

Significance
Human respiratory syncytial virus (hRSV) is the leading cause of bronchiolitis and pneumonia in children worldwide. The induction of poor T-cell immunological memory causes a high susceptibility to reinfections, which contributes to hRSV spread. Previously, we showed that hRSV inhibits T-cell activation by impairing the assembly of the dendritic cell (DC)−T-cell immunological synapse (IS). Here, we show that the nucleoprotein (N) of hRSV—a canonical cytosolic protein—is expressed on the surface of infected DCs. Further, using the supported-lipid-bilayer system (that mimics the DC/ antigen-presenting cells-membrane composition), we observed that the hRSV N interfered with pMHC−T-cell receptor interactions and inhibited IS assembly. We conclude that hRSV N may therefore be instrumental in impairing the host immune response during infection with this virus.
Keywords: T lymphocyte priming, nucleocapsid protein, cSMAC, pSMAC
Abstract
Human respiratory syncytial virus (hRSV) is the leading cause of bronchiolitis and pneumonia in young children worldwide. The recurrent hRSV outbreaks and reinfections are the cause of a significant public health burden and associate with an inefficient antiviral immunity, even after disease resolution. Although several mouse- and human cell-based studies have shown that hRSV infection prevents naïve T-cell activation by antigen-presenting cells, the mechanism underlying such inhibition remains unknown. Here, we show that the hRSV nucleoprotein (N) could be at least partially responsible for inhibiting T-cell activation during infection by this virus. Early after infection, the N protein was expressed on the surface of epithelial and dendritic cells, after interacting with trans-Golgi and lysosomal compartments. Further, experiments on supported lipid bilayers loaded with peptide-MHC (pMHC) complexes showed that surface-anchored N protein prevented immunological synapse assembly by naive CD4+ T cells and, to a lesser extent, by antigen-experienced T-cell blasts. Synapse assembly inhibition was in part due to reduced T-cell receptor (TCR) signaling and pMHC clustering at the T-cell−bilayer interface, suggesting that N protein interferes with pMHC−TCR interactions. Moreover, N protein colocalized with the TCR independently of pMHC, consistent with a possible interaction with TCR complex components. Based on these data, we conclude that hRSV N protein expression at the surface of infected cells inhibits T-cell activation. Our study defines this protein as a major virulence factor that contributes to impairing acquired immunity and enhances susceptibility to reinfection by hRSV.
Human respiratory syncytial virus (hRSV) is the leading cause of bronchiolitis and pneumonia in infants and a major economic and public health burden worldwide (1). Throughout the course of a lifetime, hRSV—a virus that has minimal antigenic variation—can repetitively infect humans and even cause reinfections during the same year (2–4). Despite repeated hRSV exposure, adult patients can develop acute respiratory tract infections with no development of protective immunity against the virus (5, 6). Because a limited T-cell response is observed in the lungs of hRSV-infected children, it is thought that T-cell-mediated immunity may be targeted by hRSV virulence mechanisms to prevent virus clearance from infected tissues (7).
Proper differentiation of naïve CD4+ T cells into T helper (Th) and memory cells is crucial to control viral infections. More specifically, memory CD4+ T cells promote viral clearance through a variety of synergizing mechanisms, including Th-mediated perforin cytotoxicity and the enhancement of B-cell and CD8+ T-cell responses (8–10). T-cell priming by dendritic cells (DCs) is a pivotal process for the acquisition of T-cell memory that largely influences the strength and efficiency of recall immune responses after subsequent virus infections (11–13). Several studies have shown that hRSV infects human and mouse DCs, impairing their capacity to activate both naïve (14) and antigen-experienced CD4+ T cells (15, 16). Furthermore, hRSV infection reduces the capacity of DCs to induce the expansion of antiviral Th1 effector cells (17–19). It has been shown that hRSV inhibits naïve T-cell priming by preventing the assembly of the immunological synapse (IS) with DCs (20), which is a supramolecular structure required for the activation of T cells by DCs and other antigen-presenting cells (APCs) (21). Nevertheless, the mechanisms responsible for the inhibition of IS assembly by hRSV infection remain unknown.
T-cell receptor (TCR) binding to cognate peptide-major histocompatibility complexes (pMHCs) is a mandatory step for the initiation of IS assembly at the APC−T-cell interface (21). The TCR−pMHC interaction triggers the rearrangement of a variety of receptor−ligand pairs and their signaling elements within centripetally migrating scaffolds, known as TCR microclusters (TCRm), which promotes signaling at synapse (22, 23). Sustained, central clustering of TCRm arranges into a TCR-enriched domain known as the central supramolecular activation cluster (cSMAC) where TCR signaling is regulated (23, 24). The massive clustering of TCRm produces a size-based exclusion of LFA-1:ICAM-1 integrin pairs from the synapse center, which arrange into a peripheral adhesion domain (or pSMAC) at the cell−cell interface (21). Importantly, the size-based segregation of surface molecules occurring during IS assembly maintains an APC−T-cell intermembrane spacing ranging 13–15 nm that promotes TCR triggering and enhances antigen sensitivity by T cells (25, 26). In fact, molecules larger than the TCR, such as CD43 and the phosphatases CD148 and CD45, jeopardize TCR triggering by either augmenting synapse spacing or exerting steric hindrance over pMHC binding (27, 28). Thus, hRSV surface proteins expressed on infected DCs, which form 11- to 20-nm-long projections (29), may therefore impede TCR−pMHC interactions at the cell−cell interface.
In the present study, we provide evidence that, in addition to the attachment (G) and fusion (F) glycoproteins, the nucleoprotein (N) of hRSV—a canonical cytosolic protein that forms ribonucleoprotein complexes for the synthesis of hRSV RNA species—can be expressed on the surface of infected cells, including DCs. Using the supported lipid bilayer model (that mimics the protein composition of the APC membrane) and TCR-transgenic CD4+ T cells, we show that bilayer-bound N impaired IS assembly of antigen-stimulated naïve—and to a lesser extent—blast T cells. Our results suggest a new function for the hRSV N as a cell surface molecule interfering with TCR−pMHC interactions, as evidenced by a reduction on pMHC central clustering, pMHC-TCR colocalization, and the phosphorylation of tyrosine residues at the T-cell−APC interface.
Results
The hRSV N Is Expressed on the Surface of Infected Cells.
To identify surface hRSV proteins that might be interfering with synapse assembly, we performed flow cytometry analyses of DCs inoculated either with noninfectious supernatant (mock) or the hRSV strain 13018-8 (hRSV13018-8). At 24 h postinfection (hpi), we observed positive staining for both hRSV fusion (F-hRSV) and attachment (G-hRSV) glycoproteins in hRSV-inoculated, but not mock-inoculated, DCs (Fig. 1A). Unexpectedly, we observed positive staining for the nucleoprotein (N-hRSV) in infected DCs under nonfixing, nonpermeabilizing conditions. DCs inoculated with the UV-hRSV13018-8 showed no significant F, G, and N signals by flow cytometry and no amplification for hRSV N transcripts by real-time PCR, which is consistent with the proper inactivation of the virus (Fig. S1A). Further, the hRSV F, G, and N proteins were also detected in hRSV-inoculated HEp-2 cells, but not in mock controls (Fig. 1B). Furthermore, N was detected on the surface of K562 (nonadherent) and Vero cells (Fig. S1B), as well as on the surface of HEp-2 cells infected with the hRSV clinical isolates JC/08 and AH/08 (Fig. S1C). Next, to corroborate these results, we performed laser confocal microscopy imaging of mouse DCs and human HEp-2 cells inoculated either with mock or hRSV13018-8. Because Orange CMTMR and BODIPY-TR dyes are transformed into cell-impermeant probes, we used these molecules for counterstaining and as indicators of membrane integrity at the moment of sample preparation. As expected, N was observed on the surface of both hRSV-infected DCs (Fig. 1C) and HEp-2 cells (Fig. 1D), but not in the respective mock controls. Further, laser confocal microscopy imaging confirmed the expression of the hRSV F and G glycoproteins on the surface of hRSV-infected DCs (Fig. S1 D and E) and HEp-2 cells (Fig. S1 F and G).
Fig. 1.

