

Abstract
Structure elucidation of biological compounds is still a major bottleneck of untargeted LC-HRMS approaches in metabolomics research. The aim of the present study was to combine stable isotope labeling and tandem mass spectrometry for the automated interpretation of the elemental composition of fragment ions and thereby facilitate the structural characterization of metabolites. The software tool FragExtract was developed and evaluated with LC-HRMS/MS spectra of both native 12C- and uniformly 13C (U-13C)-labeled analytical standards of 10 fungal substances in pure solvent and spiked into fungal culture filtrate of Fusarium graminearum respectively. Furthermore, the developed approach is exemplified with nine unknown biochemical compounds contained in F. graminearum samples derived from an untargeted metabolomics experiment. The mass difference between the corresponding fragment ions present in the MS/MS spectra of the native and U-13C-labeled compound enabled the assignment of the number of carbon atoms to each fragment signal and allowed the generation of meaningful putative molecular formulas for each fragment ion, which in turn also helped determine the elemental composition of the precursor ion. Compared to laborious manual analysis of the MS/MS spectra, the presented algorithm marks an important step toward efficient fragment signal elucidation and structure annotation of metabolites in future untargeted metabolomics studies. Moreover, as demonstrated for a fungal culture sample, FragExtract also assists the characterization of unknown metabolites, which are not contained in databases, and thus exhibits a significant contribution to untargeted metabolomics research.
The combination of electrospray ionization (ESI)–liquid chromatography (LC)–high-resolution mass spectrometry (HRMS) offers the potential to measure hundreds to thousands of metabolites contained in complex biological samples in a single analytical run.1 Although LC-HRMS(/MS) also enables the generation of structure-related information for the measured substances, the unambiguous elucidation of the elemental composition and detailed chemical structure of unknown metabolites remains one of the most challenging tasks in untargeted metabolomics studies.
In LC-HRMS-based techniques, compound annotation usually starts with the prediction of molecular formulas and matching accurately measured masses against comprehensive databases such as AntiBase2 in the case of microbial metabolites, chemical substance databases such as ChEBI3 or PubChem,4 or metabolite pathway databases such as KEGG5 or MetaCyc.6 Unfortunately, as a result of the limited resolving power and mass accuracy of MS instruments, the knowledge of a metabolite’s accurate mass is generally not sufficient to determine its elemental composition unambiguously.7,8 To reduce the number of potential molecular formulas and to eventually determine a metabolite’s molecular formula, chemical logics in combination with heuristic rules may be used9 as well as the annotation of heteroatoms (e.g., S, Fe, Cl) from isotopic fine structures.10−12
Usually many structural isomers may correspond to a single molecular formula, which further complicates the detailed elucidation of chemical structures. Consequently, definitive metabolite identification by LC-HRMS can only be achieved by comparing two or more orthogonal properties such as retention time and accurate mass with those obtained for an authentic standard under identical measurement conditions.13 Because in many cases, however, authentic compounds are not available, different strategies for the interpretation and annotation of product ion spectra have been suggested. The most common approach, if no authentic standard compound is available, is to try to putatively identify a compound by comparing the measured MS/MS fragments against spectra of tandem MS databases that are publicly available. For this purpose MassBank14 (http://www.massbank.jp), METLIN15 (http://metlin.scripps.edu/), or NIST MS/MS16 database can be used, for example. This approach can further benefit from prior data processing steps for deconvolution of recorded product ion spectra, as has been described recently.17 A major limitation of compound annotation by MS/MS spectrum matching consists in the size and content of the respective databases, because many compound (classes) may not be contained, and often only sparse information on the biological context of the metabolites is available.13
Alternatively, different computationally assisted techniques have been described to annotate metabolites of interest. Several software tools have been developed which try to generate in silico fragment spectra on the basis of different rule sets18 or combinatorial approaches19 and compare the predicted fragments to the measured product ion spectra. In addition, very recently, fragmentation-tree-based approaches have been described.20,21 These methods construct hierarchical mass spectral trees, in which measured fragments or their molecular formulas become traceable to the precursor mass or its elemental composition.22 These approaches can further be used to automatically detect similarities between the generated fragmentation trees.23
Complementary to these “classical” approaches, stable isotope-assisted techniques, with 13C, 34S, or 15N being the most frequently used, are becoming increasingly popular, because they offer powerful tools to conquer major challenges of untargeted metabolomics studies.7,11 The general concepts and applications of stable istotope labeling (SIL)-assisted approaches for improved global feature detection, tracer metabolization, more accurate comparative quantitation, and metabolite annotation24−27 have been summarized in several recent review articles.7,28,30,39 Typically, isotope-enriched metabolites or globally labeled biological samples are mixed with their native analogues prior to LC-HRMS measurements and the resulting labeling-specific mass spectral patterns are systematically used for data analysis. Although sophisticated algorithms and software tools have already been developed to automatically recognize labeling-specific isotopic patterns in GC-MS31 and LC-HRMS full scan data,28,29,32−34 to the best of the authors’ knowledge, no processing tool for the automated annotation of labeling-specific LC-HRMS/MS spectra has been published until today.
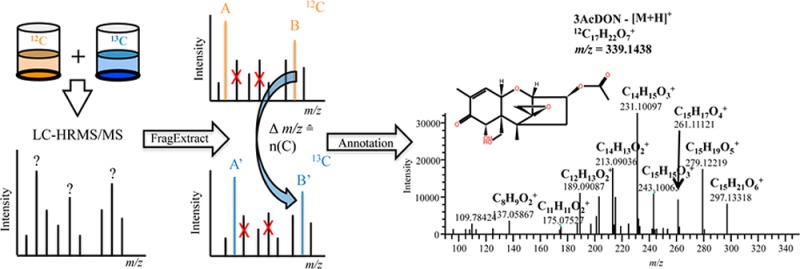
Here, we present FragExtract, a novel algorithm that resulted in a software tool, which allows the efficient filtering and unbiased assignment of MS/MS fragment signals including the correct number of carbon atoms derived from SIL-assisted metabolomics experiments. The presented approach ultimately performs a spectral clean up by extracting relevant MS/MS fragments based on pairs of corresponding native (i.e., 12C) and labeled (i.e., 13C) ions. Moreover, the precursor ions are inspected for the presence of heteroatoms, and both fragment ions and precursor ions are evaluated automatically regarding the consistency of their elemental composition. Thus, the number of possible molecular formulas can be reduced significantly, which in turn assists characterization of both known and unknown metabolites, discovered in untargeted metabolomics studies.
