

Abstract
Aspartyl aminopeptidase (DNPEP) has been implicated in the control of angiotensin signaling and endosome trafficking, but its precise biologic roles remain incompletely defined. We performed a high-throughput screen of ∼25,000 small molecules to identify inhibitors of DNPEP for use as tools to study its biologic functions. Twenty-three confirmed hits inhibited DNPEP-catalyzed hydrolysis of angiotensin II with micromolar potency. A counter screen against glutamyl aminopeptidase (ENPEP), an enzyme with substrate specificity similar to that of DNPEP, identified eight DNPEP-selective inhibitors. Structure-activity relationships and modeling studies revealed structural features common to the identified inhibitors, including a metal-chelating group and a charged or polar moiety that could interact with portions of the enzyme active site. The compounds identified in this study should be valuable tools for elucidating DNPEP physiology.
Introduction
Aminopeptidases are a heterogeneous group of enzymes that catalyze the hydrolysis of N-terminal residues from peptide substrates. Aspartyl aminopeptidase (DNPEP; EC 3.4.11.21) and glutamyl aminopeptidase (ENPEP or aminopeptidase A; EC 3.4.11.7) are the two known acidic residue-specific aminopeptidases present in mammals (Glenner et al., 1962; Wilk et al., 1998; Goto et al., 2006). Mostly found in kidney, lung, and immune cells, ENPEP is a membrane-associated ecto-enzyme belonging to the M1 metallopeptidase family (Wu et al., 1990; Nanus et al., 1993; Goto et al., 2006). ENPEP catalyzes the hydrolysis of angiotensin II (Ang II) to form angiotensin III (Ang III) and is involved in the regulation of systemic blood pressure (Reaux et al., 1999; Mitsui et al., 2003; Wright et al., 2003; Bodineau et al., 2008a) and cancer-associated angiogenesis (Marchio et al., 2004). Whereas the function of ENPEP has been well-studied, the biologic and pathologic roles of DNPEP remain poorly understood.
DNPEP belongs to the M18 metallopeptidase family, the members of which are found in all kingdoms of life (Rawlings et al., 2014). The genomes of mammals and most other vertebrate species contain only one M18 metallopeptidase-encoding gene. Sequence identity among mammalian DNPEP orthologs is generally greater than 90%. This strong conservation suggests that DNPEP may play an essential role in cellular metabolism that has remained conserved throughout evolution. DNPEP is a self-compartmentalized, binuclear zinc-containing enzyme that forms a tetrahedron-shaped homododecameric complex (Chaikuad et al., 2012; Chen et al., 2012; Sivaraman et al., 2012). The active site-containing nano-compartment enclosed by the DNPEP tetrahedron is accessible through four ∼20 Å-wide selectivity pores that allow entrance of short peptides. In mammals, DNPEP is expressed in many organ systems with especially high activity in the brain and testis (Wilk et al., 1998). This enzyme is commonly described as cytosolic, although it also exists in a membrane-associated form in some tissues (Cai et al., 2010; Mayas et al., 2012b).
A role for DNPEP in regulation of the renin-angiotensin system has been proposed on the basis of its substrate specificity (Wilk et al., 1998; Chen et al., 2012), although its involvement in the renin-angiotensin system has not been examined in vivo. Changes in DNPEP expression and/or activity have been noted in neoplastic disorders such as colon and breast cancers, squamous cell carcinoma, and gliomas (Perez et al., 2009; Martinez-Martos et al., 2011; Mayas et al., 2012a; Larrinaga et al., 2013). In mice, DNPEP was shown to be a major target of the chondrocyte-specific microRNA, Mir140, loss of which led to overexpression of DNPEP and consequent defects in skeletal development (Nakamura et al., 2011). A recent suppressor mutant study of a Caenorhabditis elegans line with endosomal trafficking defects caused by a null mutation in phosphatidylserine flippase (tat1) revealed that loss of DNPEP activity corrected the blockade in endosome cargo sorting and recycling, but not degradation (Li et al., 2013). These disparate discoveries have not yet allowed a clear, unified picture of DNPEP physiology to be developed.
Among the various approaches to studying enzyme physiology, manipulation of biologic systems through the use of selective pharmacological agents allows examination of enzyme activity loss in an acute setting before the onset of homeostatic compensation. Additionally, loss of enzyme function can be readily studied in adult subjects in situations when genetic ablation of enzyme function is not feasible due to consequent developmental defects or embryonic lethality. This concern is pertinent to DNPEP due to its highly conserved nature, broad expression pattern, and the lethality observed in Plasmodium falciparum after genetic knockdown of its M18 aspartyl aminopeptidase (PfM18AAP) (Teuscher et al., 2007). Nonselective metal chelators, reducing agents, and a substrate analog, aspartic acid hydroxamate (Asp-NHOH) (IC50 = 200 µM), have been identified as DNPEP inhibitors (Wilk et al., 1998; Stoermer et al., 2003; Hoffman et al., 2009; Chaikuad et al., 2012; Chen et al., 2012). However, the lack of selectivity and/or suboptimal potency of these agents limits their utility for biologic studies. A high-throughput screen (HTS) for inhibitors of PfM18AAP was previously reported (Pubchem Bioassay: AID1855) (Schoenen et al., 2010). However, structural differences in the substrate-binding pockets of mammalian DNPEP and Pfm18AAP could limit the efficacy of the identified hit compounds as inhibitors of mammalian DNPEP (Chen et al., 2012; Sivaraman et al., 2012).
In this study, we report the identification and characterization of a set of small-molecule inhibitors of DNPEP. These compounds displayed low micromolar inhibitory activity toward DNPEP-catalyzed hydrolysis of the biologically relevant substrate, Ang II. Some of the compounds were more selective for DNPEP than the functionally related enzyme ENPEP, whereas others were potent inhibitors of both enzymes. Structure-activity relationship (SAR) analyses and molecular modeling of the inhibitor-enzyme interactions provided insights into their mechanism(s) of DNPEP inhibition.
Materials and Methods
Chemicals.
L-aspartic acid 7-amido-4-methylcoumarin (Asp-AMC), L-glutamic acid 7-amido-4-methylcoumarin (Glu-AMC), and Ang II were purchased from Bachem (Torrance, CA).
DNPEP Expression and Purification.
Bovine DNPEP was expressed in T7 Express BL21 Escherichia coli (New England Biolabs, Ipswich, MA) and purified, as previously described (Chen et al., 2012). The concentration and purity of DNPEP were measured by the Bradford assay, Asp-AMC hydrolysis activity assay, Coomassie Brilliant Blue–stained SDS-PAGE gels, and immunoblotting (Supplemental Fig. 1, A and C).
HTS Assay Optimization.
The HTS assay was modified from a cuvette-based, fluorometric Asp-p-nitroaniline hydrolysis assay (Chen et al., 2012). Because the excitation/emission profile of p-nitroaniline was not compatible with the Plate::Vision detector (Perkin Elmer, Waltham, MA) used for the HTS, we employed the alternative synthetic substrate, Asp-AMC, which can be hydrolyzed to produce an AMC fluorophore suitable for the HTS plate reader (Supplemental Fig. 1B). To separate signal from substrate and product, excitation and emission spectra of both Asp-AMC and AMC were measured in a Flexstation3 microplate reader (Molecular Devices, Sunnyvale, CA). Optimal excitation/emission wavelengths were judged to be 380/460 nm. Dimethylsulfoxide (DMSO), the compound solvent, did not inhibit DNPEP up to a final concentration of 1% (v/v) (Supplemental Fig. 1D). Asp-AMC concentrations (50–1000 µM), DNPEP concentrations (0.01–0.22 μM), and reaction time (0–50 minutes) were individually tested to optimize the assay while minimizing the HTS costs (Supplemental Fig. 1, E and F). Day-to-day and plate-to-plate variability of the HTS assay were within 40%, which is smaller than the threefold difference suggested by the National Institutes of Health HTS guidelines (http://www.ncats.nih.gov/research/reengineering/ncgc/assay/criteria/criteria.html), thus demonstrating the reliability of the HTS assay (Supplemental Fig. 1G). The stability of the enzyme under conditions of the HTS assay was also confirmed (Supplemental Fig. 1H).
Virtual Screen.
