

Abstract
Type 2 diabetes is characterized by a decreased ability of insulin to facilitate glucose uptake into insulin sensitive tissue, i.e., skeletal muscle. The mechanism behind this is at the moment unresolved. It has been suggested that increased amount of lipids inside the skeletal muscle (intramuscular triglyceride, diacylglycerol and ceramides) will impair insulin action in skeletal muscle, but data are not consistent in the human literature. It has also been hypothesized that the impaired insulin sensitivity is due to a dysfunction in the mitochondria resulting in an impaired ability to oxidize lipids, but the majority of the literature is not supporting this hypothesis. Recently it has been suggested that the production of reactive oxygen species play an essential role in skeletal muscle insulin sensitivity. It is well accepted that physical activity (endurance, strength and high intensity training) improves insulin sensitivity in healthy humans and in patients with type 2 diabetes. Whether patients with type 2 diabetes have the same beneficial effects (same improvement) as control subjects, when it comes to regular physical activity in regard to mitochondrial function, is not established in the literature. This review will focus only on the effect of physical activity on skeletal muscle (mitochondrial function) in patients with type 2 diabetes.
Keywords: Mitochondria, Exercise, Type 2 diabetes
Core tip: It is well described that exercise interventions improves insulin sensitivity and maximal oxygen uptake in patients with type 2 diabetes as well as in control subjects. When it comes to adaptations in mitochondrial function after an exercise intervention the literature is more sparse especially in patients with type 2 diabetes. Furthermore the medication that patients with type 2 diabetes are using, are often not described well in the papers, and it is known that the different medication (statins and antihypertensive agents) have a major effect on mitochondrial function and insulin sensitivity.
INTRODUCTION
The pathophysiology of type 2 diabetes involves the secretion and the action of insulin. The prevailing view[1] is that an inability of insulin to exert its action on the central target tissues, skeletal muscle (mediate glucose uptake initialized by the binding of insulin to its receptor), adipose tissue (mediate glucose uptake and inhibit lipolysis) and hepatic tissue (inhibit glucose output), results in increasing concentrations of glucose in the blood. In response, insulin secretion from the pancreatic beta-cells is increased, and hyperinsulinemia prevails. Only in those patients in whom the enforced production of insulin from the pancreas fails, hyperglycemia develops and overt type 2 diabetes becomes apparent. The mechanism for the failing pancreatic insulin production is not resolved, while the development of impaired insulin action (insulin resistance) is linked to the development of obesity (in particular visceral fat) and a physical inactive lifestyle and the molecular mechanism is being unraveled these years[2]. Type 2 diabetes is also frequently seen in a cluster of pathologies, including hypertension, endothelial dysfunction, and obesity. Complications to type 2 diabetes include macrovascular complications (atherosclerosis), but also microvascular complications such as neuropathy, nephropathy, retinopathy and angiopathy are known to occur in these patients.
In the past decade mitochondrial dysfunction in skeletal muscle has been linked to insulin resistance[3-7], but an agreement has not been reached and the majority of data does not support this notion[8-17]. It has been shown that patients with type 2 diabetes have 30% lower mitochondrial content in their skeletal muscle compared to healthy control subjects[12,18], and yet the intrinsic mitochondrial function (i.e., respiratory rates normalized for mitochondrial content) is similar in these two groups[12-14]. As such the suggested scenario with insulin resistance being induced by mitochondrial dysfunction via accumulation of lipids and lipid intermediates, interfering with insulin signaling[19], is probably only partly correct. It is a consistent finding that lipids accumulate in insulin resistant muscle[3,20] and this is not a qualitative phenomenon (impaired mitochondrial respiration), but rather a quantitative phenomenon (decreased mitochondrial mass). The obvious question is therefore why the mitochondrial mass seems to be lower in the patients with type 2 diabetes? One explanation could be that the matching of the subjects is not optimal in the studies[12,18,21] where a lower mitochondrial content was reported. If the healthy control group and the patients with type 2 diabetes are carefully matched for physical activity level and maximal oxygen uptake, no differences exist in mitochondrial content, or intrinsic mitochondrial function[16]. Another question is the likelyhood of a marked decrease in mitochondrial content in the skeletal muscle of patients with type 2 diabetes. If a 30% decreased mitochondrial mass was indeed present in type 2 diabetes with a marked effect on respiratory capacity at rest (ex vivo), then one would expect that the in vivo exercise capacity would be severely impaired, because the mitochondrial respiratory rates increases more than ten-fold with the transition from rest to exercise. Although there may be some exercise intolerance in patients with type 2 diabetes[22], most can be explained by altered oxygen uptake kinetics[23,24] on the background of impaired peripheral blood flow distribution/microvascular function. If a reduction in the mitochondrial content in the exercising skeletal muscle was a major limitation, then one would expect that skeletal muscle arterio-venous oxygen extraction would be impaired in type 2 diabetes. This is not the case[25].
It is well known that physical exercise increases skeletal muscle insulin sensitivity in patients with type 2 diabetes[26]. Furthermore, it has been reported that improvements in insulin sensitivity is accompanied by improvements in in vivo mitochondrial function[27]. It has been suggested that insulin resistant people may have an attenuated response to exercise training, compared with healthy control subjects[28]. Furthermore it has been reported that the response to an acute bout of exercise is attenuated in insulin-resistant compared with lean control subjects[29], when investigating genes coding for mitochondrial biogenesis (PGC-1α mRNA and protein abundance), which could explain the lack of a training effect in patients with type 2 diabetes in some studies[30-32]. It has been reported that different molecular signals in the skeletal muscle are responsible for the activation of mitochondrial biogenesis after exercise. These signals include elevated levels of cytosolic Ca2+[33,34], AMP[33] and reactive oxygen species (ROS)[35]. All these studies are conducted in animals or cells, and have to our knowledge never been performed in patients with type 2 diabetes after an acute bout of exercise. An increased ROS production has also been linked to type 2 diabetes, but few human studies have actually investigated this and with conflicting results[8,10,36,37]. It has been reported in bovine aortic endothelial cells that hyperglycemia (30 mmol/L) increases ROS production[38].
This review will focus on adaptations in skeletal muscle mitochondria in patients with type 2 diabetes and healthy control participants after different exercise modalities (endurance, strength, high intensity training or a combination). Furthermore, we will attempt to clarify if the pharmacological treatment in patients with type 2 diabetes may blunt the training adaptations seen in non-diabetic people.
EFFECT OF MEDICATION ON EXERCISE ADAPTATIONS
Patients with type 2 diabetes are often treated with other medication to prevent high cholesterol and/or hypertension. In Denmark approximately 75% of all patients with diabetes are treated for hypertension, and approximately 64% are treated for hypercholesterolemia primarily with statins[39]. In Denmark approximately 90% of patients with type 2 diabetes are treated with metformin[39].
Antidiabetic agents
If a lifestyle intervention (diet and exercise) is not sufficient, metformin is the first drug of choice in the newly diagnosed patient with type 2 diabetes. Sulfonylurea may be added, and with poor glycemic control insulin treatment may be initiated. The adaptations to exercise are inadequately investigated when combined with these different medications. The mechanisms behind the glucose lowering effect of metformin is not known in detail, but a decrease in hepatic glucose production[40] and an increase in glucose disposal in skeletal muscle via activation of AMPK[41] contributes to this. Metformin does not stimulate insulin secretion. In contrast, the hypoglycaemic effect of sulfonylureas is mediated via activation of the insulin producing beta cells[42], and these drugs have no direct effect on liver or skeletal muscle.
It has been suggested that the glucose lowering effect of metformin takes place via an inhibition of complex I in the electron transport chain in the mitochondria[43-45]. One study conducted in patients treated with metformin (2000 ± 200 mg/d) reported no effect on complex I in the electron transport chain[15]. A therapeutic dose of metformin of 1000 mg in humans corresponds to a plasma metformin concentration of approximately 0.1 mmol/L[46,47], and the peak metformin concentration in skeletal muscle is much lower than in the plasma[48]. In the studies were an inhibition was seen on complex I after metformin treatment, the concentrations used were high and supraphysiological[43-45].
