

Abstract
Late-onset Alzheimer’s disease (LOAD) is the most common neurodegenerative disorder in older adults, affecting over 50% of those over age 85. Aging is the most important risk factor for the development of LOAD. Aging is associated with the decrease in the ability of cells to cope with cellular stress, especially protein aggregation. Here we will describe how the process of aging affects pathways that control the processing and degradation of abnormal proteins including amyloid-β (Aβ). Genetic association studies in LOAD have successfully identified a large number of genetic variants involved in the development of the disease. However, there is a gap in understanding the interconnections between these pathomolecular events that prevent us from discovering therapies. We will compile pertinent links to elucidate how the biology of aging affects the sequence of events in the development of LOAD. Specifically, in this review we analyze the molecular-pathologic-clinical correlations of the aging process, the HSF1 and FOXO family pathways, Aβ toxicity, Aβ degradation, and the different clinical stages of LOAD.
Introduction
Late-onset Alzheimer’s disease (LOAD) affects up to one-third of individuals by the age of 80 [1–2]. There is a strong interest in identifying pathways involved in the pathogenesis of AD in order to find new therapeutic targets for clinical trials. Evidence suggests that the pathophysiological process of AD begins years, if not decades, prior to the diagnosis of clinical dementia [3]. In this study, we will examine the development of AD and its relationship with the longevity pathways on the Aβ clearance and antiproteotoxicity systems. Clinical progression of AD evolves (presumably) from presymptomatic neurodegeneration, through mild cognitive impairment (MCI), ultimately to symptomatic AD [4–5]. MCI patients with primary memory deficits, aka “amnestic MCI”, have a significantly higher likelihood to progress to probable AD, with a conversion rate of 10–15% per year [2]. AD is associated with abnormal deposition aggregates consisting primarily of extraneuronal amyloid-β peptide (Aβ) (plaques) or intraneuronal hyperphosphorylated τ proteins (tangles) in the brain [6]. As the preclinical phase begins is presumed to begin decades before diagnosis of MCI, it would provide a critical opportunity for potential intervention with disease-modifying therapy [3]. This makes it a priority to elucidate links between the pathomolecular process that start during the preclinical phase and the onset of the clinical syndrome. A hypothetical intervention that delays the onset of AD by 5 years would result in a 57% reduction in the number of AD patients, and reduce the projected Medicare costs of AD from $627 to $344 billion dollars [7] (presuming no overall extension of lifespan due to reduction of AD).
Aging is the strongest risk factor in the development of LOAD [8]. It seems that the earliest events in the development of AD are controlled by longevity pathways [9]. These pathways control Aβ degradation and aggregation processes in the intra and extra-cellular space. These aging/longevity pathways can in theory be manipulated to prevent or delay the development of AD. Recent studies with transgenic mouse models suggest that amyloid-modifying therapies may have beneficial effects even once neuronal degeneration has begun [10]. Interestingly, recent clinical trials in the stages of mild to moderate dementia have failed to demonstrate clinical benefit, even in the setting of biomarker or autopsy evidence of decreased amyloid-burden [11]. These results may be an evidence of a failure of the amyloid hypothesis or a lack of understanding of the pathomolecular events in the development of AD; we believe the latter is a more reasonable explanation. In response to late-stage treatment failures, AD research is moving towards early stage clinical trials such as amnestic mild cognitive impairment (MCI) or even in patients with conditions currently deemed pre-clinical, such as subjective memory impairment (SMI) [12] or cognitive complaints (CC) [13]. The overarching therapeutic objective of “preclinical studies” would be to discover treatments for early pathologic processes (e.g. lower Aβ burden) in order to prevent subsequent neurodegeneration and eventual cognitive decline. For these reasons, we demonstrate new molecular targets for therapies that will focus on the preclinical stages of AD by slowing down its main risk factor, elements of the aging process itself.
Pathomolecular-Clinical Correlations
Early LOAD is associated with abnormal protein degradation and aggregation within specific areas of the brain, and this specificity helps with differential diagnosis. The Aβ deposition often begins in the temporal cortex particularly on the hippocampus [14], which causes cognitive impairment associated with MCI. Frontal and occipital brain regions are spared from early neurodegenerative pathology. Ensuing specific cognitive deficits and strengths in early stages suggest the pathologic process and help differentiate between AD and other types of dementia including Fronto-temporal (FTD) and Subcortical Ischemic Vascular Dementia (SIVD).
AD Molecular Biology and Markers
Amyloid Plaques
The distinguishing histopathological features of AD consist of cerebral plaques that consist primarily of Aβ [15], dystrophic neurites in neocortical terminal fields [16], and prominent neurofibrillary tangles in medial temporal-lobe structures [17]. Loss of neurons and white matter, congophilic (amyloid) angiopathy, inflammation, and oxidative damage are also present. Aβ peptides are natural products of metabolism consisting of 36 to 43 amino acids. Monomers of Aβ are much more prevalent than the aggregation-prone and damaging Aβ42 species [18]. Aβ originates from proteolysis of AβPP by the sequential enzymatic actions of β-site AβPP-cleaving enzyme 1 (BACE1), the primary β-secretase, and the γ-secretase protein complex, which has presenilin 1 at its catalytic core [19]. Carboxy-terminal cleavage of Aβ by γ-secretase results in the generation of multiple peptides, the two most common being 40- and 42-amino acid Aβ (Aβ40 and Aβ42, respectively). Aβ40 comprises 90–95% of secreted Aβ and is the predominant species recovered from cerebrospinal fluid [20]. In contrast, less than 10% of secreted Aβ is Aβ42. Despite the relative paucity of its production, Aβ42 is the predominant species found in plaques and is deposited initially [21], perhaps due to its ability to form insoluble amyloid aggregates more rapidly than Aβ40 [22–23]. An imbalance in intracellular Aβ clearance causes Aβ to accumulate, and this decreased degradation may be an initiating factor in LOAD [24]. This pathomolecular process starts in the preclinical stage and our hypothesis considers it likely to require the onset of the aging process.
Intracellular Aβ
Accumulation of Aβ, as seen in AD, could be due to increased generation, decreased clearance, or a combination of both events. Recently, decreased clearance has been recognized as the cause of Aβ accumulation in LOAD [24]. Accumulation of intracellular Aβ (iAβ) is an early event in AD, synaptic loss and iAβ detection occurred before the appearance of Aβ plaques [25]. The most current hypothesis states that extracellular Aβ (eAβ) causes toxicity and cell death [26]. This fails to explain why cultures of human primary neurons are resistant to the administration of eAβ [27] In this study, they used 100 nM fibrillar Aβ 1-42, which triggered upregulation of apoptotic protein Bax but not neuronal death. Since current findings suggest a more important role of oligomeric Ab1-42 compared to fibrillar Ab1-42, it could be the reason. In a recent study we observed 40% loss of cell viability in rat primary neurons upon 100 nM oligomeric Aβ 1-42 treatment for 72 h [28]. Nevertheless directly injecting intracellular Aβ is much more effective. Compared to 100 nM eAβ, direct microinjecting 1 pM iAβ induces 50–70% cell death in human neurons [29]. Most importantly, in AD transgenic mouse models, deposition of iAb precedes eAb and NFT formation and synaptic dysfunction [30]. Thus eAβ deposition alone is inadequate to account for the LOAD pathology. A complementary explanation is that iAβ causes toxicity and neuronal dysfunction. Aβ accumulates intracellularly in AD patients, it localizes in multivesicular bodies, an organelle originating from the endosomal pathway [31]. Changes in the endosomal-lysosomal pathway are amongst the earliest changes in AD [32]. Aβ can be degraded from the intracellular compartment by activation of clearance pathways [33]. Therefore, new therapeutics targeting iAβ could be an effective treatment for LOAD [34].
Neurofibrillary Tangles
Neurofibrillary tangles (NFT) are mainly aggregates of abnormally hyperphosphorylated τ protein [35]. The soluble τ protein is abundant in axons; it promotes assembly and stability of microtubules and vesicle transport [36]. Hyperphosphorylated τ is insoluble, lacks affinity for microtubules, and self-associates into paired helical filament structures [37]. Like Aβ oligomers, intermediate aggregates of abnormal τ molecules are cytotoxic [38] and impair cognition [39]. NFTs also contain significant percentage of hyperphosphorylated neurofilaments (NF) [40–41] and collapsin response mediator protein 2 (CRMP2) [42]. Neurofilaments promote an increase in axonal diameter in myelinated fibers, thereby increasing nerve conduction velocity. NF is phosphorylated physiologically in axonal compartments; however, they are hyperphosphorylated in AD and form aggregates in cell bodies. In AD, hyperphosphorylation of medium NF (13 sites) and heavy NF (10 sites) occurs at conserved KSXXP motifs located on the C-terminal tail domain, and is ~4–8 fold higher than in age-matched controls [41]. CRMP2 protein is also hyperphosphorylated in AD and co-localizes with NFT in human AD brains. Hyperphosphorylated CRMP2 is highly resistant to dephosphorylation and is used as a marker of NFT [42].
