

Abstract
Objective
New genetic investigation techniques, including next-generation sequencing, epigenetic profiling, cell lineage mapping, targeted genetic manipulation of specific neuronal cell types, stem cell reprogramming and optogenetic manipulations within epileptic networks are progressively unravelling the mysteries of epileptogenesis and ictogenesis. These techniques have opened new avenues to discover the molecular basis of epileptogenesis and to study the physiological impacts of mutations in epilepsy-associated genes on a multilayer level, from cells to circuits.
Methods
This manuscript reviews recently published applications of these new genetic technologies in the study of epilepsy, as well as work presented by the authors at the genetic session of the XII Workshop on the Neurobiology of Epilepsy in Quebec, Canada.
Results
Next-generation sequencing is providing investigators with an unbiased means to assess the molecular causes of sporadic forms of epilepsy and have revealed the complexity and genetic heterogeneity of sporadic epilepsy disorders. To assess the functional impact of mutations in these newly identified genes on specific neuronal cell-types during brain development, new modeling strategies in animals, including conditional genetics in mice and in utero knockdown approaches, are enabling functional validation with exquisite cell-type and temporal specificity. In addition, optogenetics, using cell-type specific Cre recombinase driver lines, is enabling investigators to dissect networks involved in epilepsy. Genetically-encoded cell-type labeling is also providing new means to assess the role of the non-neuronal components of epileptic networks such as glial cells. Furthermore, beyond its role in revealing coding variants involved in epileptogenesis, next-generation sequencing can be used to assess the epigenetic modifications that lead to sustained network hyperexcitability in epilepsy, including methylation changes in gene promoters and non-coding RNAs involved in modifying gene expression following seizures. In addition, genetically-based bioluminescent reporters are providing new opportunities to assess neuronal activity and neurotransmitter levels both in vitro and in vivo in the context of epilepsy. Finally, genetically rederived neurons generated from patient iPS cells and genetically-modified zebrafish have become high-throughput means to investigate disease mechanisms and potential new therapies.
Significance
Genetics has considerably changed the field of epilepsy research and is paving the way for better diagnosis and therapies for patients with epilepsy.
Keywords: -‘omics’, cytoskeleton, calcium channels, systems biology, interneurons, EEG-monitoring
Recent advances in genetic investigation techniques have considerably enhanced our ability to analyse the molecular pathways involved in epileptogenesis, as well as to dissect the neuronal networks participating in seizure generation with great cellular and temporal precision. These techniques, including next generation sequencing, epigenetic profiling, cell lineage mapping, targeted genetic manipulation of specific neuronal cell types, stem cell reprogramming and optogenetic manipulations within epileptic networks are progressively unravelling the mysteries of epileptogenesis and ictogenesis. The power of these genetic techniques in epilepsy research will be illustrated with selected examples from recent publications and presentations at the WONEP 2013 meeting held in L’Esterel, Québec, Canada.
1. Next-generation sequencing in epilepsy: gene discovery
Next-generation sequencing has recently emerged as a powerful and unbiased method to investigate the genetic basis of rare and genetically heterogeneous sporadic epilepsies not amenable to traditional genetic investigation techniques. Traditional approaches, such as linkage analysis and targeted sequencing of genes within specific metabolic pathways, required large kindreds of affected individuals. These have been instrumental in identifying genes involved in certain forms of autosomal dominant forms of epilepsy, such as generalised epilepsy with febrile seizure plus (GEFS+: SCN1A, GABRD), juvenile myoclonic epilepsy (JME: GABRA1, CACNB4), benign familial neonatal seizures (BFNS: KCNQ2, KCNQ3), autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE: CHRNA4, CHRNB2, CHRNA2), and rare recessive forms of epilepsy (Unverricht-Lundborg and CSTB mutations, GLUTI deficiency and generalised epilepsy associated with SLC2A1 mutations, pyridoxal-dependant epilepsy due to PNPO mutations, etc) to name only a few examples. However, these techniques were poorly suited to the investigation of rare genetically heterogeneous disorders such as the sporadic epileptic encephalopathies in which only one affected proband is identified per family.
Molecular karyotyping approaches, such as array comparative genomic hybridisation techniques (aCGH), have been quite informative in identifying common copy number variants (CNVs) associated with various forms of epilepsy including epileptic encephalopathies (reviewed in 1) (Figure 1). Indeed, pathogenic CNVs can be identified in 10–15% of patients with severe early-onset epileptic encephalopathies (reviewed in 1). The identification of rare and recurrent CNVs in epilepsy led to the identification of new epileptic encephalopathy genes included in these genomic intervals such as PCDH192, CDKL53, STXBP14, GRIN2A5, CACNA1A.6 Recent studies demonstrate that targeted re-sequencing of some of these well documented epilepsy genes identifies a molecular diagnosis in approximately 10% of patients with epileptic encephalopathies.7 Nonetheless, a majority of patients with epileptic encephalopathies remain without an identifiable molecular etiology.
Figure 1. Comparative Genomic Hybridization (CGH) assays frequently reveal copy number variants (CNVs) in patients with unexplained epileptic encephalopathy.

(A) Comparative Genomic Hybridization (CGH) assays are conducted by comparing a patient’s genomic DNA, labeled with a fluorescent dye such as fluorescein (green), to a control DNA, labeled with another fluorescent dye such as rhodamine (red), both applied on a microarray chip in which each well contains a probe specific for a given genomic interval. Deletions (red) and amplifications (green) in the proband’s DNA can be differentiated from areas with balanced (normal) DNA. The sensitivity of CGH analysis is inversely proportional to the spacing of consecutive probes, usually around 500Kb. (B) Examples of microdeletions (red) and microduplications (blue) presenting with neurodevelopmental phenotypes and reported in the Angelman’s syndrome interval 15q11-13, encompassing the UBE3A gene (generated using the UCSC Genome Browser on March 17th, 2014).
Next-generation sequencing techniques, such as whole-exome and whole-genome sequencing, are now providing unique opportunities to identify new epilepsy genes in unexplained cases of epileptic encephalopathies. Recent publications using whole-exome sequencing (see Figure 2) have identified de novo mutations in a variety of genes in a large fraction of patients with sporadic epileptic encephalopathies8–10). These studies demonstrate the great genetic heterogeneity of these disorders, with recurrent mutations occurring in only a minority of identified genes. For instance, a recent large-scale genetic study led by the EPI4K consortium uncovered 329 de novo mutations in 320 genes in a cohort of 264 patients with epileptic encephalopathies, with only 9 genes carrying mutations in at least two probands.8 It is difficult to predict which of these genes are truly involved in the pathogenesis of epileptic encephalopathies, when private mutations are identified in isolated cases. However, genes with recurrent de novo mutations in patients with similar clinical phenotypes are good candidates for future biological validation. Whole-exome sequencing has been particularly powerful in identifying new genes with recurrent mutations in selected clinical epilepsy disorders with strictly defined clinical criteria, such as migrating partial epilepsy of infancy (KCNT111), Otahara syndrome (CASK,12 KCNQ213), rolandic epilepsy (RBFOX1, RBFOX314), etc. Furthermore, exome sequencing studies have expanded the phenotypic spectrum associated with some of the well-known epilepsy genes. For instance, KNCQ2 mutations initially associated with benign familial myoclonic epilepsy15 were subsequent associated with severe epileptic encephalopathy and spasticity.16
Figure 2. Whole exome sequencing often reveals detrimental de novo mutations in patients with sporadic epileptic encephalopathies.

(A) Schematic representation of the procedure involved in conducting whole exome sequencing. The proband’s genomic DNA is first sheared in ≈200bp fragments which are protected with end-adaptors. Secondly, the patient’s DNA is hybridized to an exome capture library consisting of specific probes designed to recognize most coding fragments of human DNA (i.e. exons and adjacent intron-exon splice-site junctions). The hybridized fragments are extracted using systems such as streptavidin-labeled beads. The patient’s exonic DNA is then retrieved and sent for sequencing (massive parallel sequencing). (B) The sequences obtained are aligned to the reference human genome sequence. Multiple reads will be obtained for each genomic interval sequenced. De novo variants that are unique to the proband and not inherited from the parents can be identified. These variants are then confirmed using Sanger re-sequencing (illustrated in bottom panel). In this particular patient presenting with early-onset epileptic encephalopathy, exome sequencing revealed a single de novo variant, where an A replaces the reference G in a heterozygous fashion (c. G875A), in the well-known epileptic encephalopathy gene STXBP1. This variant was predicted pathogenic by different bioinformatic scores, such as SIFT and PolyPHEN, which consider the variant’s impact on protein structure and domain conservation.