It has been investigated whether metformin has an effect of exercise adaptations in young healthy subjects. One study measured maximal oxygen uptake in a double-blinded, placebo-controlled, cross-over study in healthy men and women and found a 2.7% reduction in maximal oxygen uptake after 7-9 d of treatment[49]. The authors suggest that this is unlikely to cause any individual impairment in exercise tolerance. Whether the same reduction is seen in patients with type 2 diabetes needs to be investigated. Even though 2.7% is not a major reduction, it could be argued that patients with type 2 diabetes would suffer more from this, due to a potential lower starting point. However, this finding was not confirmed in a similar study, where solely males participated[50]. Rosiglitazone (thiazolidinedione) is another antiglycemic agents, which has been reported to increase maximal oxygen uptake after 4 mo of treatment[51], the mechanisms behind the improvement is unknown.
A large proportion of patients diagnosed with type 2 diabetes have other co-morbidities, such as obesity, hypertension and dyslipidemia, i.e., components of the metabolic syndrome. The pharmacological treatment of these may interfere with skeletal muscle and mitochondrial adapation to exercise training, and the literature regarding this issue will briefly be reviewed.
Lipid-lowering agents (statins)
Is has recently been reported that statins impairs the beneficial adaptations (increased maximal oxygen uptake and mitochondrial content) normally gained after a training intervention[52]. Different studies (longitudinal and cross-sectional) have reported an impaired mitochondrial function after statin therapy[53,54], which may compromise the OXPHOS capacity of the skeletal muscle. This would, in turn, further cause exercise intolerance. It has been suggested[54] that the culprit behind the impaired mitochondrial function, maybe a reduced coenzyme Q10 content in the skeletal muscle (Figure 1). It must be mentioned that not all studies have found a negative effect of statin therapy in combination with exercise[55]. In the study by Meex et al[55] many different statins were used, which could influence the result, since it is known that statins differs in lipophilicity[56], and thereby the ability to cross cell membranes. Is has also been reported that statins impaires complex I respiration in the electron transport chain[57]. Another group reported that simvastatin increased ROS production in human skeletal myotubes in combination with an impaired mitochondrial respiratory capacity[58]. To make it even more complex, it has been demonstrated that statins have opposite effects on mitochondria from cardiac and skeletal muscle[59]. Furthermore, studies have reported that statins have an effect (impairment or improvement) on insulin sensitivity (for review see[60]).
Figure 1.
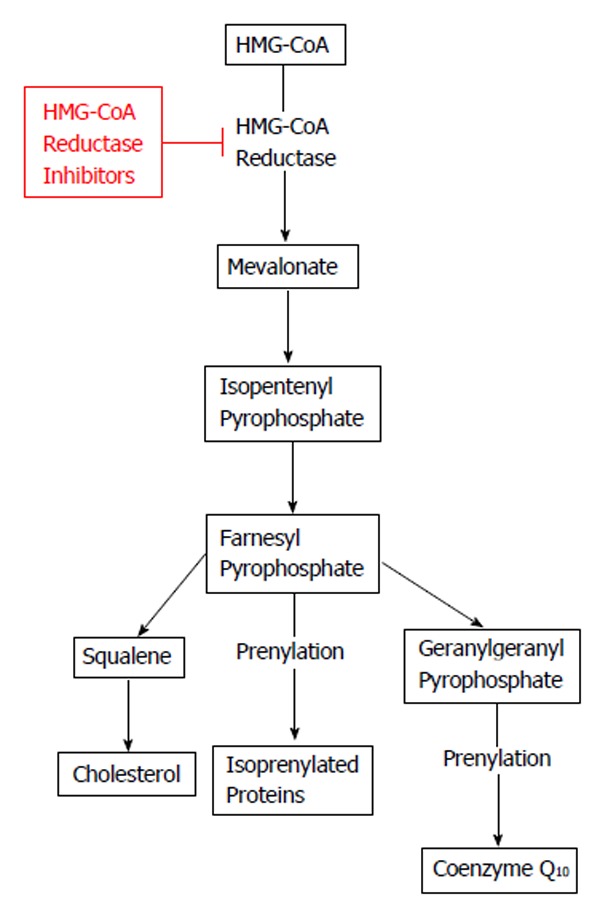
Effect of HMG-CoA reductase inhibitors (statins) on cholesterol and coenzyme Q10. HMG-CoA: 3-hydroxy-3-methylglutaryl-coenzyme A.
Antihypertensive agents
Diuretics, beta-blockers, calcium antagonists, ACE-inhibitors and angiotensin receptor blockers (ARBs) are all suitable for the initiation and maintenance of antihypertensive treatment, either as monotherapy or in combination therapy.
There are different kinds of β-blockers known as selective or nonselective. The selective can either block β1 (cardiac) or β2 (skeletal muscle) receptors, the nonselective ones blocks both receptors. The adaptations to exercise training can be influenced by using either kind[61]. Ades et al[62] investigated 10 wk of endurance training (4 times a week) in hypertensive patients, taking either metoprolol (β1 selective β-adrenergic blocker), propranolol (β1 nonselective β-adrenergic blocker) or placebo. They reported an improvement in maximal oxygen uptake and an increase in mitochondrial content (succinyl dehydrogenase activity) in the placebo and the metoprolol group, whereas no improvements after training was seen with propranolol[62]. Another group investigated 6 wk of endurance training in healthy young subjects[63]. The subjects were randomized to either a selective (atenolol) or nonselective (nadolol) β-adrenergic blocker or placebo. Subjects receiving placebo improved maximal oxygen uptake to a higher extent than the two groups receiving medication, but all groups improved maximal oxygen uptake from baseline. Furthermore mitochondrial content increased in all three groups after training, but again the placebo-group improved to a higher extent[63]. Similar results were reported by Svedenhag et al[64] in young healthy subjects after 8 wk of endurance training.
Drexler et al[65] investigated the short- and long-term effect of ACE inhibition on patients with congestive heart failure at rest and during exercise. They reported an improvement in oxygen extraction of the working muscle after ACE inhibition, and they speculate that this could be due to an increased mitochondrial content, but unfortunately muscle biopsies were not obtained to elucidate this[65]. In addition, studies have investigated the effect of ACE inhibitors on insulin sensitivity and found divergent results, with either no effect[66] or an improvement[67].
It has previously been reported that Angiotensin II receptor blockers (ARBs) have a positive effect on reactive oxygen species production and mitochondrial function in animals (for review see[68]). It has been reported that ARBs have different effects on glucose homeostasis in hypertensive patients with the metabolic syndrome[69]. Patients treated with telmisartan showed an improvement in HOMA-IR and HbA1c (surrogate measures of insulin sensitivity[70]), whereas patients treated with losartan showed no improvement[69]. Whether these improvements can be explained by improvements in mitochondrial function is at the moment impossible to say.
These results highlight the importance of controlling the medication when mitochondrial function and insulin sensitivity are measured before and after a training intervention. Otherwise the results obtained will be hard to explain. Furthermore, the interaction between the different drugs is also unknown, and would off course also be a confounding factor when results are interpretated.
MUSCULAR ADAPTATION TO DIFFERENT TRAINING MODALITIES IN PATIENTS WITH TYPE 2 DIABETES
It is well known that exercise interventions improve maximal oxygen uptake, mitochondrial content and insulin sensitivity in healthy subjects[71-75].
Different training modalities have been investigated in patients with type 2 diabetes and control subjects, to see if the training adaptation is similar in patients compared with control participants. Unfortunately many of the studies investigating the effect of exercise in patients with type 2 diabetes are lacking a healthy matched control group, which makes it impossible to compare the response between patients and control participants. Furthermore, the medication used is often not described in detail. In this review we primarily report the studies that have measured maximal oxygen uptake, mitochondrial function and insulin sensitivity (clamp, OGTT, HbA1c or fasting glucose and insulin concentrations).