Synaptic Failure
The cognitive symptoms of AD may be primarily due to synaptic failure [18]. Memory deficits in clinical LOAD are associated with degeneration of the hippocampus, which extends tissue changes associated with MCI, in which there is an early decreased hippocampal synaptic plasticity [43]. Hippocampal synapses begin to decline in patients with MCI, in whom remaining synaptic profiles show compensatory increases in size [44]. In mild AD, there is a reduction of the presynaptic vesicle protein synaptophysin [45]. With advancing disease, synapses are disproportionately lost relative to neurons, and this loss is the best correlate with dementia [46]. Aβ oligomers mediate synaptic dysfunction by binding and self-association to structures (membrane receptors, mitochondria, etc) leading to synaptic dysfunction and memory impairment [47]. While Aβ accumulation is the earliest event in the development, it is not sufficient to produce the clinical AD syndrome. The cognitive decline of AD occurs only in the setting of synaptic dysfunction and/or additional neurodegeneration, including paired helical filament τ formation and neuronal loss [48]. Evidence also suggests that additional factors, such as cognitive reserve, white matter alterations, dopaminergic depletion, cerebrovascular disease, and Lewy bodies, alter the relationship between the neuropathological and clinical manifestations of AD. We also recognize that a substantial subset of individuals can have all of the classic neuropathologic features of AD at autopsy but never show signs of dementia during life [49]. It remains unknown whether these individuals would have manifested clinical symptoms should they have lived longer or their individual adaptation mechanisms to loss of synaptic plasticity would have indefinitely prevented clinical LOAD.
Neurodegeneration
In Alzheimer’s disease the pathomolecular sequence of events show that neuronal loss is a relatively late event, typically following synaptic dysfunction, synaptic loss, neurite retraction, and the appearance of other abnormalities such as axonal transport defects [50]. Evidence suggests that although cell death itself occurs late in the degenerative process, the pathways involved in cell death signaling play critical roles in neurodegeneration, both in sub-apoptotic events such as synapse loss and in the ultimate neuronal loss itself [51–53]. Neuroinflammation, typified by the accumulation of activated microglia and reactive astrocytes, plays an initial role in neurodegeneration, and Aβ activates microglia and astrocytes localized around Aβ plaques. The severity of glial activation correlates with the extent of brain atrophy [54] and cognitive decline [55]. Glial biochemical markers are elevated in the brains of patients with AD [56]. Initially, phagocytic microglia engulf and degrade Aβ. However, chronically activated microglia release chemokines and a cascade of damaging cytokines such as interleukin-1, interleukin-6, and tumor necrosis factor α (TNF-α) [57]. In common with vascular cells, microglia express receptors for advanced glycation end products, which bind Aβ, thereby amplifying the generation of cytokines, glutamate, and nitric oxide [58]. Aβ and glial activation also stimulate the classic complement pathway [59]. Tangles and plaques contain complement cleavage products, C1q and C5b-9, indicating that opsonization and autolytic attack are under way [57]. Stimulated astroglia also release acute-phase reactants, alpha1-antichymotrypsin, alpha2-macroglobulin, and C-reactive protein, which can both aggravate and ameliorate AD.
Cytokines stimulate a variety of intracellular signalling pathways by receptor binding. These cytokines have been implicated in AD, including the activation of protein kinase C, c-Jun N-terminal kinase (JNK), p38 mitogen-activated protein kinase (p38/MAPK), PI3 kinase, and extracellular signalling-related kinase (ERK) as well as activation of caspase-1 and -3 [60]. Elevated Aβ induces apoptosis-related events in synapses and dendrites in AD through caspase activation [61]. In addition, Aβ-induced caspase-3 activation causes abnormal processing of the microtubule-associated protein τ in models of AD, and these fragments directly reduce neuronal viability [62].
An important factor in degeneration is the loss of calcium regulation in Alzheimer’s disease, elevated concentrations of cytosolic calcium stimulate Aβ aggregation and amyloidogenesis [63]. Aβ42 also increases calcium stores in the endoplasmic reticulum and the release of calcium into the cytoplasm [64]. Also increased glutamate level leads to increase cytosolic calcium, which in turn stimulates calcium-release channels in the endoplasmic reticulum. It has been shown that a chronic state of excitatory amino acid (glutamatergic) receptor activation aggravates neuronal damage in LOAD [65]. However, the evidence of excessive excitatory amino acid mechanisms in AD is modest. Aβ forms voltage-independent cat ion channels in lipid membranes [66], resulting in calcium uptake and degeneration of neurons [67].
Pathomolecular Sequence of Events of the Aging Process on Clinical AD
Aβ is generated by the sequential enzymatic cleavage of the amyloid-β protein precursor (AβPP) in the endoplasmic reticulum (ER)/intermediate compartment [68], the medial Golgi saccules [69], and the trans-Golgi network (TGN) [70–71]. Furthermore, Aβ40 is generated exclusively within the TGN and packaged into post-TGN secretary vesicles [70–71], while Aβ42 could be made and retained within the ER in an insoluble state, before entering the TGN and being packaged into secretary vesicles [72]. Aβ is recognized as an abnormal protein and reversely translocated back to the cytosol [34]. Aβ is ubiquitinated and sent to the proteasomes [73] or to the chaperone-mediated autophagy (CMA) system for degradation [74].
The ubiquitin-proteasome and autophagy-lysosome pathways are the two main routes of protein clearance in eukaryotic cells. Since the proteasome activity decreases with aging [75], inefficient degradation and clearance of Aβ would result in iAβ accumulation. This chronic proteasome inhibition causes an excessive activation of the lysosomal system demonstrated by an increase in autophagy [76]. Alternately, iAβ is internalized into Autophagic Vacuoles (AVs) for degradation [77].
Age-related decline in overall proteolytic activity has been observed in almost all organisms studied, and specific age-related defects in the different proteolytic systems have been reported [78]. The molecular basis of this age-related decline offers an explanation for the late-onset observed for many neurodegenerative diseases [79]. The earliest event in the aging process is the collapse of proteostasis, produced by a sharp decline of the heat shock response (HSR) in cell cultures and animal studies [80–83]. Therefore, it is important to correlate these initial molecular events with the development of AD (Fig. 1). For example, in humans, the HSR attenuation begins after the third or fourth decade of life [84]. Thereafter, abnormal protein accumulation occurs [83] in the form Aβ accumulation in plaques after the age of 30 years [85]. Interestingly, this occurs after the onset of the attenuation of the HSR (Fig. 2A). Then, after a couple of decades of Aβ accumulation, the clinical signs of dementia are more commonly observed when individuals are over 50 years of age [86]. These various situations are interrelated and tend to correlate with age (Table 1), although strict age causation is likely to be an overly simplistic model.
Fig. 1. Intermolecular Effects of HSF and FOXO3 in Non-Pathogenic Aβ Metabolism.
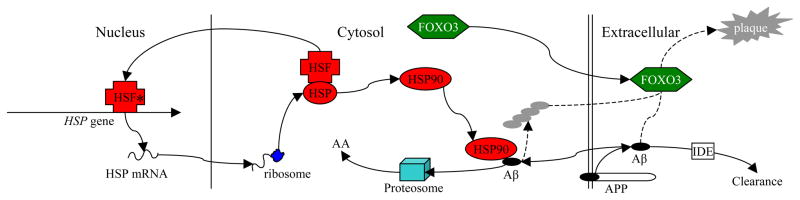
Schematic representation of interlocking roles for heat shock factors and FOXO3 in Aβ metabolism. Healthy cell. HSF, HSP, and FOXO3 are all at normal expression and activity. A minority of AβPP is processed via the β-secretase pathway, producing Aβ, at least some of which is directed intracellularly for functions such as transcription factor activity. HSF binds to HSP and is activated. Active HSF (*) is directed to the nucleus where it upregulates (among others) HSP expression. HSP activation (HSP90) directs excess intracellular Aβ toward proteasomes, where it is degraded to amino acids (AA). FOXO3, among its multiple functions, catalyzes formation of amyloid plaque from Aβ. In addition, extracellular non-plaque Aβ is cleared by enzymes such as insulin-degrading enzyme (IDE).
Fig. 2. Intermolecular Effects of HSF and FOXO3 in Pathogenesis of AD.