The hRSV N localizes on the surface of infected cells. (A) Flow cytometry analyses of mouse DCs inoculated with mock (noninfectious supernatant), UV-inactivated hRSV or infectious hRSV (MOI = 1) and stained at 24 hpi against the hRSV F (Left), G (Center Left), and N (Center Right) proteins. Mean values ± SEM are shown (n = 3). (Right) Histogram overlays representative for the F, G, and N flow cytometry analyses (blue, mock; gray, UV-hRSV; red, hRSV). (B) Histogram overlays of flow cytometry analyses for surface expression of hRSV F, G, and N proteins as in A. HEp-2 cells inoculated with mock (blue) and infectious hRSV13018-8 (red) (MOI = 1) were analyzed at 24 hpi. (C and D) Laser confocal microphotographs of mock or hRSV13018-8-infected mouse DCs and HEp-2 cells stained at 24 hpi using nonpermeabilizing conditions. In B−D, data are representative of at least three independent experiments. **P < 0.005; ***P < 0.001.
Cell-Surface Localization of hRSV N Occurs Early During the hRSV Replication Cycle.
To address whether localization of the hRSV N at the cell surface requires active viral replication, we used a recombinant hRSV A2 strain encoding the green fluorescent protein (hRSVGFP). Because GFP translation from the viral genome is regulated by the hRSV large RNA-dependent RNA polymerase, GFP synthesis serves as reporter for viral replication (17, 30). Also, because the GFP displays a half-life of 26 h (31), accumulation of GFP over time [and the increase in its mean fluorescence intensity (MFI)] serves to track the progression of the viral replication cycle in infected cells. We used HEp-2 cells, which are more permissive than DCs in sustaining viral replication, to evaluate the putative mechanisms used by N to reach the cell surface. First, we performed flow cytometry analyses of HEp-2 cells left uninfected or inoculated with UV light-inactivated hRSV (UV-hRSVGFP), fully infectious hRSV (hRSVGFP), or a neutralized immunocomplexed hRSV (IC-hRSVGFP) formed by incubating viral inoculums with an anti-hRSV F protein mAb (clone RS-348) before cell inoculation. The anti-hRSV N antibody (clone 1E9/D1), which is not expected to neutralize hRSV, was incubated with the virus as an additional control (α-N-hRSVGFP). As shown in Fig. 2A, at 48 hpi, hRSVGFP-infected HEp-2 cells, but not uninfected controls, showed significant fluorescence for both N and GFP. In contrast, UV-hRSV-inoculated HEp-2 cells at 48 hpi displayed no significant fluorescence for either N or GFP, which is indicative of proper virus inactivation (Fig. 2A, Center Left). Similarly, neither the nucleoprotein nor the GFP were detected on HEp-2 cells inoculated with IC-hRSVGFP. As expected, the anti-N-hRSV antibody failed at preventing infection of HEp-2 cells, which displayed similar N and GFP fluorescence to cells infected with untreated hRSVGFP (Fig. 2A, Right). These findings suggest that by 48 hpi, active viral replication is required for the localization of the nucleoprotein at the cell surface.
Fig. 2.

The hRSV N reaches the cell surface early during the replication cycle. (A) Contour plots of HEp-2 cells uninfected or inoculated (MOI = 1) either with UV-inactivated hRSVGFP (UV-hRSVGFP), infectious hRSVGFP, anti-F-neutralized hRSVGFP (IC-hRSVGFP) or hRSV incubated with a nonneutralizing, anti-N hRSV antibody (α-N-hRSVGFP). HEp-2 cells were harvested and stained 48 hpi with propidium iodide (for dead-cell exclusion) and an anti-N-hRSV antibody (clone 1E9/D1). (B) Kinetics (1−72 hpi) of surface N protein expression in hRSVGFP-infected HEp-2 cells. (C) (Left) Quantitative real-time RT-PCR for the detection of N transcripts in four different HEp-2 subpopulations, which were purified by FACS under a four-way purity precision schema (>90% purity) as N+/GFPneg, N+/GFPlo, Nlo/GFP+, and Nneg/GFPneg. Uninfected cells and cells inoculated with UV-hRSVGFP were used as controls. (Right) Representative purity controls for the subpopulations evaluated by qRT-PCR. (D) Nucleoprotein ELISA of cell-cleared supernatants obtained at 48 hpi from control and hRSV-infected HEp-2 and DCs. Recombinant N-hRSV was used as a standard for quantification. (E and F) Virus adsorption assays measuring surface N- and F-hRSV expression in HEp-2 cells incubated for 1 h with either untreated hRSV or hRSV pretreated with UV, anti-F, heparin (400 UI/mL), or anti-F/heparin. (G) Representative plots showing double staining for the hRSV F and N proteins in HEp-2 cells inoculated for 1 h with hRSV13018-8. (H) TEM analyses of HEp-2 cells inoculated for 1 h with hRSVGFP and then prepared as described in SI Material and Methods (scale bar: 300 nm; V, virus) (n = 2). In A−F, mean values ± SEM are shown (n = 3). For statistical analyses, one-way ANOVA and Dunnett´s multiple comparison test were used to compare all treatments against uninfected cells (B−D) or to hRSV-untreated, inoculated cells (1 hpi, E and F). *P < 0.05; ***P < 0.001. n.s., nonsignificant.
Further, flow cytometry analyses showed an inverse association between surface N and GFP expression (Fig. 2A): At higher GFP geometric MFIs, the expression of the nucleoprotein on the cell surface was lower. Therefore, although viral replication was required for surface localization of the N protein (compare UV-hRSVGFP with hRSVGFP), most N+ cells were those synthetizing low levels of viral GFP (i.e., those at early stages of hRSV replication). To further test this notion, we determined by flow cytometry the kinetics of N expression on the surface of HEp-2 cells. As soon as 1 hpi and 12 hpi, we observed a significant surface signal for N in hRSV-infected cells (Fig. 2B). Furthermore, we observed a significant increase for both the MFI derived from N protein expression and the percentage of N positive cells from 12 hpi to 72 hpi [at a multiplicity of infection (MOI) of 1 plaque-forming unit (PFU)/cell], when most HEp-2 cells expressed GFP (Fig. 2B and Fig. S2E). However, we observed that from 12 hpi to 72 hpi, most N positive cells belonged to the GFPlo population (Fig. S2 B−E).
The absence of N signal in hRSVGFP-infected cells stained at different time points postinfection with a mouse IgG2a-Alexa Fluor 647 isotype control supports the staining specificity for the anti-N-hRSV 1E9/D1 antibody (Fig. S2 A−E). Further, the reduction of the N-derived signal following staining of infected cells in the presence of either recombinant N protein or a polyclonal anti-N serum also supports the specificity of the 1E9/D1 mAb (Fig. S2 G and H, respectively). As an additional control, we evaluated whether another canonical cytosolic hRSV protein, the Matrix 2-ORF 1 protein (M2-1, ∼22 kDa), and the hRSV F protein (an integral membrane protein) could also be detected on the surface of infected HEp-2 cells. As expected, surface staining was observed for hRSV F, but not for hRSV M2-1 (Fig. S3 A and B, respectively), suggesting that surface detection of hRSV N is not due to loss of membrane integrity on hRSV-infected cells. Finally, to further corroborate the presence of N at the cell surface, we performed stripping of cell surface proteins using trypsin. As expected, pretreatment of HEp-2 cells with trypsin significantly reduced the MFIs derived from cell surface N protein at 48 hpi (Fig. S3C).
Because a N+ GFPnull population was observed at different times postinfection (upper left quadrant in N/GFP contour plots in Fig. 2A and Fig. S2 C−F), we performed fluorescence-activated cell sorting coupled to quantitative RT-PCR (using primers specific for N) to corroborate that this cell population was in fact infected. More specifically, four different populations were purified (>90% purity) according to their fluorescence for N and GFP (i.e., N+/GFPneg, N+/GFPlo, Nlo/GFP+, and Nneg/GFPneg) (Fig. 2C). Importantly, compared with uninfected or UV-inactivated hRSVGFP-treated cells, N RNA transcripts were detected in all cells inoculated with hRSVGFP, despite the level of GFP and N expression detected by flow cytometry. These results suggest that the majority of cells in the culture were infected (Fig. 2C). Low levels of N surface expression correlated with lower levels of N RNA transcripts in both Nneg/GFP+ and double-negative cells, suggesting that low levels of surface N expression may be due to reduced N neosynthesis in GFPhi cells (Fig. 2C). Because transcription (synthesis of viral mRNAs) decreases at later stages of the hRSV infectious cycle to promote virus replication (32), our data suggest that the N protein is majorly expressed on the surface of cells during early stages of the hRSV replication cycle.