Endurance training
Hey-Mogensen et al[8] investigated if 10 wk of endurance training affected mitochondrial function, maximal oxygen uptake and insulin sensitivity. The patients with type 2 diabetes in this study were treated with either metformin or sulfonylurea, other kinds of medication were not mentioned in the manuscript. A similar improvement in VO2max was seen in patients (12%) and control participants (16%). Insulin sensitivity was significantly increased after training in both control participants (22%) and patients (13%). Mitochondrial OXPHOS capacity and intrinsic mitochondrial function was measured in isolated mitochondria, with no differences between patients and control subjects, except for the increased capacity to oxidize long chain fatty acids after training in patients with type 2 diabetes which was not apparent in the control participants. This finding is in contrast to the hypothesis about reduced ability to oxidize lipids in patients with type 2 diabetes[76,77], and therefore it indicates that impaired insulin sensitivity is not caused by a reduced mitochondrial capacity for lipid oxidation. Furthermore, CS activity also increased similarly in the groups. Interestingly no differences were seen in PGC-1α (mRNA) after training in either patients or control participants. PGC-1α is a major regulator for mitochondrial biogenesis[78]. Mitochondrial ROS production was similar in the two groups and did not change significantly with training. An increased UCP3 protein content was seen, but only in the control participants[8]. It has previously been suggested that UCP3 is acting as a protective mechanism against ROS production[79]. No difference was seen in intrinsic mitochondrial respiratory function between patients and control participants in this study (both before and after training)[8], this finding is contradictory to another study (cross-sectional) from the same group, where a lower intrinsic mitochondrial function was seen in patients with type 2 diabetes[4]. Another study investigated the effect of a combination of endurance and strength training (12 wk)[17] and similar to the study by Hey-Mogensen et al[8] no information is available in the manuscript regarding other kinds of medication except for the glucose lowering agents (metformin or sulfonylurea). An increased VO2max was seen after training in the patients with type 2 diabetes, where only a tendency was seen in the control participants. An increased mitochondrial content (mtDNA) was seen after training in both groups, accompanied by a similar intrinsic mitochondrial function before and after training in both groups[17], indicating that mitochondrial OXPHOS capacity was increased to a similar extent in both groups (data not shown in the manuscript). It has recently been reported that mtDNA is not a good marker for mitochondrial content, at least not in healthy young subjects[80]. Phielix et al[5] has previously reported impaired intrinsic mitochondrial function in patients with type 2 diabetes, a finding that contradicts their own finding from 2010[17]. Meex et al[27] used the same training protocol as Phielix et al[17] with a combination of endurance and strength training for 12 wk. Again only glucose lowering medication is mentioned in the manuscript and thus not the pharmacological specification. Mitochondrial function was measured by magnetic resonance spectroscopy, and a difference was seen before training between the two groups with no difference present after training. Mitochondrial content was measured as complex I-V protein content (average of the complexes), both groups increased the average of the five complexes, but the increase tended to be more pronounced in the patients with type 2 diabetes. Maximal oxygen uptake increased significantly with training in both groups, whereas only patients with type 2 diabetes improved insulin sensitivity (clamp) after training[27]. Mogensen et al[81] conducted another study in which the effect of endurance training on skeletal muscle was studied. Again they showed a similar response in regard to maximal oxygen uptake, insulin sensitivity and mitochondrial content CS activity, where both groups improved in all parameters after training[81]. Nine months of aerobic training (150 min/wk at 50%-80% of VO2peak) in patients with type 2 diabetes did surprisingly not improve either mitochondrial content, maximal oxygen uptake or insulin sensitivity, but an increased lipid oxidation was present after training[31]. The patient’s medical records were not included in the manuscript, and no healthy control group was included. So the lack of improvement in mitochondrial content after 9 mo of aerobic training could be explained by the medication used (statins most likely). The study was an ancillary study to the HART-D study where the patients medical records are included, and a high percentage of the patients were in statin therapy[82]. Shaw et al[83] investigated 6 mo of endurance exercise (corresponding to approximately 77% of VO2peak), they found an increased maximal oxygen uptake and mitochondrial content (COX activity), but no difference in insulin sensitivity. They did not report the medication used, but states that medication was stopped three days prior to the test days, indicating that the patients were on medication during the training period, and in addition an appropriate control group was not investigated. Another group investigated lean, obese and patients with type 2 diabetes before and after 10 d of 60 min exercise at 70% of VO2peak[32]. No differences were seen in muscle oxidative capacity between groups before and after training, which is quite intriguing taking into consideration that the lean subjects had a higher maximal oxygen uptake (approximately 50%) compared with the two other groups. Insulin sensitivity was unfortunately not measured[32]. Mitochondrial volume (by TEM) was investigated after 10 wk of endurance training (approximately 70% of VO2max) and a similar increase in mitochondrial volume was seen in patients with type 2 diabetes and control participants, accompanied by improvements in maximal oxygen uptake and insulin sensitivity (clamp)[84]. Table 1 gives an overview over the published literature in regard to endurance training.
Table 1.
Effect of endurance training on maximal oxygen uptake, mitochondrial function and insulin sensitivity
| Ref. | Subjects | Training | Duration | VO2max | Mito | IS |
| Mogensen et al[81] | T2DM and CON | ET | 10 wk (2-3 times/wk) | ↑ T2DM | ↑ T2DM | ↑ T2DM |
| ↑ CON | ↑ CON | ↑ CON | ||||
| Hey-Mogensen et al[8] | T2DM and CON | ET | 10 wk (4-5 times/wk) | ↑ T2DM | ↑ T2DM | ↑ T2DM |
| ↑ CON | ↑ CON | ↑ CON | ||||
| Phielix et al[17] | T2DM and CON | ET and ST | 12 wk (3 times/wk) | ↑ T2DM | ↑ T2DM | ↑ T2DM |
| → CON | ↑ CON | → CON | ||||
| Meex et al[27] | T2DM and CON | ET and ST | 12 wk (3 times/wk) | ↑ T2DM | ↑ T2DM | ↑ T2DM |
| ↑ CON | ↑ CON | → CON | ||||
| Shaw et al[83] | T2DM | ET | 6 mo (3 times/wk) | ↑ T2DM | ↑ T2DM | → T2DM |
| Sparks et al[31] | T2DM and | ET | 9 mo (150 min/wk) | → T2DM | → T2DM | → T2DM |
| Bajpeyi et al[32] | T2DM and CON (L and O) | ET | 10 d (every day) | ND | → T2DM | ND |
| → CON (L and O) | ||||||
| Nielsen et al[84] | T2DM and CON | ET | 10 wk (4-5 times/wk) | ↑ T2DM | ↑ T2DM | ↑ T2DM |
| ↑ CON | ↑ CON | ↑ CON |
CON: Control participants; ET: Endurance training; IS: Insulin sensitivity [or surrogate measures of insulin sensitivity (HbA1c, HOMA)]; L: Lean; O: Obese; VO2max: Maximal oxygen uptake; Mito: Mitochondrial function (mitochondrial respiratory capacity, mitochondrial content); ND: Not determined; T2DM: Patients with type 2 diabetes.
High intensity training
The last five to ten years a renewed interest has been directed towards a different training method, where high intensity training is performed for shorter durations. It has been reported that high intensity training (HIT) leads to similar metabolic adaptations compared to regular endurance training when it comes to improvement in maximal oxygen uptake and increase in mitochondrial content in healthy human skeletal muscle[72,73]. This has not been investigated thoroughly in patients with type 2 diabetes.
Two weeks of HIT has been reported to increase mitochondrial content (CS activity) and improve 24 h blood glucose profile (measured 48-72 h after last training bout)[85] in patients with type 2 diabetes. Unfortunately no control group was included by Little and colleagues[85], but in another study a similar improvement in CS activity was observed in overweight women using the same training protocol[86].