The DNPEP-aspartic acid hydroxamate coordinate file (PDB accession code 3L6S) was downloaded from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (Chaikuad et al., 2012). Docking was performed with the Schrödinger software suite (Schrödinger Suite 2011: Maestro, version 9.2.109; Schrödinger, New York, NY). Coordinates were prepared for docking with the Protein Preparation Wizard (Epik version 2.2); the conformer library was generated using LIGPREP (version 2.4); and the docking was performed using GLIDE (version 5.7 with SP, followed by XP Precision). The small database of compounds for the virtual screen was constructed from the larger University of Cincinnati (UC) compound library by the following: 1) execution of similarity searches using Accelrys’ Pipeline Pilot (Ver 8.0.1.500) on the aspartic acid hydroxamate ligand from the 3L6S coordinate file; and 2) execution of a range of substructure searches for typical chelating moieties (e.g., β-hydroxycarbonyls, hydroxamates, catechols, etc.) and/or aspartyl and glutamyl scaffolds. Because the S1 pocket of 3L6S was relatively small, compounds with mol. wt. greater than 375 Da were excluded from the virtual screen. The three-dimensional structures of this library, including tautomers, alternative protonation states, and isomers, were generated in Pipeline Pilot prior to conformation generation and incorporation into the virtual screen, as detailed above. Thirty-three compounds from this screen were experimentally evaluated (Supplemental Fig. 2), and the most active compounds served as seed structures for similarity searches in Pipeline Pilot with the top 266 most similar compounds added to the UC Diversity screening set.
Small-Molecule High-Throughput Screening with a Fluorescence-Based Biochemical Assay.
Using Asp-AMC as the substrate, we monitored the effect of test compounds on DNPEP-catalyzed release of AMC by measuring the fluorescence change over time. In a 384-well, black-wall, clear-bottom plate, each well was sequentially loaded with 20 µl 50 mM Tris-HCl (pH 7.5) and 40.65 nl (12.3 µM final concentration) of test compound in DMSO and 5 µl 50 µg/ml purified DNPEP. After incubation at 37°C for 15 minutes, 25 µl 500 µM Asp-AMC substrate in 50 mM Tris-HCl (pH 7.5) buffer was added to each well. Fluorescence intensity (Eex/Eem = 380/460 nm) was read with the Plate::Vision detector immediately after substrate was added (FI0 minute) and again after 30 minutes of incubation at 37°C (FI30 minute). The relative fluorescence intensity (RFI) was defined as
Each plate included four columns of controls, as follows: 1) untreated control with maximum enzyme activity, scored as 0%; 2) maximum inhibition control treated with 0.5 mM ZnCl2, scored as −100%; 3) maximum activation control treated with 0.5 mM MnCl2, scored as +100%; and 4) blank control with no enzyme addition. The relative activity of each compound was calculated using these controls as references:
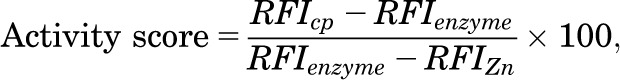 |
where RFIenzyme represents the relative fluorescence intensity of untreated control, RFIcp the relative fluorescence intensity of test compound-containing sample, and RFIZn the relative fluorescence intensity of the maximum inhibition control. A total of 26,120 compounds from the UC 25,000 Diversity Set in addition to 266 compounds selected from the UC compound collection based on virtual screening and similarity searches was tested at final concentrations of 12.3 µM in the primary HTS. Hits were selected with activity scores ≤−60% (inhibitors), or ≥+30% (activators). These hits were retested in triplicate at final concentrations of 12.3 µM. The potencies of confirmed hits were evaluated in dose-response assays performed in triplicate. Dose-response curves were generated using Genedata Screener Condoseo (Ver. 9.0.0 Standard) Software. EC50 is denoted as 50% effective concentration, that is, 50% inhibitory concentration (IC50) or 50% activation concentration (AC50). Compounds with IC50 or AC50 values ≤20 µM were selected for further evaluation. The primary, triplicate, and dose-response screens were monitored by two quality control parameters: signal to background (S/B) ratio and the Z′-factor defined, respectively, as:
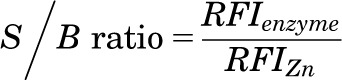 |
and
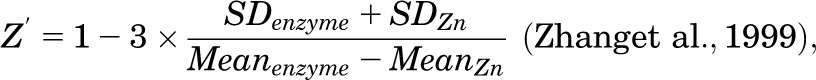 |
where S.D. and Mean are the standard deviations and means of the RFI readings.
Orthogonal Assay for Hit Confirmation.
To differentiate true hits from false positives, we tested the active compounds from the HTS in a mass spectrometry (MS)-based Ang II hydrolysis assay. Each well in a 384-well plate was loaded with 10 µl diluted compound in 50 mM Tris-HCl (pH 7.5) (to achieve a final compound concentration of 10 µM) and 10 µl 10 µg/ml purified DNPEP. Mixtures were incubated for 15 minutes at 37°C, and then the reaction was initiated by addition of 20 µl 0.5 mM Ang II in 50 mM Tris-HCl (pH 7.5). After 30 minutes of incubation at 37°C, the reaction was terminated by addition of 40 µl 40% acetic acid. Mixtures in each well were transferred to sample tubes and subjected to liquid chromatography-mass spectrometry (LC-MS) for Ang II and Ang III quantification, as described previously (Chen et al., 2012). An 1100 series Agilent (Santa Clara, CA) high-performance liquid chromatography instrument equipped with an XBridge BEH300 C4 column (Waters, Milford, MA) was used to fractionate samples, and eluates were injected into a LXQ linear ion trap mass spectrometer (Thermo Scientific, Waltham, MA) at a flow rate of 0.2 mL/min. Ang II and Ang III eluted at about the same time from the C4 column in a 50% acetonitrile and 50% H2O mobile phase. Ang III concentration was quantified by measuring the chromatogram peak areas corresponding to the +1 and +2 ions of Ang II (AreaAng II) and Ang III (AreaAng III):
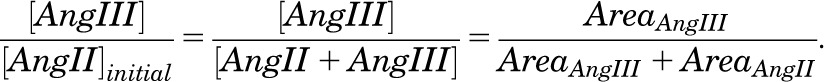 |
Reactions were optimized to ensure Ang II hydrolysis was linear over time, so that the endpoint product/initial substrate ratio represented the initial reaction velocity of the enzyme. As in the fluorogenic HTS assay, the DMSO control was scored 0%, the 0.5 mM ZnCl2-treated control was scored as −100%, and the 0.5 mM MnCl2-treated control was scored as +100%. Confirmed hits were identified as those with activity scores ≤−50% in triplicate readings and IC50 ≤ 20 µM in dose-response assays. Quality of the orthogonal assays was monitored by the S/B ratio, Z′-factor, and
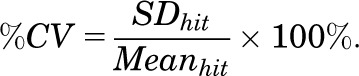 |
ENPEP Expression, Purification, and Activity Assay.
Human ENPEP was expressed in Sf9 insect cells essentially as previously described (Goto et al., 2006). The ENPEP sequence used in this work contained an A473G mutation that resulted in a Q213R amino acid substitution. The A473G change in ENPEP is a naturally occurring polymorphism found in ∼6% of the human population. The Q213R amino acid substitution was previously shown to have no effect on ENPEP catalytic properties or expression (Tonna et al., 2008). One liter of Sf9 cells at a density of 1–2 × 106 cells/ml was infected with 20–30 ml P3 baculovirus and then cultured at 28°C with 120 rpm shaking for 48–72 hours. The culture was harvested, and cells and debris were sedimented by centrifugation. ENPEP was purified from the supernatant by ammonium sulfate fractionation and Ni-affinity and gel filtration chromatography. The resulting preparation was >95% pure as judged by Coomassie-stained SDS-PAGE gels. About 2 mg purified ENPEP was obtained from a 1 L culture.
ENPEP enzymatic activity was assayed using the fluorogenic substrate Glu-AMC (Bachem) in the presence of 1 mM CaCl2, as previously described (Goto et al., 2006). The excitation and emission wavelengths were 380 nm and 460 nm, respectively. The Km value for the preparation was ∼0.4 mM consistent with previous reports (Goto et al., 2006). kcat was ∼57 seconds−1, which is somewhat higher than that previously described, possibly due to differences in the protein purification methods employed.
The counter screen of DNPEP inhibitors against ENPEP was conducted in an assay solution consisting of 10 ng/ml purified ENPEP, 1 mM CaCl2, 0.36 mM Glu-AMC, and 10 μM test or control compound (final concentrations) in a total volume of 50 or 100 μl. ZnCl2, a strong inhibitor of ENPEP activity, was used as a positive control, whereas a DMSO control was used to measure baseline ENPEP activity. The enzyme was preincubated with the test compound for 10 minutes at 37°C prior to initiation of the reaction by addition of substrate. The change in fluorescence was measured every 30 seconds for 5 minutes. The absolute quantity of AMC product generated was determined from AMC standards measured alongside the experimental samples. The degree of inhibition is presented as the difference in activity between the experimental and DMSO control samples. Inhibition by 10 μM ZnCl2, which was essentially complete, was set to −100% activity. The DMSO control was scored as 0% activity S/B ratio and Z′-factor were used to evaluate the assay quality.