Therefore, the emergence of different genetic investigation techniques in recent years have led to the identification of a multitude of new confirmed or putative epilepsy candidate genes. However, in a majority of cases, the molecular, cellular and physiological consequences of these mutations and the mechanisms by which they cause epilepsy remain largely unknown. New genetic techniques now offer great opportunities to study the cellular and network consequences of these mutations in vivo in new animals models.
2. Mice genetic approaches to study the cellular and network mechanisms of epileptogenesis
As the genetic basis of several forms of epilepsy are currently being unravelled, modelling of these mutations in animals has become a prerequisite to delineate the cellular, molecular, physiological and network mechanisms underlying epileptogenesis in these genetic forms of epilepsy. Genetic models of epilepsy have long been used to study network phenomena underlying particular forms of epilepsy. For instance, many rodent models have been traditionally used to investigate the mechanisms underlying spike-wave absence seizures, including mice carrying spontaneous mutations in different calcium channel genes, as reviewed in17 (Cacna1atg/tg, Cacna1atg-rol/tg-rol, Cacnb4lh/lh, Cacng2stg/stg, etc) or rat strains carrying multigenic variants (GAERS and WAG/rij). Furthermore, genetically-engineered mice strains carrying targeted mutations of human epilepsy genes have been particularly useful in studying various cellular and network mechanisms leading to epilepsy (ex: Arx18; 19, Scn1a20, Scn8a and Scn9a as phenotypic modifiers21; 22). However, these animal models did not provide cellular specificity and could not be used to specifically address the cell-autonomous consequences of specific genetic mutations.
2.1. Conditional genetics to assess the cellular and network consequences of epilepsy-associated mutations in specific neuronal populations
Although traditional knock-out models represent good proxies for genomic mutations found in humans, they do not permit the selective assessment of the cell-autonomous consequences of such mutations in particular cell-types and their relevance to the overall clinical phenotype. For instance, various anomalies have been identified in constitutive Cacna1a and Cacnb4 mutants with spike-wave absence seizures, including reduced cortical feed-forward inhibition,23 reduced thalamic excitatory currents,24 thalamocortical hyperexcitability due to upregulation of thalamic low-threshold calcium currents25 and gain of aberrant thalamic tonic GABAergic currents.26 However, the contribution of these phenomena to the overall epileptic phenotype was uncertain. Furthermore, investigators have shown that preventing thalamic Cav3.1 upregulation, by crossing the Cacna1g−/− mutant mice with the Cacna1a−/− mice, prevents spike-wave seizures, suggesting that low-threshold T-type calcium current upregulation may not be necessary for the hypersynchronisation underlying absence seizures in Cacna1a mutants.
To dissect the impact of Cacna1a loss-of-function mutations on different components of the thalamocortical circuitry involved in generating spike-wave seizures, Rossignol et al. used a conditional genetic strategy to selectively ablate Cacna1a in specific sub-sets of cortical GABAergic interneurons or in cortical pyramidal cells, while sparing thalamic neurons (Figure 3).27 The targeted loss of Cacna1a in MGE-derived neocortical and hippocampal GABAergic interneurons (Nkx2.1Cre;Cacna1ac/c) leads to a severe form of generalised epilepsy in mice. Furthermore, Rossignol et al. demonstrated that this mutation selectively impaired GABA release from parvalbumin-positive fast-spiking basket-cells, leading to unreliable transmission with high failure rates and perturbed kinetics (Figure 3). 27 By contrast, the loss of Cacna1a in the somatostatin-positive neuronal population was efficiently compensated by up-regulation of N-type calcium channels which preserved neurotransmission from these cells, as demonstrated through optogenetic stimulation of this population in SSTCre; Cacna1ac/c mice injected with a Cre-dependant AAV-ChR2 virus. Finally, it was demonstrated that the generalised spike-wave seizures observed in the Nkx2.1Cre;Cacna1ac/c mutants did not involve the up-regulation of thalamic calcium T-type currents. However, the additional ablation of Cacna1a in cortical pyramidal cells (Nkx2.1Cre;Emx1Cre;Cacna1ac/c) considerably lessened the seizure severity, leading to isolated absence seizures by reducing cortical excitability in the face of cortical inhibitory dysfunction.27 This study by Rossignol et al. therefore demonstrates the usefulness of targeted genetic modifications of specific neuronal cell-types within epileptic networks to clarify the basic mechanisms underlying epileptogenesis and to study the cell-type specific consequences of particular mutations in genetic forms of epilepsy.
Figure 3. Conditional genetic strategies to generate mutant mice carrying cell-type selective mutations.

(A) Conditional genetic strategies allow the generation of mutant mice carrying a specific loss-of-function mutation in a gene of interest in a cell-type and tissue specific manner. Driver mouse lines are selected based on their expression of Cre recombinase driven by promoters expressed selectively in the cell-types and tissues of interest. This line is then bred on a conditional mutant mouse line carrying a floxed allele of the gene of interest, in which Lox P sites have been inserted around specific exons. The floxed allele is expressed properly in all tissues except in cells that express the Cre recombinase. In these mutated cells, the lox P sites will be recombined, effectively generating a deletion between the 2 sites, often leading to a loss-of-function allele. A conditonal reporter mouse line can be bred unto these mutants, allowing for the specific labeling of cells expressing the Cre recombinase. The mutated cells can then be tracked in a reliable fashion for their entire life-time as EGFP will be expressed stably over time. In the example illustrated here, an Nkx2.1Cre driver line was used to selectively ablate the 4th exon of the Cacna1a gene, leading to a loss-of-function allele. (B) The Nkx2.1Cre line was selected as it efficiently recombines the majority of cortical and limbic GABAergic interneurons derived form the medial ganglionic eminence, including the parvalbumin (PV) and the somatostatin (SST) expressing populations, while sparing the GABAergic cell populations in the thalamus reticular nucleus (RN). (C) Mutant cells can then be assessed using a variety of techniques, such as immunohistochemistry and in vitro physiology. In the example illustrated here, paired recordings between cortical fast-spiking (FS) GABAergic interneurons (identified using the RCEEGFP conditional labeling) and connected pyramidal cells revealed a significant alteration in the synaptic release properties of mutated FS interneurons in conditional Nkx2.1Cre; Cacna1ac/c;RCEEGFP mutant mice compared to littermate controls. These conditional mutants developed severe early-onset generalized seizures (adapted from Rossignol et al., Annals of Neurology, 2013).
This study further supported recent work by many groups highlighting the role of GABAergic interneuron dysfunction in epilepsy (reviewed in 28). Similar conditional genetic approaches revealed GABAergic interneurons dysfunctions in Dravet-associated Scn1a mutations,29 as well as in Rett-syndrome associated Mecp2 mutations.30 Furthermore, genetic studies in mutant mice carrying targeted deletions in genes involved in cortical GABAergic interneuron specification, migration and maturation (Dlx5/6, Dlx1/2, Nkx2.1, Lhx6, Sox6, Sip1) have been shown to result in epilepsy in mice and might be involved in rare forms of epilepsy in humans (as reviewed in 28). Together, these studies highlight the importance of specific subsets of GABAergic interneurons in preventing seizures within neuronal circuits and support the idea of cell-based therapies in specific forms of epilepsy associated with GABAergic interneuron dysfunction.