We have recently investigated mitochondrial substrate sensitivity in patients with type 2 diabetes and control participants after two weeks (eight training sessions) of one legged HIT [pilot study (type 2 diabetes; n = 5-7; control subjects; n = 3-5)]. Each training session consisted of ten one minute bouts of high intense one-legged bicycle exercise interspersed with one minute recovery. The training load corresponded to minimum 60% of the maximal workload obtained during a one-legged maximal oxygen uptake test. Due to the low number of subjects investigated we did not perform any statistical analysis on the dataset. We used high resolution respirometry and measured the mitochondrial ability to use either octanoyl carnitine (medium chain fatty acid), palmitoyl coenzyme A (long chain fatty acid, using carnitine palmitoyltransferase I (CPT I) to enter the mitochondrion) and palmitoyl carnitine (long chain fatty acid, using CPT II to enter the mitochondrion). The results from the pilot study (respirometric measurements) are provided in Figures 2-4. The method we used has been described previously[12,16]. No differences were seen in mitochondrial substrate sensitivity for octanoyl carnitine between the groups and both groups increased their sensitivity for octanoyl carnitine in the trained leg (Figure 2A). It has been reported previously that no differences are present in mitochondrial subtrate sensitivity with octanoyl carnitine between patients with type 2 diabetes and obese participants[16]. No effect was seen after training in regard to maximal mitochondrial oxidative capacity with octanoyl carnitine as a substrate, but is seems as the patients with type 2 diabetes have a higher capacity to oxidize medium chain fatty acids (Figure 2B). Mitochondrial substrate sensitivity for palmitoyl coenzyme A (Figure 3A) and palmitoyl carnitine (Figure 4A) showed the same tendency where patients with type 2 diabetes had a decreased (apparent Km increased) sensitivity for long chain fatty acid (palmitoyl coenzyme A and carnitine) and control participants an increased (apparent Km decreased) sensitivity after training. No major differences were seen in maximal mitochondrial oxidative capacity with palmitoyl coenzyme A (Figure 3B) or palmitoyl carnitine (Figure 4B) between the groups and after the training intervention. From these pilot data, it seems as if no differences are present between patients and control participants in regard to maximal mitochondrial oxidative capacity with fatty acids as substrate either at baseline or after the training intervention. The improved sensitivity for CPT I and CPT II in the control participants, could be explained by an increased activity of CPT I (and maybe CPT II) which have been reported previously[87]. Why patients with type 2 diabetes show an opposite adaptation is difficult to explain, but it has been reported that CPT I activity is reduced in skeletal muscle from obese compared to lean participants[88]. To our knowledge the effect of training on CPT I and II activity has never been investigated in patients with type 2 diabetes, and it is thus impossible to say whether this can explain our results. Little et al[85] gives an overview over the published literature in regard to high intensity training.
Figure 2.
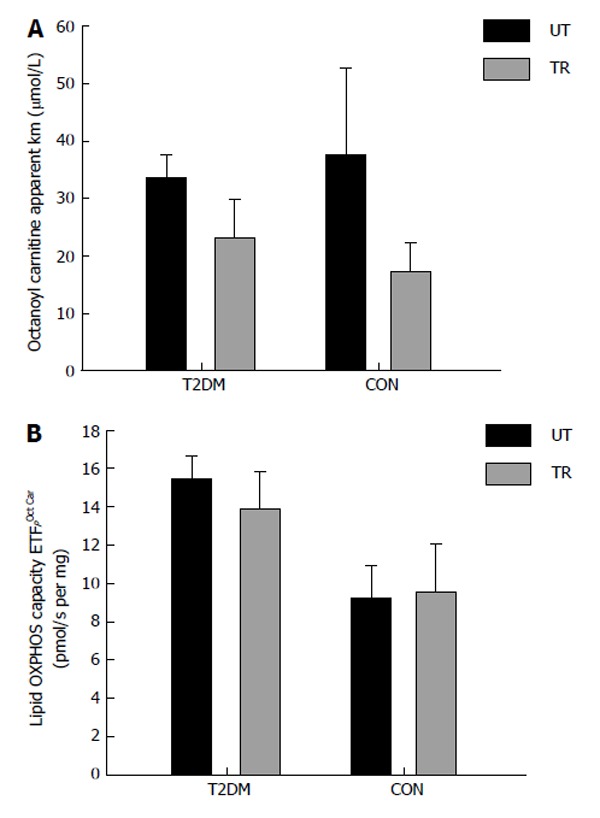
Patients with type 2 diabetes (n = 5) and healthy control subjects (n = 5) performed eight sessions of one-legged high intensity training in two weeks. Each session consisted of ten one-minute exercise bouts at 60% of one-legged maximal oxygen uptake and > 80% of maximal heart rate, interspersed by one min rest. After completion of the training muscle biopsies (vastus lateralis) were obtained from the untrained (black bars) and the trained (grey bars) leg. The measurement mitochondrial OXPHOS capacity and substrate sensitivity was performed with malate, ADP and octanoyl carnitine (titration: 5-2000 μmol/L). A: Apparent Michaelis Menten constant Km for octanoyl carnitine; B: Maximal OXPHOS capacity with the mentioned substrates. T2DM: Type 2 diabetes; CON: Control subjects; UT: Untrained; TR: Trained.
Figure 4.
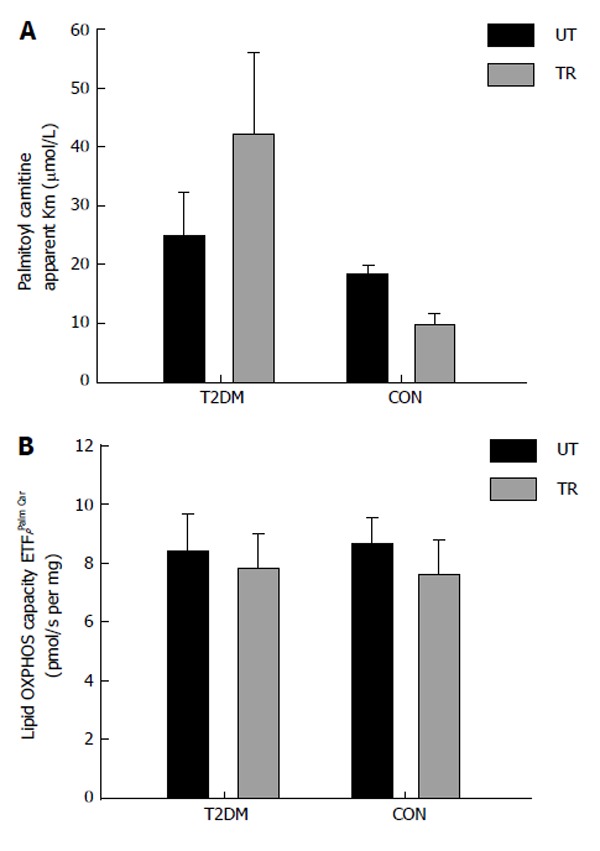
Patients with type 2 diabetes (n = 7) and healthy control subjects (n = 5) performed eight sessions of one-legged high intensity training in two weeks. Each session consisted of ten one-minute exercise bouts at 60% of one-legged maximal oxygen uptake and > 80% of maximal heart rate, interspersed by one min rest. After completion of the training muscle biopsies (vastus lateralis) were obtained from the untrained (black bars) and the trained (grey bars) leg. The measurement of mitochondrial OXPHOS capacity and substrate sensitivity was performed with malate, ADP and palmitoyl carnitine (titration: 5-200 μmol/L). A: Apparent Michaelis Menten constant Km for palmitoyl carnitine; B: Maximal OXPHOS capacity with the mentioned substrates. T2DM: Type 2 diabetes; CON: Control subjects; UT: Untrained; TR: Trained.
Figure 3.