Experimental Section
Chemicals and Reagents
Methanol (MeOH, LiChrosolv, LC gradient grade) was purchased from Merck (Darmstadt, Germany); acetonitrile (ACN, HiPerSolv Chromanorm, HPLC gradient grade) was purchased from VWR (Vienna, Austria); formic acid (FA, MS grade) was obtained from Sigma-Aldrich (Vienna, Austria). Water was purified successively by reverse osmosis and an ELGA Purelab Ultra-AN-MK2 system (Veolia Water, Vienna, Austria).
Analytical standards of both natural isotope composition and uniformly 13C (U-13C)-labeled analogues, of 3-acetyldeoxynivalenol (3AcDON), diacetoxyscirpenol (DIAS), fumonisin B1, B2, and B3 (FB1, FB2, FB3), griseofulvin (GRIS), HT-2 toxin (HT-2) and T-2 toxin (T-2), sterigmatocystin (STER), and zearalenone (ZEN) were obtained from Biopure Referenzsubstanzen GmbH (Tulln, Austria). All of these standard compounds were dissolved in ACN except for the fumonisins, which were dissolved in ACN/water = 1:1 (v/v).
Preparation of Multianalyte Standard Solutions
Analytical standard solutions were mixed to obtain two multianalyte stock solutions. Those contained identical concentrations of nonlabeled and the corresponding U-13C-labeled analytes. The multianalyte standard stock solutions were diluted with water to achieve a solvent composition of ACN/water = 1:1 (v/v). The first multianalyte standard solution was composed of 3AcDON, DIAS, FB3, HT-2, T-2, ZEN (native and U-13C-labeled substances at a concentration of 1.1 mg/L each). The second multianalyte solution consisted of 0.9 mg/L of both FB1 and FB2, 1.4 mg/L of GRIS, and 1.7 mg/L of STER.
Cultivation of Fusarium graminearum Samples
Culture filtrates of F. graminearum PH-1 were prepared in Fusarium Minimal Medium (FMM) as described earlier29 using either nonlabeled or U-13C glucose as sole carbon source. F. graminearum was grown in a UNIFILTER 24-well 10 mL filtration microplate equipped with a Whatman GF/C filter (VWR, Vienna, Austria). In each well, a 1 mL aliquot of either nonlabeled or U-13C-labeled glucose was inoculated with 2000 spores. After 7 days, the 24-well microtiter plate was centrifuged for 10 min at 2000 rpm to separate the supernatant from the mycelium. Immediately after centrifugation, acetonitrile was added to quench the culture filtrates, resulting in a final relative acetonitrile concentration of 30% (v/v).
Preparation of F. graminearum Samples Spiked with Multianalyte Standard Stock Solutions
For spiking experiments, only nonlabeled supernatants were employed. Two multianalyte standard stock solutions were prepared each of which contained both native and U-13C-labeled analogues at the same concentration level. The first stock solution contained 3AcDON, FB3, DIAS, HT-2, T-2 and ZEN standard each at a concentration of 2.2 mg/L. The second stock solution comprised 3.1 mg/L of GRIS, 3.6 mg/L of STER, 2.1 mg/L of FB1, and 2 mg/L of FB2 standards. Varying amounts of the stock solutions were evaporated to dryness at room temperature under a gentle stream of nitrogen. Dried analytes were redissolved in a mixture of fungal culture filtrate and ACN (2 + 1, v/v) to yield 1:1, 1:3, 1:5, 1:20, 1:100, and 1:300 dilutions. The 1:300 dilution level was further diluted with culture filtrate and ACN (2 + 1, v/v) 1:5 and 1:10. This dilution series led to analytical standard concentrations of 0.7 μg/L up to 1.8 mg/L.
Preparation of a Mixture of Nonlabeled and U-13C-Labeled F. graminearum Samples
The developed FragExtract algorithm was applied to LC-HRMS/MS data derived from an untargeted metabolomics experiment. For this purpose, quenched aliquots of nonlabeled and U-13C-labeled supernatants were mixed together, resulting in a final ratio of 1:1 (v/v), and measured by LC-HRMS, as described in the following.
LC-MS and LC-MS/MS Analysis
All types of samples (standards and F. graminearum samples) were analyzed as described earlier.29 In brief, a UHPLC system (Accela, Thermo Fisher Scientific, San Jose, CA, U.S.A.) equipped with a reversed-phase XBridge C18 analytical column, 150 × 2.1 mm i.d., 3.5 μm particle size (Waters, Vienna, Austria) was employed at a flow rate of 250 μL/min. Eluent A was water, eluent B was MeOH, both containing 0.1% formic acid (FA). The initial mobile phase composition (90% A) was held constant for 2 min, followed by a linear gradient to 100% B in 30 min. This final condition was held for 5 min, followed by 8 min column re-equilibration at 90% A.
The specified UHPLC system was coupled to an LTQ Orbitrap XL (Thermo Fisher Scientific) equipped with an electrospray ionization (ESI) interface, which was operated in positive ionization at 4 KV electrospray voltage and a capillary temperature of 300 °C. All other source parameters were automatically tuned for a maximum MS signal intensity of reserpine (Sigma-Aldrich (Vienna, Austria)) solution (10 mg/L). For selection of metabolic features, nonlabeled and U-13C-labeled mixture of F. graminearum samples were measured in MS full scan mode (resolving power setting of 60 000 fwhm at m/z 400, scan range of m/z 100–1000, profile mode).