The detection of N protein, but not GFP, at 1 hpi in hRSV-inoculated cells stained under nonpermeabilizing conditions (Fig. 2 B and E) suggests that exogenous N protein either soluble or virion-associated may be adsorbed on the surface of inoculated cells. To evaluate this possibility, we first performed ELISAs to detect the N protein in cell supernatants derived from hRSV-infected and control cells. As expected, N protein was detected in supernatants of hRSV-infected HEp-2 cells, but not for hRSV-infected DCs (Fig. 2D). Because DCs are poorly permissive for hRSV replication and produce only limited viral progeny (ref. 17 and Fig. S3D), our data suggest that inoculated viral particles attached to the cell surface could be responsible for the N+ signal observed in HEp-2 as soon as 1 hpi by flow cytometry.
To elucidate the contribution of either cell-attached or fused virions to the N+ signal observed at 1 hpi, virus adsorption experiments were performed in HEp-2 cells incubated for 1 h with either mock, UV-hRSV, or hRSV (MOI = 10). Some cells were treated with virus either neutralized with anti-F mAb (that prevents virus−cell fusion) or treated with heparin (400 UI/mL, which prevents virus attachment mediated by the hRSV G protein). As expected, HEp-2 cells incubated 1 h with viable hRSV, but not mock, showed N+ staining under nonpermeabilizing conditions. The observation that HEp-2 cells incubated 1 h with nonreplicative UV-inactivated hRSV displayed N protein at cell surface (Fig. 2E and Fig. S3E) suggests that the signal could derive from exogenous binding/fusion of hRSV particles to HEp-2 cells, rather than N neosynthesis. This notion was further supported by the observation that pretreatment of both hRSV and UV-hRSV with either anti-F or heparin significantly reduced the N-derived MFI in HEp-2 after 1 h postinoculation (Fig. 2E and Fig. S3E, respectively). Importantly, a similar pattern of staining for F was observed on the surface of HEp-2 cells inoculated with hRSV treated with anti-F, heparin, or both (Fig. 2F). These data suggest that the observed N+ signal was due to either cell-attached or cell-fused virus particles. As expected, we observed a reduced F-derived MFI (measured with an Alexa Fluor-labeled anti-F palivizumab) in cells challenged with hRSV pretreated with neutralizing anti-F RS-348 mAb. Furthermore, we observed that at 1 hpi, ∼30% of inoculated HEp-2 cells were double positive for N and F (MOI 10, Fig. 2G), supporting the notion that attachment/fusion of exogenous viral particles may contribute to cell surface N-derived signal. Lastly, EM analyses of HEp-2 cells inoculated for 1 h with hRSVGFP showed that most cell-attached virus particles displayed well-defined electron-dense envelopes. These data are consistent with previous studies (33) and further support the notion that N signal on the surface of infected cells at 1 hpi is due to adsorbed, structurally intact, viral particles (Fig. 2H). Altogether, these results suggest that (i) N is expressed on the surface of infected cells and (ii) the attachment and fusion of newly released hRSV particles contributes to cell surface delivery of N, presumably because hRSV virions have envelope-associated N.
Because hRSV F and G glycoproteins use the trans-Golgi network to reach the plasma membrane of hRSV-infected cells, we performed transport inhibition assays with brefeldin A (BFA) to evaluate whether the hRSV N uses a similar pathway to reach the cell surface (34–36). Based on the virus adsorption experiments described above, the putative mechanism of surface N delivery was explored starting at early infection times (i.e., 24−48 hpi) to reduce noise derived from the attachment of newly released virions that are produced at large amounts from 48 hpi to 72 hpi (Fig. S3D).
At 24 hpi, and 5 h after treatment with 10 μg/mL of BFA, a slight decrease of surface N expression in HEp-2 cells was observed (P < 0.05) (Fig. 3A, Left). As expected, no significant differences were observed between untreated or vehicle-treated hRSV-infected cells (Fig. 3A). At 48 hpi, a more significant decrease on nucleoprotein MFIs (P < 0.01) was observed in hRSV-infected cells that were pretreated with BFA, compared with untreated or vehicle-treated hRSV-infected cells (Fig. 3A, Right). These results suggest that surface localization of N depends on trans-Golgi trafficking (Fig. 3A, Right). In addition, because hRSV uses both the apical recycling endosome system (37) and the phosphatidylinositol 3-kinase (PI3K) for particle maturation and budding (38), we performed inhibition assays using wortmannin (WM) to evaluate whether virus retention increases N at cell surface. Although no differences were observed at 24 hpi, a significant increase of N protein surface expression was detected at 48 hpi after pretreatment of cells with 500 nM of WM (Fig. 3A). These data suggest that N protein can also be delivered to the cell surface during the process of virus budding.
Fig. 3.

Brefeldin A reduces surface nucleoprotein expression and increases N protein accumulation within the Golgi compartment. (A) Flow cytometry analyses of BFA- and WM-inhibition assays. At 24 hpi, control and hRSV-infected (MOI = 5) HEp-2 cells were incubated for 5 h with either BFA (10 μg/mL) or WM (500 nM), washed twice, and analyzed by flow cytometry for expression of surface hRSV N. One group for each drug (along its vehicle control) was washed and left in culture to determine N expression in the cell surface at 48 hpi (n = 3). (B) HEp-2 cells were coinfected with hRSV (MOI = 5) and a baculovirus encoding a fusion RFP-GALNT2 (particles per cell = 30). At 24 hpi, HEp-2 cells were incubated for 5 h with BFA (10 μg/mL), DMSO (vehicle), or media (untreated) and then fixed with 4% paraformaldehyde (PFA) in PBS, permeabilized with 0.2% Triton X-100 in PBS, and stained using a directly conjugated anti-N 1E9/D1 Alexa Fluor 647 (depicted in green) (n = 3). Open arrowheads (white) indicate N-hRSV–RFP colocalization in untreated and vehicle-treated cells. Thin arrows indicate accumulation of N within the RFP-GALNT2+ compartment in BFA-treated cells. *P < 0.05; ***P < 0.001. n.s., nonsignificant.
Considering the significant decrease of surface expression of N protein caused by BFA, additional experimental analyses were performed to evaluate whether this protein colocalizes or associates with Golgi vesicles. With this aim, a baculovirus encoding the red fluorescent protein (RFP) fused with the human Golgi-resident enzyme N-acetylgalactosaminyl transferase 2 (GALNT2, a secreted and membrane-anchored Golgi enzyme, CellLight Golgi-RFP) was used in coinfection with hRSV to evaluate N distribution with regard to GALNT2. As shown in Fig. 3B, a fraction of the nucleoprotein shows a discrete colocalization with GALNT2 in hRSV-infected cells (Fig. 3B, Upper Left and Center Left, white arrowheads). Importantly, after BFA treatment, we observed perinuclear redistribution of GALNT2, which is consistent with retrograde transport to the endoplasmic reticulum (39), and a significant accumulation of N within the redistributed GALNT2+ compartment (Fig. 3B, Center Right). These observations suggest that N protein is delivered to the cell surface using the trans-Golgi network. Importantly, a similar pattern of localization was observed for the F protein with GALNT2, both in vehicle- and BFA-treated cells (Fig. S3F). However, in the latter condition, a higher colocalization frequency between F and the GALNT2+ compartment was found. These data suggest that even if there was a similar association between hRSV N and F proteins with the trans-Golgi network, the interaction of N with Golgi cisternae is different than that observed for integral membrane proteins, such as the hRSV F.
Because reduction of cell surface expression of N protein after BFA treatment could be due to a trafficking blockade from Golgi to either the late endocytic pathway or lysosomes, laser confocal imaging analyses were carried out for N and both the Ras-related protein (Rab7) and the lysosomal membrane glycoprotein-1 (Lamp-1) (Fig. S3 G and H). Compared with vehicle-treated cells, a significant increase in the association and colocalization of N with Lamp-1+ vesicles (but not Rab7+) was observed after BFA treatment. This result suggests that trafficking involving the Lamp-1+ compartment (i.e., lysosomal exocytosis) may also account for the delivery of N protein to the cell surface (Fig. S3 G and H). These data suggest that hRSV N protein may reach the cell surface by interacting as an itinerant protein with different machineries involved in protein trafficking, including the trans-Golgi network and the Lamp-1+ compartment.
Surface hRSV N Prevents T-Cell Activation by DCs.
