

Abstract
Reversible protein phosphorylation on serine, threonine, and tyrosine (Ser/Thr/Tyr) residues plays a critical role in regulation of vital processes in the cell. Despite of considerable progress in our understanding of the role of this modification in bacterial physiology, the dynamics of protein phosphorylation during bacterial growth has rarely been systematically addressed. In addition, little is known about in vivo substrates of bacterial Ser/Thr/Tyr kinases and phosphatases. An excellent candidate to study these questions is the Gram-positive bacterium Bacillus subtilis, one of the most intensively investigated bacterial model organism with both research and industrial applications. Here we employed gel-free phosphoproteomics combined with SILAC labeling and high resolution mass spectrometry to study the proteome and phosphoproteome dynamics during the batch growth of B. subtilis. We measured the dynamics of 1666 proteins and 64 phosphorylation sites in five distinct phases of growth. Enzymes of the central carbon metabolism and components of the translation machinery appear to be highly phosphorylated in the stationary phase, coinciding with stronger expression of Ser/Thr kinases. We further used the SILAC workflow to identify novel putative substrates of the Ser/Thr kinase PrkC and the phosphatase PrpC during stationary phase. The overall number of putative substrates was low, pointing to a high kinase and phosphatase specificity. One of the phosphorylation sites affected by both, PrkC and PrpC, was the Ser281 on the oxidoreductase YkwC. We showed that PrkC phosphorylates and PrpC dephosphorylates YkwC in vitro and that phosphorylation at Ser281 abolishes the oxidoreductase activity of YkwC in vitro and in vivo. Our results present the most detailed phosphoproteomic analysis of B. subtilis growth to date and provide the first global in vivo screen of PrkC and PrpC substrates.
Protein phosphorylation on serine, threonine and tyrosine (Ser/Thr/Tyr) is rapidly becoming a prominent avenue of research in microbiology. Hanks-type Ser/Thr kinases and BY-kinases (bacterial Tyr kinases) were shown to have implications in vital processes such as pathogenicity (1, 2), DNA repair, heat shock response (3), cell morphology, and separation (4). In certain pathogenic species like Salmonella, Listeria and Shigella they play a vital role in virulence (5). Functions of Ser/Thr/Tyr phosphorylation have been extensively studied in Bacillus subtilis, a Gram-positive model bacterium widely used in basic research and industrial applications. It was shown that B. subtilis Ser/Thr kinases are involved in regulation of catabolic repression via phosphorylation of the CcpA co-repressor HPr (6). They are also involved in spore development via phosphorylation of a recombinase RecA (7), in spore germination (8), and in regulation of the general stress sigma factor SigB via phosphorylation of Rsb-proteins (9). Importantly, Ser/Thr kinases can also regulate complementary signal transduction systems as shown by phosphorylation of the two-component kinase DegS (10). In addition, B. subtilis tyrosine kinase PtkA plays an important role in DNA replication by phosphorylating SSB proteins (11, 12). It is involved in exopolysaccharide synthesis via phosphorylation of UDP-glucose dehydrogenases (13), and it plays a role in transcriptional regulation via phosphorylation of the fatty acid-displaced repressor FatR (14).
The best studied Ser/Thr kinase in B. subtilis is PrkC, a Hanks-type Ser/Thr kinase encoded by the same operon as PrpC, a Ser/Thr PPM phosphatase. During spore germination (15), PrkC, which contains PASTA repeats responsible for peptidoglycan binding, phosphorylates the essential translation factor EF-G (16). EF-G is a bacterial elongation factor that catalyzes the translocation of the tRNA and mRNA during polypeptide elongation. Because PrpC was shown to dephosphorylate EF-G, this kinase/phosphatase pair has been shown to have opposing functions in the stationary phase (16). In vitro studies have indicated that PrkC can phosphorylate enzymes involved in carbohydrate metabolism, for example the transaldolase YwjH, the glutamine synthetase GlnA, the isocitrate dehydrogenase Icd and the acetolacto-decarboxylase AlsD (17). However, an in vivo study of PrkC and PrpC substrates has not been reported so far.
Despite the low stoichiometry of protein phosphorylation events, improved sample preparation and high resolution mass spectrometry have made it possible to identify hundreds of bacterial phosphorylation events in a single study. Comprehensive phosphoproteomics analysis has been conducted in various bacterial systems such as E. coli (18), Latococcus lactis (19), Pseudomonas species (20), Mycobacterium tuberculosis (21), to name a few. Dynamics of Ser/Thr/Tyr cellular phosphorylation events have been further investigated in the Gram-positive model organism Bacillus subtilis by employing 2D-gel electrophoresis mass spectrometry (22, 23) or a global, gel-free, site-specific quantitative analysis (24, 25). Recently a large transcriptomic, proteomic and metabolomic study was conducted in B. subtilis to gain an understanding of the molecular changes occurring on glucose starvation (26). In this study, using various membrane fractionation and enrichment techniques, the authors were able to quantify 2142 proteins and cover 52% of the predicted proteome, which was the highest coverage of B. subtilis proteome reported so far in a single study.
In this study we performed a global analysis of B. subtilis proteome and phosphoproteome dynamics during batch growth in minimal medium. Using stable isotope labeling by amino acids in cell culture (SILAC)1 and high accuracy mass spectrometry we were able to detect 2264 proteins and 177 phosphorylation sites, and to quantify 1666 proteins and 64 phosphorylation sites in five distinct phases of growth. We extended the SILAC strategy to perform an in vivo screen of potential substrates to the kinase PrkC and phosphatase PrpC. We report that an uncharacterized oxidoreductase, YkwC, is a substrate of both, PrkC and PrpC, and we present in vivo as well as in vitro data to support our findings.
EXPERIMENTAL PROCEDURES
Gene Truncation using pG+host8
Markerless gene inactivation in Bacillus subtilis strain 168 was performed using the pG+host system (27, 28). pG+host8 vector contains a thermosensitive replicon that replicates at 28 °C but not at 37 °C and confers tetracycline resistance (tetr). Regions ∼500 base pairs upstream and downstream of the lysA ORF were cloned using primer pair's lysAko_fwd (P1) - lysAko_rvs (P2) and lysAko_fwd (P3) - lysAko_rvs (P4). The resulting fragments were inserted into pG+host8 vector and the recombinant plasmid was used to transform E. coli NM522 and was verified by sequencing. The constructed vector was then used to transform B. subtilis strain 168, and was selected on LBtet and incubated at 37 °C. Transformants were next grown in LB without tetracycline with vigorous shaking for around 4 days to allow for loss of pG+host8 vector. Gene inactivation was checked by colony PCR. The same strategy was employed to generate ΔprkC and ΔprpC in the ΔlysA background. Primers used are listed in supplemental Table S1. Tetracycline was added where appropriate: 8 μg/ml in E. coli and 15 μg/ml in B. subtilis.
SILAC Labeling of Bacterial Cells
B. subtilis 168 ΔlysA (strain auxotrophic for lysine), referred to as wild-type (WT) henceforth, was grown in the minimal medium consisting of 15 mm (NH4)2SO4, 2 mm CaCl2, 1 μm FeSO4.7H2O, 8 mm MgSO4, 10 μm MnSO4, 27 mm KCl, 0.6 mm KH2PO4, 7 mm C6H5Na3O7·2H2O (Merck), 50 mm Tris-HCl pH 7.5 (Sigma) supplemented with 0.5% glucose (AppliChem, New Haven, CT), 0.67 mm glutamic acid (Merck) and 490 μm tryptophan (Sigma). For SILAC labeling the medium was supplemented with 0.025% of the respective isotopically labeled l-lysine - Light, Lys0: 12C6 14N2 (Sigma-Aldrich); Medium, Lys4: 4,4,5,6-D4 or Heavy, Lys8: 13C6 15N2 (Euriso-Top). Cells were grown at 37 °C at 200 rpm and harvested at different stages of growth. A pre-inoculum at an OD600 of 0.6 was used to inoculate a fresh 200 ml media with a 1:20 dilution and was grown to an OD corresponding to their respective stages namely, T1 - Exponential Growth Phase (OD600 = 0.07); T2 - Entry into Retardation Phase (OD600 = 0.6); T3 - Exit from Retardation Phase (OD600 = 0.9); T4 - Early Stationary Phase (OD600 = 1.1); T5 - Late Stationary Phase (OD600 = 1.3). The nomenclature of the growth phases was based on the terms stated by Monod (29). Two triple SILAC experiments (one with T1, T2, and T3; other with T2, T4 and T5) were conducted, with T2 phase being the common point to integrate the two experiments and thus encompass the entire growth curve profile of B. subtilis. Cultures grown to T1 and T4 stages were labeled with “Light” lysine, T2 with “Medium” and T3 and T5 with “Heavy” lysine. Cells were harvested by centrifugation at 7000 × g for 10 min. They were flash frozen in liquid nitrogen and stored at −80 °C. Fig. 1 indicates the different stages of growth that were sampled and the respective isotopic labels used.