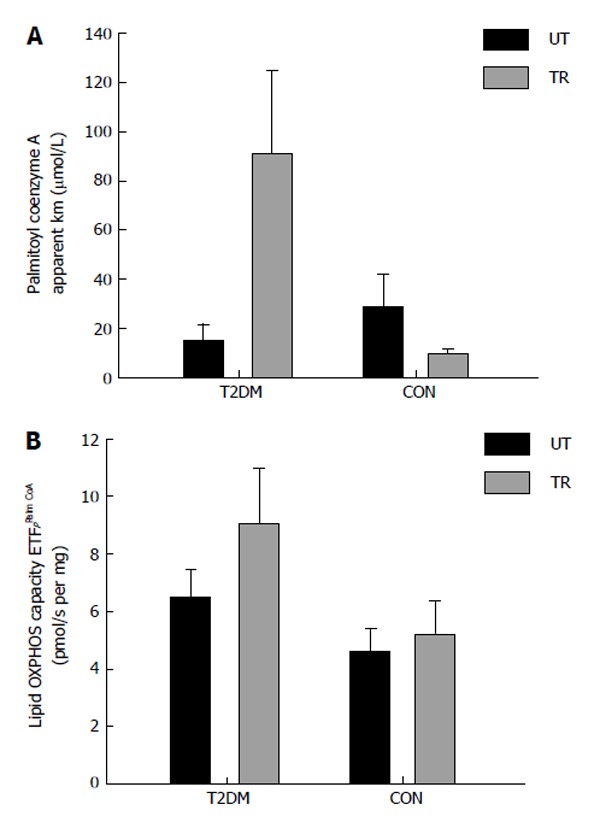
Patients with type 2 diabetes (n = 5) and healthy control subjects (n = 3) performed eight sessions of one-legged high intensity training in two weeks. Each session consisted of ten one-minute exercise bouts at 60% of one-legged maximal oxygen uptake and > 80% of maximal heart rate, interspersed by one min rest. After completion of the training muscle biopsies (vastus lateralis) were obtained from the untrained (black bars) and the trained (grey bars) leg. The measurement mitochondrial OXPHOS capacity and substrate sensitivity was performed with malate, ADP and palmitoyl coenzyme A (titration: 5-100 μmol/L). A: Apparent Michaelis Menten constant Km for palmitoyl coenzyme A; B: Maximal OXPHOS capacity with the mentioned substrates. T2DM: Type 2 diabetes; CON: Control subjects; UT: Untrained; TR: Trained.
Strength training
Is has been suggested previously that strength training represents an attractive training modality, due to the fact that many patients with type 2 diabetes are obese and have difficulties performing endurance exercise.
Few studies have been performed where adaptations in skeletal muscle have been investigated. Holten et al[30] investigated 6 wk (3 times per week) of leg strength training (one leg, other leg served as control) and found improvement in the trained leg in both groups regarding insulin sensitivity (clamp technique). Maximal oxygen uptake was not measured, but mitochondrial content (by CS activity) showed no difference between the legs in the patients but an increase was observed in the control participants. Nine months of resistance training increased mitochondrial content in patients with type 2 diabetes, but no difference was seen in maximal oxygen uptake and HbA1c[31]. This study contradicts the findings by Holten et al[30], and this may be due to a difference in duration and application of different methods to evaluate insulin sensitivity. Table 2 gives an overview over the published literature in regard to strength training.
Table 2.
Effect of strength training on maximal oxygen uptake, mitochondrial function and insulin sensitivity
| Ref. | Subjects | Training | Duration | VO2max | Mito | IS |
| Holten et al[30] | ST (one leg) | 6 wk (3 times/wk) | ND | → T2DM | ↑ T2DM | |
| ↑ CON | ↑ CON | |||||
| Sparks et al[31] | T2DM | ST | 9 mo (3 times/wk) | → T2DM | ↑ T2DM | → T2DM |
CON: Control participants; IS: Insulin sensitivity [or surrogate measures of insulin sensitivity (HbA1c, HOMA)]; VO2max: Maximal oxygen uptake; Mito: Mitochondrial function (mitochondrial respiratory capacity, mitochondrial content); ND: Not determined; ST: Strength training; T2DM: Patients with type 2 diabetes.
CONCLUSION
From the literature currently available it is difficult to recommend a training intervention to patients with type 2 diabetes where success is well documented when it comes to improvement in mitochondrial function. The problem with many of the studies available is that medicine usage is not reported, and therefore potential significant medication effects on the outcome can not be excluded, when adaptations to physical activity are investigated. Furthermore, many of the studies lack a real control group, making it impossible to determine if adaptations are the same in patients and control participants.
The literature is at current lacking well conducted controlled longitudinal studies investigating the effect of exercise on mitochondrial function, where medication is controlled and an appropriate control group is included. These studies are difficult to conduct given the ethical problem in how you control the medication without compromising and disrupting the health of the patients. One approach could be to recruite newly diagnosed patients, where medication is not started yet. A study like this needs to be conducted in the future where mitochondrial function is investigated.
Footnotes
P- Reviewer: Davison GW S- Editor: Song XX L- Editor: A E- Editor: Liu SQ
References
- 1.Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58:773–795. doi: 10.2337/db09-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abdul-Ghani MA, DeFronzo RA. Pathogenesis of insulin resistance in skeletal muscle. J Biomed Biotechnol. 2010;2010:476279. doi: 10.1155/2010/476279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mogensen M, Sahlin K, Fernström M, Glintborg D, Vind BF, Beck-Nielsen H, Højlund K. Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes. 2007;56:1592–1599. doi: 10.2337/db06-0981. [DOI] [PubMed] [Google Scholar]
- 5.Phielix E, Schrauwen-Hinderling VB, Mensink M, Lenaers E, Meex R, Hoeks J, Kooi ME, Moonen-Kornips E, Sels JP, Hesselink MK, et al. Lower intrinsic ADP-stimulated mitochondrial respiration underlies in vivo mitochondrial dysfunction in muscle of male type 2 diabetic patients. Diabetes. 2008;57:2943–2949. doi: 10.2337/db08-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schrauwen-Hinderling VB, Roden M, Kooi ME, Hesselink MK, Schrauwen P. Muscular mitochondrial dysfunction and type 2 diabetes mellitus. Curr Opin Clin Nutr Metab Care. 2007;10:698–703. doi: 10.1097/MCO.0b013e3282f0eca9. [DOI] [PubMed] [Google Scholar]
- 7.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 8.Hey-Mogensen M, Højlund K, Vind BF, Wang L, Dela F, Beck-Nielsen H, Fernström M, Sahlin K. Effect of physical training on mitochondrial respiration and reactive oxygen species release in skeletal muscle in patients with obesity and type 2 diabetes. Diabetologia. 2010;53:1976–1985. doi: 10.1007/s00125-010-1813-x. [DOI] [PubMed] [Google Scholar]
- 9.Nair KS, Bigelow ML, Asmann YW, Chow LS, Coenen-Schimke JM, Klaus KA, Guo ZK, Sreekumar R, Irving BA. Asian Indians have enhanced skeletal muscle mitochondrial capacity to produce ATP in association with severe insulin resistance. Diabetes. 2008;57:1166–1175. doi: 10.2337/db07-1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chanseaume E, Barquissau V, Salles J, Aucouturier J, Patrac V, Giraudet C, Gryson C, Duché P, Boirie Y, Chardigny JM, et al. Muscle mitochondrial oxidative phosphorylation activity, but not content, is altered with abdominal obesity in sedentary men: synergism with changes in insulin sensitivity. J Clin Endocrinol Metab. 2010;95:2948–2956. doi: 10.1210/jc.2009-1938. [DOI] [PubMed] [Google Scholar]
- 11.De Feyter HM, Lenaers E, Houten SM, Schrauwen P, Hesselink MK, Wanders RJ, Nicolay K, Prompers JJ. Increased intramyocellular lipid content but normal skeletal muscle mitochondrial oxidative capacity throughout the pathogenesis of type 2 diabetes. FASEB J. 2008;22:3947–3955. doi: 10.1096/fj.08-112318. [DOI] [PubMed] [Google Scholar]
- 12.Boushel R, Gnaiger E, Schjerling P, Skovbro M, Kraunsøe R, Dela F. Patients with type 2 diabetes have normal mitochondrial function in skeletal muscle. Diabetologia. 2007;50:790–796. doi: 10.1007/s00125-007-0594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Larsen S, Ara I, Rabøl R, Andersen JL, Boushel R, Dela F, Helge JW. Are substrate use during exercise and mitochondrial respiratory capacity decreased in arm and leg muscle in type 2 diabetes? Diabetologia. 2009;52:1400–1408. doi: 10.1007/s00125-009-1353-4. [DOI] [PubMed] [Google Scholar]