LC-MS/MS measurements were carried out for the following sample types: pure multianalyte standard solutions, spiked F. graminearum samples, and mixtures of nonlabeled and U-13C-labeled F. graminearum cultures (preselected features). Each of the tested samples was analyzed with an LC-HRMS/MS method employing three successive scan events: First, a survey full scan (resolving power setting of 30 000 fwhm at m/z 400, scan range of m/z 100–1000, profile mode) was followed by two successive product ion MS/MS measurements of nonlabeled and U-13C-labeled precursor ions, respectively. Centroid product ion spectra were recorded in collision-induced dissociation (CID) mode with a resolving power setting of 7500 fwhm at m/z 400 and a varying m/z range adapted to the analyte mass (higher m/z: ca. m/z of precursor ion; lower m/z: ca. 1/3 of precursor m/z). The isolation width for the precursor ion was set to 2 (target m/z ± 1). For eight standard compounds, the protonated molecule was chosen for fragmentation. For DIAS, HT-2, and T-2 toxin, the sodium adducts were used as precursor ions. In the case of nine selected feature pairs from mixtures of nonlabeled and U-13C-labeled F. graminearum samples, six protonated ions, two sodium adducts, and one unknown ion species were chosen as precursor ions for fragmentation (see Table S-3). The normalized collision energies (CE in %) were optimized by flow injection analysis with single standards in pure solvents. Ten microliters per minute of the respective standard solution 1–10 mg/L in ACN/water = 1:1 (v/v)) was infused via syringe pump into the mobile phase, which had a flow rate of 240 μL/min. The mobile phase composition was adjusted to the composition at chromatographic elution from the HPLC column of the respective compound. This optimization resulted in CE settings of 24% for 3AcDON, 37% for DIAS, 25% for FB1, 23% for FB2, 23% for FB3, 30% for GRIS, 29% for HT-2, 40% for STER, 32% for T-2, and 34% for ZEN. For the FT-Orbitrap, the automatic gain control was set to a target value of 5 × 105 and a maximum injection time of 500 ms was chosen for both full scan and tandem MS measurements. Data were generated using Xcalibur 2.1.0 (Thermo Fisher Scientific).
Automated Data Processing by FragExtract
The presented FragExtract algorithm uses three successively recorded MS(/MS) spectra within the LC-HRMS run (1) full-scan spectrum, (2) product ion MS/MS spectrum of the monoisotopic 12C, and (3) MS/MS spectrum of the U-13C precursor masses. Employing this information, the algorithm is capable of unambiguously annotating fragment signals of the respective precursor ions without the need of spectral comparisons to tandem mass spectra libraries or the need of in silico fragmentations of substances under investigation.
The algorithm uses a brute force approach for the MS/MS fragment annotation and the calculation of its respective number of carbon atoms. It was developed in Python (version 2.7) using the Qt 4-SDK for the graphical user interface and is available for the operating systems Windows and Mac OSX. The program is capable of processing LC-HRMS(/MS) data in the common data formats mzML35 and mzXML, which were suggested by the Metabolomics Standards Initiative (MSI). The program comprises a set of processing steps, which will be described in the following.
MS/MS Spectrum Selection
On the basis of predefined MS/MS precursor masses of both the native 12C and U-13C-labeled target substance and their approximate retention time extracted by MetExtract, the program searches the full scan data to find the most intense signals of the native target precursor ion within a certain user-defined retention time window. The two successive product ion spectra of the native and U-13C-labeled substance immediately after the peak maximum of the predefined precursor in the full scan exceeding a user-defined mininimum intensity threshold are selected for further processing.
Calculation of Carbon Atoms in Precursor Mass
The maximum number of carbon atoms (x(C)) for any MS/MS fragment is calculated by dividing the difference of the measured m/z values of the native and the corresponding U-13C-labeled precursor ions by the exact mass difference of 12C and 13C (e.g., 1.00335, Δ m/z (12C, 13C) for singly charged ions).
Fragment Signal Annotation and Calculation of Carbon Atoms Per Fragment Ion
In order to gain sufficiently high fragment ion intensities, limited selectivity of precursor selection has to be addressed (e.g., isolation width = 2 or 3 u). Therefore, depending on the isolation width setting, the first and/or second isotopolog of the target compound may also be isolated in the mass analyzer and subsequently fragmented, occurring as an isotopolog signal in the product ion spectrum. These putative isotopolog signals should not be included in further calculation steps and need to be removed from the product ion spectra. For this purpose, m/z values larger than the target mass of the respective precursor are not considered further. Then, m/z values of all fragments in each 12C and corresponding 13C MS/MS spectrum are sorted in descending order. If the mass increment between two adjacent MS/MS signals corresponds to the mass difference between 12C and 13C (i.e., 1.00335 u), the less intense signal will be marked as a putative isotopolog (F + 1). With a mass error tolerance of ±5 ppm relative to the mass of the fragment ion under investigation, F + 1 isotopologs can clearly be differentiated from adjacent fragments differing in a single hydrogen atom up to a fragment mass of 900 u.
For every remaining fragment ion observed in the MS/MS spectrum of the 12C precursor, the masses of possible corresponding U-13C fragments are calculated using the formula below, where n(C) denotes the potential number of carbon atoms for the fragment signal and 12C m/zmeas denotes the measured mass of the fragment ion in the 12C MS/MS spectrum.
The measured LC-MS/MS spectra of the U-13C substance are inspected for the presence of these corresponding 13C m/zcalc fragment masses within a user-defined mass window. For the used LTQ Orbitrap XL instrument, this mass window was set to ±10 ppm. The range-scaled relative intensities (set to a range between 1 and 100) in both spectra are calculated separately and compared for each putative fragment ion pair. Because the measured relative abundance of fragment ions in product ion spectra is largely independent of the absolute precursor abundance, the range-scaling of intensities will yield similar values for correctly matched fragment pairs. By finding the 13C fragment ion that exhibits a mass of 13C m/zcalc ± 10 ppm and a comparable relative intensity to the 12C m/zmeas fragment ion, the number of carbon atoms for the particular fragment ion is calculated using the equation above. Fragment ions without corresponding 13C m/z are not considered in further processing steps.