MS Analysis of Compound Mass.
Selected compounds were obtained as powders from the UC compound library and dissolved in water at concentrations of 1 mM for MS analysis. Solutions were directly injected into a LXQ linear ion trap mass spectrometer (Thermo Scientific) using an ESI source operating in positive ion mode.
Results
Overview.
The discovery of DNPEP modulators began with a fluorescence-based HTS of ∼25,000 chemically diverse compounds in addition to 266 compounds selected based on virtual screening. Hits from the primary screen were then tested by an orthogonal, MS-based assay to quantify their inhibition toward DNPEP-catalyzed Ang II hydrolysis. Confirmed hits were then tested for their selectivity in a counter screen against ENPEP, which has substrate specificity similar to that of DNPEP and is a therapeutic target for hypertension and angiogenesis. SAR and molecular modeling studies were performed on the hits to understand their essential chemical features and probable mode of binding to DNPEP (Fig. 1A).
Fig. 1.

Overview of DNPEP inhibitor discovery. (A) Flowchart of DNPEP inhibitor discovery and characterization. (B) The peptidomimetic, fluorogenic substrate Asp-AMC was used for the HTS assay. Hydrolysis of this compound by DNPEP yields a fluorescent product, AMC, which can be readily quantified in high-throughput format. (C) The orthogonal assay used MS to quantify enzymatic production of Ang III from Ang II.
Virtual Screen, Compound Selection, and Primary HTS.
To identify pharmacological regulators of DNPEP by HTS, a biochemical HTS assay was developed using fluorescence emission as a readout of the enzyme activity. Due to the preference of DNPEP for N-Asp–containing peptides, we employed L-aspartic acid 7-amido-4-methylcoumarin (Asp-AMC) as the substrate for the HTS (Fig. 1B). DNPEP-catalyzed hydrolysis of this compound generates the fluorescent compound AMC, which can be distinguished from intact Asp-AMC by its red-shifted excitation/emission profile. The known DNPEP inhibitor and activator, ZnCl2 (0.5 mM) and MnCl2 (0.5 mM), were used as the −100% and +100% controls, respectively, and the enzyme-only condition was set as the 0% change in activity control. The final assay quality was characterized by a Z′-factor of 0.87, a signal-to-background ratio (S/B) of 247, and coefficient of variation of 3.9%.
To enrich the HTS library with potential DNPEP inhibitors, a virtual screen was undertaken based on known DNPEP crystal structures. By comparing the structures of ligand-free DNPEP and DNPEP in complex with the weak inhibitor/substrate analog Asp-NHOH (Chaikuad et al., 2012; Chen et al., 2012), we found that the substrate-binding pocket was larger than Asp-NHOH and could potentially accommodate bulkier groups. The active site also contained two positively charged regions, the metal center where hydrolysis is catalyzed and Lys370, which is located in the S1 pocket of DNPEP and conserved in all acidic residue-specific M18 aminopeptidases. A search of the entire >350,000 UC compound library revealed 33 inhibitor candidates. Some of these molecules contained hydroxamic, sulfonic/phosphonic, or aspartic/glutamic acid groups as potential metal-chelating moieties. Nine of the 33 compounds were hits from the Pfm18AAP inhibitor screen (Schoenen et al., 2010) (Pubchem Bioassay: AID1855). These 33 compounds then were tested in the proposed HTS assay system to assess its reliability as well as the quality of the virtual screening procedure (Supplemental Fig. 2). The small-scale screen of these 33 compounds at 10 µM identified three hits with greater than 50% inhibition toward DNPEP activity (Fig. 2A; Supplemental Fig. 2). These three hits together with Asp-NHOH (Fig. 2A) were then used as seeds for a similarity and three-dimensional structure-based search of the UC compound library, which yielded 266 compounds with high docking scores to the DNPEP active site.
Fig. 2.

The primary HTS for DNPEP inhibitors. (A) DNPEP inhibitors identified in a small-scale Asp-AMC hydrolysis screen with predicted high affinity for the DNPEP active site. These compounds were used as search seeds to identify related compounds in the 350,000 UC chemical library with potentially greater efficacy and potency. This search yielded 266 compounds that were added to the 25,000 UC Diversity Set for the HTS. (B) Sorted activity plot of the 26,368 compounds from the 25,000 UC Diversity Set (black dots) and virtual screen (red dots) in the Asp-AMC hydrolysis assay. Each compound was tested at 12.3 µM. The inset table summarizes the primary HTS results. (C) Sorted activity plot of 586 hits, including 367 DNPEP inhibitors and 219 activators identified in the primary HTS, tested in triplicate at 12.3 µM in the Asp-AMC hydrolysis assay. Mean activity score (Mean) and S.D. (residual) are shown as black and red dots, respectively. Results are summarized in the inset table. (D) A sorted EC50 (i.e., IC50 and AC50 for inhibitors and activators) plot of 285 hits, including 282 DNPEP inhibitors and 3 activators, confirmed in the dose-response Asp-AMC hydrolysis screen. Summarized results are presented in the inset table.
The primary HTS was performed using the UC 25,000 compound Diversity Set (26120 compounds actually tested) and 266 compounds selected from the virtual screen. Compounds were tested at a final concentration of 12.3 µM. Compounds with activity scores less than −60% and more than +30% were identified as DNPEP inhibitors and activators, respectively (Supplemental Table 1). The hit rates for inhibitors and activators were 1.09% and 0.84%. Quality control parameters for the primary HTS are shown in Supplemental Table 2. Comparison of the distributions of compound activities from unbiased screening using the 25,000 compound Diversity Set and the pool from virtual screen revealed a 7.6-fold enriched hit rate of 8.3% for the latter group of compounds (Fig. 2B). There were 367 of 397 DNPEP inhibitors and 219 of 222 DNPEP activators available in the UC compound collection for triplicate and dose-response confirmation tests. Hits were first re-screened in triplicate at 12.3 µM with independent aliquots of the test compounds, which confirmed 282 DNPEP inhibitors and 3 activators (Fig. 2C; Supplemental Tables 3 and 4). The compound activity distribution of the triplicate screen showed a dramatic enrichment of hits (76.8% and 1.4% for inhibitors and activators), demonstrating the high quality of the primary HTS screen. Confirmed hits were next analyzed by dose-response assays performed in triplicate with 10 inhibitor concentrations obtained by two-fold serial dilutions. From this screen, 281 inhibitors and 2 activators of DNPEP were confirmed with EC50 values ≤20 µM, representing 99.6% and 66.7% retention rates, respectively (Supplemental Tables 5 and 6). The EC50 distribution of these hits is shown in Fig. 2D. In total, 36 inhibitors were identified that displayed submicromolar potency. Z′-factors for the primary, triplicate, and dose-response screens using the Asp-AMC hydrolysis assay were 0.74 ± 0.03, 0.79 ± 0.05, and 0.78 ± 0.03, which demonstrated the reliability and quality of these screens (Supplemental Tables 2, 4, and 6).
MS-Based Orthogonal Screen.