- 14.Rabøl R, Larsen S, Højberg PM, Almdal T, Boushel R, Haugaard SB, Andersen JL, Madsbad S, Dela F. Regional anatomic differences in skeletal muscle mitochondrial respiration in type 2 diabetes and obesity. J Clin Endocrinol Metab. 2010;95:857–863. doi: 10.1210/jc.2009-1844. [DOI] [PubMed] [Google Scholar]
- 15.Larsen S, Rabøl R, Hansen CN, Madsbad S, Helge JW, Dela F. Metformin-treated patients with type 2 diabetes have normal mitochondrial complex I respiration. Diabetologia. 2012;55:443–449. doi: 10.1007/s00125-011-2340-0. [DOI] [PubMed] [Google Scholar]
- 16.Larsen S, Stride N, Hey-Mogensen M, Hansen CN, Andersen JL, Madsbad S, Worm D, Helge JW, Dela F. Increased mitochondrial substrate sensitivity in skeletal muscle of patients with type 2 diabetes. Diabetologia. 2011;54:1427–1436. doi: 10.1007/s00125-011-2098-4. [DOI] [PubMed] [Google Scholar]
- 17.Phielix E, Meex R, Moonen-Kornips E, Hesselink MK, Schrauwen P. Exercise training increases mitochondrial content and ex vivo mitochondrial function similarly in patients with type 2 diabetes and in control individuals. Diabetologia. 2010;53:1714–1721. doi: 10.1007/s00125-010-1764-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asmann YW, Stump CS, Short KR, Coenen-Schimke JM, Guo Z, Bigelow ML, Nair KS. Skeletal muscle mitochondrial functions, mitochondrial DNA copy numbers, and gene transcript profiles in type 2 diabetic and nondiabetic subjects at equal levels of low or high insulin and euglycemia. Diabetes. 2006;55:3309–3319. doi: 10.2337/db05-1230. [DOI] [PubMed] [Google Scholar]
- 19.Kelley DE, Goodpaster B, Wing RR, Simoneau JA. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am J Physiol. 1999;277:E1130–E1141. doi: 10.1152/ajpendo.1999.277.6.E1130. [DOI] [PubMed] [Google Scholar]
- 20.Petersen KF, Dufour S, Shulman GI. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med. 2005;2:e233. doi: 10.1371/journal.pmed.0020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- 22.Regensteiner JG, Sippel J, McFarling ET, Wolfel EE, Hiatt WR. Effects of non-insulin-dependent diabetes on oxygen consumption during treadmill exercise. Med Sci Sports Exerc. 1995;27:875–881. [PubMed] [Google Scholar]
- 23.Bauer TA, Reusch JE, Levi M, Regensteiner JG. Skeletal muscle deoxygenation after the onset of moderate exercise suggests slowed microvascular blood flow kinetics in type 2 diabetes. Diabetes Care. 2007;30:2880–2885. doi: 10.2337/dc07-0843. [DOI] [PubMed] [Google Scholar]
- 24.Regensteiner JG, Bauer TA, Reusch JE, Brandenburg SL, Sippel JM, Vogelsong AM, Smith S, Wolfel EE, Eckel RH, Hiatt WR. Abnormal oxygen uptake kinetic responses in women with type II diabetes mellitus. J Appl Physiol (1985) 1998;85:310–317. doi: 10.1152/jappl.1998.85.1.310. [DOI] [PubMed] [Google Scholar]
- 25.Dela F, Mikines KJ, Larsen JJ, Galbo H. Glucose clearance in aged trained skeletal muscle during maximal insulin with superimposed exercise. J Appl Physiol (1985) 1999;87:2059–2067. doi: 10.1152/jappl.1999.87.6.2059. [DOI] [PubMed] [Google Scholar]
- 26.Dela F, Larsen JJ, Mikines KJ, Ploug T, Petersen LN, Galbo H. Insulin-stimulated muscle glucose clearance in patients with NIDDM. Effects of one-legged physical training. Diabetes. 1995;44:1010–1020. doi: 10.2337/diab.44.9.1010. [DOI] [PubMed] [Google Scholar]
- 27.Meex RC, Schrauwen-Hinderling VB, Moonen-Kornips E, Schaart G, Mensink M, Phielix E, van de Weijer T, Sels JP, Schrauwen P, Hesselink MK. Restoration of muscle mitochondrial function and metabolic flexibility in type 2 diabetes by exercise training is paralleled by increased myocellular fat storage and improved insulin sensitivity. Diabetes. 2010;59:572–579. doi: 10.2337/db09-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fritz T, Krämer DK, Karlsson HK, Galuska D, Engfeldt P, Zierath JR, Krook A. Low-intensity exercise increases skeletal muscle protein expression of PPARdelta and UCP3 in type 2 diabetic patients. Diabetes Metab Res Rev. 2006;22:492–498. doi: 10.1002/dmrr.656. [DOI] [PubMed] [Google Scholar]
- 29.De Filippis E, Alvarez G, Berria R, Cusi K, Everman S, Meyer C, Mandarino LJ. Insulin-resistant muscle is exercise resistant: evidence for reduced response of nuclear-encoded mitochondrial genes to exercise. Am J Physiol Endocrinol Metab. 2008;294:E607–E614. doi: 10.1152/ajpendo.00729.2007. [DOI] [PubMed] [Google Scholar]
- 30.Holten MK, Zacho M, Gaster M, Juel C, Wojtaszewski JF, Dela F. Strength training increases insulin-mediated glucose uptake, GLUT4 content, and insulin signaling in skeletal muscle in patients with type 2 diabetes. Diabetes. 2004;53:294–305. doi: 10.2337/diabetes.53.2.294. [DOI] [PubMed] [Google Scholar]
- 31.Sparks LM, Johannsen NM, Church TS, Earnest CP, Moonen-Kornips E, Moro C, Hesselink MK, Smith SR, Schrauwen P. Nine months of combined training improves ex vivo skeletal muscle metabolism in individuals with type 2 diabetes. J Clin Endocrinol Metab. 2013;98:1694–1702. doi: 10.1210/jc.2012-3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bajpeyi S, Reed MA, Molskness S, Newton C, Tanner CJ, McCartney JS, Houmard JA. Effect of short-term exercise training on intramyocellular lipid content. Appl Physiol Nutr Metab. 2012;37:822–828. doi: 10.1139/h2012-051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McConell GK, Ng GP, Phillips M, Ruan Z, Macaulay SL, Wadley GD. Central role of nitric oxide synthase in AICAR and caffeine-induced mitochondrial biogenesis in L6 myocytes. J Appl Physiol (1985) 2010;108:589–595. doi: 10.1152/japplphysiol.00377.2009. [DOI] [PubMed] [Google Scholar]
- 34.Baar K, Song Z, Semenkovich CF, Jones TE, Han DH, Nolte LA, Ojuka EO, Chen M, Holloszy JO. Skeletal muscle overexpression of nuclear respiratory factor 1 increases glucose transport capacity. FASEB J. 2003;17:1666–1673. doi: 10.1096/fj.03-0049com. [DOI] [PubMed] [Google Scholar]
- 35.Irrcher I, Ljubicic V, Hood DA. Interactions between ROS and AMP kinase activity in the regulation of PGC-1alpha transcription in skeletal muscle cells. Am J Physiol Cell Physiol. 2009;296:C116–C123. doi: 10.1152/ajpcell.00267.2007. [DOI] [PubMed] [Google Scholar]
- 36.Abdul-Ghani MA, Jani R, Chavez A, Molina-Carrion M, Tripathy D, Defronzo RA. Mitochondrial reactive oxygen species generation in obese non-diabetic and type 2 diabetic participants. Diabetologia. 2009;52:574–582. doi: 10.1007/s00125-009-1264-4. [DOI] [PubMed] [Google Scholar]
- 37.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW, Kang L, Rabinovitch PS, Szeto HH, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 39.Statens Serum Institut. 2010. Available from: http: //www.ssi.dk/Sundhedsdataogit/Analyser og rapporter/Lagemiddelforbrugsanalyser/Laegemiddelgrupper/~/media/Indhold/DK - dansk/Sundhedsdata og it/NSF/Analyser og rapporter/Laegemiddelforbrugsanalyser/2010/Polyfarmaci i diabetesbehandlingen.ashx.