Fig. 1.

A, Growth curve profile of B. subtilis in minimal medium. A composite growth curve of B. subtilis grown in batch culture, in minimal medium under laboratory conditions. In each SILAC experiment cultures were labeled with the respective isotope of lysine and cells were harvested at the denoted time points. B, Phosphoproteomics workflow. Cells were harvested and lysed at the respective stages in each SILAC experiment. Proteins were extracted, mixed in equal ratios and digested with endoproteinase Lys-C. Digested peptides were fractionated by Offgel isoelectric focusing (for whole proteome analysis) and, separately, by SCX and TiO2 chromatographies (for phosphopeptide enrichment). All samples were analyzed on LTQ-Orbitrap mass spectrometer.
An approach similar to that described above was used to identify novel in vivo substrates for the kinase PrkC and the phosphatase PrpC. A single triple SILAC experiment, wherein proteins from the WT strain were labeled with Lys0, ΔprkC strain with Lys4 and ΔprpC strain with Lys8, was carried out (Fig. 4A). Cells were harvested at a midpoint between T4 and T5 phase. All experiments were conducted in biological replicates.
Fig. 4.

SILAC screen of potential PrkC and PrpC substrates. A, Overview of the proteomics workflow adopted for the identification of novel substrates for the Ser/Thr kinase PrkC and Ser/Thr phosphatase PrpC. Briefly, cells were harvested at mid stationary phase. Extracted proteins were digested and fractionated by Offgel or GeLC. Portion of the digested peptides was analyzed for phosphorylation sites as before. WT acts as a control and all ratios were normalized to this state. Proteins with a down regulated M/L ratio and/or an up-regulated H/L ratio were considered as potential candidates for PrkC and PrpC respectively. B, C, Scatter plot showing; ΔprkC/WT SILAC ratios and ΔprpC/WT SILAC ratios respectively. Log2 ratios of the phosphorylation sites are normalized to the corresponding protein and plotted against peptide intensities in Log10 scale. Significantly changing (p = 0.05) SILAC ratios marked in red, present potential substrates of PrkC or PrpC respectively and those marked in green are differentially regulated but not direct substrates of these enzymes.
Protein Extraction and Digestion
Cells were first thawed on ice. For cell lysis, the cell pellets were resuspended in 1 ml Y-PER Reagent (Thermo Scientific) supplemented with 50 μg/ml of lysozyme (Sigma-Aldrich), 5 mm of phosphatase inhibitors—sodium fluoride and glycerol-2-phosphate (Sigma-Aldrich) and protease inhibitor mixture (Roche). Cells were lysed by incubation at 37 °C for 20 min followed by sonication for 30 s at 40% amplitude to degrade DNA. The cell debris was removed by centrifugation at 13.4 rpm for 30 min. The crude protein extract was cleaned up by chloroform/methanol precipitation and the proteins were dissolved in denaturation buffer containing 6 m urea and 2 m thiourea in 10 mm Tris-HCl pH 8.0. Protein concentration was measured by Bradford assay (Bio-Rad Laboratories, Hercules, CA). As a quality control step, protein samples labeled with Lys4 and Lys8 were reviewed for levels of SILAC label incorporation. For this purpose 10 μg of the labeled protein extract was digested (as described below) and analyzed on the mass spectrometer (MS). MS spectra were processed, as described further on in the text, and only an incorporation of 98% and above was accepted and taken further for additional analysis.
For each SILAC experiment, protein extracts labeled with Lys0, Lys4 and Lys8 were mixed in a 1:1:1 ratio to a total of 12 mg and subjected to in-solution digestion. In-solution digestion was carried out as described previously (24) with minor modifications. The protein extract was reduced with 1 mm dithiothreitol (DTT) and alkylated with 5.5 mm iodoacetamide (IAA) in the dark, for 1 h each at room temperature (RT). Proteins were predigested with endoproteinase Lys-C (Waco) (1:100, w/w) for 3 h followed by further overnight digestion at RT with Lys-C (1:100, w/w) after a dilution of 4 times the volume with deionized water. One hundred micrograms of the resulting peptide mixture was taken for isoelectric focusing, while the rest was acidified to a pH of 2.5 with 10% trifluoroacetic acid (TFA) and subjected to strong cationic exchange and titanium dioxide chromatographies.
Protein Fractionation by Offgel Isoelectric Focusing
Peptides obtained from in-solution digestion were separated based on differences in their pI or isoelectric point using the 3100 Offgel Fractionator (Agilent Technologies, Santa Clara, CA). Peptides were separated into 12 fractions using 13 cm Immobiline Drystrips (pH gradient 3–10) (GE Healthcare) using default settings of a maximum current of 50 μA and potential difference of 20 kVh. Fractionated peptides were acidified using 30% acetonitrile (ACN), 5% acetic acid, and 10% TFA in water and further purified by using C18 stage-tips (30). Briefly, C18 discs (EmporeTM) were activated with 100 μl methanol and equilibrated with 200 μl solvent A* (2% ACN/1% TFA). Sample was loaded onto the membrane and washed with 200 μl solvent A (0.5% acetic acid). Peptides were eluted in 50 μl solvent B (80% ACN/0.5% acetic acid), concentrated in a vacuum centrifuge, and subjected to nano-LC-MS/MS measurements on the LTQ-Orbitrap Elite (Thermo Fischer Scientific).
In Gel Digestion (GeLC)
Samples were separated on a NuPage Bis-Tris 4–12% gradient gel (Invitrogen) based on the manufacturer's instructions. The gel was stained with Coomassie blue and subsequently cut into 16 slices. Resulting gel slices were destained by washing three times with 10 mm ammonium bicarbonate (ABC) and ACN (1:1, v/v). Proteins were then reduced with 10 mm DTT in 20 mm ABC for 45 min at 56 °C and alkylated with 55 mm IAA in 20 mm ABC for 30 min at room temperature in the dark. After washing two times with 5 mm ABC and once with ACN, the gel slices were dehydrated in a vacuum centrifuge. Proteins were either digested Lys-C (Wako) (12.5 ng/μl in 20 mm ABC) at 37 °C over night. Resulting peptides were extracted in three subsequent steps with the following solutions: (1) 3% TFA in 30% ACN; (2) 0.5% acetic acid in 80% ACN; (3) 100% ACN. Samples were next evaporated in a vacuum centrifuge and peptides were desalted using stage-tips and analyzed on the LTQ-Orbitrap Elite (Thermo Fischer Scientific).