Molecular Formula Calculation of Fragment Ions and Precursors
First, a mass (m) for the noncharged fragment ions (m/z) is calculated by considering the mass of an electron. For each m, putative sum formulas are generated, and those with an incorrect number of carbon atoms (n(SumFormula) ≠ n(C)) are discarded. The user can define elements that shall be included in the calculation of the elemental composition for each selected fragment signal. On the basis of the publication of Kind and Fiehn9 rule no. 1 (restriction for element numbers), no. 5 (heteroatom ratio check), and no. 6 (element probability check) are also included in the presented workflow.
For chlorine and sulfur, the algorithm automatically searches for the naturally occurring isotopic signals (e.g., 37Cl, 34S) in the MS full scan spectrum of the native and the U-13C-labeled precursor to verify the presence of those elements before inclusion for the molecular formula assignment. If either Cl of S is part of a metabolite’s elemental composition, isotopologs containing 37Cl or 34S will appear at m/z values higher than the principal ion of the 13C isotopic cluster and can thus be recognized easily regardless of the resolving power of the instrument.
Furthermore, to check the elemental consistency and reduce the number of possible molecular formulas for the precursor and the fragment masses, an approach, thereafter named elemental composition filter, based on the fragment consistency rule and the combinatorial consistency rules stipulated by Rojas-Cherto et.al.22 are applied. To this end, CID product ion spectra are inspected to test if the fragment ion under investigation with its annotated elemental composition together with the mass of the neutral loss and its annotated molecular formula can be traced to the precursor ion and its formula. In a first step, putative elements and atom counts of each molecular formula of the fragment ions are compared to the putative molecular formulas of its precursor ion (postulated as described above) to find the elemental composition of the precursor for which most of the fragment formulas can be annotated. Once the best-fitting candidate formula of the precursor is found, a second iteration is started, in which the elements and element numbers of all molecular fragment formulas together with the elements and element numbers of their respective neutral loss formulas have to be traceable to the formulas of the precursor from the first iteration.
Application of the Algorithm to Multianalyte Standard Solutions Spiked into Fusarium Culture Samples
Raw proprietary LC-MS/MS data files were converted to mzML data format using msconvert of ProteoWizard.36 The user-defined positive list included the 12C and 13C precursor masses of all 10 tested fungal metabolites. The minimum base peak intensity of the MS/MS spectrum of the native compound had to exceed 100 counts. The inspected retention time window was adjusted on the basis of chromatographic separation used in the LC-MS/MS measurements from 3 to 30 min. To detect corresponding 12C/13C fragment ion pairs, a mass deviation of ±10 ppm to account for the interspectrum tolerance was allowed (Figures S-1–S-3). For the intensity ratio of the 12C fragment ion to the corresponding 13C fragment ion a maximum error of 30% was allowed, and only fragment ions with a relative intensity ≥2% were considered. For molecular formula annotation of the precursor and the MS/MS fragments C, H, N, O, Cl, S, and P were initially allowed. The maximum atom count of those seven elements was derived from Kind and Fiehn9 (m/z < 500 Da: max C, 39; max H, 72; max N, 20; max O, 20; max P, 9; max S, 10; m/z < 1000 Da: max C, 78; max H, 126; max N, 20; max O, 27; max P, 9; max S, 14). If either Cl or S was detected, the tolerated atom count for the precursor mass was set to at least one and the maximum to either 10 or 14, as described above. Based on the mass accuracy achieved for the standard compounds in MS full scan mode, a mass deviation of ±3 ppm was tolerated for the evaluation of molecular formulas of the precursor ion. Furthermore, Na was included for molecular formula calculation of HT-2, T-2, and DIAS, because Na adducts were used as precursors for MS/MS measurements of these three metabolites.
Application of the Algorithm to Selected Unknown Metabolites of a Fusarium Culture Sample
To demonstrate the suitability of the presented approach for untargeted metabolomics experiments, samples of F. graminearum grown on either 12C or U-13C enriched glucose were mixed and subsequently measured with an LTQ Orbitrap XL in full scan mode. The acquired data was processed with the MetExtract software according to the workflow for SIL-assisted untargeted metabolomics experiments recently presented by Bueschl et.al.29 with the aim to extract 12C/U-13C feature pairs. Both the native and the U-13C precursor ions had to exhibit a minimum abundance of 105 counts in at least three recorded scans for being selected for successive LC-HRMS/MS measurements and evaluation by FragExtract. Subsequently, the ion species (i.e., type of adduct) of such extracted metabolic feature pairs was manually annotated if possible. From this list of nine metabolic features, six [M + H]+, two [M + Na]+, and one unknown ion species (Table S-3) were selected for MS/MS measurements at three different collision energies (25%, 35%, and 45%). For annotation of the molecular formula of precursor ions, the maximum tolerated mass deviation was set to ±3 ppm. In addition, after manual evaluation of the MS full scan data, Fe was also included for molecular formula calculations.
Results and Discussion
Overview of Analytical Standards of Fungal Metabolites Used in This Study and Results Overview
We used a total of 10 analytical standards of fungal metabolites (see Table 1) to develop an algorithm for the automated evaluation of product ion MS/MS spectra from LC-HRMS data of mixtures of native and uniformly 13C (U-13C)-labeled substances. The algorithm is capable of unambiguously annotating fragment signals of the respective precursor ions without the need of spectral comparisons to tandem mass spectra libraries or the need of in silico fragmentations of substances under investigation.