The primary fluorescence-based assay allowed simple and robust HTS of the large chemical library selected for this study. However, compounds that are strongly fluorescent, colored, poorly soluble, or redox-active could give false-positive readings due to their ability to interfere with the fluorescence emission readout of the assay. These compounds needed to be distinguished from true hits before further investigation (Simeonov et al., 2008; Baell and Holloway, 2010; Soares et al., 2010). To identify true modulators of DNPEP activity, we employed a LC-MS–based method that directly measures products of DNPEP-catalyzed hydrolysis of the physiologic peptide substrate, Ang II (Chen et al., 2012) (Figs. 1C; 3A). As shown in Fig. 3B, 281 inhibitors and 2 activators of DNPEP were tested in triplicate at 10 μM by the Ang II hydrolysis assay. Due to the large quantity of hits to be tested and the limitation of LC-MS capacity, the test was separated into nine individual experiments with eight repeats of each control in each experiment. A total of 35 DNPEP inhibitors was confirmed by the first orthogonal screen, whereas no activators were active in this assay, which is not surprising given the inherent general difficulty in finding compounds that stimulate enzyme activity (Bishop and Chen, 2009; Mast et al., 2014) (Supplemental Tables 7 and 8). The low enrichment of hits confirmed from the orthogonal screen can be attributed to two main factors. First, there are false positives arising from various assay interference effects, as described above. Second, the specificity constant for Ang II hydrolysis by DNPEP (kcat/Km = 76.3 × 103 M−1×S−1) is much higher than that of Asp-AMC (kcat/Km = 1.3 × 103 M−1×S−1) so that either a higher inhibitor concentration or higher DNPEP-binding affinity could be required for the inhibitors to exert detectable effects. Indeed, the activity distribution chart revealed that 98 of the 248 inactive compounds retained inhibitory activities in the range of −50% to −20%, which is significantly above the level of error in the assay, consistent with the latter explanation (Fig. 3B). To eliminate systematic errors, the DNPEP inhibitors identified in the nine Ang II hydrolysis experiments were retested in a single experiment shown in Fig. 3C that yielded 26 compounds with repeatable activity scores ≤−50%, representing an 81.3% confirmation rate. The average S.D. for this screen was 5.9 ± 4.6%, and the Z′-factor was 0.63, both of which confirmed the assay reliability (Supplemental Table 9). Potencies of these confirmed 26 inhibitory compounds were then tested using a 10-concentration dose-response assay performed in triplicate. Twenty-three compounds showed IC50 values ≤20 µM (Fig. 3D; Table 1), thus confirming their activities as true DNPEP inhibitors. Quality control parameter values for the dose-response assays are shown in Supplemental Table 10.
Fig. 3.

Quantification of product of Ang II hydrolysis by LC-MS–based orthogonal assay. (A) Mass spectra of peptides present in the reaction mixtures after the 30-minute incubation. Two sets of +1, +2, and +3 ions present in the top panel correspond to Ang II (substrate, labeled black) and Ang III (cleavage product, marked in red). Production of Ang III was completely abolished in a sample treated with 0.5 mM ZnCl2 (bottom spectrum). (B) The sorted activity plot of 284 hits (282 inhibitors and 2 activators) identified in the primary HTS and retested by the Ang II hydrolysis assay at 10 µM in triplicate. Results are summarized in the inset table. (C) The repeat of Ang II hydrolysis assay to confirmed hits (32 available of 35 total) selected from triplicate screen shown in (B) by one-plate experiment, at 10 µM in triplicate. Results are summarized in the inset table. (D) The sorted IC50 plot for 23 of the most potent inhibitors. Each compound was tested at 10 different doses. Curve fitting was performed using the Hill equation.
TABLE 1.
Summary of hit activities from the primary high-throughput screen, orthogonal, and counter screen assays
| Chemical Groups | Number | Compound Identification | DNPEP | ENPEP | Δ Activity Score a | |||
|---|---|---|---|---|---|---|---|---|
| Asp-AMC Hydrolysis |
Ang II Hydrolysis |
Glu-AMC Hydrolysis |
||||||
| Activity Score (12.3 µM) | IC50 (µM) | Activity Score (10 µM) | IC50 (µM) | Activity Score (10 µM) | ||||
| A | 1 | 93293b | −95.0 ± 1.4 | 1.4 | −93.9 ± 0.6 | 0.1 | −77.8 ± 9.7 | −16.1 |
| 2 | 392425b | −87 ± 1.4 | 5.0 | −89.6 ± 0.3 | 0.81 | −87.4 ± 2.5 | −2.2 | |
| B | 3 | 897833c | −83 ± 0.2 | 1.7 | −55.6 ± 11.3 | 12 | 6.5 ± 11.1 | −62.1 |
| C | 4 | 98897b | −95.7 ± 0.4 | 0.54 | −94.3 ± 0.9 | 1.3 | −71.9 ± 2.2 | −22.4 |
| 5 | 769078b | −63.8 ± 2.4 | 5.8 | −92.7 ± 2.4 | 0.40 | −84.4 ± 1.1 | −8.3 | |
| 6 | 702610 | −84.4 ± 1.0 | 2.4 | −93.3 ± 0.6 | 2.0 | −45 ± 7.2 | −48.3 | |
| D | 7 | 241320c | −74.5 ± 5.2 | 4.4 | −91.8 ± 5.2 | 0.26 | −28.8 ± 8.7 | −63.0 |
| 8 | 705521c | −82.5 ± 0.9 | 0.16 | −78.9 ± 4.5 | 5.0 | 4.5 ± 3.9 | −83.4 | |
| 9 | 936427b | −83 ± 0.7 | 0.83 | −93.0 ± 1.3 | 1.7 | −89 ± 0.8 | −4.0 | |
| 10 | 374081c | −87.4 ± 1.2 | 2.7 | −92.2 ± 2.3 | 4.7 | 5.5 ± 3.4 | −97.7 | |
| 11 | 405279 | −73.6 ± 1.4 | 2.3 | −91.0 ± 2.4 | 3.6 | −45.8 ± 4.9 | −45.2 | |
| 12 | 914971 | −81.8 ± 0.2 | 3.1 | −50.1 ± 6.5 | 8.3 | −22.4 ± 3.1 | −27.7 | |
| 13 | 920521b | −68.1 ± 2.5 | 10.2 | −59.3 ± 8.9 | 18 | −43.2 ± 2.9 | −16.1 | |
| E | 14 | 897927c | −74.4 ± 3.1 | 5.7 | −91.7 ± 3.2 | 5.7 | −17.3 ± 2.5 | −74.4 |
| 15 | 251431c | −74.7 ± 0.7 | 1.6 | −80.5 ± 4.6 | 3.6 | 55.5 ± 9.6 | −136.0 | |
| F | 16 | 347749b | −72.3 ± 0.8 | 2.0 | −90.7 ± 3.6 | 1.4 | −72.8 ± 8.9 | −17.9 |
| 17 | 252185c | −81.7 ± 0.7 | 3.2 | −56.0 ± 9.7 | 17 | −0.2 ± 7.5 | −55.8 | |
| G | 18 | 505901b | −93.4 ± 0.2 | 0.84 | −94.0 ± 1.1 | 2.1 | −83.9 ± 2.0 | −10.1 |
| 19 | 923254 | −74.6 ± 1.2 | 7.7 | −93.0 ± 2.4 | 4.2 | −44.8 ± 5.3 | −48.2 | |
| 20 | 773587 | −62.8 ± 2.3 | 8.0 | −88.9 ± 2.5 | 6.2 | −61 ± 4.2 | −27.9 | |
| H | 21 | 406365c | −78.8 ± 0.8 | 5.7 | −75.3 ± 4.0 | 3.0 | −0.8 ± 6.6 | −74.5 |
| I | 22 | 899189 | −77.4 ± 1.5 | 3.2 | −90.7 ± 4.3 | 6.0 | −64.9 ± 3.8 | −25.8 |
| 23 | 312792b | −77.8 ± 1.0 | 3.2 | −87.2 ± 8.7 | 2.8 | −92.7 ± 1.0 | 5.5 | |
Ang II, angiotensin II; Asp-AMC, L-aspartic acid 7-amido-4-methylcoumarin; DNPEP, aspartyl aminopeptidase; ENPEP, glutamyl aminopeptidase; Glu-AMC, L-glutamic acid 7-amido-4-methylcoumarin.
Δ Activity score = activity scoreDNPEP − activity scoreENPEP, where activity scoreDNPEP is for Ang II hydrolysis.
Dual inhibitors of DNPEP and ENPEP.
DNPEP-selective inhibitors.
Counter Screen against ENPEP.
ENPEP exhibits similar substrate preferences to those of DNPEP. However, there are fundamental differences in the active site structures of these two enzymes that could make selective chemical inhibition of one enzyme over the other achievable (Chen et al., 2012; Yang et al., 2013). Selective inhibitors of DNPEP will be useful probes to study the biology of this enzyme. Additionally, dual inhibitors of both DNPEP and ENPEP could also be useful for further development potentially as therapeutic agents for treatment of hypertension ( Bodineau et al., 2008b; Marc et al., 2012; Balavoine et al., 2014). Therefore, we performed a counter screen to test the confirmed 23 DNPEP inhibitors for their activities toward ENPEP. Human ENPEP containing a C-terminal 6-His tag was expressed in Sf9 insect cells as a secreted protein (Goto et al., 2006). The purified protein exhibited catalytic parameters similar to those described previously (Goto et al., 2006) (Fig. 4, A and B). Activities of the 23 DNPEP inhibitors toward ENPEP were plotted in comparison with their activities toward DNPEP in Fig. 4C. The S/B ratio and Z′-factor for this assay were 313 and 0.56, respectively. This counter screen revealed a number of compounds that displayed strong inhibition toward both enzymes, whereas others were quite selective for DNPEP. To semiquantitatively describe their specificity, the 23 compounds were grouped as DNPEP-selective ActivityDNPEP − ActivityENPEP (≤ −50%, 8 compounds), partially DNPEP-selective (−50% < ActivityDNPEP − ActivityENPEP < −20%, 6 compounds), and dual inhibitors (ActivityDNPEP − ActivityENPEP ≥ −20%, 9 compounds), according to the difference in activity scores between the two enzymes (Table 1; Fig. 5).
