 686978 and AID# 686979. Both Pubchem sites list the most cytotoxic compounds identified in the absence (AID# 686978) and in the presence (AID# 686979) of CPT and do not report the positive hits selected for confirmation and characterization. Positive hits were selected based on their IC50 value and inhibition curve quality (curve class) [43]. Compounds showing more than 2-fold decreased in IC50 value for the 20 nM CPT-treated cells (CPT20) compared to untreated cells were selected as positive hits. Compounds that exhibited a class 4 curve (non responsive class) in the absence of CPT and a curve in the presence of CPT categorized as class 1, 2 or 3 (responsive class with various degrees), were selected as primary hits because some compounds may only exhibit their cytotoxicity when combined with CPT. Compounds meeting the above criterions but showing an IC50 value greater than 20 μM in the presence of CPT were not retained based on their lack of potency. Based on these criterions, 500 best compounds were selected and retested in quadruplicate using the primary qHTS assay in the absence and the presence of CPT using hTDP1 cells (See Fig. 1B). Five positive hits were selected for further characterization and the mean of their IC50 values in the absence or presence of CPT are reported in Table 1.
Table 1.
| Cpd | Name | Structure | IC50 (μM)
|
||
|---|---|---|---|---|---|
| HTS
|
Poly (ADP-ribose) lation | ||||
| NO CPT | CPT | ||||
| 1 | MLS002706582 |
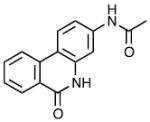
|
25.42 ± 6.07 | 0.15 ± 0.18 | <0.02 |
| 2 | NCGC00345095 |
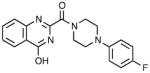
|
25.72 ± 2.90 | 1.45 ± 0.62 | 0.68 |
| 3 | MLS000552049 |
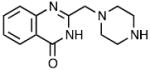
|
>42 | 3.96 ± 0.48 | 0.68 |
| 4 | NCGC00119675 |
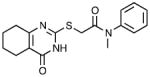
|
>42 | 5.19 ± 1.03 | 0.42 |
| 5 | MLS003171605 |
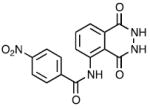
|
>42 | 11.67 ± 2.65 | 4.6 |
| Veliparib |
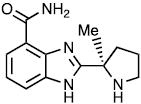
|
28.42 ± 8.58 | 0.06 ± 0.06 | <0.02 | |
3.3. Secondary Screen
The five selected compounds from the primary screen were tested in the secondary screen (Fig. 1B) in the absence and the presence of CPT in hTDP1 and TDP1-/- cells (15 and 20 nM CPT for hTDP1 cells and 5 and 10 nM CPT for TDP1-/- cells). The cell viability curves for the five compounds are presented in Table 1, Figure 2A and Supplemental Figure S2. Cpd1 showed the greatest potentiating effect in the presence of CPT in hTDP cells (Fig. 2A) without showing any apparent potentiating effect with CPT in TDP1-/- cells, which, as expected were exquisitely hypersensitive to CPT compared to the hTDP1 cells [23, 39] (Fig. 2B). The synergistic effect observed in hTDP1 cells for MLS002706582 (Cpd1) is very similar that for veliparib (Fig. 2C) with IC50 values of 150 nM and 6 nM for Cpd1 and veliparib, respectively (Table 1). Cpd1 also behaves similarly to veliparib regarding its lack of synergistic effect with CPT in TDP1-/- cells (Fig. 2D). All five compounds were not cytotoxic as single agents at concentrations below 25 μM (Table 1) but in the presence of CPT, Cpd1 exhibited a 166-fold potentiation of cytotoxicity (466-fold for veliparib). These results suggested that Cpd1 was either a direct inhibitor of TDP1 or a TDP1 repair pathway inhibitor (acting indirectly on TDP1 in cells).
Figure 2.