Table 1. Overview of Analytical Standards and Summary of Results for Fungal Metabolites Used in This Studya.
| rank of
correct molecular formula |
||||||
|---|---|---|---|---|---|---|
| name | molecular formula of Mb | ion species | m/zmeasc | no. detected/ no. annotated | Id | IIe |
| 3AcDON | C17H22O7 | [M + H]+ | 339.1436 | 37/18 | 1 | 4 |
| DIAS | C19H26O7 | [M + Na]+ | 389.1571 | 28/2 | 1 | 1 |
| HT-2 | C22H32O8 | [M + Na]+ | 447.1989 | 32/15 | 1 | 1 |
| T-2 | C24H34O9 | [M + Na]+ | 489.2093 | 72/47 | 2 | 5 |
| ZEN | C18H22O5 | [M + H]+ | 319.1538 | 89/17 | 1 | 5 |
| FB3 | C34H59NO14 | [M + H]+ | 706.4014 | 77/27 | 7 | 152 |
| GRIS | C17H17O6Cl | [M + H]+ | 353.0782 | 57/13 | 1 | 19 |
| STER | C18H12O6 | [M + H]+ | 325.0703 | 33/12 | 1 | 11 |
| FB1 | C34H59NO15 | [M + H]+ | 722.3960 | 61/15 | 4 | 74 |
| FB2 | C34H59NO14 | [M + H]+ | 706.4015 | 61/15 | 10 | 180 |
The number of initially measured fragment signals (“detected”) in the 12C derived LC-MS/MS spectrum vs the number of annotated fragments automatically found by FragExtract. The rank indicates the correct elemental formula for the precursor ions (sorted by mass deviation in ppm) calculated with Xcalibur Software (version 2.1.0.1139), which allows a max of 400 possible molecular formulas. Seven allowed elements: C, H, N, O, Cl, S, and P (for HT-2, T-2, and DIAS: additionally one Na). For standard concentration please refer to Experimental section.
M = intact neutral molecule of fungal metabolite.
m/zmeas = measured m/z value.
With the restriction of carbon atom count.
Without the restriction of carbon atom count.
The reliability of the fragment ion extraction was tested with multianalyte standards spiked into F. graminearum culture filtrate, and the algorithm was applied to nine unknown features derived from a F. graminearum culture filtrate sample of an untargeted metabolomics experiment.
The presented approach aims at the automated evaluation of high-resolution tandem mass spectra, is based on the use of highly U-13C-enriched labeled compounds or labeled biological samples, and relies on the successive LC-MS/MS recordings of 12C and U-13C-labeled substances. As native and U-13C-labeled compounds show the same fragmentation behavior in tandem MS, the resulting fragmentation pattern in the product ion spectra ultimately looks the same, only shifted toward higher masses of the U-13C-labeled compound, as for example shown for 3AcDON in Figure 1b.
Figure 1.
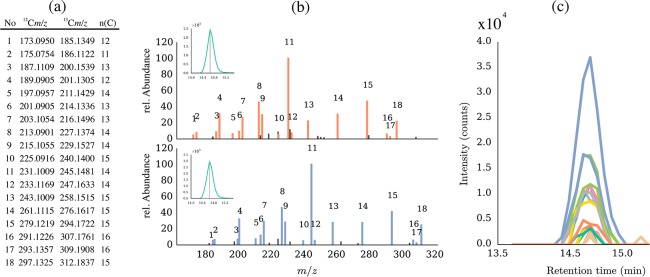
FragExtract results for LC-MS/MS spectra of native and U-13C 3AcDON standard. The numbers in the table (a) correspond to the annotated fragment signals highlighted in orange and blue in the original product ion spectra of the native and the U-13C-labeled precursor masses (m/z 339.1438 and m/z 356.2009) (b). For verification of chromatographic peak shape and coelution of the fragment ions, the extracted ion chromatograms of fragments that were annotated by FragExtract are plotted by the software (c).
As a consequence of the highly similar fragmentation patterns, evaluation of the m/z value difference between the corresponding native and U-13C-derived mass signals directly yields the number of carbon atoms present in a selected fragment ion.
When the derived carbon atom count per fragment ion was considered together with the methods described for molecular formula calculation and the accurate mass of the respective ions, FragExtract unambiguously annotated the correct elemental composition for all standard compounds in both pure solvent and the spiked fungal culture samples. For the true unknowns evaluated in the biological samples, which were analyzed with the same settings as the standard solutions and F. graminearum samples, FragExtract’s algorithm led to a maximum of two possible molecular formulas for precursor and fragment ions. Readers who are interested in the algorithm and use of FragExtract, which had been developed and is presented in this study, are asked to contact the corresponding author.
Exemplification of FragExtract-Derived Results with 3AcDON
A typical results output generated by automated MS/MS spectrum annotation is exemplified with 3AcDON (Figure 1). When using FragExtract, the user can decide which elements/elemental compositions to allow or to exclude for generation of putative fragment formulas. Moreover, because only those product ion signals are kept in the annotated LC-MS/MS spectrum, which exhibit the required 12C and corresponding U-13C pattern, the algorithm helps to efficiently filter noise and background signals and to extract meaningful fragment ions from the inspected LC-MS/MS spectra. For 3AcDON, the developed algorithm was able to annotate 18 fragment ion pairs, all of which were uniquely assigned and also manually verified (Table S-1 and Figure S-4). In the case of 3AcDON, restricting the number of possible carbon atoms to that derived by the algorithm and the additional application of the elemental composition filter, led to unambiguously assigned molecular formulas, which corresponded to the manually assigned molecular formulas (Table S-1). However, even if the number of carbon atoms is known, the higher the mass of an ion, the higher the probability for obtaining ambiguous elemental compositions. By the automated checking of the isotopic pattern of the full scan MS spectrum of the U-13C precursor for the presence of heteroatoms (see Experimental section), Cl and S could be excluded by the algorithm leaving only one possible molecular formula for each fragment ion and the precursor mass.
A possibility regarding further manual results refinement is to take the isotopic fine structure of nitrogen- and oxygen-containing precursor ions in the high-resolution full scan data into account. Especially for low molecular weight compounds in combination with FTMS instruments enabling a resolving power ≥100.000, this can help to determine the correct elemental composition, as suggested, for example, by Kaufmann37 or Pluskal et al.38 Moreover, the application of the elemental composition filter helped to determine the formula of the neutral intact ion, and furthermore, characteristic mass increments were highlighted by the algorithm, which can help in elucidation of a compounds structure. Thus, additional manual inspection of the automatically generated results can be used to further confirm the correctness of the obtained fragment formulas.