Phosphopeptide Enrichment
A 11.8 mg sample of digested proteins was subjected to phosphopeptide enrichment in two stages. In the first stage, peptides were separated by SCX (Strong Cationic Exchange) (24). The sample was loaded onto a 1 ml Resource S column (GE Healthcare) in 5 mm KH2PO4, 30% ACN, and 0.1% TFA (buffer pH 2.7) (Merck) with a flow rate of 1 ml/min. Bound peptides were eluted over a linear gradient of 0–35% in 350 mm KCl, 5 mm KH2PO4, 30% ACN and 0.1% TFA (buffer pH 2.7) over 30 min resulting in 16 2 ml fractions. Multiply phosphorylated peptides that do not bind to the column are retained in the flow-through that is collected separately. The 16 SCX fractions were pooled together according to estimated peptide amounts to form a total of nine fractions on which a second stage of phosphopeptide enrichment was performed, along with the flow-through, using TiO2 (Titanium dioxide) chromatography (18). TiO2 spheres of 10 μm (MZ Analysetechnik) were pre-incubated with 2,5-dihydrobenzoic acid in 80% ACN (final concentration 30 mg/ml). Five milligrams of TiO2 beads were added to each of the fractions including the flow-through and incubated for 30 min at RT by end-over-end rotation. The TiO2 enrichment was repeated twice for each of the pooled SCX fractions and five times in case of the flow-through. Incubated beads were washed once with 1 ml of 30% ACN/80% TFA and a second time with 80% ACN/0.1% TFA for 10 min each. The phosphopeptides were eluted from the beads with 3 × 100 μl of 40% NH4OH solution in 60% ACN, pH >10.5. The sample volume was reduced in a vacuum centrifuge at RT, acidified to a pH of 1.5 and desalted using C18 stage-tips and injected on the LTQ-Orbitrap XL (Thermo Fischer Scientific).
Mass Spectrometric Analysis
Samples were measured on an Easy-LC nano-HPLC (Proxeon Biosystems) coupled to an LTQ-Orbitrap XL (or an LTQ-Orbitrap Elite) MS, as described previously (31, 32). Chromatographic separation was performed on a 15 cm fused silica emitter with an inner diameter of 75 μm and a tip diameter of 8 μm, packed in-house with reversed-phase ReproSil-Pur C18-AQ 3 μm resin (Dr. Maisch GmbH). Peptides were injected onto the column with solvent A at a flow rate of 500 nL/min and 280 bars. Peptides were then eluted using a 130 min (or 90 min on the Orbitrap Elite) segmented gradient of 5–90% solvent B at a constant flow rate of 200 nL/min. Separated peptides were ionized by ESI (Proxeon Biosystems). The mass spectrometer was operated on a data-dependent mode. Survey full-scans for the MS spectra were recorded between 300–2000 Thompson at a resolution of 60,000 (or 120,000 on the Orbitrap Elite) with a target value of 106 charges in the Orbitrap mass analyzer. The five (or top 20 on the Orbitrap Elite) most intense peaks from the survey scans were selected for fragmentation with collision induced dissociation (CID) with a target value of 5000 charges in the linear ion trap analyzer in each scan cycle. For phosphoproteomics analysis, ions were fragmented by multi stage activation or MSA with neutral loss occurring at - 97.97, - 48.98, and - 32.66. Dynamic exclusion was set for 90 s. In addition, lock masses option was enabled on the Orbitrap-XL for internal calibration (33).
Data Processing and Bioinformatic Analysis
Acquired MS spectra were processed with MaxQuant software package version 1.2.2.9 (34, 35), integrated with Andromeda search engine. Database search was performed against a target-decoy database of Bacillus subtilis 168 obtained from Uniprot (taxonomy ID 1423), containing 4,195 B. subtilis protein entries and 248 commonly observed laboratory contaminant proteins. Endoprotease Lys-C was fixed as the protease with a maximum missed cleavage of two. Three isotopic forms of lysine (light, medium, and heavy) were stated in the search space. Oxidation of methionines, N-terminal acetylation, and phosphorylation on serine, threonine, and tyrosine residues was specified as a variable modification. Initial maximum allowed mass tolerance was set to six ppm (for the survey scan) and 0.5 Da for CID fragment ions. Carbamidomethylation on cysteines was defined as a fixed modification. Requantify was enabled. A false discovery rate of 1% was applied at the peptide, protein, and phosphorylated site level individually and only fragments with a minimum length of seven amino acids were used for SILAC peptide quantification. A minimum of two unmodified peptide counts were required for the respective protein quantification. For the quantification of the modified phosphorylated site precedence was given to the least (preferably singly) modified peptide. Downstream bioinformatics analysis was performed using the R version 3.0.1 (36). Stringent acceptance criteria were applied for modified peptides identified and quantified by the above described proteomics analysis. Phosphorylation events with a localization probability of ≥0.75 were considered localized on the respective S/T/Y residue. Modified peptides were also filtered for a posterior error probability (PEP) score of ≤ 0.001. MS/MS spectra of phosphorylated peptides were manually validated for good b- and y- ion series coverage using MaxQuant Viewer (v1.2.2.9). All ratios from each SILAC experiment are relative to the Lys4 labeled T2 phase because this was the common reference point chosen. The SILAC ratios of the phosphorylation sites were further normalized with the respective protein ratios to eliminate a bias that could be introduced based on the changing protein abundance. Phosphorylation sites were considered as differentially regulated based on intensity-weighed “Significance B” (p = 0.05) (34) calculated after normalization by corresponding protein ratios.
Clustering Analysis
Hierarchical clustering analysis was employed for the purpose of grouping similar (phosphorylated) protein expression profiles. Standard deviations of log2 transformed ratios of proteins and phosphorylation sites were calculated across the five growth stages. Only proteins belonging to the fourth quartile (topmost 25%) of calculated standard deviations were taken into account for clustering analysis to ensure significant expression change. Hierarchical clustering analysis was performed using “hclust” R-function on Z-score transformed profiles using Euclidean as a distance measure and Ward's method for linkage analysis.
GO Enrichment Analysis
We retrieved Gene Ontology (GO) annotation of Bacillus subtilis from Uniprot-GOA (downloaded on 31/03/2012). To test whether specific annotation terms are enriched or depleted within a set of proteins of interest we applied Fisher's exact test using the theoretical B. subtilis proteome as background. Derived p values were further adjusted to address multiple hypothesis testing using the method proposed by (37).
Synthesis and Purification of 6x-His Tagged Proteins
The genes ykwC, prkC, and prpC were PCR amplified on the genomic DNA extracted from B. subtilis 168 using the primers with specific restriction sites listed in supplemental Table S1. The C terminus of prkC encodes for an insoluble transmembrane region and hence it was deleted. The phosphomimetic mutant of ykwC was constructed by PCR with partially overlapping mutagenic primers (supplemental Table S1). The mutations led to the replacement of Ser281 with aspartic acid. All PCR products were ligated with pQE30-Xa (Qiagen). The sequences were verified by sequencing (GATC Biotech). The recombinant plasmids were used to transform E. coli NM522 and the transformants were selected on LB medium containing ampicillin. Cells were routinely grown in LB medium, with the addition of ampicillin (100 μg/ml) and kanamycin (25 μg/ml) when appropriate. One mm IPTG was used to induce expression of cloned genes.
6xHis-PrpC (30 kDa), -YkwC, and -YkwC S281D (33 kDa) were overxpressed in E. coli NM522. 6xHis-PrkC (41 kDa) was over-expressed in E. coli M15-pREP4::groESL because it was less soluble. Protein purification was performed using Ni-NTA beads by the standard Qiagen protocol with slight changes. Cells were lysed by sonication in a buffer containing 50 mm Tris-HCl pH 7.5, 100 mm NaCl and 10% glycerol, 5 μg/ml DNase and 2 mg/ml lysozyme (Sigma) instead of the phosphate buffer system. Beads were subsequently washed in the presence of 30 mm imidazole and proteins were eluted in 300 mm imidazole. Proteins were desalted on PD-10 columns (GE Healthcare).
In vitro Phosphorylation and Dephosphorylation Assay
Protein phosphorylation reactions were performed with 1 μm PrkC and 10 μm YkwC. In addition to the appropriate proteins, a typical 30 μl reaction mixture contained 50 μm ATP with 20 μCi/mmol [γ-32P]-ATP, 5 mm MgCl2, and 50 mm Tris-HCl pH 7.5, 100 mm NaCl, and 10% glycerol. Samples were incubated for 10, 20, and 40 min respectively at 37 °C after which the reactions were stopped by adding sample buffer for SDS-PAGE and heating at 100 °C for 5 min. Proteins were separated by electrophoresis on denaturing 0.1% SDS/15% polyacrylamide gels, which were subsequently treated with boiling 0.5 m HCl for 12 min. After drying the gels, radioactive bands were visualized with a PhosphorImager (Fujifilm FLA-7000). Exposure time was 16 h.