Because the molecular size of hRSV N (∼43 kDa) is comparable to TCR and pMHC (∼58–60 kDa), it is possible for this molecule to segregate into the IS. Thus, we evaluated whether the hRSV N protein could interfere with T-cell activation. First, we determined if soluble N protein could bind to the cell membrane of uninfected cells. As shown in Fig. S4 A and B, we observed that after 3 h of incubation, purified N protein was able to bind to the surface of DCs and HEp-2 cells in a dose-dependent manner. Next, we explored whether the surface-attached N protein might exert an inhibitory effect over the capacity of DCs to prime naïve CD4+ T cells (OT-II). Soluble, histidine-tagged recombinant N and M2-1 (used as a control) proteins of hRSV were added into the media of cocultured DCs and OT-II CD4+ T cells. As shown in left side of Fig. S4C, 24 h poststimulation, we observed that various concentrations of N protein (ranging from 0.5 nM to 50 nM), but not M2-1, impaired naïve T-cell activation by DCs, as evidenced by reduced expression of CD25 and the TCR-driven activation marker CD69. Remarkably, the level of T-cell inhibition by N protein was comparable to the suppression of T-cell priming caused by DC infection by hRSV (see right of Fig. S4C). Given that triggering of TCRs during the first hours of T-cell−DC interaction leads to the expression of CD69 (40−42), these data suggest that N may interfere with pMHC−TCR interactions at the IS.
Next, to evaluate whether modulation of N protein expression on the surface of infected cells can influence the inhibition of T-cell priming by this molecule, T-cell activation assays using DCs transduced with lentiviruses (LV) encoding either the hRSV N (LV:N-hRSV) or the GFP protein (LV:GFP, as a control) were performed. We observed that T cells stimulated by LV:N-hRSV-infected DCs showed reduced expression of CD69 and reduced secretion of IL-2 and IFN-γ, compared with the controls. This result supports the notion that hRSV N is a negative modulator of T-cell priming (Fig. S4 E−G). In addition, modulation of N protein expression was attempted by using WM- or BFA-treated DCs. However, BFA and WM treatment impaired to a similar extent the activation of T cells primed by control or hRSV-infected DCs (Fig. S4D).
The hRSV N Keeps Naïve CD4+ T Cells from Assembling Mature ISs.
Considering that hRSV-infected DCs expressed N protein on the cell surface (Fig. 1A), we next evaluated the contribution of this molecule to the inhibition of IS assembly when bound to the APC membrane. To approach this in a more physiological manner, we first estimated the approximated density of N molecules [as a range of molecules per square micrometer (molec/μm2)] on the surface of hRSV-infected DCs (HEp-2 cells and K562 cells were included as controls). We used the hRSVGFP strain to determine densities of the N protein on the surface of infected cells (i.e., GFP+). We subsequently used a fluorometric bead assay, in which the anti-N hRSV 1E9/D1 mAb labeled with Alexa Fluor 647 (at known fluorochromes/antibody) was used to measure N protein-derived MFIs in nonfixed, nonpermeabilized cells. Using the same acquisition settings, five different silica beads containing known quantities of Alexa Fluor 647 were used to estimate the amount of N protein per cell. Then, using confocal microscopy, we determined the cell surface areas (CSAs) of infected cells and calculated the density of N as molecules per square micrometer (Fig. S5). The estimated range of N densities on DCs (48 hpi), HEp-2, and K562 cells (both at 72 hpi, as controls) are summarized in Table 1.
Table 1.
N-hRSV density on different cell types
| Cell type | |||
| DCs | HEp-2 | K562 | |
| Mean density, molecules/μm2 (min−max) | 11.39 (2.59–49.61) | 20.87 (12.39–33.18) | 72.90 (34.72–176.77) |
Next, we used the in vitro supported lipid bilayer (SLB) system (which mimics an infected APC surface) to stimulate antigen-specific, TCR-transgenic (AND) CD4+ T cells. The T-cell−bilayer contacts forming cSMACs (and pSMAC) were quantified as mature ISs. Naïve CD4+ T cells were incubated for 20 min on SLB loaded with physiological densities of the I-Ek-MCC (pMHC-II, at 5 molec/μm2), ICAM-1 (at 200 molec/μm2) and 10 molec/μm2 of either N protein or the control proteins CD58 and M2-1. CD58 is the human ligand for the T-cell adhesion molecule CD2 and is not expected to bind mouse CD2; therefore it would not affect IS assembly by mouse T cells (43). As shown in Fig. 4 A and C, naïve T cells stimulated by N protein-loaded bilayers displayed a reduced frequency of mature ISs compared with controls. Interestingly, inhibition of mature IS assembly was also observed in experiments using varying N protein densities, ranging from 2 molec/μm2 to 60 molec/μm2 (Fig. 4B).
Fig. 4.

The hRSV N protein prevents mature IS assembly by naïve CD4+ T cells. (A) Total internal reflection fluorescence (TIRF) microscopy analyses showing the frequencies of mature IS assembled by naïve CD4+ T cell stimulated on SLB containing 5 molec/μm2 of I-Ek-MCC, 200 molec/μm2 of Alexa Fluor 405-labeled ICAM-1, and 10 molec/μm2 of either hRSV N, hRSV M2-1, or CD58. Bilayers with no additional proteins (untreated) and preincubated with vehicle were used as additional controls. (B) Frequencies of mature ISs assembled by naïve T cells stimulated with SLB loaded with increasing densities of N-hRSV (none/vehicle and 2–60 molec/μm2). (C) Representative pictures of T cells stimulated with untreated (Upper) and N-hRSV-loaded (Lower) SLB. White arrows show cSMACs (ICAM-1 exclusion and TCR-central clustering). (D) TFI for TCR were measured as readout for centripetal TCRm accumulation at the T-cell−SLB interface. (E) MFIs were measured for ICAM-1 at interfaces formed by AND naïve CD4+ T cells and untreated or N-hRSV-loaded SLB. Mean values ± SEM are shown in A−E (n = 3). One-way ANOVA and Dunnett´s multiple comparison test were used for statistical analyses. *P < 0.05; **P < 0.005; ***P < 0.001. n.s., nonsignificant.
Upon TCR engagement by antigen, the central clustering of TCRm leads to cSMAC formation. Hence, we measured TCR total fluorescent intensity (TFI) as readout for TCRm clustering within the cell−bilayer junction. As expected, impairment of IS assembly was associated with reduced TFIs for TCR at T-cell−bilayer contacts (Fig. 4D). Because TCR signaling leads to an increase of LFA-1 binding affinity to ICAM-1, we also measured the amount of bound ICAM-1 at contacts (as MFI). We observed that inhibition of synapse assembly was accompanied with a reduced binding of ICAM-1 (Fig. 4E).
Reduced IS Assembly Is Accompanied by Reduced Proximal TCR Signaling.
Because (i) central clustering of TCR occurs mostly as pMHC bound pairs and (ii) synapse assembly is largely dependent on the level of proximal TCR signaling (23), we evaluated whether the reduction in cSMAC frequencies was associated with reduced TCR-proximal signaling. To address this question, as a readout for TCR signaling implicating the involvement of kinases and proximal downstream phosphorylation events, we measured the extent of overall tyrosine phosphorylation in naïve CD4+ T cells stimulated 20 min on SLB loaded with either N protein or control proteins. Compared with M2-1 and CD58, 10 molec/μm2 of hRSV N protein significantly reduced the phosphorylation of tyrosine residues in the stimulated T cells (Fig. 5A). Strikingly, although we observed reduced phosphotyrosine staining with N protein densities as low as 2 molec/μm2, no significant differences were observed at densities equal to 40 molec/μm2 and 60 molec/μm2 (Fig. 5B). The subtle increase in tyrosine phosphorylation observed with 60 molec/μm2 of N protein from hRSV suggests that this molecule may bind a signaling receptor on T cells (not related necessarily with cSMAC formation) or work as a superantigen binding an element of the TCR complex (see Discussion). Because the reduction in tyrosine phosphorylation may be explained by a reduction in pMHC engagement by the TCR, the relative amounts of fluorescent I-Ek molecules recruited at the cell−bilayer interface was evaluated. Interestingly, naïve T cells incubated with bilayers containing 10 molec/μm2 of N protein displayed a slight, but significant, decrease in the amount of fluorescent pMHC recruited to the T-cell−bilayer interface, compared with untreated controls (Fig. 5C). Fluorescence recovery after photo-bleaching (FRAP) of fluorescent ICAM-1 and tracking of fluorescent N protein ruled out the possibility that decreased pMHC clustering was due to reduced bilayer fluidity, reduced ICAM-1 lateral mobility, or massive N protein aggregation (Fig. S6A and Movie S1). Further, most fluorochrome-labeled N protein displayed high lateral mobility on ICAM-1 and I-Ek-MCC-loaded SLB (Movie S1). These results suggest that inhibition of mature IS assembly was due to reduced TCR−pMHC engagement, and hence, by an impaired downstream signaling.