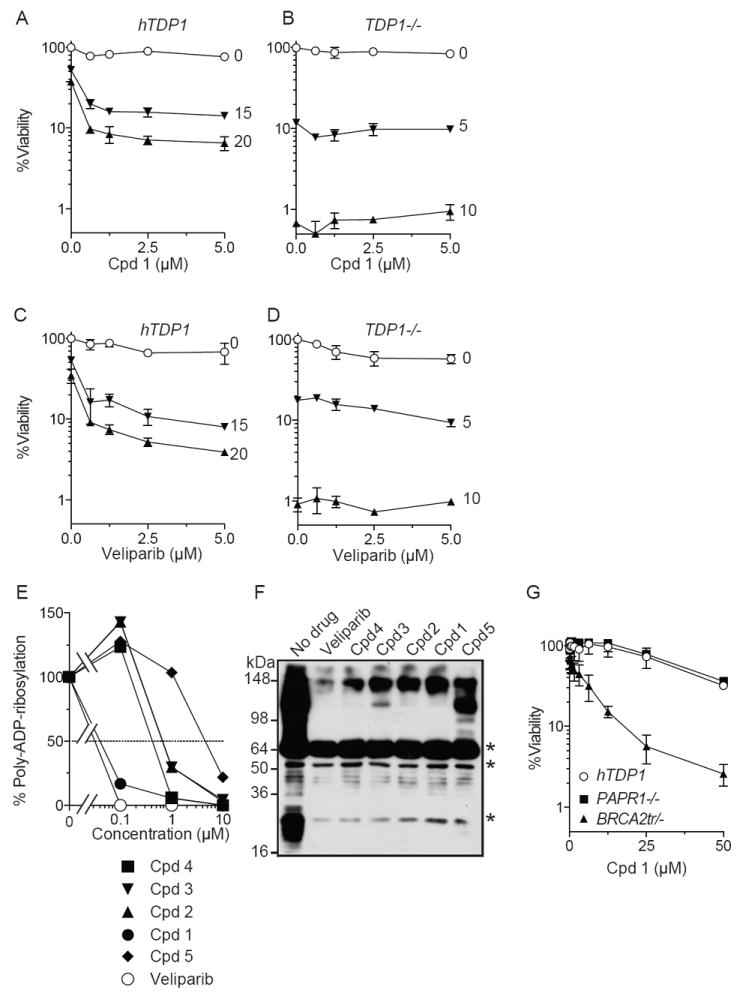
Inhibition of PARP1 by compound 1 (Cpd1). A & B: Cellular viability in the presence of Cpd1 and various concentrations of CPT (indicated beside each curve in nanomolar unit) in hTDP1 (A) and TDP1-/- cells (B). C & D: Cellular viability curves in the presence of veliparib and various concentrations of CPT (indicated beside each curve in nanomolar unit) in hTDP1 (C) and TDP1-/- cells (D). E & F: Inhibition of PARP1 by compounds 1-5 in comparison to the PARP inhibitor veliparib measured by ELISA (E) and by Western blotting (F). Asterisks in (F) indicate non-specific bands. G: Cpd1-dependent viability curves in PARP1-/-, BRCA2tr/- and hTDP1 cells.
3.4. TDP1 catalytic inhibition
To determine whether the five selected compounds from both the primary and secondary screens were direct TDP1 inhibitors, we ran them against recombinant TDP1 in our gel-based biochemical assays [23, 26]. When tested in comparison with a published TDP1 inhibitor [44, 45], none of the five compounds could inhibit efficiently recombinant TDP1 (Supplemental Figure S3). Although, we found a weak TDP1 inhibition for Cpd5 at a dose above 300 μM, these results suggested that the five compounds did not directly target TDP1.
3.5. PARP catalytic inhibition
Because these five compounds contained nicotinamide mimicking moieties (see structures in Table 1) and behave like veliparib (see Fig. 2), we next evaluated their potency as PARP inhibitors. All five compounds inhibited PARP. Moreover, Cpd1 showed PARP catalytic inhibition comparable to veliparib in ELISA assay [40, 41] (Fig. 2E) with sub-nanomolar IC50 values for both compounds (Table 1). These results were confirmed by immunoblotting assay [37]. Thus, all five compounds derived from the cell-based TDP1 screen turned out to be PARP inhibitors (Fig. 2F).