A) Initial breakdown of HSF processes. HSF activity is reduced due to age or environmental stressors. Part of the result is the reduction of the feedback loop that provides HSP90 for intracellular Aβ degradation. Excess intracellular Aβ is able to aggregate into toxic oligomers, producing reactive oxidizing species (ROS). B) “Recruitment” of FOXO3 into the cytotoxic cascade. ROS stimulates FOXO3, directing it to the nucleus, wherein it participates in apoptosis. Redirection of FOXO3 reduces production of plaque, permitting greater formation of toxic extracellular Aβ oligomers and interfering with enzymatic clearance of eAβ, permitting plaque buildup even though overall plaque catalysis has been reduced. Combination of neuronal cell death and extracellular events lead to clinical AD symptoms.
Table 1.
Interlocking Age-Related Effects of HSF1, FOXO3, Aβ, and AD Risk
| Age Group | Years | HSF1-DNA binding | FOXO3a | Aβ | AD Risk | ||
|---|---|---|---|---|---|---|---|
| cyto | nucl | plaque | oligo | ||||
| Adult | 20–40 | ||||||
| Mid Life | 30–50 | ↓ | ↑ | ||||
| Late Mid Life | 40–60 | ↓ | ↓ | ↑ | ↑ | ↑ | |
| Early Old | 50–70 | ↓↓ | ↓ | ↑ | ↑↑ | ↑↑ | ↑ |
| Middle Old | 60–90 | ↓↓↓ | ↓↓ | ↑↑ | ↑↑ | ↑↑ | ↑↑ |
| Oldest Old | 90+ | ↓↓↓ | ↓↓ | ↑↑(↑)a | ↑↑(↑) | ↑↑(↑) | ↑↑(↑) |
Location of FOXO3 would be either cytoplasmic (cyto—neuroprotective) or nuclear (nucl—neurodegenerative)
Aβ forms of note would be either plaque or higher-toxicity oligomers. Monomeric Aβ is not presented in this table.
Prevalence of AD may level off among the oldest old [157].
The general picture that emerges from these observations is a model that favors the following pathomolecular sequence of events: firstly, HSR attenuation, which leads to proteasome and CMA dysfunction with increased iAβ, resulting in an inefficient compensatory activation of AV, and this AV accumulates in the cytoplasm and ends up releasing ubiquitin tagged eAβ that forms toxic oligomers (Fig 2A). At this point, multifunctional factors, such as FOXO3, are activated, which form non-toxic neuritic plaques and thus prevent neurodegeneration from Aβ oligomers. Later, the age-related attenuation of FOXO3 is initiated and inflammatory/oxidative stress directs FOXO3 toward the nucleus, an apoptotic pathway. FOXO3 stops forming plaques with the subsequent increase in extracellular toxic oligomers which causes neurodegeneration and the onset of clinical AD (Fig. 2B).
It is important that HSR is regulated at the transcriptional level by HSF1 [87]. At the molecular level a decreased HSF1-DNA binding is responsible for the age-related attenuation of the HSR and heat shock proteins (HSP) expression, especially HSP70 [88–89]. The chaperone HSP70 is very important for the intracellular degradation of Aβ [90]. Also, the regulation of aging and the maintenance of proteostasis in invertebrates require the activation of the transcription factor FOXO [91]. In a recent study, the overexpression of HSP70 preserved proteostasis during aging but additional FOXO-dependent pathway responses were required [92]. In the same study, the overexpression of FOXO delayed protein accumulation and increased lifespan in Drosophila [92]. Therefore, we believe that in humans an age-related decline in activation of a member or members of the mammalian FOXO family is required to allow proteotoxicity and neuronal degeneration (Fig. 1). This is supported by the fact that, in invertebrates FoxO regulates [93] the expression of several autophagy genes which progressively decline during aging in muscles [92, 94]. This is also supported by the extrapolation of data from C. elegans (flatworm) on the age-related effects of the HSF1 and FOXO pathways [9]. This C. elegans transgenic model for AD revealed that FOXO and HSF1 are required for Aβ degradation and proteotoxicity prevention. They used HSF1 and FOXO (DAF16) interfering RNA (RNAi) in worms to demonstrate the function of these pathways. HSF1 regulated disaggregation, whereas the FOXO mediated the formation of less-toxic high-molecular weight aggregates. This process may be analogous to the human disease, when organisms would be young and both pathways are functional, but there are no signs of disease. Later when humans develop age-related attenuation of HSF1 analogous to the inhibition of worm HSF1 by RNAi, the formation of less-toxic high-MW aggregates would be inhibited and formation of toxic oligomers and clinical symptoms would appear. It should be noted that C. elegans APL-1, the native AβPP homologue, does not, itself produce Aβ, and this aggregate-inducing activity has not yet been specifically replicated in mammals.
Interestingly, activation of FOXO family members can also lead to cell death via upregulation of Bcl-2 interacting mediator of cell death (Bim) and FAS ligand (FasL) under neurotoxic conditions. Although gaps exist in our understanding of the mechanisms by which FOXO family proteins tilt the balance in favor of cell death, SIRT1-mediated regulation of FOXO family proteins offers an intriguing possibility. SIRT1 deacetylates invertebrate FOXO, which induces transcription of stress-resistance genes [95] and prevents induction of apoptosis genes [96]. Thus, it appears that SIRT1 plays a crucial role in tipping the balance of FOXO functions away from cell death towards stress resistance. Importantly, SIRT1 levels are significantly reduced in AD patients as compared to controls and correlate well with toxic Aβ and NFT accumulation [97]. These findings further support our model that age-related attenuation and/or altered FOXO signaling (due to reduced deacetylation) are critical in promoting proteotoxicity in LOAD pathogenesis.
It is therefore of interest to understand the functions of the HSF1 and FOXO pathways and their role in regulating HSP expression during ubiquitin–proteasome, CMA degradation, and defense against proteotoxicity. The key regulatory role of HSF1 in the ubiquitin proteolytic pathway was established using HSF1 null cells and the capability of exogenous HSF1 to restore the HSF DNA binding activity and inducible HSP70 expression upon proteasome inhibition [98]. HSF1 activation pathway leads to enhanced expression of HSP70 and other molecular chaperones. These molecular chaperones work in tandem with the ubiquitin-proteasome pathway for protein quality control to prevent the accumulation of abnormal proteins, including Aβ [99–100]. Interestingly, intracellular Aβ also prompts the rapid induction of stress-inducible HSP70 protein in neurons [101], most likely due to activation of HSF1 [102].
Chaperones such as HSP70 limit the accumulation of damaged proteins by suppression of misfolding and enhancing chaperone mediated folding [103–104], promoting the enzymatic removal of covalent modifications [105], and stimulating clearance by ubiquitin-mediated, proteosomal [106], and autophagic processes [9, 107]. The ubiquitin-proteasome system is the primary mechanism for disposal of stress-denatured proteins [108–109] as well as being responsible for the majority of normal protein turnover [110]. The fact that the HSF1, FOXO, and proteasome pathways are implicated in several neurodegenerative diseases [111–114], including Aβ degradation [106], raises a potential therapeutic role for HSF1 and FOXO in AD and other neurodegenerative diseases.
Therefore, the enhancement of the HSF1 and appropriate FOXO pathways simultaneously as a new therapeutic strategy to improve the proteasome-mediated and CMA degradation of Aβ has been suggested recently [115]. Interestingly, the Rpn4, ubiquitin, an E3 ligase, and a ubiquitin-conjugating enzyme were identified in a genome-wide screen for HSF1-regulated genes in yeast [116], raising the possibility that HSF1 could affect expression of components of the ubiquitin-proteasome system in stressed cells, in addition to the well-characterized upregulation of HSP gene expression [98, 117]. This raises the question if reversing the attenuation of the HSR would reverse the age-related ubiquitin-proteasome decline.
HSF1 has variable effects on expression of proteasome subunits in mammalian cells, depending on the experimental conditions and cell type being investigated. Oxidative stress had no effect on proteasome expression in liver epithelial cells [108] but did increase expression of proteasome subunits in neuroblastoma cells [87]. Repeated, mild heat shock resulted in significant upregulation of proteasomal catalytic activities and 11S subunit levels in human fibroblasts [118], but acute, mild heat shock led to decreased proteasome messenger RNA (mRNA) levels, inhibition of proteasome complex assembly, decrease in chymotrypsin-like activity, and reorganization of the intracellular distribution of proteasomes [119]. Overexpressing a constitutively active form of HSF1 in mouse embryonic fibroblasts (MEF) did not increase the level of proteasomal proteins expression or proteasomal activity [120]. When the ubiquitin–proteasome network is down-regulated, certain heat shock proteins such as HSP70, among other molecular chaperones, are induced [117]. Proteasome inhibition leads to the activation of all members of the heat-shock-factor family [121].