2.2. Gene repression via in utero electroporations to study the early developmental consequences of specific mutations during embryogenesis
Generating new mice lines to study the impact of particular mutations on neuronal development can be time-consuming and expensive. To accelerate the process of functional validation of newly identified epilepsy genes, knock-down approaches using miRNA and shRNA are increasingly being used. These approaches must be carefully controlled as non-specific effects can occur and can bias the interpretation of findings. For instance, an shRNA targeting a specific channel could result in unexpected cellular findings due to its non-specific effects on other genetically related ionic channels. Therefore, it is customary to combine knock-down experiments with rescue experiments using shRNA-resistant plasmids to confirm the specificity of these findings. When carefully conducted, experiments using knock-down strategies enable rapid screening of different genes identified through clinical studies, with great spatial and temporal precision. For instance, genetic repression of the Dravet-associated Scn1a gene within septal neuronal populations recently allowed investigators to study the role of this gene in septal GABAergic neurons governing their regulation of hippocampal theta rhythms during cognitive tasks.31
The delivery of experimental and scramble control shRNA to neuronal cell-types can be achieved using viral vectors (i.e. AAV, lentiviruses, etc), which will ensure widespread expression within neuronal populations in a given location, with high temporal and spatial resolution. Although these viral-based strategies offer good recombination capabilities, they are difficult to titrate to smaller cell numbers in order to address cell-autonomous phenotypes without affecting the global environment. Furthermore, these strategies cannot be use to study the impact of particular mutations in early embryonic processes, such as neuronal migration, due to the delay of expression of virally transfected plasmids. For such purposes, in utero electroporations offer greater flexibility with genetic repression observed within 24–48 hours, compared to the usual 1–2 weeks required for virus-based recombination. In utero electroporation offers significant advantages over traditional knock-out approaches to study the cell-autonomous consequences of mutations in disease-associated genes at a cell-specific level during brain development.32 For instance, this mosaic genetic approach offers unique opportunities to study the role of specific cytoskeletal molecules on neuronal migration as a fundamental process of neurodevelopment disrupted in epileptogenesis. Such shRNA-based approaches using in utero electroporations have been widely used to study the molecular mechanisms regulating neuronal migration. For instance, deficits and/or rescue of cortical pyramidal cell division and migration defects associated with Lis1, Dcx, Dab2ip, Tubg1, Kif5C, Kif2A mutations have been demonstrated using in utero electroporations. 33–36
Grisar and colleagues have used such approaches to study the role of a new epilepsy gene. Heterozygous mutations in Myoclonin1/EFHC1 have recently been shown to co-segregate with juvenile myoclonic epilepsy (JME) phenotypes. In adolescent JME patients, these genetic alterations produce subtle malformations of cortical and subcortical architecture, whereas homozygous F229L mutation in infancy induces severe brain pathology and death. However, the underlying pathological mechanisms for these observations remain unknown. Grisar and colleagues first demonstrated that EFHC1 is a microtubules-associated protein (MAP) involved in cell division and radial migration during cerebral corticogenesis.37 JME-mutations, including F229L, do not alter the ability of EFHC1 to co-localize with the centrosome and the mitotic spindle but act in a dominant-negative manner to impair mitotic spindle organization.38 Using both in utero and ex vivo electroporation technologies, Grisar et al. also found that mutant EFHC1 expression disrupted radial and tangential migration by affecting morphology of radial glia and migrating neurons (Figure 4).38 These results illustrate how Myoclonin1/EFHC1 mutations disrupt brain development, potentially leading to structural brain abnormalities as a basis for epileptogenesis.
Figure 4. Invalidating myoclonin1 disrupts radial neuronal migration.
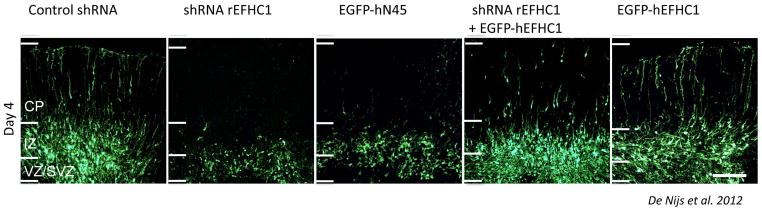
Downregulation of myoclonin1 using an shRNA delivered through in utero electroporation reveals a substantial impairement in neuronal migration and confirms the role of this gene in cortical development (CP: Cortical plate, IZ: intermediate zone, VZ/SVZ: (sub)ventricular zone).37
3. Optogenetics to dissect network components in the epileptic brain
The advent of optogenetics with it’s ability to selectively modulate specific neuronal populations involved in epileptic networks has provided unique opportunities to unravel some of the basic mechanisms of seizure generation. These techniques are taking advantage of recent advances in genetic mouse engineering and neurobiology to enable scientists to selectively modulate different components of epileptic networks as briefly illustrated here. Recent advances in our understanding of the genetic determinants controlling the specification of various subsets of GABAergic interneurons (reviewed in28), led to the generation of selective Cre-recombinase mouse driver lines39 that can now be used to selectively modulate neuronal activity using optogenetic approaches. The use of Cre-dependant AAV-based viral vectors to selectively express opsins (ChR2, Arch, etc) in specific neuronal populations allows the dissection of neuronal networks involved in epilepsy and the study of the cell-type specific impact of particular genetic mutations. For instance, using cell-type specific GFP expression in somatostatin-positive GABAergic interneurons (GINEGFP mouse), researchers recently identified cortical GABAergic network reorganisation leading to somatostatin-cell sprouting in the hippocampus of pilocarpine-exposed mice.40 Furthermore, by using ChR2 expressed selectively in these cells, this sprouting was demonstrated to be biologically relevant as the cells form functional synapses onto neighbouring projection neurons.40 Optogenetic approaches also enabled the precise dissection of corticothalamic networks involved in seizure generation following focal cortical strokes and illustrated that the silencing of ventrobasal thalamocortical projection neurons can be used to abort cortically-induced seizures using a closed-loop approach.41 Closed-loop interruption of seizures can also be achieved by optogenetical silencing of excitatory neurons or optogenetical activation of parvalbumin-positive basket cells in temporal lobe epilepsy models.42
4. Non-neuronal populations contribute extensively to network homeostasis: role of neuroglial interactions and microglia in epilepsy
Emerging evidence has revealed the fundamental roles of glial cells, and particularly astrocytes, in regulating neuronal excitability and synaptic transmission.43 The ability to visualise these glial populations through targeted genetic labelling, as well as to monitor their activity in neuronal circuits through targeted calcium imaging,44 both in vitro and in vivo, has revealed the capital role of these cells in cortical circuits. In the context of epilepsy, glial cells have been shown to regulate glutamate levels and to restrain cortical excitability.45 Furthermore, astrocytes have been shown to regulate neurovascular coupling46, which is sometimes perturbed in the context of metabolic failure or hyperlactatemia within damaged or chronically hyperexcitable tissues.
Microglia have also been recently implicated in the pathophysiology of chronic epilepsy. Indeed, TLE is characterized not only by hippocampal neuronal cell death and reactive astrogliosis, but also by the presence of activated microglia,47 which is thought to be detrimental for neuronal survival. However, recent findings suggest that microglia display neuroprotective properties in various CNS pathologies. Vinet and colleagues determined the effect of microglia depletion on excitotoxicity-induced neurodegeneration using mouse organotypic hippocampal slice cultures. Treatment of slice cultures with 10–50 μMN-methyl-D-aspartic acid (NMDA) induced a region specific increase in neuronal cell death with CA1 neurons being most vulnerable to NMDA-exposure, followed by CA3 and DG neurons, respectively.48 This selective neuronal vulnerability strongly correlated with activation of local microglia. Ablation of microglia by treatment of slice cultures with liposome-encapsulated clodronate, and subsequent exposure to NMDA, resulted in severely enhanced neuronal cell death in the CA3 and DG region. Replenishment of microglia-free slices with microglia restored the original resistance of CA3 and DG neurons towards NMDA. These data suggests that ramified microglia contribute to the protection of neurons under excitotoxic conditions and that activated microglia might serve to remove dead neuronal debris by phagocytosis. Thus, to better understand the role of microglia in TLE, Vinet and colleagues are acutely isolating microglia from the hippocampi of pilocarpine-treated animals. For this purpose, antibodies raised against specific membrane proteins are used to discriminate microglia from infiltrating macrophages through FACS sorting. This enables further experiments of specific genetic determinants of activated microglia by analyzing the transcriptional and proteomic profile of these cells using a combination of qPCR and Western blotting focusing on a set of markers involved in inflammatory processes and cell survival.49 This approach has been used successfully in animal models of neurological disease, such as multiple sclerosis. 50 Moreover, such approaches have been optimized for human tissue. 51 Such approaches could therefore eventually be used in human biopsy specimens of patients with pharmacoresistant TLE to further dissect the role of microglia in chronic epilepsy.
5. Epigenetic modifications in epilepsy
Beyond genomic mutations, epigenetic modifications are known to contribute to disease states in chronic neurological disorders, including in epilepsy. For instance, in various animal models of epilepsy, including the status-induced epilepsy, the pharmacologically-induced chronic epilepsies (pilocarpine, kainate, etc) and in genetic models of epilepsy, circuits exposed to chronic hyperactivity have been shown to undergo epigenetic modifications that considerably alter neuronal excitability and further aggravate the seizure phenotype. For instance, decreased HCN channels (reviewed in52), reorganised GABAA receptor subunits (reviewed in53), and aberrant tonic GABA currents54 have all been shown to maintain an hyperexcitable state in chronic epilepsy models. Many more epigenetic modifications are likely to take place in chronically hyperactive networks. The recent advances in massive parallel sequencing technologies coupled with improved molecular and computational techniques55 now allow researchers to profile the entire transcriptome, genome, targeted exome, as well as all different layers of the epigenome at very high resolutions.56
5.1. Study of the “methylome” in epilepsy
Taking advantage of these new technologies to assess potential epigenetic changes in known and unknown genes in an unbiased fashion, Kobow and colleagues used a massive parallel sequencing approach to map genome-wide alterations in DNA methylation in a rat model of chronic temporal lobe epilepsy (TLE; Figure 5). Sequencing of mRNA was further used in identical specimens for complementary gene expression profiling and integration with methylome data.57 Unsupervised clustering of an epigenetic mark, i.e. DNA methylation, separates epileptic from non-epileptic animals. Furthermore, DNA methylation was found to be inversely correlated with gene expression, suggesting that epigenetic regulation of gene expression may be critical in epileptogenesis and in the maintenance of the chronic disease state. Aberrant locus-specific DNA methylation may also be of interest as a potential biomarker for early detection of disease onset, prognosis or monitoring of disease after therapy. These experiments therefore highlight multiple new pathways of epigenetic mechanisms contributing to chronic epilepsy. These results will be particularly important as some of these epigenetic modifications could possibly be prevented by using selective medications aimed at controlling these epigenetic processes in patients with refractory epilepsy, for instance by using valproate acid with its potential as an HDAC inhibitor. Such approaches might be particularly important in cases where the underlying molecular mechanism is one that affects epigenetic control of gene expression, such as with UBE3A mutations in Angelman syndrome or MECP2 and CDKL5 mutations in Rett syndrome.