3.6. Hypersensitivity of BRCA2-deficient cells
Because PARP inhibitors are known to be selectively cytotoxic to homologous recombination-deficient cells [34, 35], MLS002706582 (Cpd1) was tested for its cytotoxic effect in BRCA2tr/- cells (homologous recombination-deficient cells) in comparison to PARP1-/- and hTDP1 cells. Cpd1, similar to veliparib [37], was selectively cytotoxic to BRCA2tr/- cells but not to PARP1-/- and hTDP1 cells (Fig. 2G).
4. Conclusions
Altogether, our results demonstrate that screening of the 400,000-compound Small Molecule Library Repository (NIH Molecular Libraries) allowed the identification of five compounds as inhibitors of the TDP1-related DNA repair pathway. These compounds do not inhibit TDP1 directly but act indirectly on the TDP1 pathway by inhibiting PARP1. These finding are in agreement with recent mechanism-based molecular studies showing that PARP is critical to stabilize and recruit TDP1 to Top1-induced DNA lesions [25]. Among the five compounds identified, one of them, Cpd1 offers characteristic close to veliparib and may represent a new scaffold for the development of PARP inhibitors.
Supplementary Material
Acknowledgments
This study was supported in part by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH (Z01 BC 006150) and by the NIH R03 Grant MH095538-01A1 (YP).
Footnotes
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dexheimer TS, Antony S, Marchand C, Pommier Y. Tyrosyl-DNA phosphodiesterase as a target for anticancer therapy. Anticancer Agents Med Chem. 2008;8:381–389. doi: 10.2174/187152008784220357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.El-Khamisy SF. To live or to die: a matter of processing damaged DNA termini in neurons. Embo Mol Med. 2011;3:78–88. doi: 10.1002/emmm.201000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dexheimer TS, Kozekova A, Rizzo CJ, Stone MP, Pommier Y. The modulation of topoisomerase I-mediated DNA cleavage and the induction of DNA-topoisomerase I crosslinks by crotonaldehyde-derived DNA adducts. Nucleic Acids Res. 2008;36:4128–4136. doi: 10.1093/nar/gkn334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pommier Y, Kohlhagen G, Pourquier P, Sayer JM, Kroth H, Jerina DM. Benzo[a]pyrene epoxide adducts in DNA are potent inhibitors of a normal topoisomerase I cleavage site and powerful inducers of other topoisomerase I cleavages. Proc Natl Acad Sci U S A. 2000;97:2040–2045. doi: 10.1073/pnas.040397497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pourquier P, Pommier Y. Topoisomerase I-mediated DNA damage. Adv Cancer Res. 2001;80:189–216. doi: 10.1016/s0065-230x(01)80016-6. [DOI] [PubMed] [Google Scholar]
- 6.Pommier Y. DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev. 2009;109:2894–2902. doi: 10.1021/cr900097c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem Biol. 2013;8:82–95. doi: 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsiang Y-H, Lihou MG, Liu LF. Arrest of DNA replication by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res. 1989;49:5077–5082. [PubMed] [Google Scholar]
- 9.Holm C, Covey JM, Kerrigan D, Pommier Y. Differential requirement of DNA replication for the cytotoxicity of DNA topoisomerase I and II inhibitors in Chinese hamster DC3F cells. Cancer Res. 1989;49:6365–6368. [PubMed] [Google Scholar]
- 10.Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5’-phosphorylated DNA double-strand breaks by replication runoff. Mol Cell Biol. 2000;20:3977–3987. doi: 10.1128/mcb.20.11.3977-3987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Khamisy SF, Katyal S, Patel P, Ju L, McKinnon PJ, Caldecott KW. Synergistic decrease of DNA single-strand break repair rates in mouse neural cells lacking both Tdp1 and aprataxin. DNA Repair (Amst) 2009;8:760–766. doi: 10.1016/j.dnarep.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, Caldecott KW. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature. 2005;434:108–113. doi: 10.1038/nature03314. [DOI] [PubMed] [Google Scholar]