For dephosphorylation experiments, [γ-32P] ATP was first removed from the 32P-labeled P-Ser/Thr protein samples by using concentrators (Pierce), before adding 10 μm PrpC. The reaction mixtures were incubated for 1 and 2 h respectively at 37 °C before dephosphorylation was stopped by adding sample buffer for SDS-PAGE and heating at 100 °C for 5 min. PAGE and autoradiography were carried out as described above.
Enzymatic Assays
The enzyme kinetics of purified YkwC was monitored in a 200 μl reaction at RT (38). The reaction mixture contained 100 mm Tris-HCl pH 8.8, 100 μm MgCl2, 500 μm NADP, 50 mm 3-HP (3-hydroxyproprionate), and 1 μg of the enzyme. Measurements were taken every 10 s for 30 min. The production of NADPH was measured at 340 nm. Similar measurements were made with 1 μg concentration of YkwC S281D, the phosphomimetic mutant.
For in vivo enzymatic assays, B. subtilis WT, ΔprkC and ΔprpC strain were grown in LB and LB supplemented with 0.3% glucose [LB + Glucose] at 37 °C with vigorous shaking in flasks. Samples were collected during the transition phase (at OD600 approx. 1.4). Cells were lysed by sonication in a buffer containing 50 mm Tris-HCl pH 8.5, 50 mm NaCl, 10% glycerol, and 1 mg/ml lysozyme (Sigma). Cell debris was removed by centrifugation and the supernatant was desalted using PD-10 columns (GE healthcare). The obtained crude extracts were used for the enzymatic assay as described above.
Circular Dichroism Spectroscopy
CD measurements were made on a Jasco J-810 instrument at RT. Suprasil cells (0.1 cm path length) were used for all the measurements. Baselines were the respective buffer solutions (50 mm NaH2PO4 pH 7.5, 100 mm NaCl) in which the proteins were dissolved. At least five repeat scans were obtained for each sample and its respective baseline. The averaged baseline spectrum was subtracted from the averaged sample spectrum. Measurements were made in the range of 240–190 nm (far UV CD). The scanning speed was set to 100 nm/min with a response time being 1 s. Bandwidth was set at 1 nm and scanning mode was continuous. CD measurements of YkwC WT and YkwC S281D were recorded.
RESULTS
Experimental Design of the Growth Curve Measurements
The proteome and phosphoproteome dynamics of the lysine-auxotroph B. subtilis ΔlysA strain (25) was investigated by carrying out two triple-SILAC experiments. Cell cultures labeled with different forms of lysine were harvested at specific time points: T1) exponential growth (Lys0); T2) entry into retardation phase (Lys4); T3) transition to stationary phase (Lys8); T4) early stationary phase (Lys0); T5) late stationary phase (Lys8) (29) (Fig. 1). A common time point, T2 (Lys4), was used to integrate the data into a five time point expression curve for each quantified protein. Correlation of intensities of Lys4-labeled peptides was high (Pearson coefficient 0.8) (supplemental Fig. S1). Proteins extracted from corresponding SILAC cultures were mixed in equal ratios and digested with endoproteinase Lys-C. The resulting peptide mixtures were subjected to phosphopeptide enrichment using SCX and TiO2 chromatography; for proteome analysis peptides were fractionated by Offgel isoelectric focusing. All samples were analyzed by nanoLC-MS/MS on Orbitrap mass spectrometers (Fig. 1). As a quality control step, Lys4-labeled cultures (T2) and Lys8-labeled cultures (T3 and T5) were assessed for the level of incorporation of the lysine label. In all cases incorporation of 98% or higher was observed indicating complete metabolic labeling of the cells (supplemental Fig. S2). Two biological replicates were performed; Pearson correlation coefficients between the replicates ranged from 0.6 (T1) to 0.74 (T4) (supplemental Fig. S3).
Proteome and Phosphoproteome Dynamics During Growth in Batch Culture
Analysis of the proteome of B. subtilis resulted in identification of 18,699 peptides mapping to 2264 proteins at false discovery rate (FDR) of 1% at the peptide and protein level; 2033 proteins were quantified in one of the five analyzed phases, whereas 1666 proteins were quantified in all analyzed phases of growth (supplemental Fig. S4). The phosphoproteome analysis resulted in identification of 177 phosphorylation events of which 156 were quantified in any of the analyzed phases and 144 were localized to a specific residue; 64 phosphorylation sites (on 56 proteins) were quantified in all analyzed phases. List of all detected proteins and phosphorylation sites is presented in supplemental Table S2. In agreement with previous studies, most of the phosphorylation events were observed on Ser residue (74.6%), followed by Thr (18.6%) and Tyr (7.3%) (supplemental Fig. S5). Normalization of phosphopeptide ratios with corresponding protein ratios showed little influence of protein dynamics on phosphorylation site dynamics, especially in early phases of growth (supplemental Fig. S6).
Dynamics of proteome and phosphoproteome across different stages of growth is shown in Fig. 2A. Fluctuation was more pronounced at the phosphorylation level, resulting in higher standard deviation of SILAC ratios in the phosphoproteome dataset. As observed previously (25), proteome dynamics was more pronounced at later stages of growth resulting in a wide spread of measured SILAC ratios. The phosphoproteome dynamics followed the same trend, but showed predominantly an increase in phosphorylation levels at later stages of growth. Interestingly, we recently observed the same trend in the phosphoproteome of E. coli grown under similar conditions (39), pointing to the fact that bacterial protein phosphorylation is predominantly occurring at later stages of growth. To define the subset of dynamic (highly fluctuating) and static proteins we separated them into four bins based on the standard deviation of ratios measured across all growth phases (Fig. 2C). Functional enrichment analysis showed that ribosomal proteins and proteins involved in ATP metabolism were highly fluctuating, whereas enzymes of basic metabolism (e.g. glycolysis) had more constant levels across analyzed growth phases (supplemental Fig. S7).
Fig. 2.

Distribution of SILAC ratios of proteins and phosphorylation sites quantified during B. subtilis growth. A, Histogram depicting measured ratios of proteins and phosphorylation sites in each stage of growth; T1, T3, T4, and T5. All distributions are from the biological replicate 1 and relative to the common time point, T2. B, Changing expression profiles of proteins and phosphorylation sites detected across all 5 stages of growth. C, Box plot depicting standard deviation across growth phases T1-T5 measured for proteins and phosphorylation sites represented in 2B. Data for second biological replicate are presented in supplemental Fig. S11.
To obtain further insights into functional classes of proteins and phosphorylation sites differentially regulated across the analyzed stages of growth we performed hierarchical clustering analysis (Fig. 3A, 3B). Differentially regulated proteins grouped broadly into four clusters: Cluster 1 consisted of proteins that had increased levels at later stages of growth and had functions related to proteolysis and degradation (e.g. d-aminopeptidase, DppA; cell wall-associated protease, WprA) and respiration (e.g. alkyl hydroperoxide reductase, AhpF; cytochrome c-550, CccA). Cluster 2 showed the opposite trend and contained functions related to translation and gene expression (e.g. 30S ribosomal protein S11, RpsK; 50S ribosomal protein L14, RplN) and ATP-binding proteins (e.g. ABC transporters such as cell division protein, FtsE; thiamine import protein, YkoD). Cluster 3 contained proteins with expression peaking in transition to stationary phase and was enriched in functions related to antibiotic biosynthesis pathways (e.g. polyketide biosynthesis protein, PksE), fatty acid or carbohydrate metabolism (e.g. teichoic acid biosynthesis protein, GgaA) and response to stress (e.g. general stress protein A, YwaG). Finally, the Cluster 4 consisted of proteins that had abruptly lower levels in the late stationary phase of growth and included transcription factors such as elongation factor Tu, TufA; transcriptional repressor, NrdR and proteins involved in amino acid biosynthesis (e.g. threonine deaminase, IlvA).
Fig. 3.

Hierarchical clustering analysis of SILAC ratios with highest fluctuation. A, Protein groups; and B, Phosphorylation sites. Fluctuation is defined based on standard deviation, as described in the text. This data concerns biological replicate 1. Data for second biological replicate are presented in supplemental Fig. S12.