- 40.Stumvoll M, Nurjhan N, Perriello G, Dailey G, Gerich JE. Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N Engl J Med. 1995;333:550–554. doi: 10.1056/NEJM199508313330903. [DOI] [PubMed] [Google Scholar]
- 41.Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P, Rooyackers O, Zhou G, Williamson JM, Ljunqvist O, Efendic S, et al. Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes. 2002;51:2074–2081. doi: 10.2337/diabetes.51.7.2074. [DOI] [PubMed] [Google Scholar]
- 42.Groop LC. Sulfonylureas in NIDDM. Diabetes Care. 1992;15:737–754. doi: 10.2337/diacare.15.6.737. [DOI] [PubMed] [Google Scholar]
- 43.Brunmair B, Staniek K, Gras F, Scharf N, Althaym A, Clara R, Roden M, Gnaiger E, Nohl H, Waldhäusl W, et al. Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes. 2004;53:1052–1059. doi: 10.2337/diabetes.53.4.1052. [DOI] [PubMed] [Google Scholar]
- 44.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348 Pt 3:607–614. [PMC free article] [PubMed] [Google Scholar]
- 45.El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 46.Scheen AJ. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 1996;30:359–371. doi: 10.2165/00003088-199630050-00003. [DOI] [PubMed] [Google Scholar]
- 47.Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334:574–579. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- 48.Wilcock C, Bailey CJ. Accumulation of metformin by tissues of the normal and diabetic mouse. Xenobiotica. 1994;24:49–57. doi: 10.3109/00498259409043220. [DOI] [PubMed] [Google Scholar]
- 49.Braun B, Eze P, Stephens BR, Hagobian TA, Sharoff CG, Chipkin SR, Goldstein B. Impact of metformin on peak aerobic capacity. Appl Physiol Nutr Metab. 2008;33:61–67. doi: 10.1139/H07-144. [DOI] [PubMed] [Google Scholar]
- 50.Johnson ST, Robert C, Bell GJ, Bell RC, Lewanczuk RZ, Boulé NG. Acute effect of metformin on exercise capacity in active males. Diabetes Obes Metab. 2008;10:747–754. doi: 10.1111/j.1463-1326.2007.00805.x. [DOI] [PubMed] [Google Scholar]
- 51.Regensteiner JG, Bauer TA, Reusch JE. Rosiglitazone improves exercise capacity in individuals with type 2 diabetes. Diabetes Care. 2005;28:2877–2883. doi: 10.2337/diacare.28.12.2877. [DOI] [PubMed] [Google Scholar]
- 52.Mikus CR, Boyle LJ, Borengasser SJ, Oberlin DJ, Naples SP, Fletcher J, Meers GM, Ruebel M, Laughlin MH, Dellsperger KC, et al. Simvastatin impairs exercise training adaptations. J Am Coll Cardiol. 2013;62:709–714. doi: 10.1016/j.jacc.2013.02.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Päivä H, Thelen KM, Van Coster R, Smet J, De Paepe B, Mattila KM, Laakso J, Lehtimäki T, von Bergmann K, Lütjohann D, et al. High-dose statins and skeletal muscle metabolism in humans: a randomized, controlled trial. Clin Pharmacol Ther. 2005;78:60–68. doi: 10.1016/j.clpt.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 54.Larsen S, Stride N, Hey-Mogensen M, Hansen CN, Bang LE, Bundgaard H, Nielsen LB, Helge JW, Dela F. Simvastatin effects on skeletal muscle: relation to decreased mitochondrial function and glucose intolerance. J Am Coll Cardiol. 2013;61:44–53. doi: 10.1016/j.jacc.2012.09.036. [DOI] [PubMed] [Google Scholar]
- 55.Meex RC, Phielix E, Schrauwen-Hinderling VB, Moonen-Kornips E, Schaart G, Schrauwen P, Hesselink MK. The use of statins potentiates the insulin-sensitizing effect of exercise training in obese males with and without Type 2 diabetes. Clin Sci (Lond) 2010;119:293–301. doi: 10.1042/CS20100153. [DOI] [PubMed] [Google Scholar]
- 56.Chong PH, Seeger JD, Franklin C. Clinically relevant differences between the statins: implications for therapeutic selection. Am J Med. 2001;111:390–400. doi: 10.1016/s0002-9343(01)00870-1. [DOI] [PubMed] [Google Scholar]
- 57.Sirvent P, Bordenave S, Vermaelen M, Roels B, Vassort G, Mercier J, Raynaud E, Lacampagne A. Simvastatin induces impairment in skeletal muscle while heart is protected. Biochem Biophys Res Commun. 2005;338:1426–1434. doi: 10.1016/j.bbrc.2005.10.108. [DOI] [PubMed] [Google Scholar]
- 58.Kwak HB, Thalacker-Mercer A, Anderson EJ, Lin CT, Kane DA, Lee NS, Cortright RN, Bamman MM, Neufer PD. Simvastatin impairs ADP-stimulated respiration and increases mitochondrial oxidative stress in primary human skeletal myotubes. Free Radic Biol Med. 2012;52:198–207. doi: 10.1016/j.freeradbiomed.2011.10.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bouitbir J, Charles AL, Echaniz-Laguna A, Kindo M, Daussin F, Auwerx J, Piquard F, Geny B, Zoll J. Opposite effects of statins on mitochondria of cardiac and skeletal muscles: a ‘mitohormesis’ mechanism involving reactive oxygen species and PGC-1. Eur Heart J. 2012;33:1397–1407. doi: 10.1093/eurheartj/ehr224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koh KK, Sakuma I, Quon MJ. Differential metabolic effects of distinct statins. Atherosclerosis. 2011;215:1–8. doi: 10.1016/j.atherosclerosis.2010.10.036. [DOI] [PubMed] [Google Scholar]
- 61.Robinson MM, Hamilton KL, Miller BF. The interactions of some commonly consumed drugs with mitochondrial adaptations to exercise. J Appl Physiol (1985) 2009;107:8–16. doi: 10.1152/japplphysiol.00343.2009. [DOI] [PubMed] [Google Scholar]
- 62.Ades PA, Gunther PG, Meyer WL, Gibson TC, Maddalena J, Orfeo T. Cardiac and skeletal muscle adaptations to training in systemic hypertension and effect of beta blockade (metoprolol or propranolol) Am J Cardiol. 1990;66:591–596. doi: 10.1016/0002-9149(90)90486-k. [DOI] [PubMed] [Google Scholar]
- 63.Wolfel EE, Hiatt WR, Brammell HL, Carry MR, Ringel SP, Travis V, Horwitz LD. Effects of selective and nonselective beta-adrenergic blockade on mechanisms of exercise conditioning. Circulation. 1986;74:664–674. doi: 10.1161/01.cir.74.4.664. [DOI] [PubMed] [Google Scholar]
- 64.Svedenhag J, Henriksson J, Juhlin-Dannfelt A. Beta-adrenergic blockade and training in human subjects: effects on muscle metabolic capacity. Am J Physiol. 1984;247:E305–E311. doi: 10.1152/ajpendo.1984.247.3.E305. [DOI] [PubMed] [Google Scholar]
- 65.Drexler H, Banhardt U, Meinertz T, Wollschläger H, Lehmann M, Just H. Contrasting peripheral short-term and long-term effects of converting enzyme inhibition in patients with congestive heart failure. A double-blind, placebo-controlled trial. Circulation. 1989;79:491–502. doi: 10.1161/01.cir.79.3.491. [DOI] [PubMed] [Google Scholar]
- 66.Reisin E, Weir MR, Falkner B, Hutchinson HG, Anzalone DA, Tuck ML. Lisinopril versus hydrochlorothiazide in obese hypertensive patients: a multicenter placebo-controlled trial. Treatment in Obese Patients With Hypertension (TROPHY) Study Group. Hypertension. 1997;30:140–145. doi: 10.1161/01.hyp.30.1.140. [DOI] [PubMed] [Google Scholar]