Fig. 4.
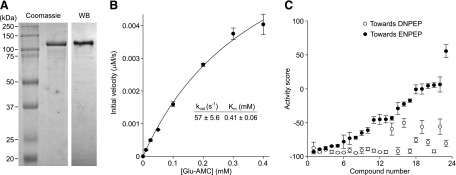
Counter screen of hits toward ENPEP. (A) Coomassie-Blue–stained SDS-PAGE gel and immunoblot of purified human ENPEP. (B) Michaelis Menten kinetics of Glu-AMC hydrolysis by ENPEP. The inset shows the calculated Km and kcat values. (C) Comparison of hits toward DNPEP in Ang II hydrolysis over ENPEP in Glu-AMC hydrolysis in activity score at 10 µM (sorted by activity scores toward ENPEP). Differences in activity score toward DNPEP over ENPEP (Δactivity score) are shown in Table 1. Hits with Δactivity scores ≤−50%, between −50% and −20%, or ≥−20% were defined as DNPEP-selective, DNPEP-preferential, and dual inhibitors, respectively.
Fig. 5.

Layout of confirmed DNPEP inhibitors clustered in groups A–I. On top are the functional groups found in the identified DNPEP inhibitors. Each compound is displayed by its chemical structure, the compound identification, and its activities toward DNPEP (Ang II hydrolysis assay) and ENPEP (Glu-AMC hydrolysis assay) at 10 µM. Concentrations are IC50 values obtained from dose-response assays. Activity scores ≤−50% are marked in red. DNPEP-selective inhibitors and dual inhibitors are marked with asterisks and double daggers, respectively.
Confirmation of Chemical Composition and Activity.
To confirm the identity of the most selective and potent hits from each promising chemical class shown in Fig. 5, we obtained compounds 93293, 98897, 241320, 705521, 897927, 374081, and 406365 as powders from the UC chemical library and analyzed them by MS. The principle observed ion exactly matched the expected mass for all of these compounds except 406365. There were no major additional peaks in their spectra, indicating their purity. The mass spectrum of compound 406365 showed an ion at the expected mass (m/z = 435) as well as additional peaks at m/z values of 432 and 437, indicating that the sample was probably not pure. Freshly made aqueous stocks of compounds 93293, 98897, 897927, and 406365 all inhibited DNPEP in a dose-dependent manner with IC50 values very similar to those shown in Fig. 5 and Table 1, thus confirming their activity toward DNPEP. Compounds 241320, 705521, and 374081 also were active, but only when dissolved in alkaline buffers possibly due to poor aqueous solubility at neutral pH.
Chemical Properties of Identified Hits.
According to their chemical similarities, the 23 confirmed DNPEP inhibitors were clustered into 9 groups, named A through I (Fig. 5; Table 1). Notably, many of these compounds contain two negatively charged or polar groups separated by four to six covalent bonds. Catechol/Benzenetriol, aminothiazole, amide, carboxylate, furan, rhodanine, polycyclic (hydro)quinones, and sulfonic acid groups were especially prevalent among the identified inhibitors (Fig. 5). Catechol and rhodanine are known metal chelators; sulfonic acid or related phosphonic acid–containing compounds have previously been identified as metallo-aminopeptidase inhibitors (Mucha et al., 2010); and carboxylate and amide groups resemble features of DNPEP substrates. Compounds 899189 and 312792 (Fig. 5, group I) possess reactive, electrophilic benzoquinone groups that most likely make them nonspecific enzyme inhibitors. Four compounds with identification numbers 93293, 392425, 769078, and 241320 belonging to groups A, C, and D displayed submicromolar potencies and maximum inhibitory effects greater than 89% (Fig. 5).
As shown in Fig. 5, DNPEP-selective inhibitors marked with an asterisk and dual inhibitors marked with a double dagger each shared broadly similar chemical characteristics, with some exceptions. Amide or amide-like hydrozones (897833, 705521, and 406365), amino carboxylic acids (241320, 374081, and 406365), sulfonic acids (897927 and 251431), and compound 252185 are DNPEP-selective inhibitors that are not rigidly planar. Conversely, most dual inhibitors were planar molecules residing in chemical groups A, C, G, and I that featured catechol-aminothiazole, rhodanine-enylfuran, fluorescein, and quinone derivatives. Exceptions to this general trend included compounds 347749 (group F) and 936427 (group D) that are nonplanar. Interestingly, compound 251431 acted as a stimulator of ENPEP boosting its activity 1.5-fold (Fig. 5E).
Among the submicromolar potent inhibitors, only compound 241320 (group D) was DNPEP-selective, whereas 93293, 392425 (group A), and 769078 (group C) inhibited both DNPEP and ENPEP. Compounds 93293 and 392425 (group A inhibitors) contain metal-chelating catechol and thiazole groups also found in E. coli methionine aminopeptidase (MetAP) inhibitors (Wang et al., 2008; Chai and Ye, 2010). Compound 769078 represents group C dual inhibitors containing a rhodanine moiety linked to furan ring by an alkene spacer. Rhodanine is a known pan assay-interference moiety (Mendgen et al., 2012) that has been reported active in a significant number of HTS assays, causing concerns about the specificity of compounds containing this chemical group (Carter et al., 2001; Voss et al., 2003; Carlson et al., 2006; Powers et al., 2006; Baell and Holloway, 2010). However, these compounds share the common features seen in other DNPEP inhibitors with two functional groups bridged by four to six covalent bonds, suggesting that they could exert a DNPEP-selective effect with further modifications. The fluorescein derivatives in group G are fluorescent compounds that are commonly used as dyes. Despite concerns about these compounds interfering with the primary screening assay, their inhibitory effect toward DNPEP was confirmed in the orthogonal, MS-based assay, thus assuring that they are genuine inhibitors. However, their bulky, planar structures and ortho-semiquinone moieties suggest that these compounds are nonspecific inhibitors and thus will not undergo further development.
SAR Analysis of DNPEP Inhibitors.
We used five strong DNPEP inhibitors (241320, 347749, 98897, 769078, and 702610) as seeds to search for related compounds in the UC compound library. Similar compounds identified by molecular fingerprint comparisons were manually inspected to ensure all selected compounds retained the putative metal-chelating functional groups expected to interact with the DNPEP metal center (92 compounds; Supplemental Figs. 3 and 4). The inhibitory activity of these compounds toward DNPEP was then tested in triplicate at a final concentration of 10 µM by the Asp-AMC hydrolysis assay. A total of 25 of 92 tested compounds was identified as DNPEP inhibitors with activity scores ≤−60 (27.2% hit rate) (Supplemental Figs. 3 and 4). The Z′-factor from the similar compound test was 0.96, demonstrating the reliability of this assay (Supplemental Table 11).
Compound 241320 was the most potent DNPEP-specific inhibitor (IC50 = 0.26 µM), and an activity test of its neighboring compounds (Supplemental Fig. 3) revealed that DNPEP inhibition by this scaffold could tolerate minor modifications on the serine group, including deletion of the 3-hydroxyl group, addition of a methyl group, deletion of the carboxylate group, or replacement of the carboxylate with a hydroxyamide moiety (compounds 503659, 247266, 247275, and 93984). However, halogenations of the catechol ring abolished DNPEP inhibitory activity (compounds 520266 and 130024), suggesting that the inhibitory effect of the catechol is due to its interaction with the DNPEP metal center. These related active compounds retain the most essential features of compound 241320 and are likely to be DNPEP-selective.
Compound 347749, the only nonplanar dual inhibitor, is comprised of threonine coupled to a rhodanine ring. Activities of its neighboring molecules (Supplemental Fig. 3) showed that compound 347749 is relatively intolerant of chemical changes for maintenance of its inhibitory activity. Modifications of the threonine group mostly ablated DNPEP inhibitory activity of the molecule. However, compound 279700 with threonine replaced with 4-bromoaniline retained weak DNPEP inhibitory activity. Despite concerns about nonspecific effects of rhodanine compounds, the SAR results indicate that compound 347749 could be selective for DNPEP and ENPEP.