Differentially regulated phosphorylation sites grouped into three clusters. The phosphorylation on the first cluster of proteins peaked at the transition phase and abruptly dropped during the stationary phase (e.g. tyrosine-protein kinase PtkB, (EpsB, YveL). The second cluster showed increasing phosphorylation from T1 to T5 and these included proteins involved in carbon metabolism, such as aconitate hydratase, CitB and fructose-1,6-bisphosphate aldolase, Fba. The third cluster had gradually decreasing phosphorylation levels (e.g. chorismate mutase, AroA).
SILAC-based Screen of In Vivo Substrates of the Kinase PrkC and Phosphatase PrpC
The triple-SILAC workflow established to monitor (phospho) proteome dynamics during growth was extended to identification of novel substrates of the kinase PrkC and phosphatase PrpC (Fig. 4A). To this end, we labeled the WT (ΔlysA) strain with Lys0, the kinase knockout (ΔprkC) strain with the Lys4 and the phosphatase knockout (ΔprpC) strain with Lys8. From our preliminary growth curve experiments, we identified stationary phase as the phase of growth where the kinase and phosphatase were most abundant. The kinase and phosphatase KO strains, along with the WT (acting as control), were inoculated in respective SILAC minimal media and the cells were harvested in mid-stationary phase (OD600 = 1.35). Proteome and phosphoproteome measurements were performed as described above. A total of 177 phosphorylation events were identified of which 124 were quantified. One hundred and forty-one phosphopeptides were localized with a probability of ≥ 0.75. Ratios obtained from the analysis of the proteome were used to normalize the quantified phospho-sites. In these measurements we identified 2131 proteins and quantified 2026 of them. In addition, we measured the incorporation of the medium and heavy label to be above 98% of in all replicates (supplemental Fig. S8). List of all detected proteins and phosphorylation sites in the PrkC/PrpC screen is listed in supplemental Table S3.
Global phosphoproteomic analysis during conditions of absence of the kinase or phosphatase led to the detection of seven differentially down regulated events in the ΔprkC strain (potential substrates of the PrkC kinase) (Fig. 4B) and eight differentially up-regulated events in the ΔprpC strain (potential substrates of the PrpC phosphatase) (Fig. 4C).
Among potential PrkC substrates were CitM, a 2-oxoglutarate dehydrogenase complex component E2 which plays an important role in the Krebs cycle; JofD, subunit of the 30s ribosomal protein complex; YabS, an uncharacterized protein whose function has not yet been established; ThiA, a thiamine biosynthesis protein; and AhpF, an alkyl hydroperoxide reductase that protects the cell against reactive oxygen species during stress. Potential PrpC substrates included CotB, a spore coat protein that helps in resistance during adverse conditions; PatA, a putative aminotransferase involved in protein biosynthesis; YmfM, an uncharacterized membrane protein implicated in having a role in cell shape determination; GapA, glyceraldehyde-3-phosphate dehydrogenase enzyme associated with glycolysis; PtkA (YwqD), a tyrosine kinase; and RplN, subunit of the 50s ribosomal protein. We also detected an autophosphorylation site of PrkC, known to be dephosphorylated by PrpC, to be highly regulated in the ΔprpC strain thus acting as a positive control. Interestingly, one phosphorylation site, Ser281 on the uncharacterized oxidoreductase YkwC, was significantly affected by both, PrkC and PrpC, making it a potential substrate of both enzymes (supplemental Table S4). Manual validation of the MS/MS spectrum of the phosphorylated YkwC peptide showed good coverage and annotation of fragment ions (Fig. 5).
Fig. 5.

MS/MS spectrum of the YkwC peptide containing phosphorylation at Ser281. This residue is a potential substrate of both, PrkC and PrpC. Inset shows the corresponding SILAC triplet in a survey scan.
Interestingly, in both the kinase and the phosphatase knockout we have identified phosphorylation sites for which the occupancy is affected in the unexpected direction, higher in the kinase knockout (CotB, GapA and PtkA) and lower in the phosphatase knockout (PhoD, GlmM and PtsH). Although these phosphorylation sites cannot be direct substrates of PrkC and PrpC, they are likely indirectly regulated or positioned further downstream in the signal transduction cascade, a phenomenon commonly observed in eukarya (40). For example, in our data set the inactivation of PrkC leads to an increase of phosphorylation of the BY-kinase PtkA at Y228 (autophosphorylation of PtkA). We have recently established that PrkC phosphorylates PtkA at S223, in the immediate vicinity of Y228 (Shi et al., unpublished results); this phosphorylation event presumably prevents autophosphorylation of PtkA at Y228, leading to an increase of PtkA autophosphorylation in ΔprkC strain that we observed here. The remaining indirectly affected sites we detected are likely to be explained by similar phenomena of cross-talk between kinases and phosphatases, which seem to be abundant in B. subtilis (Shi et al. unpublished results).
Validation of YkwC Ser281 as a Substrate of PrkC and PrpC
To validate Ser281 on YkwC as a substrate of PrkC and PrpC we conducted further biochemical experiments. We first over-expressed and purified YkwC, PrkC and PrpC in E. coli and performed in vitro phosphorylation and dephosphorylation assays using γ-32P ATP. The assays showed that YkwC could not autophosphorylate; that PrkC was able to phosphorylate YkwC; and that PrpC was able to dephosphorylate phosphorylated YkwC in vitro (Fig. 6A). This confirmed that YkwC is indeed a substrate of both enzymes. We next carried out enzymatic assays to test the influence of phosphorylation on Ser281 on YkwC activity. For this purpose we synthesized the phosphomimetic mutant protein YkwC S281D. As a putative oxidoreductase, YkwC was proposed to act in the valine metabolism pathway by performing NAD+-dependent conversion of 3-hydroxy isobutanoate to methyl-malonate semialdehyde (Fig. 6B). To test the oxidoreductase activity of YkwC we used a previously described assay for conversion of 3-hydroxyproprionate to malonate semialdehyde (38). YkwC and YkwC S281D were incubated separately with 3-hydroxyproprionate in the presence of NADP, and the rate of formation of NADPH was measured as an indication of oxidoreductase activity. The WT form of the enzyme was active, demonstrating that YkwC is indeed a true oxidoreductase. By contrast, the phosphomimetic mutant was inactive (Fig. 6C), pointing to the fact that phosphorylation at Ser281 may inhibit enzymatic activity of YkwC. To ensure that the effect shown by the mutant was not because of protein misfolding induced by the mutation, we compared circular dichroism (CD) spectra of Ykwc WT and YkwC S821D. The spectra of the two proteins were very similar, indicating that the mutation did not disrupt the protein structure (supplemental Fig. S9). To validate our in vitro findings in vivo, we repeated the same enzymatic assay with dialyzed total protein extracts obtained from the WT, ΔprkC and ΔprpC strains. The results showed increased oxidoreductase activity of YkwC in ΔprkC strain and loss of oxidoreductase activity in ΔprpC strain (Fig. 6D). This was fully consistent with in vitro findings, and confirmed that PrkC and PrpC control the occupancy of this phosphorylation site in vivo. As a negative control, cells were grown without glucose (conditions under which YkwC is not expressed), and YkwC activity could not be detected (Fig. 6D).
Fig. 6.