- 67.Galletti F, Strazzullo P, Capaldo B, Carretta R, Fabris F, Ferrara LA, Glorioso N, Semplicini A, Mancini M. Controlled study of the effect of angiotensin converting enzyme inhibition versus calcium-entry blockade on insulin sensitivity in overweight hypertensive patients: Trandolapril Italian Study (TRIS) J Hypertens. 1999;17:439–445. doi: 10.1097/00004872-199917030-00018. [DOI] [PubMed] [Google Scholar]
- 68.de Cavanagh EM, Inserra F, Ferder L. Angiotensin II blockade: a strategy to slow ageing by protecting mitochondria? Cardiovasc Res. 2011;89:31–40. doi: 10.1093/cvr/cvq285. [DOI] [PubMed] [Google Scholar]
- 69.Vitale C, Mercuro G, Castiglioni C, Cornoldi A, Tulli A, Fini M, Volterrani M, Rosano GM. Metabolic effect of telmisartan and losartan in hypertensive patients with metabolic syndrome. Cardiovasc Diabetol. 2005;4:6. doi: 10.1186/1475-2840-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lorenzo C, Haffner SM, Stancáková A, Laakso M. Relation of direct and surrogate measures of insulin resistance to cardiovascular risk factors in nondiabetic finnish offspring of type 2 diabetic individuals. J Clin Endocrinol Metab. 2010;95:5082–5090. doi: 10.1210/jc.2010-1144. [DOI] [PubMed] [Google Scholar]
- 71.Richards JC, Johnson TK, Kuzma JN, Lonac MC, Schweder MM, Voyles WF, Bell C. Short-term sprint interval training increases insulin sensitivity in healthy adults but does not affect the thermogenic response to beta-adrenergic stimulation. J Physiol. 2010;588:2961–2972. doi: 10.1113/jphysiol.2010.189886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Burgomaster KA, Howarth KR, Phillips SM, Rakobowchuk M, Macdonald MJ, McGee SL, Gibala MJ. Similar metabolic adaptations during exercise after low volume sprint interval and traditional endurance training in humans. J Physiol. 2008;586:151–160. doi: 10.1113/jphysiol.2007.142109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gibala MJ, Little JP, van Essen M, Wilkin GP, Burgomaster KA, Safdar A, Raha S, Tarnopolsky MA. Short-term sprint interval versus traditional endurance training: similar initial adaptations in human skeletal muscle and exercise performance. J Physiol. 2006;575:901–911. doi: 10.1113/jphysiol.2006.112094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tonkonogi M, Krook A, Walsh B, Sahlin K. Endurance training increases stimulation of uncoupling of skeletal muscle mitochondria in humans by non-esterified fatty acids: an uncoupling-protein-mediated effect? Biochem J. 2000;351 Pt 3:805–810. [PMC free article] [PubMed] [Google Scholar]
- 75.Walsh B, Tonkonogi M, Sahlin K. Effect of endurance training on oxidative and antioxidative function in human permeabilized muscle fibres. Pflugers Arch. 2001;442:420–425. doi: 10.1007/s004240100538. [DOI] [PubMed] [Google Scholar]
- 76.Kelley DE, Simoneau JA. Impaired free fatty acid utilization by skeletal muscle in non-insulin-dependent diabetes mellitus. J Clin Invest. 1994;94:2349–2356. doi: 10.1172/JCI117600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kelley DE, Mandarino LJ. Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes. 2000;49:677–683. doi: 10.2337/diabetes.49.5.677. [DOI] [PubMed] [Google Scholar]
- 78.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev. 2008;88:611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- 79.Nabben M, Hoeks J, Briedé JJ, Glatz JF, Moonen-Kornips E, Hesselink MK, Schrauwen P. The effect of UCP3 overexpression on mitochondrial ROS production in skeletal muscle of young versus aged mice. FEBS Lett. 2008;582:4147–4152. doi: 10.1016/j.febslet.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 80.Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, Schroder HD, Boushel R, Helge JW, Dela F, et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol. 2012;590:3349–3360. doi: 10.1113/jphysiol.2012.230185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mogensen M, Vind BF, Højlund K, Beck-Nielsen H, Sahlin K. Maximal lipid oxidation in patients with type 2 diabetes is normal and shows an adequate increase in response to aerobic training. Diabetes Obes Metab. 2009;11:874–883. doi: 10.1111/j.1463-1326.2009.01063.x. [DOI] [PubMed] [Google Scholar]
- 82.Church TS, Blair SN, Cocreham S, Johannsen N, Johnson W, Kramer K, Mikus CR, Myers V, Nauta M, Rodarte RQ, et al. Effects of aerobic and resistance training on hemoglobin A1c levels in patients with type 2 diabetes: a randomized controlled trial. JAMA. 2010;304:2253–2262. doi: 10.1001/jama.2010.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shaw CS, Shepherd SO, Wagenmakers AJ, Hansen D, Dendale P, van Loon LJ. Prolonged exercise training increases intramuscular lipid content and perilipin 2 expression in type I muscle fibers of patients with type 2 diabetes. Am J Physiol Endocrinol Metab. 2012;303:E1158–E1165. doi: 10.1152/ajpendo.00272.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nielsen J, Mogensen M, Vind BF, Sahlin K, Højlund K, Schrøder HD, Ortenblad N. Increased subsarcolemmal lipids in type 2 diabetes: effect of training on localization of lipids, mitochondria, and glycogen in sedentary human skeletal muscle. Am J Physiol Endocrinol Metab. 2010;298:E706–E713. doi: 10.1152/ajpendo.00692.2009. [DOI] [PubMed] [Google Scholar]
- 85.Little JP, Gillen JB, Percival ME, Safdar A, Tarnopolsky MA, Punthakee Z, Jung ME, Gibala MJ. Low-volume high-intensity interval training reduces hyperglycemia and increases muscle mitochondrial capacity in patients with type 2 diabetes. J Appl Physiol (1985) 2011;111:1554–1560. doi: 10.1152/japplphysiol.00921.2011. [DOI] [PubMed] [Google Scholar]
- 86.Gillen JB, Percival ME, Ludzki A, Tarnopolsky MA, Gibala MJ. Interval training in the fed or fasted state improves body composition and muscle oxidative capacity in overweight women. Obesity (Silver Spring) 2013;21:2249–2255. doi: 10.1002/oby.20379. [DOI] [PubMed] [Google Scholar]
- 87.Bruce CR, Thrush AB, Mertz VA, Bezaire V, Chabowski A, Heigenhauser GJ, Dyck DJ. Endurance training in obese humans improves glucose tolerance and mitochondrial fatty acid oxidation and alters muscle lipid content. Am J Physiol Endocrinol Metab. 2006;291:E99–E107. doi: 10.1152/ajpendo.00587.2005. [DOI] [PubMed] [Google Scholar]
- 88.Kim JY, Hickner RC, Cortright RL, Dohm GL, Houmard JA. Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol Endocrinol Metab. 2000;279:E1039–E1044. doi: 10.1152/ajpendo.2000.279.5.E1039. [DOI] [PubMed] [Google Scholar]