Fig. 5.

Impairment of IS assembly by hRSV N protein is associated with a reduction in TCR signaling. (A) TIRF microscopy analyses of tyrosine phosphorylation in naïve AND CD4+ T cells stimulated over SLB containing I-Ek, ICAM-1, and 10 molec/μm2 of CD58, hRSV N, or hRSV M2-1. Following fixation, T cells were permeabilized and stained with an anti-pTyr antibody (clone PY20) (n = 3). (B) MFIs for pTyr in naïve AND T cells stimulated with SLB containing increasing nucleoprotein densities (none/vehicle and 2–60 molec/μm2). (C) TFIs for I-Ek loaded with DR-640-labeled MCCp in the interface of T cells stimulated with untreated or N-loaded (10 molec/μm2) SLB. (D) Frequencies of mature ISs assembled by naïve CD4+ T cells stimulated with SLB containing 20 molec/μm2 of an Alexa Fluor 568-labeled anti-CD3ε antibody (clone 2C11), and none or 10 molec/μm2 of either hRSV N- or M2-1. (E) TFIs for anti-CD3ε at contacts of naïve T cells for the experiments shown in D. Mean fluorescent AUs ± SEM are shown from >100 cells (n = 3). *P < 0.05; ***P < 0.001. n.s., nonsignificant.
To better understand the effect of hRSV N protein on the IS, T cells were stimulated with anti-CD3ε instead of cognate pMHC. SLB were loaded with anti-CD3ε (clone 2C11, at 20 molec/μm2) alongside 10 molec/μm2 of either hRSV N or M2-1 proteins. Compared with untreated and M2-1-loaded bilayers, no differences were observed in the percentage of mature IS (∼80%) assembled in the presence of N protein (Fig. 5D). Furthermore, whereas no significant differences were observed for the recruitment of 2C11 TFI at contacts (Fig. 5E), the N protein seems unable to interfere with the assembly of IS induced by anti-CD3ε stimulation. This was more evident when comparing the frequency of IS assembly by T cells stimulated with SLB loaded with N protein/pMHC versus N protein/anti-CD3ε (Fig. S6B). These data suggest that the hRSV N may inhibit IS assembly via mechanisms that take advantage of the low affinity of the physiological TCR−pMHC interaction. For example, N may (i) impair bending of the T-cell membrane necessary to form a close contact (44), or (ii) impair physiological microclustering of newly formed monovalent pMHC−TCR pairs.
To test whether the impairment in mature IS assembly caused by the N protein was due to reduced TCR−pMHC binding, experiments were performed with antigen-experienced blast CD4+ T cells. As expected, AND T-cell blasts were more resistant to inhibition of mature IS synapse after stimulation by bilayers containing 10 molec/μm2 of N protein, compared with naïve AND T cells (Fig. S7A). This observation could be due to an increased sensitivity of T-cell blasts to pMHC, as evidenced by increased frequencies of mature ISs in antigen-experienced cells (∼70%), compared with naïve cells (∼50%) at equal pMHC density (Fig. 4A and Fig. S7A). Consistently, T-cell blasts showed no differences in tyrosine phosphorylation after 15 min of stimulation with control and N-loaded SLB (Fig. S7B). As expected, impairment of IS was less pronounced when T-cell blasts were stimulated with bilayers containing 20 molec/μm2 instead of 5 molec/μm2 of I-Ek-MCC (Fig. S7C). The increased tyrosine phosphorylation caused by human CD58 in the bilayers was not expected and could be due to either a weak interaction of human CD58 with mouse CD2 or the up-regulation on activated mouse T cells of a distinct receptor that binds human CD58.
The hRSV N Accumulates at the T-Cell−Bilayer Interface Independently of pMHC Binding.
The reduced pMHC accumulation at contacts and the recovery of synapse assembly by high pMHC densities suggest that either a reduction of pMHC binding or accessibility could be responsible for synapse impairment. Given that N protein may interfere with TCR−pMHC binding kinetics by interacting with pMHC, we performed single-particle tracking (SPT) experiments to evaluate whether fluorescent I-Ek-MCC (at 0.3 molec/μm2) interacts with fluorescently labeled hRSV N protein (at 40 molec/μm2 and 100 molec/μm2). Interestingly, neither colocalization nor comovement was observed between N protein and I-Ek-MCC, even when the former aggregated (at 100 molec/μm2) (Fig. S8A and Movie S2). Furthermore, no changes on I-Ek-MCC lateral mobility were observed between control and N protein-loaded bilayers (containing also ICAM-1 and I-Ek-MCC) (Fig. S8B). These data suggest that impairment of synapse assembly and pMHC clustering were not mediated by in cis interactions between N protein and the pMHC. In addition, considering that the central clustering of pMHC at the IS depends largely on TCR binding, we evaluated whether reduction in pMHC segregation might be explained by physical interactions between N protein and the TCR, or other T-cell proteins. Using a fluorochrome-labeled N protein, we evaluated whether this protein formed clusters at T-cell−bilayer interfaces and, more specifically, at the cSMAC. As shown in Fig. 6 A and B, N protein accumulated at the interface between SLB and either naïve or antigen-experienced T-cell blasts. Furthermore, the N protein accumulated at the synapse center, forming cSMAC-like structures sometimes (Fig. S9A). A discrete correlation was observed between TCR and N protein MFIs when measured at the interface of naïve and blast T cells (Fig. S9 B and C, respectively). These data suggest that to reach that central location, N protein could be interacting in trans with some T-cell molecules present in TCR microclusters, such as CD2, CD3, CD4, CD28, and TCR, among others. To evaluate this possibility, bilayers with no pMHC were used (and therefore bilayers not expecting to trigger TCRm assembly) to determine whether the N protein could be dragged into the center of cell contacts. Indeed, central clustering of hRSV N protein at cell−bilayer contacts was observed, regardless of pMHC presence (Fig. 6C). Strikingly, in some contacts, the nucleoprotein colocalized with the TCR and triggered the central segregation of this molecule in the absence of I-Ek-MCC (Fig. 6C, Lower). Therefore, N protein and TCR cosegregation would be consistent with an in trans interaction of the former with an element of the TCR complex. Such an interaction could explain most of the altered T-cell responses triggered by the N protein, which include a reduction of CD69 expression in DC−T-cell cocultures, impaired synapse assembly, decreased pMHC binding, clustering, and tyrosine phosphorylation at T-cell−bilayer contacts.
Fig. 6.
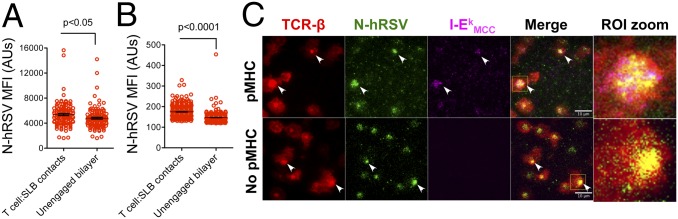
The hRSV N protein accumulates within cell contacts and cosegregates with the TCR. MFI for hRSV N at both contacts and cell-unbound bilayer from naïve (A) and blast (B) AND CD4+ T cells (n = 3). (C) TCR−N colocalization analyses at T-cell−bilayer interfaces formed over SLB loaded with 10 molec/μm2 of hRSV N (green) and either 20 (Upper) or 0 (Lower) molec/μm2 of I-Ek-MCC (magenta). Arrows show colocalizing pixels for nucleoprotein, TCR-β, and I-Ek (white pixels) or colocalization of N and TCR-β (yellow pixels). Image zooms are shown for each ROI highlighted in merge (n = 3).
Discussion
The health burden caused by hRSV infection is characterized by recurrent epidemics and subsequent reinfections, which are established throughout life. Because DCs play a crucial role in the initiation and perpetuation of T-cell immune memory, some studies have assessed whether hRSV impairs DC function as a mechanism to restrain T-cell immunity. These studies have shown that infection of DCs by hRSV renders these cells inefficient at priming naïve T cells, in both humans and mice (14, 20). While one of these studies suggests that naïve T cells are more susceptible than effector/memory T cells to hRSV-mediated inhibition, the other study shows that hRSV-infected DCs fail at assembling IS with naïve T cells (14, 20). Because receptor−ligand interactions lead to a size-segregation patterning at the synapse that promotes TCR triggering, it is possible that hRSV surface proteins expressed on the DC could directly impair TCR−pMHC interactions and IS function.