Results after Application of FragExtract to Fungal Metabolite Standards Spiked into Fusarium Culture Samples
In view of future applications of the algorithm to unknown substances that can be detected in untargeted metabolomics experiments, we evaluated the performance of the algorithm under more realistic conditions. At the example of spiked culture filtrates of F. graminearum, we evaluated whether signals from the matrix (culture medium) or other signals of nonbiological relevance would disturb the software algorithm. Therefore, analytical standard compounds were spiked into culture filtrates of F. graminearum in decreasing concentrations, as low abundant precursor ions are a particular issue in the structure elucidation process of metabolites in any biological study. The F. graminearum cultures were grown with a native carbon source and the filtrates contained none of the compounds that were spiked for verification purposes; the only exception was 3AcDON, which in the full scan mode exhibited a maximum signal height of 1 × 103 counts for the nonspiked culture filtrates, which is too low for generating MS/MS spectra.
Figure 2 shows LC-HRMS/MS spectra of native 3AcDON standard in spiked F. graminearum culture filtrates at the highest concentration tested (1 mg/L, Figure 2a) and the lowest concentration for which at least one fragment signal could still be annotated (0.1 mg/L, Figure 2b). In Figure 2c, the extracted ion chromatogram (EIC) of the precursor ion 3AcDON and EICs of selected MS/MS signals are presented. For the LC-MS/MS spectra at 1 mg/L, we found similar results compared to pure standards (i.e., no unspecific MS/MS signals fit the predefined criteria). At a concentration of 0.1 mg/L, only one fragment signal (m/z 231.0996) was automatically annotated, which in contrast to the other fragments found at this concentration showed a similar chromatographic peak shape and retention time compared to the full scan EIC of the precursor ion 3AcDON (m/z 339.1438). The selected other fragments however could be classified as background signals or pseudo ions (m/z 224.848 and 109.767) on the basis of their chromatographic behavior or represented “spike” signals (m/z 320.055 and 271.779), observed only once in a single MS/MS spectrum. Lowering the compound’s concentrations obviously leads to lower precursor intensities and hence, less fragment signals, which can be automatically recognized by the software (Table S-2). It was shown for all 10 analytical standards spiked to the culture filtrate that even at the lowest tested concentration levels neither the presence of matrix compounds nor pseudo ions altered the algorithm’s ability to filter unspecific MS/MS signals (Figures S-5 to S-56).
Figure 2.
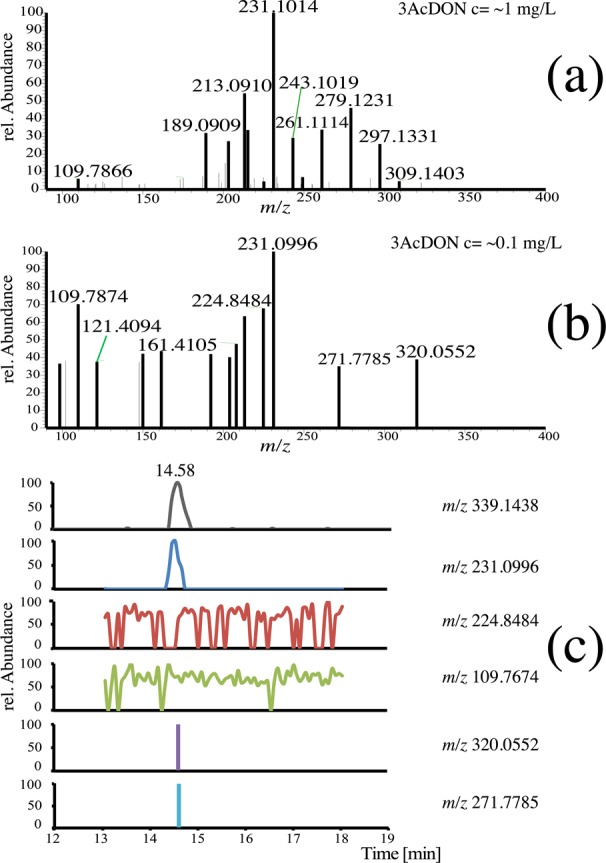
MS/MS spectra of 3AcDON standard spiked into culture filtrates. The upper spectrum shows the fragment signals of the native 3AcDON (m/z 339.1438) at the initial concentration of 1 mg/L (a) and at approximately 0.1 mg/L (b). Combined view of the EIC [M + H]+ of 3AcDON in the full scan mode and EICs of selected MS/MS signals (0.1 mg/L) (c).
Application of the Algorithm to Selected Unknown Metabolites of a F. graminearum Culture Filtrate Sample
The established automated algorithm was applied to a culture filtrate sample of a F. graminearum strain that was grown on liquid minimal medium containing either native or U-13C-labeled glucose. Nine automatically detected feature pairs (Table S-3) with a minimum abundance of 105 counts were selected for subsequent MS/MS experiments. Detailed results on the biological relevance will be published elsewhere.
For eight of the nine tested metabolites, each of the 12C fragments were unequivocally assigned to a single corresponding 13C fragment. A summary of the results for all unknown metabolites together with the annotated molecular formula for the precursor mass can be found in Table S-3. Moreover, all LC-HRMS/MS spectra and FragExtract derived fragment ions are listed (Figure S-57 to S-65) in the Supporting Information. For one metabolite (m/z 761.3612), a total of eight multiple assignments at all three different collision energies was annotated (e.g., one 13C signal could be assigned to two different 12C signals), which translates to a multiple assignment rate of approximately 7% (for 115 signals annotated in total for one metabolite at three different collision energies). All of those fragment ions exhibited a relative intensity of below 5% compared to the most intense MS/MS signal. Therefore, the user can set a relative intensity threshold for extraction and annotation of fragment ions. For four of the unknown metabolites, unambiguous elemental compositions were annotated (m/z 647.3724 at 23.9 and 26.74 min, m/z 651.5653 and m/z 787.5031). Interestingly, for most of the tested precursor ions, annotated molecular formulas indicated that they probably contained phosphorus. However, on the basis of accurate m/z, number of carbon atoms derived by the algorithm, prior exclusion of Cl and S and number of phosphorus atoms per formula none of the metabolites could be annotated when matched against Antibase. Nevertheless, for all analyzed unknown metabolites, FragExtract performed a spectral cleanup and restricted the number of possible molecular formulas to one or two possibilities.