Molecular chaperones of the 70-kDa heat shock protein family (HSP70) are involved in protein folding, protein translocation, and protein degradation [122–124]. Other proteins are necessary for HSP70 function, and the protein BAG-1 associates with the proteasome in an ATP dependent manner and promote binding of constitutive heat shock protein 70 (HSC70) and HSP70 to the proteolytic complex. BAG-1 can apparently act as a coupling factor between the HSP70 chaperone system and the protein degradation machinery [125]. Also, the membrane-stabilizing effect of HSP70 and its related molecules are involved in lysosomal function. HSP70 can be efficiently endocytosed and localized to the lysosomes, and binding of HSP70 to a lysosome-specific lipid stabilizes lysosomes [126]. HSP70 has a positive effect on the lysosomal integrity by binding specifically to BMP [127]. In cancer cells, [126], Kirkegaard et al demonstrated that in acidic environments, HSP70 binds with high affinity and specificity to BMP, facilitates an activity of ASM, and thereby stabilizes lysosomes. HSP70 also serves cytoprotective roles as a guardian of the lysosomal membrane integrity by assisting sphingomyelin degradation or maintaining proper protein folding and recycling as a chaperone. However, calpain mediated cleavage of HSP70, especially after its carbonylation due to oxidative stress can induce lysosomal rupture. Furthermore, HSP70 dysfunction activates nuclear factor-kappaB (NF-κB) signaling that can also promote neurodegeneration [128].
Lysosomes are involved in protein degradation through autophagy. Autophagy is the cellular process that mediates the degradation of iAβ, and studies in aging models have revealed that a failure of the autophagic system results in marked accumulation of abnormal proteins leading to dysfunction and cell death [107, 129]. Different types of autophagy have been described, depending on the mechanism that mediates the delivery of cytosolic cargo to lysosomes for degradation. The three main types of autophagy in mammals are macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [130–131]. CMA and proteasome defective cells maintain normal rates of long-lived protein degradation by up-regulating macroautophagy, the major form of autophagy. Macroautophagy is an active pathway for turning over AβPP and generating Aβ peptide [132]. Constitutive upregulation of macroautophagy is unable, however, to compensate for all CMA and proteasome functions [133–134]. Thus, this limitation leads to long term protein accumulation such as Aβ plaque formation. In human neurons, serum starvation, which strongly induces autophagy, elevates Aβ levels threefold [135].
The Aβ generated in AVs is presumably delivered principally to lysosomes and degraded by cathepsins, which have the necessary cleavage specificity [136]. Cathepsin B deletion in mice, for example, elevates Aβ levels in brain, whereas increasing cathepsin B expression has the opposite effect [137]. The presence of undigested proteins in lysosomes could be responsible for their impaired ability to fuse and/or degrade the autophagosome contents. A decrease in the lysosomal levels of the CMA receptor (LAMP-2A) is the primary defect responsible for the diminished CMA activity during aging [138]. Normal CMA activity is initially maintained (during middle age) by increasing the amount of luminal chaperone. At advanced ages, the levels of the receptor are so low that compensation by the chaperone is no longer possible [139].
CMA has shown to be defective in several neurodegenerative diseases, and it involves the facilitated direct entry into lysosomes of selected cytosolic proteins containing a chaperone-mediated autophagy-targeting motif [133]. This also denotes the existence of crosstalk among different forms of autophagy [134]. In CMA, cytosolic proteins containing a KFERQ motif are selectively targeted to the lysosomal lumen for degradation. Notably AβPP and synuclein, a protein implicated in Parkinson disease, contain this targeting sequence [140] and impaired CMA-mediated degradation contributes to accumulation of Aβ in AD [141].
Interestingly, the age-related FOXO attenuation leads to an increase in oxidative stress which causes an age-related lysosomal dysfunction. FOXO controls the SOD and catalase genes [142], so its attenuation during aging causes increased oxidative injuries. This oxidative stress damages the lysosome and may lead to macroautophagy inhibition if the result of sustained oxidative stress is a net compromise in lysosome function [143].
Translating invertebrate FOXO activity to an appropriate human analogue should be done with some care. Just as Drosophila APPL and C. elegans APL-1 are reflected in not a single mammalian protein but a three-protein family, consisting of AβPP and AβPP-like proteins 1 (APLP1) and 2 (APLP2), the invertebrate FOXO has a four-member family homologue in mammals, consisting of FOXO1, FOXO3, FOXO4, and FOXO6, with both overlapping and distinct functions, and function of an individual FOXO family member in terms of neuroprotection can reverse depending upon circumstance [144]. The effects of individual family members may be counter our model. For example, FOXO1 expression is positively correlated to AD and associated cognitive decline in humans [145]. Likewise, exclusion of FOXO3 from the nucleus due to calorie restriction attenuated memory loss and other deficiencies in the AD transgenic mouse model AβPPSWE-Tg2576 [146]. On the other hand, FOXO3 protects quiescent cells from oxidative stress [142], and it is our contention that it is not FOXO3’s nuclear activity but its cytosolic or extracellular activities that would be neuroprotective. Likewise, while nuclear FOXO3 is apoptotic, and overall restriction of FOXO3 levels attenuates the BACE1 processing pathway [146], it may be an oversimplification to equate Aβ with AD, particularly since Aβ neurotoxicity is as much a factor of its tertiary and quaternary structures as its mere presence. Likewise, Aβ has been found to potentially have multiple non-pathogenic functions, including acting as a transcription factor [147]. The specific biochemical stages of “pre-AD” have not been adequately elucidated, and relative contributions of Aβ, oligomeric Aβ, and amyloid plaque to the disorder are not yet characterized.
Decreased autophagic activity is associated with neurodegenerative disorders, such as Parkinson’s [148] and AD [149]. In addition to the well-documented lysosomal dysfunction in AD [150], a decrease in autophagy has recently been found [132]. The hallmark feature of AD pathology is the presence of enormously swollen “dystrophic” neuritis in which autophagic vacuoles (AV) progressively accumulate and become the predominant organelle [132]. In normal brain cells, autophagosomes are usually found in low numbers. However, in a large number of brain cells from AD patients, autophagosomes are very prominent. In AD brains, diminished autophagosome-lysosome fusion results in accumulation of Aβ-filled autophagosomes, which constitute a major intracellular storage of the toxic protein [151]. This probably indicates activation of autophagy but with impaired maturation and/or impaired autophagosome-lysosome fusion [152], which eventually leads to apoptosis. Excessive generation of Aβ in AD brain cells is also driven by increased levels of the γ-secretase complex within autophagosomes [153]. eAβ can also be cleared from the brain through interaction with lipoprotein receptors [154]. ApoE, a lipid transporter, also has a role in the clearance of Aβ from the brain, and ApoEε4 is shown to be less effective, compared to ApoEε2 and ApoEε3 variants [155].
Conclusions
Although the current model of AD views Aβ peptide accumulation as etiologic and suggests that abnormal accumulation of Aβ42, is an early event in the pathophysiologic cascade, we would like to add that age-related attenuation in the HSF1 and FOXO pathways play an even earlier role than Aβ accumulation and proteotoxicity respectively in the predisposition to LOAD (Fig. 3). Recently, it has been shown that the clearance of Aβ42 is in fact the primary inciting event in LOAD. Both autopsy and biomarker studies similarly suggest that Aβ42 accumulation increases with advanced aging, the greatest risk factor for developing LOAD. Therefore the aging process is required for the development of LOAD. The initial event in the aging process should be correlated to the initiation of LOAD, first initiated by the attenuation of HSF1 leading to decreased degradation of Aβ, facilitated by decreased chaperone production, CMA degradation, and proteasome activity. This will cause increased iAβ levels which will activate autophagy systems. At this stage we can see increased AV in pathologic studies.
This increase in autophagy cannot be sustained and the iAβ is released into the extracellular space (ECS). In the ECS Aβ forms toxic oligomers, but the activation of certain FOXO family members would convert these oligomers in non-toxic plaques. This process explains the temporary lag between plaque formation and clinical symptoms. As long as certain of the FOXO family pathways prevent the formation of oligomers, there would be no AD clinical symptomatology. Later, attenuation (e.g., age-related) of appropriate FOXO family members would appear, and clinical symptoms develop.
Longevity pathways are crucial to the development of AD. Aβ clearance and antiproteotoxic pathways have been unequivocally associated with LOAD. However, the complex features of LOAD, especially its convoluted molecular biology and (epi)genetics, have prevented the resolution of the age-related specific associations within AD. In order to clarify the role of the aging process on the Aβ clearance and antiproteotoxic pathways, we postulate a new model in which the aging process would precede Aβ accumulation, and attenuation of HSF1 is an “upstream” event in the cascade that results in excess iAβ and synaptic dysfunction, which may lead directly to cognitive impairment and/or trigger “downstream” neurodegeneration and cell loss. Specific host factors, such as the activity of FOXO family pathways, would mediate the response to Aβ toxicity and the pace of progression towards the clinical manifestations of AD.