Figure 5. Analyzing epigenomic signatures in epilepsy using Next Generation Sequencing.

Epigenetic marks can be analyzed on a genome level using massive parallel sequencing technologies also referred to as Next Generation Sequencing (NGS). To study DNA methylation, preparation of genomic DNA is required followed by a sonication step to fragment DNA (300–400bp). Methylated DNA can then be enriched using either 5-mC specific antibodies (i.e. Methylated DNA immunoprecipitation, MeDIP) or methyl-binding domain (MBD) proteins. 5-mC capture associated NGS can identify genomic regions with medium to high 5-mC content. However, for methylation analysis down to single base pair resolution genomic bisulfite sequencing is required (not shown). Kobow and colleagues recently mapped global DNA methylation patterns in a rat model of TLE. They provide the first report of unsupervised clustering of an epigenetic mark being used in epilepsy research to separate epileptic from non-epileptic animals. NGS can also be combined with Chromatin Immunoprecipitation (ChIP) or selective preparation methods for coding and non-coding RNAs (mRNA, microRNAs, long ncRNAs) to analyze histone modifications or RNA expression patterns respectively. Currently no such epigenomic data sets are available from human or experimental epilepsy tissue (N/A).
5.2. miRNA in epilepsy
miRNA are small endogenous non-coding RNAs which exert crucial roles in regulating gene expression and particular genetic programs by regulating the expression of target mRNAs at a post-transcriptional level (Figure 6). To date, more than 1000 human miRNAs have been identified. Specific miRNAs have recently been shown to regulate cell-fate specification and brain development (reviewed in 58; 59). Interestingly, mutations and polymorphisms involving selected miRNAs are increasingly being associated with a variety of neuropsychiatric disorders including schizophrenia, Huntington’s disease and Alzheimer’s disease.60 miRNAs have also been shown to modify transcript dynamics in a fundamental manner in epileptogenesis and represent key target structures for new therapy developments. Specifically, selected miRNAs are involved in various cellular processes known to be disregulated in chronic epilepsy, including different deregulation of cell death, neurogenesis and synaptic plasticity.61 Thus, understanding which specific miRNAs are differentially expressed in epilepsy may help to identify some mechanisms underlying the disease. Moreover, these miRNAs may represent biomarkers with prognostic value that identify specific subpopulations of epileptic patients.
Figure 6. MicroRNA biogenesis and function.

miRNA genes are transcribed to generate primary miRNA (pri-miRNA) transcripts with a hairpin secondary structure. Pri-miRNAs are cleaved by a multiprotein complex that includes Drosha, generating pre-miRNAs that are exported into the cytoplasm by Exportin 5. In the cytoplasm, Dicer cleaves the hairpin loop, producing miRNA duplexes that are then unwound to yield single-strand mature miRNAs. Finally, mature miRNAs are incorporated into the RNA-induced silencing complex (RISC) in a sequence of events involving several proteins, including Argonaute proteins like Ago2. Once incorporated into RISC, miRNAs guide the complex to specific mRNAs through complementary base-pairing, leading to their cleavage or repression. Respective mechanisms appear to be active in epileptic brain tissue.
Simonato and colleagues (article under review) examined the expression of more than 1.000 human miRNAs in the dentate gyrus granular cell layer of resected surgical specimen from 10 patients who underwent surgery for intractable TLE. By profiling the miRNA expressed in surgical specimens with or without granule cell dispersion, Simonato and colleagues identified specific miRNA expressed selectively in tissue with granule cell dispersion, and characterized the downstream targets of these identified miRNAs. Given that granule cell dispersion is a frequent pathological hallmark in the hippocampus of TLE patients,62 the identification of a miRNA signature can improve our understanding of this pathogenetic process with potential diagnostic and therapeutic implications.63
5.3. Neuroanatomical correlates to relative seizure disposition and comorbid behavioral profiles: Lessons from ‘FAST’ and ‘SLOW’ rats
Epilepsy and autism spectrum disorder (ASD) share several primary and comorbid symptoms, particularly when acquired during childhood.64 Common traits often associated with both disorders include seizures, developmental delay, hyperactivity, impulsivity, aggression and intellectual impairment. Such extensive clinical overlap is believed to signify a ‘spectrum of vulnerability’ that arises out of an early common dysfunction in central nervous system development.64
The seizure-prone (FAST) and seizure-resistant (SLOW) rat strains represent an animal model to address respective comorbidities. FAST and SLOW rat strains were derived from parent populations of Long Evans Hooded (LEH) and Wistar rats, using selective breeding processes based on a differential vulnerability to amygdala kindling.65 Remarkably, as kindling sensitivity increased over generations in the FAST strain, additional traits evolved that are highly reminiscent of those observed in ASD, including hyperactivity, impulsivity, learning deficits, repetitive behaviors and delays in visuomotor and social development. Given rising interest in the identification of neuroanatomical correlates able to predict vulnerability towards these interrelated disorders, Gilby and colleagues first used T2 weighted magnetic resonance (MR) imaging to investigate any gross neuroanatomical differences that might exist between these strains. MR tractography was then used to more closely examine differences in white matter tracts and connectivity. Both imaging techniques demonstrated clear differences in the brains of FAST versus SLOW rats, including larger white matter and ventricular volumes in FAST rats. These findings provide evidence of structural correlates with the potential to serve as a biomarker for individuals that have a predisposition towards or against the seizure-prone condition and associated comorbid behavioral traits. The ability of these and other non-invasive imaging techniques to identify individuals at risk is potentially of great value for epilepsy and ASD, given that early intervention has proven beneficial for both of these disorders. Given the substantial relevance of genetic and epigenetic changes in these animals,66 next-generation sequencing may eventually be applied to these models to identify the genetic modifiers predisposing to different aspects of the comorbidities observed. Furthermore, molecular imaging could be used in these models subsequently.
6. Genetically-encoded In vivo bioluminescent reporters to study neuronal activity, excitability, neurotransmitter homeostasis and specific promoter activation in the context of epilepsy
6.1. Neuronal activity monitoring via calcium imaging
Live imaging of neuronal activity via voltage sensor dyes and genetically-encoded calcium biosensors has gained widespread popularity in the field of neuroscience, both in vitro and in vivo.67 These techniques enable scientists to image neuronal activity in a variety of contexts, for instance to study the maturation of brain networks68 and identify the generators of particular oscillatory activities during development,69 to dissect the connectivity within given networks when specific neuronal populations are activated,70 to study the activation of specific networks or entire cortical columns during task-related behaviour71; 72 and to assess synapses unto specific dendrites within 3D networks.73 In the field of epilepsy research, calcium imaging has been used to monitor the propagation of epileptic discharges within limbic74 and neocortical structures75; 76 and to study the activation of various neuronal clusters77 and specific GABAergic interneurons75 during ictal discharges.