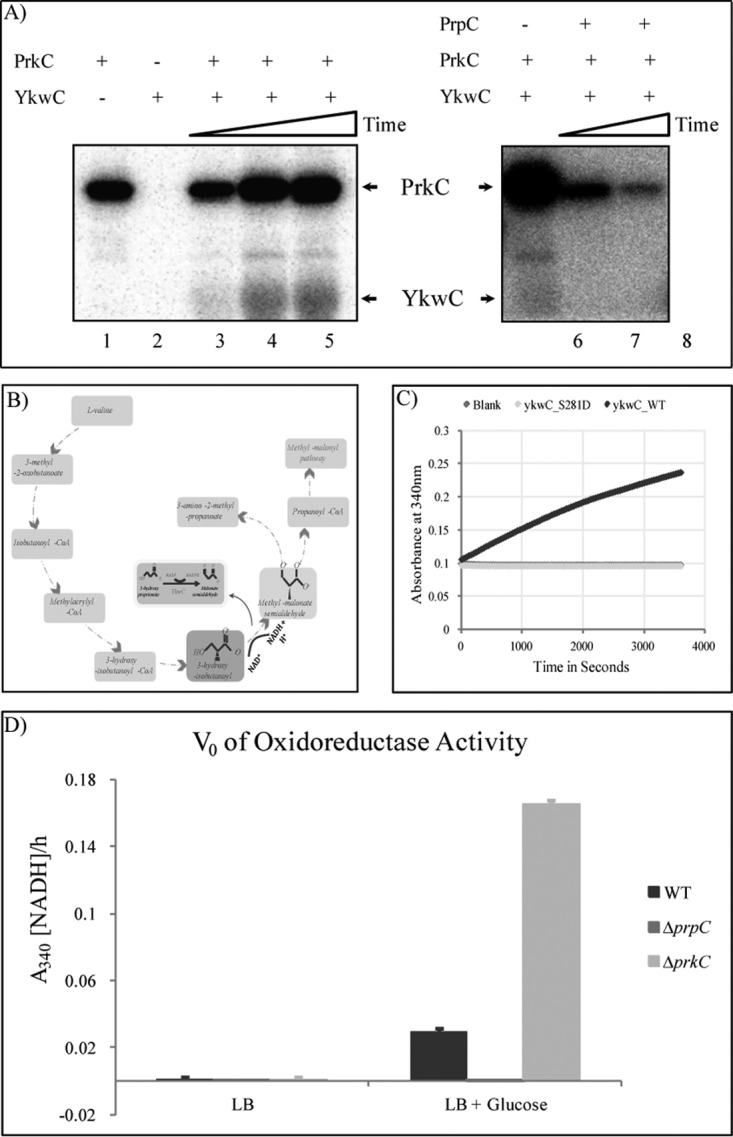
Validation of YkwC Ser281 as a substrate of PrkC and PrpC. A, In vitro phosphorylation of YkwC by PrkC and dephosphorylation by PrpC. Autoradiography showing in vitro phosphorylation/dephosphorylation assays of YkwC labeled with 32P issued from 32P-gamma ATP. Autophosphorylated PrkC and phosphorylated YkwC are indicated by arrows. For the phosphorylation assay, PrkC alone and YkwC alone were incubated for 40 min (lane 1 and 2); YkwC was incubated with PrkC for 10, 20, and 40 min (lane 3, 4 and 5). For the dephosphorylation assay, YkwC was incubated with PrkC for 1 h (lane 6), and then with PrpC for 1 and 2 h (lane 7 and 8). B, Valine metabolism pathway in B. subtilis and the role of YkwC. C, Enzyme activity of YkwC WT (1 μg) and YkwC S281D (1 μg), the phosphomimetic mutant, incubated with its substrate (3-HP) was monitored over 1 h at intervals of 30 s at 340 nm. The rate of formation of NADPH was measured in a 200 μl reaction. D, V0 of oxidoreductase activity of dialyzed total protein extract from B. subtilis WT, ΔprpC and ΔprkC cells cultured in LB and LB supplemented with 0.3% glucose [LB + Glucose] during transition phase. NADPH was detected at A340.
DISCUSSION
The main objective of this study was to analyze the global dynamics of proteins and phosphorylation events as the B. subtilis culture undergoes different stages of growth. In the course of this study we detected 55.2% of the B. subtilis theoretical proteome, which represents one of the most exhaustive proteomic datasets of this model organism. In addition, in both experiments we detected a total of 272 phosphorylation events, significantly increasing the number of previously reported phosphorylation events in B. subtilis.
To validate the quantitative proteome dataset, we analyzed the expression correlation of subunits of known protein complexes, as they are expected to be regulated in a similar fashion (supplemental Fig. S10). Expression of proteins involved in sporulation initiation (SpoOKD - SpoOKB - SpoOKE - SpoOKC - SpoOKA) and proteases belonging to the same proteasomal complex (ClpC - ClpX - ClpP) were not significantly altered from T1 through T5, and remained highly correlated. Proteins belonging to the flagellar hook assembly system (FlgE - FlgL - FlgK) were highly correlated and up-regulated in T5. This is coherent with nutrient deprivation at T5, which induces chemotaxis to look for an additional source of subsistence. The respiratory complex (CtaC - CtaF) was up-regulated during T3 after which it steadily dropped down toward T5 to conserve energy during adverse conditions.
As expected, the phosphoproteome of B. subtilis was highly dynamic during growth. Phosphorylation of glycolytic enzymes and other enzyme of the central carbon metabolism increased strongly in early and late stationary phase. These highly phosphorylated enzymes included: Eno (enolase), Fba (aldolase), Pgi (glucose-6-phosphate isomerase), GapA (glyceraldehyde-3-phosphate dehydrogenase), CitK (2-oxoglutarate dehydrogenase), YkjI (6-phosphogluconate dehydrogenase), CitB (citrate hydro-liase), SerA (3-phosphoglycerate dehydrogenase), CitC (isocytrate dehydrogenase) SucC (succinyl-CoA ligase), and TktA (transketolase). It has been reported by Pietack et al. (17) that phosphorylation of several glycolytic enzymes did not affect their enzyme activity directly. However, glycolytic flux is known to be controlled in some cases at the level of enzyme assemblies that channel substrates (41). Such multi-enzyme assemblies have been shown to exist in humans, and their assembly is regulated by protein phosphorylation (42). The high correlation of phosphorylation dynamics between different glycolytic enzymes in B. subtilis opens up the possibility that a similar regulation may exist in bacteria. Phosphorylation of a number of house-keeping proteins involved in translation also increased toward the stationary phase. These include ribosomal proteins (JofD, RpsH, RplL, RplN, RplR, RpmD), chaperones (GroEL) and elongation factors (FusA, Tsf, TufA). The competence protein ComJ (Nin) known to inhibit the DNase activity of NucA (43) exhibited a significant drop of phosphorylation occupancy in the early stationary phase, corresponding to the “time window” for competence development. This suggests that phosphorylation may have a functional consequence. An interesting differential phosphorylation pattern was detected in the purine synthesis pathway, where PurA and PurM were more strongly phosphorylated toward stationary phase, whereas phosphorylation of PurH diminished. Because these proteins act in concert, and PurH and PurM are encoded by the same operon, these phosphorylation events may be expected to have opposite effects on the target enzymes. Proteins involved in amino acid biosynthesis were strongly phosphorylated in the late stationary phase. Among them, MetC, and ThrC are involved in biosynthesis of methionine and threonine and IlvA, IlvC, LeuA, and LeuB are involved in the biosynthesis of branched-chain amino acids and are induced during amino acid starvation (44). IlvC has already been found to be phosphorylated at residue Ser338 in our previous study (25); here we detected new phosphorylated residues. Of particular interest, proteins involved in phosphate metabolism exhibited strong differential phosphorylation. Phosphate binding ABC transporter protein PstS has previously been identified in the phosphoproteome of B. subtilis (22), and is known to be strongly induced by phosphate starvation (45). Our data set indicates that PstS is very strongly dephosphorylated in early and late stationary phase (where phosphate depletion is expected), suggesting that the dephosphorylated state may be more active in phosphate import. Similarly, in the case of PhoA, alkaline phosphatase 4, it is strongly induced on phosphate starvation (46) and dephosphorylated in stationary phase in our data set, indicating that the nonphosphorylated form of this enzyme may be more active. Phosphorylation of the recombinase RecA at Ser2, which has been shown to contribute to a check-point in spore development (7), was depleted in the transition phase and nondetectable throughout the stationary phase. This is consistent with the fact that this phosphorylation is catalyzed by the kinase YabT, whose expression is tightly shut until the onset of spore development (7). All these examples testify to the capacity of quantitative phosphoproteomics to detect relevant signaling and regulatory events in vivo.