As a result of these observations, a genetic and molecular model is proposed here, in which assessment of the activity of the HSF1 and FOXO3 (as a suggested family member) would give information of sequential preclinical biomarkers of AD pathology. This biomarker model parallels the pathomolecular sequence of events in AD, and is particularly relevant to tracking the preclinical stages of LOAD of patients at high risk for developing this disease.
Any preclinical biomarkers should be complemented by clinical biomarkers of brain Aβ amyloidosis, such as reductions in CSF Aβ42 and increased amyloid PET tracer retention. Elevated CSF τ is thought to be a biomarker of τ-mediated neuronal injury and is not specific for AD pathology [156]. At a more advanced disease, a decreased [18F]-fludeoxyglucose (FDG) uptake on PET in a characteristic temporal-parietal pattern is a biomarker of AD-related synaptic dysfunction [14]. When neurodegeneration is initiated, brain atrophy on structural MRI in a characteristic pattern involving medial temporal, paralimbic and temporal-partial isocortex serves as a biomarker of AD-related neurodegeneration.
This new preclinical biomarker model has the following features: 1) HSF1-DNA binding attenuation would risk abnormality due to effects of the aging process; 2) Aβ oligomer and amyloid plaque biomarkers would become abnormal, and a substantial Aβ load accumulates prior to the appearance of clinical symptoms. So far as this model of brain amyloid deposition is necessary but not sufficient to produce the clinical symptoms of MCI and dementia, it requires an additional step to produce clinical symptoms, specifically 3) Attenuation of FOXO family pathways causing Aβ oligomer formation and neurodegeneration; 4) Finally, biomarkers of dysfunction, neuronal injury, and degeneration would be confirmatory later in the disease. The severity and change over time in these biomarkers correlate with clinical symptoms. Structural MRI would be the last biomarker to become abnormal.
Definitive studies to determine if asymptomatic individuals with HSF1 and FOXO family pathway attenuation associated with Aβ accumulation are indeed destined to develop AD dementia, and whether new therapies targeting these pathways will prevent cognitive decline are a needed strategy to prevent this epidemic disease.
References
- 1.Tanzi RE, Bertram L. Twenty Years of the Alzheimer’s Disease Amyloid Hypothesis: A Genetic Perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56:303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- 3.Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 4.Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- 5.Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Archives of neurology. 1999;56:303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- 6.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 7.Jack CR, Jr, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC, Thies B, Phelps CH. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:257–262. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corrada MM, Brookmeyer R, Berlau D, Paganini-Hill A, Kawas CH. Prevalence of dementia after age 90: results from the 90+ study. Neurology. 2008;71:337–343. doi: 10.1212/01.wnl.0000310773.65918.cd. [DOI] [PubMed] [Google Scholar]
- 9.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 10.Arendash GW, Sanchez-Ramos J, Mori T, Mamcarz M, Lin X, Runfeldt M, Wang L, Zhang G, Sava V, Tan J, Cao C. Electromagnetic field treatment protects against and reverses cognitive impairment in Alzheimer’s disease mice. J Alzheimers Dis. 2010;19:191–210. doi: 10.3233/JAD-2010-1228. [DOI] [PubMed] [Google Scholar]
- 11.Panza F, Frisardi V, Solfrizzi V, Imbimbo BP, Logroscino G, Santamato A, Greco A, Seripa D, Pilotto A. Immunotherapy for Alzheimer’s disease: from anti-beta-amyloid to tau-based immunization strategies. Immunotherapy. 2012;4:213–238. doi: 10.2217/imt.11.170. [DOI] [PubMed] [Google Scholar]
- 12.Jessen F, Wiese B, Bachmann C, Eifflaender-Gorfer S, Haller F, Kolsch H, Luck T, Mosch E, van den Bussche H, Wagner M, Wollny A, Zimmermann T, Pentzek M, Riedel-Heller SG, Romberg H-P, Weyerer S, Kaduszkiewicz H, Maier W, Bickel H, Abholz H-H, Angermeyer MC, Blank W, Buchwald M, Colditz M, Daerr M, Dichgans M, Finckh U, Frenzen A, Fuchs A, Eisele M, Heinrich S, Kaufeler T, Konig H-H, Luppa M, Mayer M, Olbrich J, Rudolph A, Sandholzer H, Sauder M, Schuermann B, Werle J German Study on Aging C, Dementia in Primary Care Patients Study G. Prediction of dementia by subjective memory impairment: effects of severity and temporal association with cognitive impairment. Archives of general psychiatry. 2010;67:414–422. doi: 10.1001/archgenpsychiatry.2010.30. [DOI] [PubMed] [Google Scholar]
- 13.Saykin AJ, Wishart HA, Rabin LA, Santulli RB, Flashman LA, West JD, McHugh TL, Mamourian AC. Older adults with cognitive complaints show brain atrophy similar to that of amnestic MCI. Neurology. 2006;67:834–842. doi: 10.1212/01.wnl.0000234032.77541.a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Leon MJ, DeSanti S, Zinkowski R, Mehta PD, Pratico D, Segal S, Clark C, Kerkman D, DeBernardis J, Li J, Lair L, Reisberg B, Tsui W, Rusinek H. MRI and CSF studies in the early diagnosis of Alzheimer’s disease. J Intern Med. 2004;256:205–223. doi: 10.1111/j.1365-2796.2004.01381.x. [DOI] [PubMed] [Google Scholar]
- 15.Liao L, Cheng D, Wang J, Duong DM, Losik TG, Gearing M, Rees HD, Lah JJ, Levey AI, Peng J. Proteomic characterization of postmortem amyloid plaques isolated by laser capture microdissection. J Biol Chem. 2004;279:37061–37068. doi: 10.1074/jbc.M403672200. [DOI] [PubMed] [Google Scholar]
- 16.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 17.Kar S, Slowikowski SP, Westaway D, Mount HT. Interactions between beta-amyloid and central cholinergic neurons: implications for Alzheimer’s disease. J Psychiatry Neurosci. 2004;29:427–441. [PMC free article] [PubMed] [Google Scholar]
- 18.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 19.Zhang M, Deng Y, Luo Y, Zhang S, Zou H, Cai F, Wada K, Song W. Control of BACE1 degradation and APP processing by ubiquitin carboxyl-terminal hydrolase L1. J Neurochem. 2012;120:1129–1138. doi: 10.1111/j.1471-4159.2011.07644.x. [DOI] [PubMed] [Google Scholar]
- 20.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C, et al. Isolation and quantification of soluble Alzheimer’s beta-peptide from biological fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 21.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 22.Jarrett JT, Berger EP, Lansbury PT., Jr The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 23.Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 24.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Echeverria V, Ducatenzeiler A, Alhonen L, Janne J, Grant SM, Wandosell F, Muro A, Baralle F, Li H, Duff K, Szyf M, Cuello AC. Rat transgenic models with a phenotype of intracellular Abeta accumulation in hippocampus and cortex. J Alzheimers Dis. 2004;6:209–219. doi: 10.3233/jad-2004-6301. [DOI] [PubMed] [Google Scholar]
- 26.De Strooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113 (Pt 11):1857–1870. doi: 10.1242/jcs.113.11.1857. [DOI] [PubMed] [Google Scholar]
- 27.Paradis E, Douillard H, Koutroumanis M, Goodyer C, LeBlanc A. Amyloid beta peptide of Alzheimer’s disease downregulates Bcl-2 and upregulates bax expression in human neurons. J Neurosci. 1996;16:7533–7539. doi: 10.1523/JNEUROSCI.16-23-07533.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang KH, Vincent F, Shah K. Deregulated Cdk5 triggers aberrant activation of cell cycle kinases and phosphatases inducing neuronal death. J Cell Sci. 2012;125:5124–5137. doi: 10.1242/jcs.108183. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, McLaughlin R, Goodyer C, LeBlanc A. Selective cytotoxicity of intracellular amyloid beta peptide1-42 through p53 and Bax in cultured primary human neurons. J Cell Biol. 2002;156:519–529. doi: 10.1083/jcb.200110119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA. Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett. 2001;306:116–120. doi: 10.1016/s0304-3940(01)01876-6. [DOI] [PubMed] [Google Scholar]