Here, we show that in contrast to what has been described regarding hRSV virion structure, the hRSV N can be expressed on the surface of a variety of infected cells, including DCs. Furthermore, our data suggest that the N protein may be expressed on the surface of viral particles as well, as evidenced by virus adsorption experiments, which showed N and F signal on the cell surface as early as 1 hpi (Fig. 2 D and E). In agreement with this notion, we observed that N protein expression was significantly reduced after preincubating infectious hRSV with either heparin (that blocks G) or neutralizing anti-F mAb (Fig. 2D). These results suggest that virus attachment and virus−cell fusion will be sufficient to deliver the N protein to the surface of APCs and therefore to impair T-cell priming, regardless of hRSV replication in these cells. The specific mechanism underlying nucleoprotein transport to the cell surface remains to be elucidated.
As concluded from flow cytometry analyses of HEp-2 cells infected with hRSVGFP, surface expression of the N protein takes place at early stages of the hRSV replication cycle (cells expressing low quantities of viral GFP throughout the assay, Fig. 2B and Fig. S2). Furthermore, cell sorting coupled to RT-PCR analyses revealed that GFPlo cells show significantly higher levels of N transcripts than GFP+/Nneg cells. These observations suggest that differences in N protein surface expression were due to variant levels of protein neosynthesis. Because at early times post-hRSV infection cells express mostly soluble N protein (not associated with RNA) in the cytosol (45), a possible mechanism for the targeting to the surface may involve the interaction of N protein with cellular membranes, such as those composing Golgi or lysosomal vesicles. This notion is supported by the observation that at 24 hpi, the hRSV N protein associates to and localizes near Golgi vesicles of infected cells, displaying a slight colocalization with GALNT2+ structures (Fig. 3B). Furthermore, the observation that BFA causes N protein to withdraw from the surface and accumulate in GALNT2+ and Lamp-1+ compartments suggests that both the trans-Golgi and the lysosomal exocytic pathway are involved in N protein trafficking to the cell surface (Fig. 3 A and B and Fig. S3 G and H). Finally, our results also suggest that soluble hRSV N is released from infected cells (Fig. 2D) and might bind to the surface of uninfected cells. Consistent with this notion is the observation that soluble recombinant N protein was capable of binding to the surface of both DCs and HEp-2 cells (Fig. S4 A and B).
Intracellular trafficking and cell surface expression of the hRSV N protein are likely to require a specific mechanism for maintaining a pool of monomeric N in the cytosol of infected cells. Similarly to other paramyxoviruses (46–48), the hRSV phosphoprotein (P) could work as a chaperone to keep a pool of soluble monomeric N protein that does not bind to RNA (49). Such a pool of monomeric N molecules could be available either for nucleocapsid assembly or membrane binding and targeting to the cell surface. Alternatively, the recent description of novel binding partners for N protein, such as the methylosome protein (WDR77) and the Heat-shock protein 70 (Hsp70) (50), raises the possibility that these proteins could contribute to preventing N protein from either being incorporated within ribonucleoprotein complexes or self-aggregating. Elucidation of the mechanism responsible for this phenomenon would require further research.
Our data, together with the observation that the measles virus nucleoprotein, also a Paramyxoviridae, is expressed on the surface of infected leukocytes (51) and inhibits B-cell activation (52), support a general role for N proteins as virulence factors that can modulate the host immune response and contribute to virus pathogenicity. Specifically, we describe here that the hRSV N protein inhibits T-cell activation when expressed on the surface of APCs. Among others, we observed a significant reduction of the TCR-driven activation marker CD69 in naïve T cells stimulated with DCs in the presence of soluble N, as well as in naïve T cells stimulated with DCs transduced with a lentivirus encoding the hRSV nucleoprotein (Fig. S4 E−G). Because the N protein size is permissive for segregation into the T-cell−APC synapse, we evaluated whether this molecule could interfere with TCR−pMHC interactions and IS assembly needed for CD69 up-regulation. To approach this question, the SLB system was used as an experimental model, which allows evaluating the effect of individual virus proteins on the synapse function without unspecific effects due to expression of other viral proteins by infected DCs. We observed that N protein prevented IS assembly and significantly decreased the level of tyrosine phosphorylation in naïve T cells stimulated with cognate antigen (Fig. 5). Additionally, these alterations were associated with reduced binding of ICAM-1 inside cell−cell contacts, which suggests that the N protein interferes with receptor−ligand interactions at the IS. Consistently, we observed that N protein segregates into forming synapses, reducing the accumulation of pMHC and TCR. Because both molecules cluster as bound elements, our data suggest that the N protein prevents IS assembly by interfering with TCR−pMHC interactions. In agreement with this notion, we circumvented N protein-mediated inhibition by replacing pMHC with anti-CD3ε, which activates T cells in an antigen (pMHC)-independent manner through engagement of CD3ε in the TCR complex. Furthermore, central clustering of the N protein alongside the TCR, even in the absence of pMHC (Fig. 6C), suggests a possible interaction between this protein and an element of the TCR complex. Additional research would be required to clarify whether the hRSV N interacts with the TCR as a mechanism to impair pMHC−TCR interactions.
Because the strength of naïve T-cell activation influences future recall responses, by impairing synapse assembly, N, a highly conserved hRSV protein, could ultimately constrain the acquisition of hRSV-specific T-cell immunity. This becomes particularly relevant when considering susceptibility to hRSV reinfections throughout life. In other words, we propose that by interfering with T-cell−APC synapse formation, the nucleoprotein could play a significant role in delaying the acquisition of T-cell memory. As a result, the adaptive immune response of the host would require several exposures to the virus to develop a somewhat limited T-cell response and succeed in controlling new viral aggressions. Such delayed/inefficient hRSV-specific T-cell responses might explain why healthy adults remain susceptible to hRSV reinfection yet suffer only mild respiratory diseases (6).
The mechanism described here is in agreement with the notion of hRSV interference with the priming of naïve CD4+ T cells, which is critical for the induction of protective antiviral adaptive immunity. Nevertheless, whether N protein may also interfere with the cytolytic function of CD8+ Cytotoxic T Lymphocytes (CTLs) remains to be evaluated. This aspect is of special relevance considering that several circulating CTL clones that are specific for hRSV antigens (including the N protein) have been identified in adults (53–56). Because circulating memory CD8+ T cells rapidly differentiate into effector CTLs (57, 58), the presence and effector function shown by these CTLs further support the notion that antigen-experienced T cells are less susceptible to inhibition by the hRSV. Consistently, APCs infected with hRSV or vaccinia viruses encoding the hRSV N protein can be lysed by human CTLs previously sensitized by DCs (53, 54). These data highlight the potential protective role against hRSV reexposures by CTLs and helper T cells primed by the appropriate immunogenic APCs. Along these lines, new vaccine strategies aimed at developing T-cell immunity against hRSV should consider the proper activation of hRSV-specific helper and CTLs and the induction of immunological T-cell memory to overcome the inhibitory constraints imposed by the nucleoprotein to the priming of naïve T cells.
The cSMAC, which is reduced by N protein, is composed of TCR-enriched extracellular vesicles (59). Therefore, an alternative explanation for the inhibition observed by the hRSV nucleoprotein may involve interference with the proper formation of these membranous structures at the cell–cell interface.
Materials and Methods
Mice.
Six- to eight-week-old C57BL/6J and the AND B10Br and OT-II transgenic mouse strains encoding specific TCRs for the I-Ek/moth cytochrome C (MCC)88-103 and the I-Ab/ovalbumin (OVA)323-339, respectively, were purchased from The Jackson Laboratory. Mice were handled according to institutional guidelines and maintained at the pathogen-free facility of the Pontificia Universidad Católica de Chile and the Skirball Institute NYU School of Medicine.
Viruses.
Two serogroup A2 hRSV strains [the 13018-8 clinical isolate—from the Public Health Institute of Chile—and a recombinant hRSV encoding the GFP—kindly provided by Mark E. Peeples (60)] were propagated over human laryngeal epidermoid carcinoma (HEp-2) and green monkey kidney (Vero) cells. Handling and titration of viral stocks, as well as the preparation of IC-hRSV (anti-F incubated hRSV) or heparin is detailed in SI Materials and Methods. Ultraviolet (UV)-light inactivation was performed by irradiating ice-packed virus for 1 h over a 302-nm, 15-W lamp transilluminator. Corroboration of virus inactivation was performed over HEp-2 monolayers either by plaque reduction assays (no detectable virus replication) or flow cytometry analysis (<1% of GFP+ cells) at 48 hpi.
Cells, Confocal Microscopy, and Flow Cytometry.