6.2. Neuronal excitability monitoring via chloride imaging
Monitoring synaptic inhibition and intracellular chloride homeostasis during network development has been made possible by the generation of genetically-encoded chloride sensors.78 The use of such chloride sensors has revealed pathological deregulation of chloride homeostasis rendering GABA paradoxically excitatory in disease conditions, including in post-traumatic epilepsy,79 ischemia80 and neonatal seizures.81
6.3. Neurotransmitter homeostasis monitoring via glutamate biosensors
Recent advances have also made it possible to directly monitor tissue levels of specific neurotransmitters in the context of genetic epilepsy. For instance, glutamate excitotoxicity is thought to contribute to the disease phenotype in Rett syndrome (RTT). This disorder, most often caused by X-linked mutations in the MECP2 gene encoding the methyl-CpG-binding protein 2,82 is often complicated by epilepsy. Clinical studies show that early-onset epilepsy leads to a more rapid neurological deterioration and a worst prognosis in RTT patients.83 Abnormal EEGs are found in a 100% of RTT cases and are associated with severe sleep dysfunction.84 RTT patients display higher glutamate levels in their spinal fluid, and this excitotoxic state has been proposed to underlie the regression and synaptic dysfunction seen in this disorder.85
Mice with Mecp2 mutations show neuropathological and behavioral deficits similar to that reported for RTT patients. Sophisticated EEG-monitoring in mice is critical in order to determine the functional impact of genetic alterations on the epileptic phenotype.86 To investigate the effects of sleep-cycle dynamics on brain glutamate homeostasis in RTT, Kadam and colleagues used 24h video-EEG/EMG recordings with synchronous in-vivo cortical glutamate biosensors in symptomatic Mecp2 KO mice (Figure 7). Spectral analysis on non-ictal EEGs correlated with synchronous fluctuations in extracellular glutamate levels in Mecp2 mutant mice as compared to controls. Intriguingly, significant dark-cycle specific biomarkers of significant alterations in sleep macro- and microstructure that resembled that of severe sleep deprivation were observed. Absolute glutamate levels were significantly higher in frontal cortices of KOs and the activity-dependent homeostasis of glutamate was also severely impaired. Therefore, similar to RTT patients, Mecp2 KO mice showed significant sleep-cycle dysfunction and the 24h recordings identified biomarkers that underlie the progressive deterioration and fatality in Mecp2 KO mice. Such biomarkers might eventually be used to evaluate the rescue efficacy of novel interventions in clinical practice. Similar approaches to monitor specific neurotransmitter dynamics in disease state (i.e. dopamine, serotonin) in conjunction with functional imaging in humans have been widely used to assess disease progression and response to treatment, for instance in depression (reviewed in87). Biosensors for glutamate and GABA are under development for clinical use and could prove very instructive in epilepsy.
Figure 7. Experimental design and biosensor glutamate specificity.
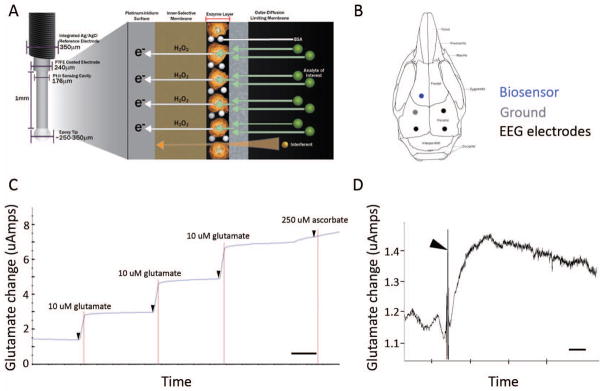
(A) Schematics of biosensor that used a glutamate specific oxidative enzyme reaction to detect every molecule of glutamate in extracellular space where implanted (image with permission from vendor website, Pinnacle Technologies Inc. Kansas, USA). (B) Schematic of location of stereotaxic implant to biosensor into frontal neocortex (blue); placement of EEG leads (2 recording and 1 reference) and mounting screw to anchor head mount to skull. (C) Representative recording trace of the post-experiment ex-vivo calibration of glutamate biosensor shows specificity to glutamate (arrow heads). Step readings for every 10uM glutamate (three repeats) added to media were averaged for each sensor in the study (time scale bar = 1min). (D) Sensitivity of the glutamate biosensor in-vivo was tested by injecting MK801 intra-peritoneal injection (5mg/kg), which elicited an immediate (< 1min) and significant rise in the glutamate reading in the frontal cortex, by the sensor. MK801 which is an NMDA receptor antagonist is known to induce increases in endogenous glutamate levels (Wyckhuys et al., 2013) and has been used to model various neurologic disorders, such as epilepsy, schizophrenia, and Parkinson disease where abnormal glutamate transmission is hypothesized to be involved (Roenker et al., 2012). Rapid increases in glutamate levels in the recorded trace after injection artifact (arrowhead) indicate potent NMDA receptor block and potent sensitivity of biosensor in-vivo (time scale bar = 5 min).
6.4. In vivo monitoring of specific promoter activation: calcium channel upregulation in chronic epilepsy
In vivo bioluminescence has so far been mainly used to address dynamics of tumor growth. However, it also allows the investigation of rapidly fluctuating molecular switches occurring in epileptic brains. Status epilepticus (SE) induced by pilocarpine in rodents causes a transformation of most CA1 pyramidal cells from regular- to burst-firing mode. By complementary electrophysiological, pharmacological and molecular approaches Becker, Yaari and colleagues observed the underlying mechanism to be the specific transcriptional upregulation of CaV3.2, a T-type Ca2+ channel pore-forming subunit, around day 3 after SE.88; 89 This subunit likely plays a key role in epileptogenesis, as its deletion in mice substantially attenuates the development of spontaneous chronic seizure activity and the degeneration of hippocampal neurons normally induced by pilocarpine elicited SE.
Seizures occur spontaneously in the chronic epileptic stage and seizure activity may have short-term influence on the mRNA levels of CaV3.2. Therefore, an approach was used that allows analysis of the CaV3.2 promoter activity as surrogate marker of corresponding gene expression in living mice, which monitored in parallel for spontaneous seizure activity. In order to monitor and quantify CaV3.2 promoter activity in a time wise manner in vivo, the CaV3.2 core promoter (CaV3.2cp90) was characterized and and bioluminescence imaging was applied (BLI; IVIS-Spectrum Optical In Vivo Imaging System) on mice injected in the hippocampus with rAAVs harboring a luciferase reporter gene under CaV3.2cp-control (Figure 8). Using this approach, specific CaV3.2cp hippocampal expression was observed, indicating sufficient basal reporter gene activation and activation only in the epileptogenesis stage. This experimental setup provides an intriguing tool in order to monitor the effects of interfering with transcriptional control mechanisms in vivo and correlate molecular genetic manipulation with behavioral and e.g. semiological consequences. The optimization of reporter molecules and a potential switch to fluorescence reporter molecules may be relevant in future approaches.91 Bioluminescence imaging can be used to repeatedly monitor and quantify gene activity in the same animal and thus can serve as an optimal tool to analyze the role of CaV3.2 in the developing epileptogenic network at various stages of epileptogenesis.
Figure 8. Imaging with in vivo bioluminescence probes reveal tissue-specific activation of candidate genes following status epilepticus.

(A) Representative time course of an in vivo bioluminescence experiment of promoter activation after brain insults. Initially, the virus harboring the promoter-bioluminescence reporter construct is injected in relevant brain structures such as the hippocampus. Days later, the first in vivo bioluminescence analysis (IVIS) is performed. The insult, e.g. status epilepticus is induced. Repetitive IVIS analyses are following before the animal is sacrificed for histology. (B) Representative increased in vivo bioluminescence reflects increased candidate promoter activity after SE.
7. Systems biology in the study of epilepsy
The integration of the wealth of genetic information obtained from tissue resected from patients with chronic epilepsy, stemming from genomic mutations, to epigenetic modifications, proteomics, and possible somatic mutations requires the implementation of a system biology approach. The easy availability of surgical specimens from patients undergoing epilepsy surgery represents a unique prerequisite to integrate different genetic approaches to study diseased tissues comprehensively on different molecular levels. Integrated analyses of respective data may differentiate between distinct pathogenetic aspects versus common downstream pathways that can result from multiple brain abnormalities in particular forms of epilepsy. While a wide variety of genetic and acquired histopathological lesions have been discovered, often no clear pathological lesions are seen in human epileptic neocortex resected for the treatment of intractable seizures. Loeb and colleagues have undertaken a systems biology approach to identify what is unique and what features are shared amongst neocortical brain regions that produce spontaneous spikes and/or seizures.92 Using a combination of functional genomics, proteomics, and metabolomics, an online dataset and a multivariate interactome are established that link all of these molecular features of the tissue to electrical and clinical features of the disease.
Recently, Loeb and colleagues have used this platform to identify differentially-expressed genes in regions of seizure onset93 and regions showing high levels of interictal spiking.94 Interestingly, the lists of genes in these two groups are highly overlapping, just as is seen electrophysiologically, where regions of high interictal spiking are often the same regions that generate seizures. Perhaps even more interesting, patterns of gene expression became evident that are unique for both spiking and seizures, with particular involvement of components of the MAPK pathway. These molecular studies have also produced a set of ‘tissue biomarkers’ that can be used to stain human epileptic cortex to identify the cellular populations involved for both interictal spiking and seizures. Loeb and colleagues found that in the case of interictal spiking, the most salient pathways were all activated in superficial cortical layers (I–III), suggesting that spiking may be generated from these cortical layers. Consistently, an animal model of interictal spiking that uses tetanus toxin in the somatosensory cortex shows activation of these same neuronal lamina with the same tissue biomarkers.95 Loeb and colleagues are also developing a novel computational method to ‘predict’ additional histological and molecular abnormalities shared by all patients with neocortical epilepsy, regardless of the underlying cause. These predictions in human neocortical tissues suggest the existence of novel microlesions in deeper neocortical layers that is associated with segmental activation of plasticity pathways in adjacent superficial layers. Taken together, systems biological studies of electrically-mapped human neocortex are powerful tools to hone in on common pathways and specific laminar involvement in patients with neocortical epilepsy and can be compared to many other genetic and epigenetic approaches.