Application of the developed algorithm resulted in the annotation of two possible elemental compositions for the precursor ion at m/z 571.0856 (C30H19O12, C30H24O5NP3) and each of the annotated fragment ions. This metabolite could be identified as aurofusarin (C30H18O12, monoisotopic mass: 570.0798 Da), as follows. The fragment ions annotated by FragExtract were compared to the product ion spectrum of the authentic standard of aurofusarin, which was measured at the same collision energy and under the same experimental conditions as the biological sample. The retention time of the putatively annotated aurofusarin and the standard matched and the dotproduct between the product ion spectra extract by FragExtract and the authentic standards was 0.9977. Many of the neutral losses and the respective fragments were typical for certain structural units, which additionally helped in the identification process of aurofusarin (e.g., Δ CH3 for methyl, Δ CH2O for methoxy, Δ CH3CO for methyl, and CO in the ring structure).
When more than one elemental formula is automatically annotated by FragExtract, the decision which molecular formula is more likely needs to be made case by case by manual inspection. With respect to all further metabolites, evaluation with FragExtract provides a good basis for detailed metabolite characterization in future studies.
Conclusions
Structure elucidation of unknown compounds still is a major bottleneck in untargeted metabolomics approaches. Our results illustrate that stable isotope labeling with 13C shows high potential for molecular formula determination of both intact molecules as well as fragment ions as the number of carbon atoms can be derived from SIL-derived LC-HRMS and LC-HRMS/MS data. The established FragExtract algorithm is capable of efficiently filtering meaningful fragment signals from MS/MS spectra of native and 13C-labeled compounds even in the presence of highly complex biological matrices. We have demonstrated that stable isotope labeling in combination with the presented algorithm for automated data analysis can be effectively used to assist in the automated characterization and elucidation of both certain structural units, as shown for aurofusarin and unknown compounds found in untargeted metabolomics experiments. The application of this novel software tool significantly reduces data processing time and also allows the automated annotation of tandem mass spectra. Moreover, the “cleaning” of MS/MS spectra from nonspecific signals derived from background or electronic noise is of particular interest for data storage in MS/MS spectral databases, especially with regard to their use as references for MS/MS spectrum similarity match and for the elucidation of unknown compounds that occur in untargeted metabolomics experiments. In addition, the software is suitable to process even MS3 or higher-order fragmentation spectra. We expect that the presented automated approach is of great interest for any researcher performing SIL-assisted metabolomics.
Acknowledgments
The Federal Country Lower Austria and the European Regional Development Fund (ERDF) of the European Union is acknowledged for financial support (grant no. GZ WST3-T-95/001-2006), which also enabled the Ph.D. studies of S.M.L. (analytical chemistry). The authors thank the Austrian Science Fund (project SFB Fusarium 3706-B11) for financial support, which enabled the Ph.D. studies of N.K.N.N. (bioinformatics).
Supporting Information Available
Product ion spectra of 3AcDON, all results of the spiked fungal culture samples, list of precursors of the unknown metabolites, and all according results generated by the application of FragExtract. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Patti G. J.; Yanes O.; Siuzdak G. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laatsch H. In AntiBase 2007: The Natural Product Identifier; Wiley-VCH GmbH: Weinheim, Germany, 2007. [Google Scholar]
- Degtyarenko K.; de Matos P.; Ennis M.; Hastings J.; Zbinden M.; McNaught A.; Alcantara R.; Darsow M.; Guedj M.; Ashburner M. Nucleic Acids Res. 2008, 36, D344–D350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PubChem. http://pubchem.ncbi.nlm.nih.gov/.
- KEGG Compound. http://www.genome.jp/kegg/compound/.
- Caspi R.; Altman T.; Billington R.; Dreher K.; Foerster H.; Fulcher C. A.; Holland T. A.; Keseler I. M.; Kothari A.; Kubo A.; Krummenacker M.; Latendresse M.; Mueller L. A.; Ong Q.; Paley S.; Subhraveti P.; Weaver D. S.; Weerasinghe D.; Zhang P.; Karp P. D. Nucleic Acids Res. 2014, 42, D459–D471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S.; Heinzle E. Wiley Interdiscip. Rev.: Syst. Biol. Med. 2012, 4, 261–272. [DOI] [PubMed] [Google Scholar]
- Kind T.; Fiehn O. BMC Bioinf. 2006, 7, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind T.; Fiehn O. BMC Bioinf. 2007, 8, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böcker S.; Letzel M. C.; Liptak Z.; Pervukhin A. Bioinformatics 2009, 25, 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakabayashi R.; Sawada Y.; Yamada Y.; Suzuki M.; Hirai M. Y.; Sakurai T.; Saito K. Anal. Chem. 2013, 85, 1310–1315. [DOI] [PubMed] [Google Scholar]
- Glaser K.; Kanawati B.; Kubo T.; Schmitt-Kopplin P.; Grill E. Plant J. 2013, 77, 31–45. [DOI] [PubMed] [Google Scholar]
- Dunn W.; Erban A.; Weber R. M.; Creek D.; Brown M.; Breitling R.; Hankemeier T.; Goodacre R.; Neumann S.; Kopka J.; Viant M. Metabolomics 2013, 9, 44–66. [Google Scholar]
- Horai H.; Arita M.; Kanaya S.; Nihei Y.; Ikeda T.; Suwa K.; Ojima Y.; Tanaka K.; Tanaka S.; Aoshima K.; Oda Y.; Kakazu Y.; Kusano M.; Tohge T.; Matsuda F.; Sawada Y.; Hirai M. Y.; Nakanishi H.; Ikeda K.; Akimoto N.; Maoka T.; Takahashi H.; Ara T.; Sakurai N.; Suzuki H.; Shibata D.; Neumann S.; Iida T.; Tanaka K.; Funatsu K.; Matsuura F.; Soga T.; Taguchi R.; Saito K.; Nishioka T. J. Mass Spectrom. 2010, 45, 703–714. [DOI] [PubMed] [Google Scholar]
- Smith C. A.; O’Maille G.; Want E. J.; Qin C.; Trauger S. A.; Brandon T. R.; Custodio D. E.; Abagyan R.; Siuzdak G. Ther. Drug Monit. 2005, 27, 747–751. [DOI] [PubMed] [Google Scholar]
- NIST MS/MS. http://www.nist.gov/mml/bmd/data/tandemmass-speclib.cfm.