This phosphoproteome dataset is particularly interesting from the perspective of B. subtilis protein-tyrosine kinases, which catalyze the least abundant of phosphorylation events detected here. We detected phosphorylation of the putative BY-kinase PtkB (EpsB, YveL), for which the kinase activity has been previously suggested but never demonstrated in vitro (13, 47). This enzyme exhibited a very interesting probable autophosphorylation profile, with the site occupancy peak in the transition phase. This could be correlated to the involvement of PtkB in biofilm development, which has been previously suggested (48). The main B. subtilis BY-kinase PtkA was found to be phosphorylated by an unknown Ser/Thr kinase and dephosphorylated by the phosphatase PrpC, which is a novel example of cross talk between different families of bacterial kinases. In terms of BY-kinase substrate phosphorylation, phosphorylation of the global gene regulator AbrB at Tyr39 was detected, and the occupancy of this phosphorylation site increased toward stationary phase. Phosphorylation site of AbrB is in its DNA-binding domain and phosphorylation could hence be expected to impair DNA binding. This would be consistent with abolishing AbrB-mediated repression concomitant to the exit from exponential phase (49). A similar mechanism has already been described for another PtkA substrate, the transcription regulator FatR (14). We also detected phosphorylation of known PtkA substrates (Eno, Asd, and YjoA (50)) on Ser or Thr residues, indicating a cross-talk between BY-kinases and Hanks-type kinases not only on the kinase but also on the substrate level.
We have argued previously that SILAC experiments with kinase and phosphatase knockouts will reveal physiological substrates for these enzymes, by pin-pointing phosphorylation sites for which the occupancy changes dramatically in the respective knockout mutants (2). Here we demonstrated for the first time the validity of this principle. The phosphorylation of YkwC Ser281 dropped significantly in ΔprkC and increased in ΔprpC- suggesting that it is a substrate for both these enzymes. We have validated this hypothesis by showing in vitro phosphorylation and dephosphorylation of YkwC by PrkC and PrpC, respectively. Moreover, we have shown that PrkC dependent phosphorylation regulates the activity of YkwC in vivo and in vitro. This demonstrates the tremendous potential of quantitative phosphoproteomics in deciphering regulatory networks in bacteria. Bacterial kinases and phosphatases are known to be promiscuous, and our SILAC experiment with ΔprkC and ΔprpC yielded perhaps less substrates than expected (seven for PrkC, nine for PrpC). However it must be mentioned that this experiment was performed only at one time point on the growth curve, and one can reasonably expect to detect more substrate by changing the growth conditions. In conclusion, this study reveals important emerging features of bacterial regulatory networks based on protein phosphorylation and opens numerous leads for physiological validation of the dataset.
Supplementary Material
Acknowledgments
We thank Dr. Birte Höcker and Kaspar Feldmeier for help with CD measurement and interpretation.
Footnotes
Author contributions: I.M. and B.M. designed research; V.R. and L.S. performed research; V.R., L.S., and K.K. analyzed data; V.R., I.M., and B.M. wrote the paper; A.D., C.J., C.C., and A.K. generated a mutant strain used in this study.
* This work was supported by the the Juniorprofessoren-Programm of the BW-Stiftung, the PRIME-XS Consortium, the German Research Foundation (DFG) and the SFB-766 (to BM); and Agence Nationale de la Recherche (2010-BLAN-1303-01) to IM.
 This article contains supplemental Figs. S1 to S12 and Tables S1 to S4.
This article contains supplemental Figs. S1 to S12 and Tables S1 to S4.
We make all data related to this manuscript available at PeptideAtlas, www.peptideatlas.org (ID: PASS00350; Password: FG3595uex). Single peptide and PMF identifications, annotated spectra are provided for each protein in supplemental data.
1 The abbreviations used are:
- SILAC
- stable isotope labeling by amino acids in cell culture
- 3-HP
- 3-hydroxyproprionate
- FDR
- false discovery rate
- KO
- Knock-out
- PEP
- posterior error probability
- SCX
- strong cationic exchange
- TiO2
- titanium dioxide
- WT
- wild-type.
REFERENCES
- 1. Pereira S. F., Goss L., Dworkin J. (2011) Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol. Mol. Biol. Rev. 75, 192–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mijakovic I., Macek B. (2012) Impact of phosphoproteomics on studies of bacterial physiology. FEMS Microbiol. Rev. 36, 877–892 [DOI] [PubMed] [Google Scholar]
- 3. Klein G., Dartigalongue C., Raina S. (2003) Phosphorylation-mediated regulation of heat shock response in Escherichia coli. Mol. Microbiol. 48, 269–285 [DOI] [PubMed] [Google Scholar]
- 4. Shapland E. B., Reisinger S. J., Bajwa A. K., Ryan K. R. (2011) An essential tyrosine phosphatase homolog regulates cell separation, outer membrane integrity, and morphology in Caulobacter crescentus. J. Bacteriol. 193, 4361–4370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cozzone A. J. (2005) Role of protein phosphorylation on serine/threonine and tyrosine in the virulence of bacterial pathogens. J. Mol. Microbiol. Biotechnol. 9, 198–213 [DOI] [PubMed] [Google Scholar]
- 6. Deutscher J., Kuster E., Bergstedt U., Charrier V., Hillen W. (1995) Protein kinase-dependent HPr/CcpA interaction links glycolytic activity to carbon catabolite repression in gram-positive bacteria. Mol. Microbiol. 15, 1049–1053 [DOI] [PubMed] [Google Scholar]
- 7. Bidnenko V., Shi L., Kobir A., Ventroux M., Pigeonneau N., Henry C., Trubuil A., Noirot-Gros M. F., Mijakovic I. (2013) Bacillus subtilis serine/threonine protein kinase YabT is involved in spore development via phosphorylation of a bacterial recombinase. Mol. Microbiol. 88, 921–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shah I. M., Laaberki M. H., Popham D. L., Dworkin J. (2008) A eukaryotic-like Ser/Thr kinase signals bacteria to exit dormancy in response to peptidoglycan fragments. Cell 135, 486–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kang C. M., Brody M. S., Akbar S., Yang X., Price C. W. (1996) Homologous pairs of regulatory proteins control activity of Bacillus subtilis transcription factor sigma(b) in response to environmental stress. J. Bacteriol. 178, 3846–3853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jers C., Kobir A., Sondergaard E. O., Jensen P. R., Mijakovic I. (2011) Bacillus subtilis two-component system sensory kinase DegS is regulated by serine phosphorylation in its input domain. PLoS One 6, e14653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mijakovic I., Petranovic D., Macek B., Cepo T., Mann M., Davies J., Jensen P. R., Vujaklija D. (2006) Bacterial single-stranded DNA-binding proteins are phosphorylated on tyrosine. Nucleic Acids Res. 34, 1588–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Petranovic D., Michelsen O., Zahradka K., Silva C., Petranovic M., Jensen P. R., Mijakovic I. (2007) Bacillus subtilis strain deficient for the protein-tyrosine kinase PtkA exhibits impaired DNA replication. Mol. Microbiol. 63, 1797–1805 [DOI] [PubMed] [Google Scholar]
- 13. Mijakovic I., Poncet S., Boel G., Maze A., Gillet S., Jamet E., Decottignies P., Grangeasse C., Doublet P., Le Marechal P., Deutscher J. (2003) Transmembrane modulator-dependent bacterial tyrosine kinase activates UDP-glucose dehydrogenases. EMBO J. 22, 4709–4718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Derouiche A., Bidnenko V., Grenha R., Pigonneau N., Ventroux M., Franz-Wachtel M., Nessler S., Noirot-Gros M. F., Mijakovic I. (2013) Interaction of bacterial fatty-acid-displaced regulators with DNA is interrupted by tyrosine phosphorylation in the helix-turn-helix domain. Nucleic Acids Res. 41, 9371–9381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Madec E., Laszkiewicz A., Iwanicki A., Obuchowski M., Seror S. (2002) Characterization of a membrane-linked Ser/Thr protein kinase in Bacillus subtilis, implicated in developmental processes. Mol. Microbiol. 46, 571–586 [DOI] [PubMed] [Google Scholar]