8. Towards novel therapies for specific genetic disorders: cell-based therapy and high-throughput drug screening
8.1. Stem cells: use of patient-derived iPS cells re-specified into neurons to study the biological relevance of new epilepsy mutations and possible avenues for stem cell therapies
Although genetic modelling of specific mutations in rodents has proved highly relevant to some aspects of epilepsy research, it is time- and energy-consuming and is somewhat limited by inter-species differences. Furthermore, the breath of new genetic findings in clinical research will prevent quick modeling in mice as the generation of a knock-in mouse takes many months, is considerably expensive, and could not address the diversity of mutations identified within any given gene. For these reasons, modeling of human mutations in vitro using genetically-engineered neurons rederived from iPS cells (induced pluripotent stem cells) obtained from skin fibroblasts of patients with epilepsy is a powerful emerging technology. Using these iPS rederivation techniques, scientists are now able to generate specific subtypes of neurons carrying the exact same mutation as that of the patient being investigated. Using such technologies, scientists have recently been able to study the impact of specific Dravet-associated SCN1A mutations on neuronal excitability in neurons obtained after rederivation of patient fibroblasts, shedding new lights on some of the mechanisms underling the clinical phenotypes in this genetic epilepsy.96
Furthermore, stem cell engineering is now offering exciting prospects for new cell-based therapies for epilepsy.97 Indeed, stem-cell derived medial ganglionic eminence progenitors of specific sub-types of GABAergic interneurons, namely parvalbumin basket cells, have recently been shown to migrate, disperse, mature and integrate properly in developing and mature networks in mice, and to revert seizure and cognitive manifestations of different genetic mutations.98; 99 Furthermore, these transplanted GABAergic progenitors have been shown to scale their number and output in proportion to surrounding network activity such that no excessive inhibition results from these transplanted cells.
iPS cells derived from human skin fibroblasts can be genetically reprogrammed to acquire the properties of specific cell-types, including GABAergic interneurons. Recent advances in stem cell research have improved protocols to generate higher numbers of particular cell types of interest using human embryonic stem cells. Using a combination of specific growth factors and genetic drivers, scientists have been able to produce large amounts of neural cell-types with GABAergic neurons phenotypes.100 One can now hope that such technologies, if applied to genetically-engineered MGE-type GABAergic interneurons re-derived from human skin iPS cells, could eventually lead to cell-based therapies in selected cases of severe refractory epilepsies not amendable to epilepsy surgery or alternative therapies such as the ketogenic diet or vagus nerve stimulator.
8.2. High true-put drug screening using non-mammalian models: genetically- modified zebra fish
Although stem cell approaches permit relatively high thru-put screening of the basic cellular mechanisms associated with particular mutations, they cannot properly address network phenomena nor the clinical correlates of specific therapies aimed at particular genetic mutations. For this purpose, investigators have recently designed high thru-put strategies to genetically modify zebra fish, assess the behavioural and electroencephalographic correlates of given mutation and screen small-molecule libraries in search for new anti-epileptic treatments.101 Using a morpholino-based Scn1a knock-down strategy in zebrafish, Baraban and al. screened a library of 320 micromolecules and identified clomizole as a potential new anticonvulsive medication. Such unbiased screening approach will likely identify many more potential therapeutic agents in the coming years.
9. Conclusions
The new genetic investigation techniques that have emerged in recent years have considerably advanced our understanding of the molecular and epigenetic etiologies underlying various forms of epilepsy in humans and have provided unparalleled tools to dissect and study the impact of these genes on various components of the neural circuits involved in seizure generation. Furthermore, these new techniques are now opening new avenues for the development of novel therapeutic approaches in epilepsy.
Supplementary Material
Acknowledgments
ER’s work is funded by the Canadian Institutes for Health research (CIHR 259491), the Fond de recherche en santé du Québec (FRQS 24445), the Scottish Rite Foundation and the Savoy Foundation in Epilepsy. The work of JL was funded by NIH/NINDS Grants R01NS045207 and R01NS058802. AJB’s work is supported by Epitarget, EuroEpinomics and DFG (SFB 1089, KFO 177), the Else Kröner-Fresenius and German Israeli Foundations and BonFor. MS has received grants from the European Community [FP7-PEOPLE-2011-IAPP project 285827 (EPIXCHANGE)] and from the Italian Ministry for the University and Research [PRIN 2010-11 project 2010N8PBAA (IN-BDNF)].
Footnotes
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Disclosure
Jonathan Vinet has received support form the Italian Foundation for Multiple Sclerosis (FISM 2011/B/7). Katja Kobow, Jefferey Loeb, Michele Simonato, Thierry Grisar, Krista L. Gilby, Shilba D. Kadam, E. Rossignol and Albert J. Becker have no disclosures of conflict of interest. Wherever necessary, permission were obtained to re-use components of figures previously published by individual authors.
References
- 1.Mulley JC, Mefford HC. Epilepsy and the new cytogenetics. Epilepsia. 2011;52:423–432. doi: 10.1111/j.1528-1167.2010.02932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dibbens LM, Tarpey PS, Hynes K, et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet. 2008;40:776–781. doi: 10.1038/ng.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scala E, Ariani F, Mari F, et al. CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J Med Genet. 2005;42:103–107. doi: 10.1136/jmg.2004.026237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saitsu H, Kato M, Mizuguchi T, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. 2008;40:782–788. doi: 10.1038/ng.150. [DOI] [PubMed] [Google Scholar]
- 5.Carvill GL, Regan BM, Yendle SC, et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet. 2013;45:1073–1076. doi: 10.1038/ng.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Auvin S, Holder-Espinasse M, Lamblin MD, et al. Array-CGH detection of a de novo 0.7-Mb deletion in 19p13. 13 including CACNA1A associated with mental retardation and epilepsy with infantile spasms. Epilepsia. 2009;50:2501–2503. doi: 10.1111/j.1528-1167.2009.02189.x. [DOI] [PubMed] [Google Scholar]
- 7.Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45:825–830. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allen AS, Berkovic SF, Cossette P, et al. De novo mutations in epileptic encephalopathies. Nature. 2013 doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Veeramah KR, Johnstone L, Karafet TM, et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia. 2013;54:1270–1281. doi: 10.1111/epi.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michaud JL, Lachance M, Hamdan FF, et al. The genetic landscape of infantile spasms. Hum Mol Genet. 2014 doi: 10.1093/hmg/ddu199. [DOI] [PubMed] [Google Scholar]
- 11.Barcia G, Fleming MR, Deligniere A, et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet. 2012;44:1255–1259. doi: 10.1038/ng.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saitsu H, Kato M, Osaka H, et al. CASK aberrations in male patients with Ohtahara syndrome and cerebellar hypoplasia. Epilepsia. 2012;53:1441–1449. doi: 10.1111/j.1528-1167.2012.03548.x. [DOI] [PubMed] [Google Scholar]
- 13.Saitsu H, Kato M, Koide A, et al. Whole exome sequencing identifies KCNQ2 mutations in Ohtahara syndrome. Ann Neurol. 2012;72:298–300. doi: 10.1002/ana.23620. [DOI] [PubMed] [Google Scholar]
- 14.Lal D, Reinthaler EM, Altmuller J, et al. RBFOX1 and RBFOX3 mutations in rolandic epilepsy. PLoS One. 2013;8:e73323. doi: 10.1371/journal.pone.0073323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh NA, Charlier C, Stauffer D, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- 16.Kato M, Yamagata T, Kubota M, et al. Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia. 2013;54:1282–1287. doi: 10.1111/epi.12200. [DOI] [PubMed] [Google Scholar]
- 17.Burgess DL, Noebels JL. Calcium channel defects in models of inherited generalized epilepsy. Epilepsia. 2000;41:1074–1075. doi: 10.1111/j.1528-1157.2000.tb00305.x. [DOI] [PubMed] [Google Scholar]