- Nikolskiy I.; Mahieu N. G.; Chen Y. J.; Tautenhahn R.; Patti G. J. Anal. Chem. 2013, 85, 7713–7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistrik R.; HighChem, Ltd.: Bratislava, Slovakia. [Google Scholar]
- Wolf S.; Schmidt S.; Muller-Hannemann M.; Neumann S. BMC Bioinf. 2010, 11, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasche F.; Svatos A.; Maddula R. K.; Böttcher C.; Böcker S. Anal. Chem. 2010, 83, 1243–1251. [DOI] [PubMed] [Google Scholar]
- Peironcely J. E.; Rojas-Cherto M.; Tas A.; Vreeken R.; Reijmers T.; Coulier L.; Hankemeier T. Anal. Chem. 2013, 85, 3576–3583. [DOI] [PubMed] [Google Scholar]
- Rojas-Cherto M.; Kasper P. T.; Willighagen E. L.; Vreeken R. J.; Hankemeier T.; Reijmers T. H. Bioinformatics 2011, 27, 2376–2383. [DOI] [PubMed] [Google Scholar]
- Rojas-Cherto M.; Peironcely J. E.; Kasper P. T.; van der Hooft J. J. J.; de Vos R. C. H.; Vreeken R.; Hankemeier T.; Reijmers T. Anal. Chem. 2012, 84, 5524–5534. [DOI] [PubMed] [Google Scholar]
- Hegeman A. D.; Schulte C. F.; Cui Q.; Lewis I. A.; Huttlin E. L.; Eghbalnia H.; Harms A. C.; Ulrich E. L.; Markley J. L.; Sussman M. R. Anal. Chem. 2007, 79, 6912–6921. [DOI] [PubMed] [Google Scholar]
- Baran R.; Bowen B. P.; Bouskill N. J.; Brodie E. L.; Yannone S. M.; Northen T. R. Anal. Chem. 2010, 82, 9034–9042. [DOI] [PubMed] [Google Scholar]
- Giavalisco P.; Hummel J.; Lisec J.; Inostroza A. C.; Catchpole G.; Willmitzer L. Anal. Chem. 2008, 80, 9417–9425. [DOI] [PubMed] [Google Scholar]
- Cano P. M.; Jamin E. L.; Tadrist S.; Bourdaud’hui P.; Pean M.; Debrauwer L.; Oswald I. P.; Delaforge M.; Puel O. Anal. Chem. 2013, 85, 8412–8420. [DOI] [PubMed] [Google Scholar]
- Chokkathukalam A.; Kim D. H.; Barrett M. P.; Breitling R.; Creek D. J. Bioanalysis 2014, 6, 511–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueschl C.; Kluger B.; Lemmens M.; Adam G.; Wiesenberger G.; Maschietto V.; Marocco A.; Strauss J.; Bödi S.; Thallinger G.; Krska R.; Schuhmacher R. Metabolomics 2013, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller K.; Metallo C. M. Curr. Opin. Biotechnol. 2013, 24, 60–68. [DOI] [PubMed] [Google Scholar]
- Wegner A.; Weindl D.; Jager C.; Sapcariu S. C.; Dong X.; Stephanopoulos G.; Hiller K. Anal. Chem. 2014, 86, 2221–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueschl C.; Kluger B.; Berthiller F.; Lirk G.; Winkler S.; Krska R.; Schuhmacher R. Bioinformatics 2012, 28, 736–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creek D. J.; Chokkathukalam A.; Jankevics A.; Burgess K. E. V.; Breitling R.; Barrett M. P. Anal. Chem. 2012, 84, 8442–8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Chen Y. J.; Cho K.; Nikolskiy I.; Crawford P. A.; Patti G. J. Anal. Chem. 2014, 86, 1632–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bald T.; Barth J.; Niehues A.; Specht M.; Hippler M.; Fufezan C. Bioinformatics 2012, 28, 1052–1053. [DOI] [PubMed] [Google Scholar]
- Chambers M. C.; Maclean B.; Burke R.; Amodei D.; Ruderman D. L.; Neumann S.; Gatto L.; Fischer B.; Pratt B.; Egertson J.; Hoff K.; Kessner D.; Tasman N.; Shulman N.; Frewen B.; Baker T. A.; Brusniak M. Y.; Paulse C.; Creasy D.; Flashner L.; Kani K.; Moulding C.; Seymour S. L.; Nuwaysir L. M.; Lefebvre B.; Kuhlmann F.; Roark J.; Rainer P.; Detlev S.; Hemenway T.; Huhmer A.; Langridge J.; Connolly B.; Chadick T.; Holly K.; Eckels J.; Deutsch E. W.; Moritz R. L.; Katz J. E.; Agus D. B.; MacCoss M.; Tabb D. L.; Mallick P. Nat. Biotechnol. 2012, 30, 918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann A. Rapid Commun. Mass Spectrom. 2010, 24, 2035–2045. [DOI] [PubMed] [Google Scholar]
- Pluskal T.; Uehara T.; Yanagida M. Anal. Chem. 2012, 84, 4396–4403. [DOI] [PubMed] [Google Scholar]
- Bueschl C.; Krska R.; Kluger B.; Schuhmacher R. Anal. Bioanal. Chem. 2013, 405, 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.