Compounds 769078 and 98897 are strong dual inhibitors containing rhodanine rings with rigid hydrophobic vinyl furan extensions (Fig. 5). Compound 702610 is a DNPEP preferred inhibitor, grouped in the same chemical cluster with 769078 and 98897, but containing a 4-amino-thioxo-1,2,4-triazinone ring in place of rhodanine. The exocyclic NH2 group and adjacent carbonyl and thiocarbonyl moieties most likely form a strong metal-chelating functionality. The neighboring search and activity test of group C compounds (Supplemental Fig. 4) showed they tolerate geometrical isomerizations and even alkene linker extension or truncation while still retaining DNPEP inhibitory activity; however, addition of alkylation of the alkene linkage of furan to rhodanine abolished activity (compounds 921127, 926399, and 921314). Introduction of polar groups on the N3 atom of the rhodanine ring, for example amines or carboxylic acids, was tolerated (compounds 910179, 966958, 521685, 943988, and 994188). Conversely, methyl, ethyl, or isobutyl alkylation generally abolished inhibitory activity (e.g., compounds 938731, 920776, 917820, 374601, and 771512). Selective modifications to and even opening of the rhodanine ring with deletion of the carbonyl group (compound 96270) did not ablate the inhibitory activity, suggesting the sulfur moiety is the immediate metal-chelating atom of the rhodanine-containing group. The furan ring could not be replaced with thiophene or pyridine (e.g., compounds 980083, 496193, 240994, and 240991). A variety of groups, including methyls (e.g., compounds 770885 and 911299), halogens (e.g., compounds 98875, 769080, and 1001360), carboxylate (compound 98871), nitrate (compound 769641), methoxyl (compound 98897), and even bulky groups such as chlorophenyl (compound 397172) or morpholine (compound 954647) could be linked at position 2 of the furan ring while maintaining reasonable inhibitory activity toward DNPEP. This indicates that the vinylfuran group may be bound in the larger S1′-S2′ pocket of the enzyme, which can accommodate bulkier groups compared with the S1 pocket. In agreement with this hypothesis, compound 897927 (Fig. 5E) contains a group C compound scaffold (typical of dual inhibitors) linked to a bulky benzylsulfonic acid moiety that confers DNPEP selectivity. Therefore, the 2-furan extension may be considered as a modification site for further development of group C compounds to increase their DNPEP selectivity.
Discussion
In this study, we identified 23 DNPEP inhibitors with IC50 values <20 µM, among which 8 exhibited selectivity for DNPEP over ENPEP. An additional 25 DNPEP inhibitors were identified from a chemical similarity search based on 5 selected inhibitors. Together, these compounds displayed significant chemical diversity and could be clustered into 9 families based on their chemical structures. Most of the inhibitors have in common a putative metal coordinating moiety linked by four to six covalent bonds to a negatively charged or polar group. The most common metal-coordinating groups found were catechol and rhodanine rings. SAR analysis of all DNPEP inhibitors from the initial and hit enrichment screens showed that DNPEP inhibitors could occupy at least two distinct positions in the active site of the enzyme in addition to coordinating the metal center. Small negatively charged groups could occupy the S1 pocket of the active site consisting of a small, positively charged pocket, whereas more bulky groups could interact with the putative S1′-S2′ sites of the peptide-binding groove (Fig. 6A). The group A and C dual inhibitors are rigid and could bind to the zinc centers of both DNPEP and ENPEP with similar affinity. These DNPEP and ENPEP dual inhibitors could be useful as pan-blockers of aspartyl aminopeptidase activity, but their lack of selectivity would be a concern in cell-based or in vivo experiments. Conversely, the group D and E DNPEP-selective inhibitors in addition to possessing strong chelator functionalities have extensions with rotatable bonds that could conform to the DNPEP active site and cause selective binding.
Fig. 6.
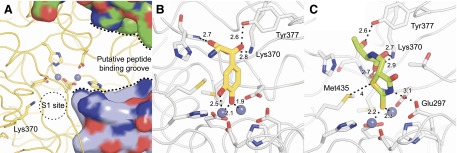
Modeling of selected inhibitors in the DNPEP active site. (A) Structure of the DNPEP active site (PDB accession code 3VAT) showing the binuclear zinc center (gray spheres) in relation to the S1 site and the peptide-binding groove of the enzyme, which is partially formed by additional subunits of the DNPEP complex (shown in surface representation). Lys370, present in the S1 site of the enzyme, confers selectivity by interacting with the negatively charged N-terminal Asp or Glu residue of DNPEP peptide substrates. Models of (B) compound 241320 and (C) compound 347749 binding to the DNPEP active site. The model in (B) was docked based on the structure of methionine aminopeptidase in complex with a catechol-containing inhibitor (Wang et al., 2008), whereas the model in (C) was placed assuming that the exocyclic double-bonded sulfur atom is the relevant metal chelator in this compound. Both models were energy minimized using the Crystallography and NMR System program (Brunger, 2007). Distances are given in angstroms.
Catechols are known to coordinate divalent metal ions by virtue of their acidic ortho-hydroxyl groups. A recent HTS screening campaign to develop inhibitors against Pfm18AAP, a potential drug target for the treatment of malaria, identified a number of catechol-containing inhibitory molecules (Schoenen et al., 2010). In fact, the best-in-class probe identified in that screen, ML369 (Supplemental Fig. 5A), features a catechol moiety linked to a piperidine-tetrahydroquinoline ring system. Catechols have also been identified as inhibitors of methionine aminopeptidase, which, like DNPEP, also contains a binuclear zinc center. A structure of E. coli methionine aminopeptidase in complex with a catechol-containing compound revealed the mode of interaction of the catechol ring with the binuclear zinc center (Wang et al., 2008). In this structure, one of the hydroxyl groups of the catechol replaced water as a bridging ligand, whereas the other interacted with one of the zinc ions enforced by the active site shape. A second mode of binding whereby both hydroxyl groups serve as metal bridging ligands with the plane of the catechol group perpendicular to the inter-metal axis might also be possible depending upon the active site structure. Modeling of the catechol-containing compound 241320 into the active site structure of mammalian DNPEP suggests that it could adopt a mode of binding similar to the methionine aminopeptidase-catechol complex (Fig. 6B). The alternative binding mode with perpendicular catechol and intermetal planes may not be feasible due to steric clashes with surrounding active site residues. Compound 241320 also features a serinyl side chain that appears to be sufficiently small in size to accommodate the S1 pocket of DNPEP, with the carboxylate and hydroxyl groups interacting with the surrounding Lys370 side chain thus mimicking the Asp side chain of peptide substrates (Fig. 6, A and B). Several compounds from the current HTS as well as the Pfm18AAP HTS are catechols linked to bulky groups that would be unable to fit in the S1 pocket. Instead, these groups most likely bind in the putative peptide-binding groove of the enzyme or one of the other small pockets located in close proximity to the metal center. Notably, residues within 8 Å of the DNPEP and Pfm18AAP metal centers, including those that make up the S1 pocket, are absolutely conserved, whereas differences can be found in the peptide-binding grooves of these two enzymes. The previously reported counter screen of Pfm18AAP inhibitors against human DNPEP (hM18AAP, AID588696) identified 25 compounds active against human DNPEP as defined by IC50 values <100 µM. Data from this screen provide additional SAR information for M18 aminopeptidase inhibitors. The compound used as a basis for this counter screen SAR analysis (CID 23724194) consisted of a catechol ring linked by an aminoethyl chain to an acridine heteroaromatic ring (Supplemental Fig. 5B). This compound was intolerant to changes in the catechol ring for maintenance of inhibitory activity toward DNPEP. An increase or decrease in the linker by one carbon atom substantially reduced or abolished activity against both DNPEP and Pfm18AAP. Interestingly, modifications to the heteroaromatic ring with or without changes in the linker portion of the molecule were observed to substantially alter the IC50 values of these compounds toward DNPEP without changing their activity toward Pfm18AAP. These data support the proposition that differences in residues lining the peptide-binding groove between these two enzymes where the bulky heterocyclic rings most likely bind can be exploited to achieve selectivity.