Mouse dendritic cells were differentiated from C57BL/6 bone marrows as described elsewhere (20). AND and OT-II naïve and blast T cells were obtained as described previously (21). DCs (fifth day of culture) or HEp-2 cells (60–80% confluence) were infected with hRSV13018-8, hRSVGFP or mock. For flow cytometry analyses, nonadherent (K562) and semiadherent cells (DCs) were harvested in RPMI 1640 (10% FBS, 10 mM Hepes) by gently pipetting. Adherent HEp-2 cells were washed with PBS and then harvested in either EDTA (10 mM in PBS) or in 0.5% trypsin-EDTA (in RPMI as a control) after 10 min incubation at 37 °C by gently pipetting detaching cell monolayers. For microscopy, staining was performed in the coverslips in which cells were grown (for further details, see SI Materials and Methods). Propidium iodide staining was used to exclude dead cells from flow cytometry analyses (see also Fig. S2). For microscopy, 1 μM of either CellTracker Orange CMTMR or CellTraceBODIPY TR, which are transformed into cell-impermeant dyes, were used to stain live cells. ELISA, virus-adsorption assays and brefeldin A (BFA) and WM-inhibition assays are detailed in the Fig. 2, Fig. 3, and SI Material and Methods. Staining of the hRSV N was performed using Alexa Fluor 647-labeled anti-N mAbs 1E9/D1 (previously described in ref. 61). The mouse IgG2a clone MOPC-173 (Biolegend) was used as isotype control. Staining of hRSV F was performed with Alexa Fluor 647-labeled palivizumab and the RS-348 mAb. Staining of hRSV M2-1 was performed with the clone 8A7/G9.
SLB and Total Internal Reflection Fluorescence Microscopy.
Either 12.5% or 5% DOGS-NTA [(vol/vol) in DOPC and POPC, respectively]--containing SLB were formed in sticky-Slide VI0.4 chambers (Ibidi) as described in SI Materials and Methods. SLB were loaded with physiological densities of fluorescent class II MHC (I-Ek-MCC88-103-12His, 5 molec/μm2) and ICAM-1−6His (200 molec/μm2). Additionally, SLB were loaded with either N-hRSV-6His (2–60 molec/μm2), or the control proteins CD58-6His or M2-1-hRSV-6His (10 molec/μm2). Naïve and blast AND CD4+ T cells were incubated over SLBs (for 20 min and 15 min, respectively), and either fixed using 2% PFA in PIPES HEPES EGTA Magnesium buffer, or live-imaged at 37 °C. TCR-β labeling was performed with fluorochrome-labeled Fab anti-TCR-β (clone H57). Phosphotyrosine staining was performed using an anti-pY (clone PY20, Biolegend) as detailed in SI Materials and Methods.
Image Analyses.
Acquisition settings were maintained constant throughout each imaging procedure and between samples. Image analyses were performed using ImageJ64 version 1.47d. Briefly, individual-cell contacts were traced with region of interest (ROI) on the surface reflection interference contrast channel, and then raw images were subtracted (corrected) for background, and the obtained fluorescence intensity values were plotted as raw values, which are referred to as fluorescence arbitrary units (AUs).
Statistical Analyses.
Statistical analyses were performed using the Graph Pad Prism v5 software (GraphPad Software, Inc.). P values were calculated using either One-way ANOVA or Student´s two-tailed unpaired t test (at 99% confidence interval). For BFA and WM inhibition assays, P values were calculated using One-way ANOVA and Dunnett´s Multiple comparison test to compare all drug/vehicle treatments against untreated hRSV-infected cells. P values < 0.05 were considered to be statistically significant.
Supplementary Material
Acknowledgments
We thank Dr. Mark E. Peeples at The Research Institute at Nationwide Children's Hospital for kindly providing the hRSVGFP. We also thank Dr. Pierre Pothier at the University of Dijon for kindly providing us the anti-F-hRSV (clone RS-348) monoclonal antibody. This work was supported by Fondo Nacional de Desarrollo Científico y Tecnológico Grants 3100090, 1070352, and 3140455; Fondo de Fomento al Desarrollo Científico y Tecnológico D061008; and the Millennium Institute on Immunology and Immunotherapy (P07/088-F) and Grant “Nouvelles Equipes-nouvelles thématiques” from the La Région Pays De La Loire and the Evaluation-Orientation of Scientific Cooperation France-Chile Grant. P.F.C., B.A.R., R.S.G., and S.A.R. are supported by the Comisión Nacional de Investigación Científica y Tecnológica (CONICYT). P.F.C. was supported by a Becas Chile scholarship. J.P.M.-O. is supported by Proyecto de Inserción de Capital Humano en la Academia no. 79100015 from CONICYT. A.M.K. is a Chaire De La Région Pays De La Loire, Chercheur Étranger D'excellence, France. D.D. and C.S. are supported by National Institutes of Health Grants AI043542 and AI080192. M.L.D. is supported by the Wellcome Trust and Kennedy Trust.
Footnotes
Conflict of interest statement: P.F.C., R.S.G., S.M.B., and A.M.K. have a pending patent request regarding the anti-N-hRSV 1E9/D1 and 8E4/A7 clones and their applications for diagnosis.
This article is a PNAS Direct Submission. J.E.C. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1400760111/-/DCSupplemental.
References
- 1.Nair H, et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet. 2010;375(9725):1545–1555. doi: 10.1016/S0140-6736(10)60206-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohuma EO, et al. The natural history of respiratory syncytial virus in a birth cohort: The influence of age and previous infection on reinfection and disease. Am J Epidemiol. 2012;176(9):794–802. doi: 10.1093/aje/kws257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henderson FW, Collier AM, Clyde WA, Jr, Denny FW. Respiratory-syncytial-virus infections, reinfections and immunity. A prospective, longitudinal study in young children. N Engl J Med. 1979;300(10):530–534. doi: 10.1056/NEJM197903083001004. [DOI] [PubMed] [Google Scholar]
- 4.Nokes DJ, et al. Respiratory syncytial virus infection and disease in infants and young children observed from birth in Kilifi District, Kenya. Clin Infect Dis. 2008;46(1):50–57. doi: 10.1086/524019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walsh EE. Respiratory syncytial virus infection in adults. Semin Respir Crit Care Med. 2011;32(4):423–432. doi: 10.1055/s-0031-1283282. [DOI] [PubMed] [Google Scholar]
- 6.DeVincenzo JP, et al. Viral load drives disease in humans experimentally infected with respiratory syncytial virus. Am J Respir Crit Care Med. 2010;182(10):1305–1314. doi: 10.1164/rccm.201002-0221OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Welliver TP, et al. Severe human lower respiratory tract illness caused by respiratory syncytial virus and influenza virus is characterized by the absence of pulmonary cytotoxic lymphocyte responses. J Infect Dis. 2007;195(8):1126–1136. doi: 10.1086/512615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKinstry KK, et al. Memory CD4+ T cells protect against influenza through multiple synergizing mechanisms. J Clin Invest. 2012;122(8):2847–2856. doi: 10.1172/JCI63689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boonnak K, Subbarao K. Memory CD4+ T cells: Beyond “helper” functions. J Clin Invest. 2012;122(8):2768–2770. doi: 10.1172/JCI65208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rasheed MA, et al. Interleukin-21 is a critical cytokine for the generation of virus-specific long-lived plasma cells. J Virol. 2013;87(13):7737–7746. doi: 10.1128/JVI.00063-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scholer A, Hugues S, Boissonnas A, Fetler L, Amigorena S. Intercellular adhesion molecule-1-dependent stable interactions between T cells and dendritic cells determine CD8+ T cell memory. Immunity. 2008;28(2):258–270. doi: 10.1016/j.immuni.2007.12.016. [DOI] [PubMed] [Google Scholar]
- 12.Blair DA, et al. Duration of antigen availability influences the expansion and memory differentiation of T cells. J Immunol. 2011;187(5):2310–2321. doi: 10.4049/jimmunol.1100363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zammit DJ, Cauley LS, Pham QM, Lefrançois L. Dendritic cells maximize the memory CD8 T cell response to infection. Immunity. 2005;22(5):561–570. doi: 10.1016/j.immuni.2005.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rothoeft T, et al. Differential response of human naive and memory/effector T cells to dendritic cells infected by respiratory syncytial virus. Clin Exp Immunol. 2007;150(2):263–273. doi: 10.1111/j.1365-2249.2007.03497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang J, Braciale TJ. Respiratory syncytial virus infection suppresses lung CD8+ T-cell effector activity and peripheral CD8+ T-cell memory in the respiratory tract. Nat Med. 2002;8(1):54–60. doi: 10.1038/nm0102-54. [DOI] [PubMed] [Google Scholar]