- 31.Agholme L, Hallbeck M, Benedikz E, Marcusson J, Kagedal K. Amyloid-beta secretion, generation, and lysosomal sequestration in response to proteasome inhibition: involvement of autophagy. J Alzheimers Dis. 2012;31:343–358. doi: 10.3233/JAD-2012-120001. [DOI] [PubMed] [Google Scholar]
- 32.Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li M, Chen L, Lee DH, Yu LC, Zhang Y. The role of intracellular amyloid beta in Alzheimer’s disease. Prog Neurobiol. 2007;83:131–139. doi: 10.1016/j.pneurobio.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Buee L, Troquier L, Burnouf S, Belarbi K, Van der Jeugd A, Ahmed T, Fernandez-Gomez F, Caillierez R, Grosjean ME, Begard S, Barbot B, Demeyer D, Obriot H, Brion I, Buee-Scherrer V, Maurage CA, Balschun D, D’Hooge R, Hamdane M, Blum D, Sergeant N. From tau phosphorylation to tau aggregation: what about neuronal death? Biochem Soc Trans. 2010;38:967–972. doi: 10.1042/BST0380967. [DOI] [PubMed] [Google Scholar]
- 36.Schaeffer EL, De-Paula VJ, da Silva ER, de ANB, Skaf HD, Forlenza OV, Gattaz WF. Inhibition of phospholipase A2 in rat brain decreases the levels of total Tau protein. J Neural Transm. 2011;118:1273–1279. doi: 10.1007/s00702-011-0619-4. [DOI] [PubMed] [Google Scholar]
- 37.Yu X, Luo Y, Dinkel P, Zheng J, Wei G, Margittai M, Nussinov R, Ma B. Cross-seeding and conformational selection between three- and four-repeat human Tau proteins. J Biol Chem. 2012;287:14950–14959. doi: 10.1074/jbc.M112.340794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khlistunova I, Biernat J, Wang Y, Pickhardt M, von Bergen M, Gazova Z, Mandelkow E, Mandelkow EM. Inducible expression of Tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. 2006;281:1205–1214. doi: 10.1074/jbc.M507753200. [DOI] [PubMed] [Google Scholar]
- 39.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cork LC, Sternberger NH, Sternberger LA, Casanova MF, Struble RG, Price DL. Phosphorylated neurofilament antigens in neurofibrillary tangles in Alzheimer’s disease. J Neuropathol Exp Neurol. 1986;45:56–64. doi: 10.1097/00005072-198601000-00005. [DOI] [PubMed] [Google Scholar]
- 41.Rudrabhatla P, Grant P, Jaffe H, Strong MJ, Pant HC. Quantitative phosphoproteomic analysis of neuronal intermediate filament proteins (NF-M/H) in Alzheimer’s disease by iTRAQ. FASEB J. 2010;24:4396–4407. doi: 10.1096/fj.10-157859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williamson R, van Aalten L, Mann DM, Platt B, Plattner F, Bedford L, Mayer J, Howlett D, Usardi A, Sutherland C, Cole AR. CRMP2 hyperphosphorylation is characteristic of Alzheimer’s disease and not a feature common to other neurodegenerative diseases. J Alzheimers Dis. 2011;27:615–625. doi: 10.3233/JAD-2011-110617. [DOI] [PubMed] [Google Scholar]
- 43.Shankar GM, Walsh DM. Alzheimer’s disease: synaptic dysfunction and Abeta. Mol Neurodegener. 2009;4:48. doi: 10.1186/1750-1326-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- 45.Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Jr, Morris JC. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology. 2001;56:127–129. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- 46.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 47.Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, Palmeri A, Arancio O. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Filipcik P, Zilka N, Bugos O, Kucerak J, Koson P, Novak P, Novak M. First transgenic rat model developing progressive cortical neurofibrillary tangles. Neurobiol Aging. 2012;33:1448–1456. doi: 10.1016/j.neurobiolaging.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 49.Mortimer JA, Snowdon DA, Markesbery WR. Head circumference, education and risk of dementia: findings from the Nun Study. J Clin Exp Neuropsychol. 2003;25:671–679. doi: 10.1076/jcen.25.5.671.14584. [DOI] [PubMed] [Google Scholar]
- 50.Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci U S A. 1990;87:1561–1565. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nikolaev A, McLaughlin T, O’Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Saganich MJ, Schroeder BE, Galvan V, Bredesen DE, Koo EH, Heinemann SF. Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require C-terminal cleavage of APP. J Neurosci. 2006;26:13428–13436. doi: 10.1523/JNEUROSCI.4180-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Galvan V, Gorostiza OF, Banwait S, Ataie M, Logvinova AV, Sitaraman S, Carlson E, Sagi SA, Chevallier N, Jin K, Greenberg DA, Bredesen DE. Reversal of Alzheimer’s-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc Natl Acad Sci U S A. 2006;103:7130–7135. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- 55.Parachikova A, Agadjanyan MG, Cribbs DH, Blurton-Jones M, Perreau V, Rogers J, Beach TG, Cotman CW. Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol Aging. 2007;28:1821–1833. doi: 10.1016/j.neurobiolaging.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dhib-Jalbut S, Arnold DL, Cleveland DW, Fisher M, Friedlander RM, Mouradian MM, Przedborski S, Trapp BD, Wyss-Coray T, Yong VW. Neurodegeneration and neuroprotection in multiple sclerosis and other neurodegenerative diseases. J Neuroimmunol. 2006;176:198–215. doi: 10.1016/j.jneuroim.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 57.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 59.McGeer EG, Yasojima K, Schwab C, McGeer PL. The pentraxins: possible role in Alzheimer’s disease and other innate inflammatory diseases. Neurobiol Aging. 2001;22:843–848. doi: 10.1016/s0197-4580(01)00288-3. [DOI] [PubMed] [Google Scholar]
- 60.Van Eldik LJ, Thompson WL, Ralay Ranaivo H, Behanna HA, Martin Watterson D. Glia proinflammatory cytokine upregulation as a therapeutic target for neurodegenerative diseases: function-based and target-based discovery approaches. Int Rev Neurobiol. 2007;82:277–296. doi: 10.1016/S0074-7742(07)82015-0. [DOI] [PubMed] [Google Scholar]
- 61.Mattson MP, Partin J, Begley JG. Amyloid beta-peptide induces apoptosis-related events in synapses and dendrites. Brain Res. 1998;807:167–176. doi: 10.1016/s0006-8993(98)00763-x. [DOI] [PubMed] [Google Scholar]
- 62.Chung CW, Song YH, Kim IK, Yoon WJ, Ryu BR, Jo DG, Woo HN, Kwon YK, Kim HH, Gwag BJ, Mook-Jung IH, Jung YK. Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol Dis. 2001;8:162–172. doi: 10.1006/nbdi.2000.0335. [DOI] [PubMed] [Google Scholar]
- 63.Isaacs AM, Senn DB, Yuan M, Shine JP, Yankner BA. Acceleration of amyloid beta-peptide aggregation by physiological concentrations of calcium. J Biol Chem. 2006;281:27916–27923. doi: 10.1074/jbc.M602061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- 65.Rothman SM, Olney JW. Excitotoxicity and the NMDA receptor--still lethal after eight years. Trends Neurosci. 1995;18:57–58. doi: 10.1016/0166-2236(95)93869-y. [DOI] [PubMed] [Google Scholar]
- 66.Arispe N, Pollard HB, Rojas E. Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [A beta P-(1-40)] in bilayer membranes. Proc Natl Acad Sci U S A. 1993;90:10573–10577. doi: 10.1073/pnas.90.22.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin H, Bhatia R, Lal R. Amyloid beta protein forms ion channels: implications for Alzheimer’s disease pathophysiology. FASEB J. 2001;15:2433–2444. doi: 10.1096/fj.01-0377com. [DOI] [PubMed] [Google Scholar]
- 68.Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwatsubo T, Lee VM, Doms RW. Alzheimer’s A beta(1-42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nature medicine. 1997;3:1021–1023. doi: 10.1038/nm0997-1021. [DOI] [PubMed] [Google Scholar]
- 69.Sudoh M, Pauletti GM, Yao W, Moser W, Yokoyama A, Pasternak A, Sprengeler PA, Smith AB, 3rd, Hirschmann R, Borchardt RT. Transport characteristics of peptidomimetics. Effect of the pyrrolinone bioisostere on transport across Caco-2 cell monolayers. Pharm Res. 1998;15:719–725. doi: 10.1023/a:1011966918959. [DOI] [PubMed] [Google Scholar]
- 70.Greenfield JP, Tsai J, Gouras GK, Hai B, Thinakaran G, Checler F, Sisodia SS, Greengard P, Xu H. Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc Natl Acad Sci U S A. 1999;96:742–747. doi: 10.1073/pnas.96.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu H, Sweeney D, Wang R, Thinakaran G, Lo AC, Sisodia SS, Greengard P, Gandy S. Generation of Alzheimer beta-amyloid protein in the trans-Golgi network in the apparent absence of vesicle formation. Proc Natl Acad Sci U S A. 1997;94:3748–3752. doi: 10.1073/pnas.94.8.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. The American journal of pathology. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee EK, Park YW, Shin DY, Mook-Jung I, Yoo YJ. Cytosolic amyloid-beta peptide 42 escaping from degradation induces cell death. Biochem Biophys Res Commun. 2006;344:471–477. doi: 10.1016/j.bbrc.2006.03.166. [DOI] [PubMed] [Google Scholar]