- 18.Colombo E, Collombat P, Colasante G, et al. Inactivation of Arx, the murine ortholog of the X-linked lissencephaly with ambiguous genitalia gene, leads to severe disorganization of the ventral telencephalon with impaired neuronal migration and differentiation. J Neurosci. 2007;27:4786–4798. doi: 10.1523/JNEUROSCI.0417-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Price MG, Yoo JW, Burgess DL, et al. A triplet repeat expansion genetic mouse model of infantile spasms syndrome, Arx(GCG)10+7, with interneuronopathy, spasms in infancy, persistent seizures, and adult cognitive and behavioral impairment. J Neurosci. 2009;29:8752–8763. doi: 10.1523/JNEUROSCI.0915-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu FH, Mantegazza M, Westenbroek RE, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- 21.Martin MS, Tang B, Papale LA, et al. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet. 2007;16:2892–2899. doi: 10.1093/hmg/ddm248. [DOI] [PubMed] [Google Scholar]
- 22.Singh NA, Pappas C, Dahle EJ, et al. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet. 2009;5:e1000649. doi: 10.1371/journal.pgen.1000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sasaki S, Huda K, Inoue T, et al. Impaired feedforward inhibition of the thalamocortical projection in epileptic Ca2+ channel mutant mice, tottering. J Neurosci. 2006;26:3056–3065. doi: 10.1523/JNEUROSCI.5422-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caddick SJ, Wang C, Fletcher CF, et al. Excitatory but not inhibitory synaptic transmission is reduced in lethargic (Cacnb4(lh)) and tottering (Cacna1atg) mouse thalami. J Neurophysiol. 1999;81:2066–2074. doi: 10.1152/jn.1999.81.5.2066. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Mori M, Burgess DL, et al. Mutations in high-voltage-activated calcium channel genes stimulate low-voltage-activated currents in mouse thalamic relay neurons. J Neurosci. 2002;22:6362–6371. doi: 10.1523/JNEUROSCI.22-15-06362.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cope DW, Di Giovanni G, Fyson SJ, et al. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. 2009;15:1392–1398. doi: 10.1038/nm.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossignol E, Kruglikov I, van den Maagdenberg AM, et al. Ca 2.1 ablation in cortical interneurons selectively impairs fast-spiking basket cells and causes generalized seizures. Ann Neurol. 2013 doi: 10.1002/ana.23913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rossignol E. Genetics and function of neocortical GABAergic interneurons in neurodevelopmental disorders. Neural Plast. 2011;2011:649325. doi: 10.1155/2011/649325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheah CS, Yu FH, Westenbroek RE, et al. Specific deletion of NaV1. 1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A. 2012;109:14646–14651. doi: 10.1073/pnas.1211591109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chao HT, Chen H, Samaco RC, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bender AC, Natola H, Ndong C, et al. Focal Scn1a knockdown induces cognitive impairment without seizures. Neurobiol Dis. 2013;54:297–307. doi: 10.1016/j.nbd.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LoTurco JJ, Bai J. The multipolar stage and disruptions in neuronal migration. Trends Neurosci. 2006;29:407–413. doi: 10.1016/j.tins.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 33.Tsai JW, Chen Y, Kriegstein AR, et al. LIS1 RNA interference blocks neural stem cell division, morphogenesis, and motility at multiple stages. J Cell Biol. 2005;170:935–945. doi: 10.1083/jcb.200505166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shu T, Tseng HC, Sapir T, et al. Doublecortin-like kinase controls neurogenesis by regulating mitotic spindles and M phase progression. Neuron. 2006;49:25–39. doi: 10.1016/j.neuron.2005.10.039. [DOI] [PubMed] [Google Scholar]
- 35.Lee GH, Kim SH, Homayouni R, et al. Dab2ip regulates neuronal migration and neurite outgrowth in the developing neocortex. PLoS One. 2012;7:e46592. doi: 10.1371/journal.pone.0046592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poirier K, Lebrun N, Broix L, et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet. 2013;45:639–647. doi: 10.1038/ng.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Nijs L, Leon C, Nguyen L, et al. EFHC1 interacts with microtubules to regulate cell division and cortical development. Nat Neurosci. 2009;12:1266–1274. doi: 10.1038/nn.2390. [DOI] [PubMed] [Google Scholar]
- 38.de Nijs L, Wolkoff N, Coumans B, et al. Mutations of EFHC1, linked to juvenile myoclonic epilepsy, disrupt radial and tangential migrations during brain development. Hum Mol Genet. 2012;21:5106–5117. doi: 10.1093/hmg/dds356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taniguchi H, He M, Wu P, et al. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 2011;71:995–1013. doi: 10.1016/j.neuron.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng Z, Zhang N, Wei W, et al. A reorganized GABAergic circuit in a model of epilepsy: evidence from optogenetic labeling and stimulation of somatostatin interneurons. J Neurosci. 2013;33:14392–14405. doi: 10.1523/JNEUROSCI.2045-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paz JT, Davidson TJ, Frechette ES, et al. Closed-loop optogenetic control of thalamus as a tool for interrupting seizures after cortical injury. Nat Neurosci. 2012;16:64–70. doi: 10.1038/nn.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krook-Magnuson E, Armstrong C, Oijala M, et al. On-demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat Commun. 2013;4:1376. doi: 10.1038/ncomms2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fellin T, Sul JY, D’Ascenzo M, et al. Bidirectional astrocyte-neuron communication: the many roles of glutamate and ATP. Novartis Found Symp. 2006;276:208–217. doi: 10.1002/9780470032244.ch16. discussion 217–221, 233–207, 275–281. [DOI] [PubMed] [Google Scholar]
- 44.Hirase H, Qian L, Bartho P, et al. Calcium dynamics of cortical astrocytic networks in vivo. PLoS Biol. 2004;2:E96. doi: 10.1371/journal.pbio.0020096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tian GF, Azmi H, Takano T, et al. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takano T, Tian GF, Peng W, et al. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- 47.Vezzani A, French J, Bartfai T, et al. The role of inflammation in epilepsy. Nature reviews Neurology. 2011;7:31–40. doi: 10.1038/nrneurol.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vinet J, Weering HR, Heinrich A, et al. Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J Neuroinflammation. 2012;9:27. doi: 10.1186/1742-2094-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Haas AH, Boddeke HW, Brouwer N, et al. Optimized isolation enables ex vivo analysis of microglia from various central nervous system regions. Glia. 2007;55:1374–1384. doi: 10.1002/glia.20554. [DOI] [PubMed] [Google Scholar]
- 50.Olah M, Amor S, Brouwer N, et al. Identification of a microglia phenotype supportive of remyelination. Glia. 2012;60:306–321. doi: 10.1002/glia.21266. [DOI] [PubMed] [Google Scholar]
- 51.Olah M, Raj D, Brouwer N, et al. An optimized protocol for the acute isolation of human microglia from autopsy brain samples. Glia. 2012;60:96–111. doi: 10.1002/glia.21251. [DOI] [PubMed] [Google Scholar]
- 52.Poolos NP. Hyperpolarization-Activated Cyclic Nucleotide-Gated (HCN) Ion Channelopathy in Epilepsy. 2012 [PubMed] [Google Scholar]
- 53.Grabenstatter HL, Russek SJ, Brooks-Kayal AR. Molecular pathways controlling inhibitory receptor expression. Epilepsia. 2012;53 (Suppl 9):71–78. doi: 10.1111/epi.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yu J, Proddutur A, Elgammal FS, et al. Status epilepticus enhances tonic GABA currents and depolarizes GABA reversal potential in dentate fast-spiking basket cells. J Neurophysiol. 2013;109:1746–1763. doi: 10.1152/jn.00891.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berger B, Peng J, Singh M. Computational solutions for omics data. Nat Rev Genet. 2013;14:333–346. doi: 10.1038/nrg3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rizzo JM, Buck MJ. Key principles and clinical applications of “next-generation” DNA sequencing. Cancer Prev Res (Phila) 2012;5:887–900. doi: 10.1158/1940-6207.CAPR-11-0432. [DOI] [PubMed] [Google Scholar]
- 57.Kobow K, Kaspi A, Harikrishnan KN, et al. Deep sequencing reveals increased DNA methylation in chronic rat epilepsy. Acta Neuropathol. 2013 doi: 10.1007/s00401-013-1168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun AX, Crabtree GR, Yoo AS. MicroRNAs: regulators of neuronal fate. Curr Opin Cell Biol. 2013;25:215–221. doi: 10.1016/j.ceb.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petri R, Malmevik J, Fasching L, et al. miRNAs in brain development. Exp Cell Res. 2013 doi: 10.1016/j.yexcr.2013.09.022. [DOI] [PubMed] [Google Scholar]
- 60.Sheinerman KS, Umansky SR. Circulating cell-free microRNA as biomarkers for screening, diagnosis and monitoring of neurodegenerative diseases and other neurologic pathologies. Front Cell Neurosci. 2013;7:150. doi: 10.3389/fncel.2013.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jimenez-Mateos EM, Henshall DC. Epilepsy and microRNA. Neuroscience. 2013;238:218–229. doi: 10.1016/j.neuroscience.2013.02.027. [DOI] [PubMed] [Google Scholar]