- 16. Gaidenko T. A., Kim T. J., Price C. W. (2002) The PrpC serine-threonine phosphatase and PrkC kinase have opposing physiological roles in stationary-phase Bacillus subtilis cells. J. Bacteriol. 184, 6109–6114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pietack N., Becher D., Schmidl S. R., Saier M. H., Hecker M., Commichau F. M., Stulke J. (2010) In vitro phosphorylation of key metabolic enzymes from Bacillus subtilis: PrkC phosphorylates enzymes from different branches of basic metabolism. J. Mol. Microbiol. Biotechnol. 18, 129–140 [DOI] [PubMed] [Google Scholar]
- 18. Macek B., Gnad F., Soufi B., Kumar C., Olsen J. V., Mijakovic I., Mann M. (2008) Phosphoproteome analysis of E. coli reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol. Cell. Proteomics 7, 299–307 [DOI] [PubMed] [Google Scholar]
- 19. Soufi B., Gnad F., Jensen P. R., Petranovic D., Mann M., Mijakovic I., Macek B. (2008) The Ser/Thr/Tyr phosphoproteome of Lactococcus lactis IL1403 reveals multiply phosphorylated proteins. Proteomics 8, 3486–3493 [DOI] [PubMed] [Google Scholar]
- 20. Ravichandran A., Sugiyama N., Tomita M., Swarup S., Ishihama Y. (2009) Ser/Thr/Tyr phosphoproteome analysis of pathogenic and non-pathogenic Pseudomonas species. Proteomics 9, 2764–2775 [DOI] [PubMed] [Google Scholar]
- 21. Prisic S., Dankwa S., Schwartz D., Chou M. F., Locasale J. W., Kang C. M., Bemis G., Church G. M., Steen H., Husson R. N. (2010) Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc. Natl. Acad. Sci. U.S.A. 107, 7521–7526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Levine A., Vannier F., Absalon C., Kuhn L., Jackson P., Scrivener E., Labas V., Vinh J., Courtney P., Garin J., Seror S. J. (2006) Analysis of the dynamic Bacillus subtilis Ser/Thr/Tyr phosphoproteome implicated in a wide variety of cellular processes. Proteomics 6, 2157–2173 [DOI] [PubMed] [Google Scholar]
- 23. Eymann C., Becher D., Bernhardt J., Gronau K., Klutzny A., Hecker M. (2007) Dynamics of protein phosphorylation on Ser/Thr/Tyr in Bacillus subtilis. Proteomics 7, 3509–3526 [DOI] [PubMed] [Google Scholar]
- 24. Macek B., Mijakovic I., Olsen J. V., Gnad F., Kumar C., Jensen P. R., Mann M. (2007) The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol. Cell. Proteomics 6, 697–707 [DOI] [PubMed] [Google Scholar]
- 25. Soufi B., Kumar C., Gnad F., Mann M., Mijakovic I., Macek B. (2010) Stable isotope labeling by amino acids in cell culture (SILAC) applied to quantitative proteomics of Bacillus subtilis. J. Proteome Res. 9, 3638–3646 [DOI] [PubMed] [Google Scholar]
- 26. Otto A., Bernhardt J., Meyer H., Schaffer M., Herbst F. A., Siebourg J., Mader U., Lalk M., Hecker M., Becher D. (2010) Systems-wide temporal proteomic profiling in glucose-starved Bacillus subtilis. Nat. Commun. 1, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maguin E., Prevost H., Ehrlich S. D., Gruss A. (1996) Efficient insertional mutagenesis in lactococci and other gram-positive bacteria. J. Bacteriol. 178, 931–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Monedero V., Kuipers O. P., Jamet E., Deutscher J. (2001) Regulatory functions of serine-46-phosphorylated HPr in Lactococcus lactis. J. Bacteriol. 183, 3391–3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Monod J. (1949) The Growth of Bacterial Cultures. Ann. Rev. Microbiol. 3, 371–394 [Google Scholar]
- 30. Ishihama Y., Rappsilber J., Mann M. (2006) Modular stop and go extraction tips with stacked disks for parallel and multidimensional Peptide fractionation in proteomics. J. Proteome Res. 5, 988–994 [DOI] [PubMed] [Google Scholar]
- 31. Franz-Wachtel M., Eisler S. A., Krug K., Wahl S., Carpy A., Nordheim A., Pfizenmaier K., Hausser A., Macek B. (2012) Global detection of protein kinase D-dependent phosphorylation events in nocodazole-treated human cells. Mol. Cell. Proteomics 11, 160–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krug K., Carpy A., Behrends G., Matic K., Soares N. C., Macek B. (2013) Deep coverage of the Escherichia coli proteome enables the assessment of false discovery rates in simple proteogenomic experiments. Mol. Cell. Proteomics [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Olsen J. V., de Godoy L. M., Li G., Macek B., Mortensen P., Pesch R., Makarov A., Lange O., Horning S., Mann M. (2005) Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics 4, 2010–2021 [DOI] [PubMed] [Google Scholar]
- 34. Cox J., Matic I., Hilger M., Nagaraj N., Selbach M., Olsen J. V., Mann M. (2009) A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 4, 698–705 [DOI] [PubMed] [Google Scholar]
- 35. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 36. Team R. D. C. (2013) R: A Language and Environment for Statistical Computing [Google Scholar]
- 37. Benjamini Y., Hochberg Y. (1995) Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. Roy. Statistical Soc. Series B 57, 289–300 [Google Scholar]
- 38. Yao T., Xu L., Ying H., Huang H., Yan M. (2010) The catalytic property of 3-hydroxyisobutyrate dehydrogenase from Bacillus cereus on 3-hydroxypropionate. Appl. Biochem. Biotechnol. 160, 694–703 [DOI] [PubMed] [Google Scholar]
- 39. Soares N. C., Spat P., Krug K., Macek B. (2013) Global dynamics of the Escherichia coli proteome and phosphoproteome during growth in minimal medium. J. Proteome Res. 12, 2611–2621 [DOI] [PubMed] [Google Scholar]
- 40. Knight Z. A., Lin H., Shokat K. M. (2010) Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer 10, 130–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kholodenko B. N., Sakamoto N., Puigjaner J., Westerhoff H. V., Cascante M. (1996) Strong control on the transit time in metabolic channelling. FEBS Lett. 389, 123–125 [DOI] [PubMed] [Google Scholar]
- 42. Campanella M. E., Chu H., Low P. S. (2005) Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proc. Natl. Acad. Sci. U.S.A. 102, 2402–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Sinderen D., Kiewiet R., Venema G. (1995) Differential expression of two closely related deoxyribonuclease genes, nucA and nucB, in Bacillus subtilis. Mol. Microbiol. 15, 213–223 [DOI] [PubMed] [Google Scholar]
- 44. Tojo S., Satomura T., Kumamoto K., Hirooka K., Fujita Y. (2008) Molecular mechanisms underlying the positive stringent response of the Bacillus subtilis ilv-leu operon, involved in the biosynthesis of branched-chain amino acids. J. Bacteriol. 190, 6134–6147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Qi Y., Kobayashi Y., Hulett F. M. (1997) The pst operon of Bacillus subtilis has a phosphate-regulated promoter and is involved in phosphate transport but not in regulation of the pho regulon. J. Bacteriol. 179, 2534–2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Antelmann H., Scharf C., Hecker M. (2000) Phosphate starvation-inducible proteins of Bacillus subtilis: proteomics and transcriptional analysis. J. Bacteriol. 182, 4478–4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mijakovic I., Petranovic D., Bottini N., Deutscher J., Ruhdal Jensen P. (2005) Protein-tyrosine phosphorylation in Bacillus subtilis. J. Mol. Microbiol. Biotechnol. 9, 189–197 [DOI] [PubMed] [Google Scholar]
- 48. Chu F., Kearns D. B., Branda S. S., Kolter R., Losick R. (2006) Targets of the master regulator of biofilm formation in Bacillus subtilis. Mol. Microbiol. 59, 1216–1228 [DOI] [PubMed] [Google Scholar]
- 49. O'Reilly M., Devine K. M. (1997) Expression of AbrB, a transition state regulator from Bacillus subtilis, is growth phase dependent in a manner resembling that of Fis, the nucleoid binding protein from Escherichia coli. J. Bacteriol. 179, 522–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jers C., Pedersen M. M., Paspaliari D. K., Schutz W., Johnsson C., Soufi B., Macek B., Jensen P. R., Mijakovic I. (2010) Bacillus subtilis BY-kinase PtkA controls enzyme activity and localization of its protein substrates. Mol. Microbiol. 77, 287–299 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.