- 13.Miao ZH, Agama K, Sordet O, Povirk L, Kohn KW, Pommier Y. Hereditary ataxia SCAN1 cells are defective for the repair of transcription-dependent topoisomerase I cleavage complexes. DNA Repair (Amst) 2006;5:1489–1494. doi: 10.1016/j.dnarep.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 14.Sordet O, Redon CE, Guirouilh-Barbat J, Smith S, Solier S, Douarre C, Conti C, Nakamura AJ, Das BB, Nicolas E, Kohn KW, Bonner WM, Pommier Y. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep. 2009;10:887–893. doi: 10.1038/embor.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies DR, Interthal H, Champoux JJ, Hol WGJ. Crystal structure of a transition state mimic for Tdp1 assembled from vanadate, DNA, and a topoisomerase I-derived peptide. Chem Biol. 2003;10:139–147. doi: 10.1016/s1074-5521(03)00021-8. [DOI] [PubMed] [Google Scholar]
- 16.Interthal H, Pouliot JJ, Champoux JJ. The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the phospholipase D superfamily. Proc Natl Acad Sci U S A. 2001;98:12009–12014. doi: 10.1073/pnas.211429198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang SW, Burgin AB, Jr, Huizenga BN, Robertson CA, Yao KC, Nash HA. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc Natl Acad Sci U S A. 1996;93:11534–11539. doi: 10.1073/pnas.93.21.11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El-Khamisy SF, Hartsuiker E, Caldecott KW. TDP1 facilitates repair of ionizing radiation-induced DNA single-strand breaks. DNA Repair (Amst) 2007;6:1485–1495. doi: 10.1016/j.dnarep.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 19.Zhou T, Akopiants K, Mohapatra S, Lin PS, Valerie K, Ramsden DA, Lees-Miller SP, Povirk LF. Tyrosyl-DNA phosphodiesterase and the repair of 3’-phosphoglycolate-terminated DNA double-strand breaks. DNA Repair (Amst) 2009;8:901–911. doi: 10.1016/j.dnarep.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dexheimer TS, Stephen AG, Fivash MJ, Fisher RJ, Pommier Y. The DNA binding and 3’-end preferential activity of human tyrosyl-DNA phosphodiesterase. Nucleic Acids Res. 2010;38:2444–2452. doi: 10.1093/nar/gkp1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Interthal H, Chen HJ, Champoux JJ. Human Tdp1 cleaves a broad spectrum of substrates, including phosphoamide linkages. J Biol Chem. 2005;280:36518–36528. doi: 10.1074/jbc.M508898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang SY, Murai J, Dalla Rosa I, Dexheimer TS, Naumova A, Gmeiner WH, Pommier Y. TDP1 repairs nuclear and mitochondrial DNA damage induced by chain-terminating anticancer and antiviral nucleoside analogs. Nucleic Acids Research. 2013;41:7793–7803. doi: 10.1093/nar/gkt483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murai J, Huang SY, Das BB, Dexheimer TS, Takeda S, Pommier Y. Tyrosyl-DNA phosphodiesterase 1 (TDP1) repairs DNA damage induced by topoisomerases I and II and base alkylation in vertebrate cells. The Journal of biological chemistry. 2012;287:12848–12857. doi: 10.1074/jbc.M111.333963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nitiss KC, Malik M, He X, White SW, Nitiss JL. Tyrosyl-DNA phosphodiesterase (Tdp1) participates in the repair of Top2-mediated DNA damage. Proc Natl Acad Sci U S A. 2006;103:8953–8958. doi: 10.1073/pnas.0603455103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Das B, Huang SN, Murai J, Rehman I, Amé J-C, Sengupta S, Das SK, Majumdar P, Zhang H, Biard D, Majumder HK, Schreiber V, Pommier Y. PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucl Acids Res. 2014 doi: 10.1093/nar/gku088. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, Pommier Y. Association of XRCC1 and tyrosyl DNA phosphodiesterase (Tdp1) for the repair of topoisomerase I-mediated DNA lesions. DNA Repair (Amst) 2003;2:1087–1100. doi: 10.1016/s1568-7864(03)00116-2. [DOI] [PubMed] [Google Scholar]
- 27.Zhang YW, Regairaz M, Seiler JA, Agama KK, Doroshow JH, Pommier Y. Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkq1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berti M, Chaudhuri AR, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, Marino F, Lucic B, Biasin V, Gstaiger M, Aebersold R, Sidorova JM, Monnat RJ, Jr, Lopes M, Vindigni A. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol. 2013;20:347–354. doi: 10.1038/nsmb.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol. 2012 doi: 10.1038/nsmb.2258. [DOI] [PubMed] [Google Scholar]