- 74.VanSlyke JK, Musil LS. Dislocation and degradation from the ER are regulated by cytosolic stress. J Cell Biol. 2002;157:381–394. doi: 10.1083/jcb.200111045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Keller JN, Huang FF, Markesbery WR. Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience. 2000;98:149–156. doi: 10.1016/s0306-4522(00)00067-1. [DOI] [PubMed] [Google Scholar]
- 76.Ding Q, Dimayuga E, Martin S, Bruce-Keller AJ, Nukala V, Cuervo AM, Keller JN. Characterization of chronic low-level proteasome inhibition on neural homeostasis. J Neurochem. 2003;86:489–497. doi: 10.1046/j.1471-4159.2003.01885.x. [DOI] [PubMed] [Google Scholar]
- 77.Selkoe DJ, Yamazaki T, Citron M, Podlisny MB, Koo EH, Teplow DB, Haass C. The role of APP processing and trafficking pathways in the formation of amyloid beta-protein. Ann N Y Acad Sci. 1996;777:57–64. doi: 10.1111/j.1749-6632.1996.tb34401.x. [DOI] [PubMed] [Google Scholar]
- 78.Martinez-Vicente M, Sovak G, Cuervo AM. Protein degradation and aging. Experimental gerontology. 2005;40:622–633. doi: 10.1016/j.exger.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 79.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 80.Volovik Y, Maman M, Dubnikov T, Bejerano-Sagie M, Joyce D, Kapernick EA, Cohen E, Dillin A. Temporal requirements of heat shock factor-1 for longevity assurance. Aging Cell. 2012;11:491–499. doi: 10.1111/j.1474-9726.2012.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Locke M, Tanguay RM. Diminished heat shock response in the aged myocardium. Cell Stress Chaperones. 1996;1:251–260. doi: 10.1379/1466-1268(1996)001<0251:dhsrit>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ben-Zvi A, Miller EA, Morimoto RI. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci U S A. 2009;106:14914–14919. doi: 10.1073/pnas.0902882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Verbeke P, Fonager J, Clark BF, Rattan SI. Heat shock response and ageing: mechanisms and applications. Cell Biol Int. 2001;25:845–857. doi: 10.1006/cbir.2001.0789. [DOI] [PubMed] [Google Scholar]
- 84.Jurivich DA, Qiu L, Welk JF. Attenuated stress responses in young and old human lymphocytes. Mech Ageing Dev. 1997;94:233–249. doi: 10.1016/s0047-6374(96)01856-8. [DOI] [PubMed] [Google Scholar]
- 85.Nistor M, Don M, Parekh M, Sarsoza F, Goodus M, Lopez GE, Kawas C, Leverenz J, Doran E, Lott IT, Hill M, Head E. Alpha- and beta-secretase activity as a function of age and beta-amyloid in Down syndrome and normal brain. Neurobiol Aging. 2007;28:1493–1506. doi: 10.1016/j.neurobiolaging.2006.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Jr, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089–1115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- 88.Campanini C, Petronini PG, Alfieri R, Borghetti AF. Decreased expression of heat shock protein 70 mRNA and protein in WI-38 human fibroblasts aging in vitro. Annals of the New York Academy of Sciences. 1992;663:442–443. doi: 10.1111/j.1749-6632.1992.tb38695.x. [DOI] [PubMed] [Google Scholar]
- 89.Heydari AR, You S, Takahashi R, Gutsmann-Conrad A, Sarge KD, Richardson A. Age-related alterations in the activation of heat shock transcription factor 1 in rat hepatocytes. Exp Cell Res. 2000;256:83–93. doi: 10.1006/excr.2000.4808. [DOI] [PubMed] [Google Scholar]
- 90.Wu Y, Cao Z, Klein WL, Luo Y. Heat shock treatment reduces beta amyloid toxicity in vivo by diminishing oligomers. Neurobiol Aging. 2010;31:1055–1058. doi: 10.1016/j.neurobiolaging.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morley JF, Morimoto RI. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Molecular biology of the cell. 2004;15:657–664. doi: 10.1091/mbc.E03-07-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Demontis F, Perrimon N. FOXO/4E-BP signaling in Drosophila muscles regulates organism-wide proteostasis during aging. Cell. 2010;143:813–825. doi: 10.1016/j.cell.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gorski SM, Chittaranjan S, Pleasance ED, Freeman JD, Anderson CL, Varhol RJ, Coughlin SM, Zuyderduyn SD, Jones SJ, Marra MA. A SAGE approach to discovery of genes involved in autophagic cell death. Current biology : CB. 2003;13:358–363. doi: 10.1016/s0960-9822(03)00082-4. [DOI] [PubMed] [Google Scholar]
- 94.Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2008;4:176–184. doi: 10.4161/auto.5269. [DOI] [PubMed] [Google Scholar]
- 95.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 96.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 97.Julien C, Tremblay C, Emond V, Lebbadi M, Salem N, Jr, Bennett DA, Calon F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol. 2009;68:48–58. doi: 10.1097/NEN.0b013e3181922348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pirkkala L, Alastalo TP, Zuo X, Benjamin IJ, Sistonen L. Disruption of heat shock factor 1 reveals an essential role in the ubiquitin proteolytic pathway. Molecular and cellular biology. 2000;20:2670–2675. doi: 10.1128/mcb.20.8.2670-2675.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 100.Fonte V, Kipp DR, Yerg J, 3rd, Merin D, Forrestal M, Wagner E, Roberts CM, Link CD. Suppression of in vivo beta-amyloid peptide toxicity by overexpression of the HSP-16.2 small chaperone protein. J Biol Chem. 2008;283:784–791. doi: 10.1074/jbc.M703339200. [DOI] [PubMed] [Google Scholar]
- 101.Magrane J, Smith RC, Walsh K, Querfurth HW. Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:1700–1706. doi: 10.1523/JNEUROSCI.4330-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Perez FP, Zhou X, Morisaki J, Jurivich D. Electromagnetic field therapy delays cellular senescence and death by enhancement of the heat shock response. Experimental gerontology. 2008;43:307–316. doi: 10.1016/j.exger.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 103.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 104.Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes & development. 2008;22:1427–1438. doi: 10.1101/gad.1657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stadtman ER. Protein oxidation and aging. Science. 1992;257:1220–1224. doi: 10.1126/science.1355616. [DOI] [PubMed] [Google Scholar]
- 106.Kumar P, Ambasta RK, Veereshwarayya V, Rosen KM, Kosik KS, Band H, Mestril R, Patterson C, Querfurth HW. CHIP and HSPs interact with beta-APP in a proteasome-dependent manner and influence Abeta metabolism. Hum Mol Genet. 2007;16:848–864. doi: 10.1093/hmg/ddm030. [DOI] [PubMed] [Google Scholar]
- 107.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Grune T, Reinheckel T, Joshi M, Davies KJ. Proteolysis in cultured liver epithelial cells during oxidative stress. Role of the multicatalytic proteinase complex, proteasome. J Biol Chem. 1995;270:2344–2351. doi: 10.1074/jbc.270.5.2344. [DOI] [PubMed] [Google Scholar]
- 109.Sitte N, Merker K, Grune T. Proteasome-dependent degradation of oxidized proteins in MRC-5 fibroblasts. FEBS Lett. 1998;440:399–402. doi: 10.1016/s0014-5793(98)01495-1. [DOI] [PubMed] [Google Scholar]
- 110.Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell. 1994;78:761–771. doi: 10.1016/s0092-8674(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 111.Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 112.Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- 113.Petrucelli L, Dawson TM. Mechanism of neurodegenerative disease: role of the ubiquitin proteasome system. Ann Med. 2004;36:315–320. doi: 10.1080/07853890410031948. [DOI] [PubMed] [Google Scholar]
- 114.Ross CA, Pickart CM. The ubiquitin-proteasome pathway in Parkinson’s disease and other neurodegenerative diseases. Trends Cell Biol. 2004;14:703–711. doi: 10.1016/j.tcb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 115.Perez FP, Moinuddin SS, ul ain Shamim Q, Joseph DJ, Morisaki J, Zhou X. Longevity pathways: HSF1 and FoxO pathways, a new therapeutic target to prevent age-related diseases. Curr Aging Sci. 2012;5:87–95. doi: 10.2174/1874609811205020087. [DOI] [PubMed] [Google Scholar]