- 62.Blumcke I, Thom M, Aronica E, et al. International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: A Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia. 2013;54:1315–1329. doi: 10.1111/epi.12220. [DOI] [PubMed] [Google Scholar]
- 63.Simonato M, Bennett J, Boulis NM, et al. Progress in gene therapy for neurological disorders. Nature reviews Neurology. 2013;9:277–291. doi: 10.1038/nrneurol.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gilby KL, O’Brien TJ. Epilepsy, autism, and neurodevelopment: kindling a shared vulnerability? Epilepsy Behav. 2013;26:370–374. doi: 10.1016/j.yebeh.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 65.Azarbar A, McIntyre DC, Gilby KL. Caloric restriction alters seizure disposition and behavioral profiles in seizure-prone (fast) versus seizure-resistant (slow) rats. Behav Neurosci. 2010;124:106–114. doi: 10.1037/a0018307. [DOI] [PubMed] [Google Scholar]
- 66.Gilby KL. Investigating epigenetic influences on seizure disposition. Can J Neurol Sci. 2009;36 (Suppl 2):S78–81. [PubMed] [Google Scholar]
- 67.Tantama M, Hung YP, Yellen G. Optogenetic reporters: Fluorescent protein-based genetically encoded indicators of signaling and metabolism in the brain. Prog Brain Res. 2012;196:235–263. doi: 10.1016/B978-0-444-59426-6.00012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu EP, Dengler CG, Frausto SF, et al. Protracted postnatal development of sparse, specific dentate granule cell activation in the mouse hippocampus. J Neurosci. 2013;33:2947–2960. doi: 10.1523/JNEUROSCI.1868-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bonifazi P, Goldin M, Picardo MA, et al. GABAergic hub neurons orchestrate synchrony in developing hippocampal networks. Science. 2009;326:1419–1424. doi: 10.1126/science.1175509. [DOI] [PubMed] [Google Scholar]
- 70.Fino E, Yuste R. Dense inhibitory connectivity in neocortex. Neuron. 2011;69:1188–1203. doi: 10.1016/j.neuron.2011.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Andermann ML, Gilfoy NB, Goldey GJ, et al. Chronic cellular imaging of entire cortical columns in awake mice using microprisms. Neuron. 2013;80:900–913. doi: 10.1016/j.neuron.2013.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen Q, Cichon J, Wang W, et al. Imaging neural activity using Thy1-GCaMP transgenic mice. Neuron. 2012;76:297–308. doi: 10.1016/j.neuron.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fernandez-Alfonso T, Nadella KM, Iacaruso MF, et al. Monitoring synaptic and neuronal activity in 3D with synthetic and genetic indicators using a compact acousto-optic lens two-photon microscope. J Neurosci Methods. 2013;222C:69–81. doi: 10.1016/j.jneumeth.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takano H, Coulter DA. Imaging of Hippocampal Circuits in Epilepsy. 2012 [PubMed] [Google Scholar]
- 75.Cammarota M, Losi G, Chiavegato A, et al. Fast spiking interneuron control of seizure propagation in a cortical slice model of focal epilepsy. J Physiol. 2012;591:807–822. doi: 10.1113/jphysiol.2012.238154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Trevelyan AJ, Sussillo D, Watson BO, et al. Modular propagation of epileptiform activity: evidence for an inhibitory veto in neocortex. J Neurosci. 2006;26:12447–12455. doi: 10.1523/JNEUROSCI.2787-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Feldt Muldoon S, Soltesz I, Cossart R. Spatially clustered neuronal assemblies comprise the microstructure of synchrony in chronically epileptic networks. Proc Natl Acad Sci U S A. 2013;110:3567–3572. doi: 10.1073/pnas.1216958110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kuner T, Augustine GJ. A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons. Neuron. 2000;27:447–459. doi: 10.1016/s0896-6273(00)00056-8. [DOI] [PubMed] [Google Scholar]
- 79.Dzhala V, Valeeva G, Glykys J, et al. Traumatic alterations in GABA signaling disrupt hippocampal network activity in the developing brain. J Neurosci. 2012;32:4017–4031. doi: 10.1523/JNEUROSCI.5139-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pond BB, Berglund K, Kuner T, et al. The chloride transporter Na(+)-K(+)-Cl− cotransporter isoform-1 contributes to intracellular chloride increases after in vitro ischemia. J Neurosci. 2006;26:1396–1406. doi: 10.1523/JNEUROSCI.1421-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dzhala VI, Kuchibhotla KV, Glykys JC, et al. Progressive NKCC1-dependent neuronal chloride accumulation during neonatal seizures. J Neurosci. 2010;30:11745–11761. doi: 10.1523/JNEUROSCI.1769-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Amir RE, Van den Veyver IB, Wan M, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 83.Pintaudi M, Calevo MG, Vignoli A, et al. Epilepsy in Rett syndrome: clinical and genetic features. Epilepsy Behav. 2010;19:296–300. doi: 10.1016/j.yebeh.2010.06.051. [DOI] [PubMed] [Google Scholar]
- 84.Young D, Nagarajan L, de Klerk N, et al. Sleep problems in Rett syndrome. Brain Dev. 2007;29:609–616. doi: 10.1016/j.braindev.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J Neurosci. 2010;30:5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kadam SD, White AM, Staley KJ, et al. Continuous electroencephalographic monitoring with radio-telemetry in a rat model of perinatal hypoxia-ischemia reveals progressive post-stroke epilepsy. J Neurosci. 2010;30:404–415. doi: 10.1523/JNEUROSCI.4093-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hoflich A, Baldinger P, Savli M, et al. Imaging treatment effects in depression. Rev Neurosci. 2012;23:227–252. doi: 10.1515/revneuro-2012-0038. [DOI] [PubMed] [Google Scholar]
- 88.Su H, Sochivko D, Becker A, et al. Upregulation of a T-type Ca2+ channel causes a long-lasting modification of neuronal firing mode after status epilepticus. J Neurosci. 2002;22:3645–3655. doi: 10.1523/JNEUROSCI.22-09-03645.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Becker AJ, Pitsch J, Sochivko D, et al. Transcriptional upregulation of Cav3. 2 mediates epileptogenesis in the pilocarpine model of epilepsy. J Neurosci. 2008;28:13341–13353. doi: 10.1523/JNEUROSCI.1421-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.van Loo KM, Schaub C, Pernhorst K, et al. Transcriptional regulation of T-type calcium channel CaV3. 2: bi-directionality by early growth response 1 (Egr1) and repressor element 1 (RE-1) protein-silencing transcription factor (REST) J Biol Chem. 2012;287:15489–15501. doi: 10.1074/jbc.M111.310763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Close DM, Xu T, Sayler GS, et al. In vivo bioluminescent imaging (BLI): noninvasive visualization and interrogation of biological processes in living animals. Sensors (Basel) 2011;11:180–206. doi: 10.3390/s110100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Loeb JA. Identifying targets for preventing epilepsy using systems biology. Neurosci Lett. 2011;497:205–212. doi: 10.1016/j.neulet.2011.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Beaumont TL, Yao B, Shah A, et al. Layer-specific CREB target gene induction in human neocortical epilepsy. J Neurosci. 2012;32:14389–14401. doi: 10.1523/JNEUROSCI.3408-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lipovich L, Dachet F, Cai J, et al. Activity-dependent human brain coding/noncoding gene regulatory networks. Genetics. 2012;192:1133–1148. doi: 10.1534/genetics.112.145128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Barkmeier DT, Senador D, Leclercq K, et al. Electrical, molecular and behavioral effects of interictal spiking in the rat. Neurobiol Dis. 2012;47:92–101. doi: 10.1016/j.nbd.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu Y, Lopez-Santiago LF, Yuan Y, et al. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann Neurol. 2013;74:128–139. doi: 10.1002/ana.23897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sebe JY, Baraban SC. The promise of an interneuron-based cell therapy for epilepsy. Dev Neurobiol. 2010;71:107–117. doi: 10.1002/dneu.20813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hunt RF, Girskis KM, Rubenstein JL, et al. GABA progenitors grafted into the adult epileptic brain control seizures and abnormal behavior. Nat Neurosci. 2013;16:692–697. doi: 10.1038/nn.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Baraban SC, Southwell DG, Estrada RC, et al. Reduction of seizures by transplantation of cortical GABAergic interneuron precursors into Kv1. 1 mutant mice. Proc Natl Acad Sci U S A. 2009;106:15472–15477. doi: 10.1073/pnas.0900141106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maroof AM, Keros S, Tyson JA, et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 2013;12:559–572. doi: 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hortopan GA, Dinday MT, Baraban SC. Zebrafish as a model for studying genetic aspects of epilepsy. Dis Model Mech. 2010;3:144–148. doi: 10.1242/dmm.002139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.