- 30.Katyal S, el-Khamisy SF, Russell HR, Li Y, Ju L, Caldecott KW, McKinnon PJ. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J. 2007;26:4720–4731. doi: 10.1038/sj.emboj.7601869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marchand C, Lea WA, Jadhav A, Dexheimer TS, Austin CP, Inglese J, Pommier Y, Simeonov A. Identification of phosphotyrosine mimetic inhibitors of human tyrosyl-DNA phosphodiesterase I by a novel AlphaScreen high-throughput assay. Mol Cancer Ther. 2009;8:240–248. doi: 10.1158/1535-7163.MCT-08-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alagoz M, Wells OS, El-Khamisy SF. TDP1 deficiency sensitizes human cells to base damage via distinct topoisomerase I and PARP mechanisms with potential applications for cancer therapy. Nucleic Acids Res. 2014 doi: 10.1093/nar/gkt1260. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nature reviews Cancer. 2012;12:801–817. doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- 34.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 35.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 36.Huang SY, Pommier Y, Marchand C. Tyrosyl-DNA Phosphodiesterase 1 (Tdp1) inhibitors. Expert Opin Ther Pat. 2011;21:1285–1292. doi: 10.1517/13543776.2011.604314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer research. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, Morris J, Teicher B, Doroshow JH, Pommier Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol Cancer Ther. 2014;13:433–443. doi: 10.1158/1535-7163.MCT-13-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maede Y, Shimizu H, Fukushima T, Kogame T, Nakamura T, Miki T, Takeda S, Pommier Y, Murai J. Differential and Common DNA Repair Pathways for Topoisomerase I- and II-Targeted Drugs in a Genetic DT40 Repair Cell Screen Panel. Mol Cancer Ther. 2014;13:214–220. doi: 10.1158/1535-7163.MCT-13-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ji J, Kinders RJ, Zhang Y, Rubinstein L, Kummar S, Parchment RE, Tomaszewski JE, Doroshow JH. Modeling pharmacodynamic response to the poly(ADP-Ribose) polymerase inhibitor ABT-888 in human peripheral blood mononuclear cells. PloS one. 2011;6:e26152. doi: 10.1371/journal.pone.0026152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kummar S, Kinders R, Gutierrez ME, Rubinstein L, Parchment RE, Phillips LR, Ji J, Monks A, Low JA, Chen A, Murgo AJ, Collins J, Steinberg SM, Eliopoulos H, Giranda VL, Gordon G, Helman L, Wiltrout R, Tomaszewski JE, Doroshow JH. Phase 0 clinical trial of the poly (ADP-ribose) polymerase inhibitor ABT-888 in patients with advanced malignancies. J Clin Oncol. 2009;27:2705–2711. doi: 10.1200/JCO.2008.19.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buerstedde JM, Takeda S. Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell. 1991;67:179–188. doi: 10.1016/0092-8674(91)90581-i. [DOI] [PubMed] [Google Scholar]
- 43.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci U S A. 2006;103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conda-Sheridan M, Reddy PV, Morrell A, Cobb BT, Marchand C, Agama K, Chergui A, Renaud A, Stephen AG, Bindu LK, Pommier Y, Cushman M. Synthesis and Biological Evaluation of Indenoisoquinolines That Inhibit Both Tyrosyl-DNA Phosphodiesterase I (Tdp1) and Topoisomerase I (Top1) J Med Chem. 2013;56:182–200. doi: 10.1021/jm3014458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nguyen TX, Morrell A, Conda-Sheridan M, Marchand C, Agama K, Bermingam A, Stephen AG, Chergui A, Naumova A, Fisher R, O’Keefe BR, Pommier Y, Cushman M. Synthesis and Biological Evaluation of the First Dual Tyrosyl-DNA Phosphodiesterase I (Tdp1)-Topoisomerase I (Top1) Inhibitors. J Med Chem. 2012;55:4457–4478. doi: 10.1021/jm300335n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.