- 116.Hahn JS, Hu Z, Thiele DJ, Iyer VR. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol Cell Biol. 2004;24:5249–5256. doi: 10.1128/MCB.24.12.5249-5256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kawazoe Y, Nakai A, Tanabe M, Nagata K. Proteasome inhibition leads to the activation of all members of the heat-shock-factor family. Eur J Biochem. 1998;255:356–362. doi: 10.1046/j.1432-1327.1998.2550356.x. [DOI] [PubMed] [Google Scholar]
- 118.Beedholm R, Clark BF, Rattan SI. Mild heat stress stimulates 20S proteasome and its 11S activator in human fibroblasts undergoing aging in vitro. Cell Stress Chaperones. 2004;9:49–57. doi: 10.1379/475.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kuckelkorn U, Knuehl C, Boes-Fabian B, Drung I, Kloetzel PM. The effect of heat shock on 20S/26S proteasomes. Biol Chem. 2000;381:1017–1023. doi: 10.1515/BC.2000.125. [DOI] [PubMed] [Google Scholar]
- 120.Taylor DM, Kabashi E, Agar JN, Minotti S, Durham HD. Proteasome activity or expression is not altered by activation of the heat shock transcription factor Hsf1 in cultured fibroblasts or myoblasts. Cell Stress Chaperones. 2005;10:230–241. doi: 10.1379/CSC-119R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim D, Kim SH, Li GC. Proteasome inhibitors MG132 and lactacystin hyperphosphorylate HSF1 and induce hsp70 and hsp27 expression. Biochem Biophys Res Commun. 1999;254:264–268. doi: 10.1006/bbrc.1998.9840. [DOI] [PubMed] [Google Scholar]
- 122.Craig EA, Weissman JS, Horwich AL. Heat shock proteins and molecular chaperones: mediators of protein conformation and turnover in the cell. Cell. 1994;78:365–372. doi: 10.1016/0092-8674(94)90416-2. [DOI] [PubMed] [Google Scholar]
- 123.Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–579. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 124.Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- 125.Luders J, Demand J, Hohfeld J. The ubiquitin-related BAG-1 provides a link between the molecular chaperones Hsc70/Hsp70 and the proteasome. J Biol Chem. 2000;275:4613–4617. doi: 10.1074/jbc.275.7.4613. [DOI] [PubMed] [Google Scholar]
- 126.Kirkegaard T, Roth AG, Petersen NH, Mahalka AK, Olsen OD, Moilanen I, Zylicz A, Knudsen J, Sandhoff K, Arenz C, Kinnunen PK, Nylandsted J, Jaattela M. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature. 2010;463:549–553. doi: 10.1038/nature08710. [DOI] [PubMed] [Google Scholar]
- 127.Petersen NH, Kirkegaard T, Olsen OD, Jaattela M. Connecting Hsp70, sphingolipid metabolism and lysosomal stability. Cell Cycle. 2010;9:2305–2309. doi: 10.4161/cc.9.12.12052. [DOI] [PubMed] [Google Scholar]
- 128.Yamashima T. Hsp70.1 and related lysosomal factors for necrotic neuronal death. J Neurochem. 2012;120:477–494. doi: 10.1111/j.1471-4159.2011.07596.x. [DOI] [PubMed] [Google Scholar]
- 129.Li W, Yang Q, Mao Z. Chaperone-mediated autophagy: machinery, regulation and biological consequences. Cell Mol Life Sci. 2011;68:749–763. doi: 10.1007/s00018-010-0565-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Klionsky DJ. The molecular machinery of autophagy: unanswered questions. J Cell Sci. 2005;118:7–18. doi: 10.1242/jcs.01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cuervo AM. Autophagy: many paths to the same end. Mol Cell Biochem. 2004;263:55–72. doi: 10.1023/B:MCBI.0000041848.57020.57. [DOI] [PubMed] [Google Scholar]
- 132.Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Naslund J, Mathews PM, Cataldo AM, Nixon RA. Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wong E, Cuervo AM. Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol. 2010;2:a006734. doi: 10.1101/cshperspect.a006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2006;103:5805–5810. doi: 10.1073/pnas.0507436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.LeBlanc AC, Xue R, Gambetti P. Amyloid precursor protein metabolism in primary cell cultures of neurons, astrocytes, and microglia. J Neurochem. 1996;66:2300–2310. doi: 10.1046/j.1471-4159.1996.66062300.x. [DOI] [PubMed] [Google Scholar]
- 136.Grbovic OM, Mathews PM, Jiang Y, Schmidt SD, Dinakar R, Summers-Terio NB, Ceresa BP, Nixon RA, Cataldo AM. Rab5-stimulated up-regulation of the endocytic pathway increases intracellular beta-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Abeta production. J Biol Chem. 2003;278:31261–31268. doi: 10.1074/jbc.M304122200. [DOI] [PubMed] [Google Scholar]
- 137.Florez-McClure ML, Hohsfield LA, Fonte G, Bealor MT, Link CD. Decreased insulin-receptor signaling promotes the autophagic degradation of beta-amyloid peptide in C. elegans. Autophagy. 2007;3:569–580. doi: 10.4161/auto.4776. [DOI] [PubMed] [Google Scholar]
- 138.Terman A, Gustafsson B, Brunk UT. Autophagy, organelles and ageing. J Pathol. 2007;211:134–143. doi: 10.1002/path.2094. [DOI] [PubMed] [Google Scholar]
- 139.Rajawat YS, Bossis I. Autophagy in aging and in neurodegenerative disorders. Hormones (Athens) 2008;7:46–61. doi: 10.14310/horm.2002.1111037. [DOI] [PubMed] [Google Scholar]
- 140.Massey A, Kiffin R, Cuervo AM. Pathophysiology of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004;36:2420–2434. doi: 10.1016/j.biocel.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 141.Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, Wyss-Coray T. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–2199. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 143.Pivtoraiko VN, Stone SL, Roth KA, Shacka JJ. Oxidative stress and autophagy in the regulation of lysosome-dependent neuron death. Antioxid Redox Signal. 2009;11:481–496. doi: 10.1089/ars.2008.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Monsalve M, Olmos Y. The complex biology of FOXO. Curr Drug Targets. 2011;12:1322–1350. doi: 10.2174/138945011796150307. [DOI] [PubMed] [Google Scholar]
- 145.Gomez Ravetti M, Rosso OA, Berretta R, Moscato P. Uncovering molecular biomarkers that correlate cognitive decline with the changes of hippocampus’ gene expression profiles in Alzheimer’s disease. PLoS One. 2010;5:e10153. doi: 10.1371/journal.pone.0010153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Qin W, Zhao W, Ho L, Wang J, Walsh K, Gandy S, Pasinetti GM. Regulation of forkhead transcription factor FoxO3a contributes to calorie restriction-induced prevention of Alzheimer’s disease-type amyloid neuropathology and spatial memory deterioration. Ann N Y Acad Sci. 2008;1147:335–347. doi: 10.1196/annals.1427.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bailey JA, Maloney B, Ge YW, Lahiri DK. Functional activity of the novel Alzheimer’s amyloid beta-peptide interacting domain (AbetaID) in the APP and BACE1 promoter sequences and implications in activating apoptotic genes and in amyloidogenesis. Gene. 2011;488:13–22. doi: 10.1016/j.gene.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 149.Cataldo AM, Hamilton DJ, Barnett JL, Paskevich PA, Nixon RA. Properties of the endosomal-lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer’s disease. J Neurosci. 1996;16:186–199. doi: 10.1523/JNEUROSCI.16-01-00186.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Nixon RA, Cataldo AM. Lysosomal system pathways: genes to neurodegeneration in Alzheimer’s disease. J Alzheimers Dis. 2006;9:277–289. doi: 10.3233/jad-2006-9s331. [DOI] [PubMed] [Google Scholar]
- 151.Glabe C. Intracellular mechanisms of amyloid accumulation and pathogenesis in Alzheimer’s disease. J Mol Neurosci. 2001;17:137–145. doi: 10.1385/JMN:17:2:137. [DOI] [PubMed] [Google Scholar]
- 152.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 153.Dorn BR, Dunn WA, Jr, Progulske-Fox A. Bacterial interactions with the autophagic pathway. Cell Microbiol. 2002;4:1–10. doi: 10.1046/j.1462-5822.2002.00164.x. [DOI] [PubMed] [Google Scholar]
- 154.Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 155.Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Irazuzta JE, de Courten-Myers G, Zemlan FP, Bekkedal MY, Rossi J., 3rd Serum cleaved Tau protein and neurobehavioral battery of tests as markers of brain injury in experimental bacterial meningitis. Brain Res. 2001;913:95–105. doi: 10.1016/s0006-8993(01)02764-0. [DOI] [PubMed] [Google Scholar]
- 157.Jellinger KA, Attems J. Prevalence of dementia disorders in the oldest-old: an autopsy study. Acta Neuropathol. 2010;119:421–433. doi: 10.1007/s00401-010-0654-5. [DOI] [PubMed] [Google Scholar]