The metal-binding capacity of rhodanine is well-known from its use as a copper-detecting agent in histologic studies (Lindquist, 1969). Direct binding of rhodanine to the zinc center of anthrax lethal factor, a metallopeptidase, was previously reported (Forino et al., 2005), and ligand-docking studies suggest that it can also bind the zinc center of matrix metalloproteases (Hu et al., 2008). In these studies, the rhodanine was observed or predicted to coordinate zinc through its endocyclic sulfur atom. Based on steric considerations, we hypothesize that rhodanine is more likely to interact with the binuclear center of DNPEP via its exocyclic rather than endocyclic sulfur atom. Density function theory calculations predict that the highest occupied molecular orbital and negative electrostatic potential of the rhodanine ring localize predominantly to the exocyclic sulfur atom (Mendgen et al., 2012), which supports the proposed role of this site in metal coordination. Compound 347749 can be modeled into the DNPEP active site in a sterically acceptable fashion with the exocyclic sulfur binding in the bridging ligand position between the zinc ions and the negatively charged theoninyl side chain occupying the S1 pocket via an ionic interaction with Lys370 and hydrogen-bonding interactions with surrounding protein groups. In this orientation, the endocyclic sulfur atom of the rhodanine ring can form a favorable interaction with the Met435 side chain sulfur atom (Fig. 6C). Rhodanine has a notorious reputation in the field of HTS drug discovery due to its tendency to display activity in a diverse range of HTS assays (Baell and Holloway, 2010; Mendgen et al., 2012). This compound is reportedly able to bind molecules in a promiscuous fashion leading to nonselective effects (Baell, 2010). Despite this potential, two of the rhodanine-containing compounds identified in our study (compounds 897927 and 252185) exhibited good selectivity for DNPEP over ENPEP. Additionally, a screen of 16 compounds similar to 347749 revealed only a single moderate DNPEP inhibitor, which demonstrates that the rhodanine group alone is not sufficient for these compounds to inhibit DNPEP. We expect that these compounds will be useful for further development as selective and potent pharmacological agents to study DNPEP biology.
Supplementary Material
Acknowledgments
The authors thank Dr. Leslie T. Webster, Jr., for critically reviewing the manuscript.
Abbreviations
- Ang II
angiotensin II
- Ang III
angiotensin III
- Asp-AMC
L-aspartic acid 7-amido-4-methylcoumarin
- Asp-NHOH
aspartic acid hydroxamate
- DMSO
dimethylsulfoxide
- DNPEP
aspartyl aminopeptidase
- ENPEP
glutamyl aminopeptidase
- Glu-AMC
L-glutamic acid 7-amido-4-methylcoumarin
- HTS
high-throughput screen
- LC-MS
liquid chromatography-mass spectrometry
- MS
mass spectrometry
- Pfm18AAP
Plasmodium falciparum M18 aspartyl aminopeptidase
- SAR
structure-activity relationship
- S/B
signal to background
- UC
University of Cincinnati
Authorship Contributions
Participated in research design: Chen, Tang, Seibel, Papoian, Palczewski, Kiser.
Conducted experiments: Chen, Tang, Seibel, Oh, Li, Golczak, Kiser.
Performed data analysis: Chen, Tang, Seibel, Zhang, Golczak, Palczewski, Kiser.
Wrote or contributed to the writing of the manuscript: Chen, Kiser.
Footnotes
This work was supported by Case Western Reserve University School of Medicine; and National Institutes of Health [Grant EY008061 (to K.P.)]. K.P. is the John H. Hord Professor of Pharmacology.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Baell JB. (2010) Observations on screening-based research and some concerning trends in the literature. Future Med Chem 2:1529–1546 [DOI] [PubMed] [Google Scholar]
- Baell JB, Holloway GA. (2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53:2719–2740 [DOI] [PubMed] [Google Scholar]
- Balavoine F, Azizi M, Bergerot D, De Mota N, Patouret R, Roques BP, Llorens-Cortes C. (2014) Randomised, double-blind, placebo-controlled, dose-escalating phase I study of QGC001, a centrally acting aminopeptidase A inhibitor prodrug. Clin Pharmacokinet 53:385–395 [DOI] [PubMed] [Google Scholar]
- Bishop AC, Chen VL. (2009) Brought to life: targeted activation of enzyme function with small molecules. J Chem Biol 2:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodineau L, Frugière A, Marc Y, Claperon C, Llorens-Cortes C. (2008a) Aminopeptidase A inhibitors as centrally acting antihypertensive agents. Heart Fail Rev 13:311–319 [DOI] [PubMed] [Google Scholar]
- Bodineau L, Frugière A, Marc Y, Inguimbert N, Fassot C, Balavoine F, Roques B, Llorens-Cortes C. (2008b) Orally active aminopeptidase A inhibitors reduce blood pressure: a new strategy for treating hypertension. Hypertension 51:1318–1325 [DOI] [PubMed] [Google Scholar]
- Brunger AT. (2007) Version 1.2 of the crystallography and NMR system. Nat Protoc 2:2728–2733 [DOI] [PubMed] [Google Scholar]
- Cai WW, Wang L, Chen Y. (2010) Aspartyl aminopeptidase, encoded by an evolutionarily conserved syntenic gene, is colocalized with its cluster in secretory granules of pancreatic islet cells. Biosci Biotechnol Biochem 74:2050–2055 [DOI] [PubMed] [Google Scholar]
- Carlson EE, May JF, Kiessling LL. (2006) Chemical probes of UDP-galactopyranose mutase. Chem Biol 13:825–837 [DOI] [PubMed] [Google Scholar]
- Carter PH, Scherle PA, Muckelbauer JK, Voss ME, Liu RQ, Thompson LA, Tebben AJ, Solomon KA, Lo YC, Li Z, et al. (2001) Photochemically enhanced binding of small molecules to the tumor necrosis factor receptor-1 inhibits the binding of TNF-alpha. Proc Natl Acad Sci USA 98:11879–11884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai SC, Ye QZ. (2010) A cell-based assay that targets methionine aminopeptidase in a physiologically relevant environment. Bioorg Med Chem Lett 20:2129–2132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaikuad A, Pilka ES, De Riso A, von Delft F, Kavanagh KL, Vénien-Bryan C, Oppermann U, Yue WW. (2012) Structure of human aspartyl aminopeptidase complexed with substrate analogue: insight into catalytic mechanism, substrate specificity and M18 peptidase family. BMC Struct Biol 12:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Farquhar ER, Chance MR, Palczewski K, Kiser PD. (2012) Insights into substrate specificity and metal activation of mammalian tetrahedral aspartyl aminopeptidase. J Biol Chem 287:13356–13370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forino M, Johnson S, Wong TY, Rozanov DV, Savinov AY, Li W, Fattorusso R, Becattini B, Orry AJ, Jung D, et al. (2005) Efficient synthetic inhibitors of anthrax lethal factor. Proc Natl Acad Sci USA 102:9499–9504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner GG, McMillan PJ, Folk JE. (1962) A mammalian peptidase specific for the hydrolysis of N-terminal alpha-L-glutamyl and aspartyl residues. Nature 194:867. [DOI] [PubMed] [Google Scholar]
- Goto Y, Hattori A, Ishii Y, Mizutani S, Tsujimoto M. (2006) Enzymatic properties of human aminopeptidase A: regulation of its enzymatic activity by calcium and angiotensin IV. J Biol Chem 281:23503–23513 [DOI] [PubMed] [Google Scholar]
- Hoffman HE, Jirásková J, Cígler P, Sanda M, Schraml J, Konvalinka J. (2009) Hydroxamic acids as a novel family of serine racemase inhibitors: mechanistic analysis reveals different modes of interaction with the pyridoxal-5′-phosphate cofactor. J Med Chem 52:6032–6041 [DOI] [PubMed] [Google Scholar]
- Hu M, Li J, Yao SQ. (2008) In situ “click” assembly of small molecule matrix metalloprotease inhibitors containing zinc-chelating groups. Org Lett 10:5529–5531 [DOI] [PubMed] [Google Scholar]
- Larrinaga G, Perez I, Ariz U, Sanz B, Beitia M, Errarte P, Etxezarraga C, Candenas ML, Pinto FM, López JI. (2013) Clinical impact of aspartyl aminopeptidase expression and activity in colorectal cancer. Transl Res 162:297–308 [DOI] [PubMed] [Google Scholar]
- Li X, Chen B, Yoshina S, Cai T, Yang F, Mitani S, Wang X. (2013) Inactivation of Caenorhabditis elegans aminopeptidase DNPP-1 restores endocytic sorting and recycling in tat-1 mutants. Mol Biol Cell 24:1163–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist RR. (1969) Studies on the pathogenesis of hepatolenticular degeneration. II. Cytochemical methods for the localization of copper. Arch Pathol 87:370–379 [PubMed] [Google Scholar]
- Marc Y, Gao J, Balavoine F, Michaud A, Roques BP, Llorens-Cortes C. (2012) Central antihypertensive effects of orally active aminopeptidase A inhibitors in spontaneously hypertensive rats. Hypertension 60:411–418 [DOI] [PubMed] [Google Scholar]
- Marchiò S, Lahdenranta J, Schlingemann RO, Valdembri D, Wesseling P, Arap MA, Hajitou A, Ozawa MG, Trepel M, Giordano RJ, et al. (2004) Aminopeptidase A is a functional target in angiogenic blood vessels. Cancer Cell 5:151–162 [DOI] [PubMed] [Google Scholar]
- Martínez-Martos JM, del Pilar Carrera-González M, Dueñas B, Mayas MD, García MJ, Ramírez-Expósito MJ. (2011) Renin angiotensin system-regulating aminopeptidase activities in serum of pre- and postmenopausal women with breast cancer. Breast 20:444–447 [DOI] [PubMed] [Google Scholar]
- Mast N, Li Y, Linger M, Clark M, Wiseman J, Pikuleva IA. (2014) Pharmacologic stimulation of cytochrome P450 46A1 and cerebral cholesterol turnover in mice. J Biol Chem 289:3529–3538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayas MD, Ramírez-Expósito MJ, Carrera MP, Cobo M, Martínez-Martos JM. (2012a) Renin-angiotensin system-regulating aminopeptidases in tumor growth of rat C6 gliomas implanted at the subcutaneous region. Anticancer Res 32:3675–3682 [PubMed] [Google Scholar]
- Mayas MD, Ramírez-Expósito MJ, García MJ, Carrera MP, Martínez-Martos JM. (2012b) Influence of chronic ethanol intake on mouse synaptosomal aspartyl aminopeptidase and aminopeptidase A: relationship with oxidative stress indicators. Alcohol 46:481–487 [DOI] [PubMed] [Google Scholar]
- Mendgen T, Steuer C, Klein CD. (2012) Privileged scaffolds or promiscuous binders: a comparative study on rhodanines and related heterocycles in medicinal chemistry. J Med Chem 55:743–753 [DOI] [PubMed] [Google Scholar]
- Mitsui T, Nomura S, Okada M, Ohno Y, Kobayashi H, Nakashima Y, Murata Y, Takeuchi M, Kuno N, Nagasaka T, et al. (2003) Hypertension and angiotensin II hypersensitivity in aminopeptidase A-deficient mice. Mol Med 9:57–62 [PMC free article] [PubMed] [Google Scholar]
- Mucha A, Drag M, Dalton JP, Kafarski P. (2010) Metallo-aminopeptidase inhibitors. Biochimie 92:1509–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Inloes JB, Katagiri T, Kobayashi T. (2011) Chondrocyte-specific microRNA-140 regulates endochondral bone development and targets Dnpep to modulate bone morphogenetic protein signaling. Mol Cell Biol 31:3019–3028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanus DM, Engelstein D, Gastl GA, Gluck L, Vidal MJ, Morrison M, Finstad CL, Bander NH, Albino AP. (1993) Molecular cloning of the human kidney differentiation antigen gp160: human aminopeptidase A. Proc Natl Acad Sci USA 90:7069–7073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez I, Varona A, Blanco L, Gil J, Santaolalla F, Zabala A, Ibarguen AM, Irazusta J, Larrinaga G. (2009) Increased APN/CD13 and acid aminopeptidase activities in head and neck squamous cell carcinoma. Head Neck 31:1335–1340 [DOI] [PubMed] [Google Scholar]
- Powers JP, Piper DE, Li Y, Mayorga V, Anzola J, Chen JM, Jaen JC, Lee G, Liu J, Peterson MG, et al. (2006) SAR and mode of action of novel non-nucleoside inhibitors of hepatitis C NS5b RNA polymerase. J Med Chem 49:1034–1046 [DOI] [PubMed] [Google Scholar]
- Rawlings ND, Waller M, Barrett AJ, Bateman A. (2014) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 42: D503–D509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaux A, Fournie-Zaluski MC, David C, Zini S, Roques BP, Corvol P, Llorens-Cortes C. (1999) Aminopeptidase A inhibitors as potential central antihypertensive agents. Proc Natl Acad Sci USA 96:13415–13420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenen FJ, Weiner WS, Baillargeon P, Brown CL, Chase P, Ferguson J, Fernandez-Vega V, Ghosh P, Hodder P, Krise JP, et al. (2010) Probe Reports from the NIH Molecular Libraries Program: Inhibitors of the Plasmodium falciparum M18 Aspartyl Aminopeptidase, National Center for Biotechnology Information US, Bethesda, MD: [PubMed] [Google Scholar]
- Simeonov A, Jadhav A, Thomas CJ, Wang Y, Huang R, Southall NT, Shinn P, Smith J, Austin CP, Auld DS, et al. (2008) Fluorescence spectroscopic profiling of compound libraries. J Med Chem 51:2363–2371 [DOI] [PubMed] [Google Scholar]
- Sivaraman KK, Oellig CA, Huynh K, Atkinson SC, Poreba M, Perugini MA, Trenholme KR, Gardiner DL, Salvesen G, Drag M, et al. (2012) X-ray crystal structure and specificity of the Plasmodium falciparum malaria aminopeptidase PfM18AAP. J Mol Biol 422:495–507 [DOI] [PubMed] [Google Scholar]
- Soares KM, Blackmon N, Shun TY, Shinde SN, Takyi HK, Wipf P, Lazo JS, Johnston PA. (2010) Profiling the NIH Small Molecule Repository for compounds that generate H2O2 by redox cycling in reducing environments. Assay Drug Dev Technol 8:152–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoermer D, Liu Q, Hall MR, Flanary JM, Thomas AG, Rojas C, Slusher BS, Tsukamoto T. (2003) Synthesis and biological evaluation of hydroxamate-based inhibitors of glutamate carboxypeptidase II. Bioorg Med Chem Lett 13:2097–2100 [DOI] [PubMed] [Google Scholar]
- Teuscher F, Lowther J, Skinner-Adams TS, Spielmann T, Dixon MW, Stack CM, Donnelly S, Mucha A, Kafarski P, Vassiliou S, et al. (2007) The M18 aspartyl aminopeptidase of the human malaria parasite Plasmodium falciparum. J Biol Chem 282:30817–30826 [DOI] [PubMed] [Google Scholar]
- Tonna S, Dandapani SV, Uscinski A, Appel GB, Schlöndorff JS, Zhang K, Denker BM, Pollak MR. (2008) Functional genetic variation in aminopeptidase A (ENPEP): lack of clear association with focal and segmental glomerulosclerosis (FSGS). Gene 410:44–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss ME, Carter PH, Tebben AJ, Scherle PA, Brown GD, Thompson LA, Xu M, Lo YC, Yang G, Liu RQ, et al. (2003) Both 5-arylidene-2-thioxodihydropyrimidine-4,6(1H,5H)-diones and 3-thioxo-2,3-dihydro-1H-imidazo[1,5-a]indol-1-ones are light-dependent tumor necrosis factor-alpha antagonists. Bioorg Med Chem Lett 13:533–538 [DOI] [PubMed] [Google Scholar]
- Wang WL, Chai SC, Huang M, He HZ, Hurley TD, Ye QZ. (2008) Discovery of inhibitors of Escherichia coli methionine aminopeptidase with the Fe(II)-form selectivity and antibacterial activity. J Med Chem 51:6110–6120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk S, Wilk E, Magnusson RP. (1998) Purification, characterization, and cloning of a cytosolic aspartyl aminopeptidase. J Biol Chem 273:15961–15970 [DOI] [PubMed] [Google Scholar]
- Wright JW, Tamura-Myers E, Wilson WL, Roques BP, Llorens-Cortes C, Speth RC, Harding JW. (2003) Conversion of brain angiotensin II to angiotensin III is critical for pressor response in rats. Am J Physiol Regul Integr Comp Physiol 284:R725–R733 [DOI] [PubMed] [Google Scholar]
- Wu Q, Lahti JM, Air GM, Burrows PD, Cooper MD. (1990) Molecular cloning of the murine BP-1/6C3 antigen: a member of the zinc-dependent metallopeptidase family. Proc Natl Acad Sci USA 87:993–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Liu C, Lin YL, Li F. (2013) Structural insights into central hypertension regulation by human aminopeptidase A. J Biol Chem 288:25638–25645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4:67–73 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.