- 16.Guerrero-Plata A, et al. Differential response of dendritic cells to human metapneumovirus and respiratory syncytial virus. Am J Respir Cell Mol Biol. 2006;34(3):320–329. doi: 10.1165/rcmb.2005-0287OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le Nouën C, et al. Infection and maturation of monocyte-derived human dendritic cells by human respiratory syncytial virus, human metapneumovirus, and human parainfluenza virus type 3. Virology. 2009;385(1):169–182. doi: 10.1016/j.virol.2008.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Nouën C, et al. Effects of human respiratory syncytial virus, metapneumovirus, parainfluenza virus 3 and influenza virus on CD4+ T cell activation by dendritic cells. PLoS ONE. 2010;5(11):e15017. doi: 10.1371/journal.pone.0015017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munir S, et al. Respiratory syncytial virus interferon antagonist NS1 protein suppresses and skews the human T lymphocyte response. PLoS Pathog. 2011;7(4):e1001336. doi: 10.1371/journal.ppat.1001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.González PA, et al. Respiratory syncytial virus impairs T cell activation by preventing synapse assembly with dendritic cells. Proc Natl Acad Sci USA. 2008;105(39):14999–15004. doi: 10.1073/pnas.0802555105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grakoui A, et al. The immunological synapse: A molecular machine controlling T cell activation. Science. 1999;285(5425):221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 22.Tseng SY, Waite JC, Liu M, Vardhana S, Dustin ML. T cell-dendritic cell immunological synapses contain TCR-dependent CD28-CD80 clusters that recruit protein kinase C theta. J Immunol. 2008;181(7):4852–4863. doi: 10.4049/jimmunol.181.7.4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity. 2006;25(1):117–127. doi: 10.1016/j.immuni.2006.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiedemann A, et al. T-cell activation is accompanied by an ubiquitination process occurring at the immunological synapse. Immunol Lett. 2005;98(1):57–61. doi: 10.1016/j.imlet.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 25.Milstein O, et al. Nanoscale increases in CD2-CD48-mediated intermembrane spacing decrease adhesion and reorganize the immunological synapse. J Biol Chem. 2008;283(49):34414–34422. doi: 10.1074/jbc.M804756200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choudhuri K, Wiseman D, Brown MH, Gould K, van der Merwe PA. T-cell receptor triggering is critically dependent on the dimensions of its peptide-MHC ligand. Nature. 2005;436(7050):578–582. doi: 10.1038/nature03843. [DOI] [PubMed] [Google Scholar]
- 27.Ilani T, Khanna C, Zhou M, Veenstra TD, Bretscher A. Immune synapse formation requires ZAP-70 recruitment by ezrin and CD43 removal by moesin. J Cell Biol. 2007;179(4):733–746. doi: 10.1083/jcb.200707199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Graf B, Bushnell T, Miller J. LFA-1-mediated T cell costimulation through increased localization of TCR/class II complexes to the central supramolecular activation cluster and exclusion of CD45 from the immunological synapse. J Immunol. 2007;179(3):1616–1624. doi: 10.4049/jimmunol.179.3.1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Giuli C, Kawai S, Dales S, Hanafusa H. Absence of surface projections of some noninfectious forms of RSV. Virology. 1975;66(1):253–260. doi: 10.1016/0042-6822(75)90195-6. [DOI] [PubMed] [Google Scholar]
- 30.Hallak LK, Spillmann D, Collins PL, Peeples ME. Glycosaminoglycan sulfation requirements for respiratory syncytial virus infection. J Virol. 2000;74(22):10508–10513. doi: 10.1128/jvi.74.22.10508-10513.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corish P, Tyler-Smith C. Attenuation of green fluorescent protein half-life in mammalian cells. Protein Eng. 1999;12(12):1035–1040. doi: 10.1093/protein/12.12.1035. [DOI] [PubMed] [Google Scholar]
- 32.Bermingham A, Collins PL. The M2-2 protein of human respiratory syncytial virus is a regulatory factor involved in the balance between RNA replication and transcription. Proc Natl Acad Sci USA. 1999;96(20):11259–11264. doi: 10.1073/pnas.96.20.11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown G, Aitken J, Rixon HW, Sugrue RJ. Caveolin-1 is incorporated into mature respiratory syncytial virus particles during virus assembly on the surface of virus-infected cells. J Gen Virol. 2002;83(Pt 3):611–621. doi: 10.1099/0022-1317-83-3-611. [DOI] [PubMed] [Google Scholar]
- 34.Miller SG, Carnell L, Moore HH. Post-Golgi membrane traffic: Brefeldin A inhibits export from distal Golgi compartments to the cell surface but not recycling. J Cell Biol. 1992;118(2):267–283. doi: 10.1083/jcb.118.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collins PL, Mottet G. Post-translational processing and oligomerization of the fusion glycoprotein of human respiratory syncytial virus. J Gen Virol. 1991;72(Pt 12):3095–3101. doi: 10.1099/0022-1317-72-12-3095. [DOI] [PubMed] [Google Scholar]
- 36.Collins PL, Mottet G. Oligomerization and post-translational processing of glycoprotein G of human respiratory syncytial virus: Altered O-glycosylation in the presence of brefeldin A. J Gen Virol. 1992;73(Pt 4):849–863. doi: 10.1099/0022-1317-73-4-849. [DOI] [PubMed] [Google Scholar]
- 37.Utley TJ, et al. Respiratory syncytial virus uses a Vps4-independent budding mechanism controlled by Rab11-FIP2. Proc Natl Acad Sci USA. 2008;105(29):10209–10214. doi: 10.1073/pnas.0712144105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeffree CE, et al. Ultrastructural analysis of the interaction between F-actin and respiratory syncytial virus during virus assembly. Virology. 2007;369(2):309–323. doi: 10.1016/j.virol.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 39.Doms RW, Russ G, Yewdell JW. Brefeldin A redistributes resident and itinerant Golgi proteins to the endoplasmic reticulum. J Cell Biol. 1989;109(1):61–72. doi: 10.1083/jcb.109.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature. 2004;427(6970):154–159. doi: 10.1038/nature02238. [DOI] [PubMed] [Google Scholar]
- 41.Skokos D, et al. Peptide-MHC potency governs dynamic interactions between T cells and dendritic cells in lymph nodes. Nat Immunol. 2007;8(8):835–844. doi: 10.1038/ni1490. [DOI] [PubMed] [Google Scholar]
- 42.Friedman RS, Beemiller P, Sorensen CM, Jacobelli J, Krummel MF. Real-time analysis of T cell receptors in naive cells in vitro and in vivo reveals flexibility in synapse and signaling dynamics. J Exp Med. 2010;207(12):2733–2749. doi: 10.1084/jem.20091201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van der Merwe PA, et al. Human cell-adhesion molecule CD2 binds CD58 (LFA-3) with a very low affinity and an extremely fast dissociation rate but does not bind CD48 or CD59. Biochemistry. 1994;33(33):10149–10160. doi: 10.1021/bi00199a043. [DOI] [PubMed] [Google Scholar]
- 44.James JR, Vale RD. Biophysical mechanism of T-cell receptor triggering in a reconstituted system. Nature. 2012;487(7405):64–69. doi: 10.1038/nature11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fearns R, Peeples ME, Collins PL. Increased expression of the N protein of respiratory syncytial virus stimulates minigenome replication but does not alter the balance between the synthesis of mRNA and antigenome. Virology. 1997;236(1):188–201. doi: 10.1006/viro.1997.8734. [DOI] [PubMed] [Google Scholar]
- 46.Horikami SM, Curran J, Kolakofsky D, Moyer SA. Complexes of Sendai virus NP-P and P-L proteins are required for defective interfering particle genome replication in vitro. J Virol. 1992;66(8):4901–4908. doi: 10.1128/jvi.66.8.4901-4908.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buchholz CJ, Spehner D, Drillien R, Neubert WJ, Homann HE. The conserved N-terminal region of Sendai virus nucleocapsid protein NP is required for nucleocapsid assembly. J Virol. 1993;67(10):5803–5812. doi: 10.1128/jvi.67.10.5803-5812.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Curran J, Marq JB, Kolakofsky D. An N-terminal domain of the Sendai paramyxovirus P protein acts as a chaperone for the NP protein during the nascent chain assembly step of genome replication. J Virol. 1995;69(2):849–855. doi: 10.1128/jvi.69.2.849-855.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Castagné N, et al. Biochemical characterization of the respiratory syncytial virus P-P and P-N protein complexes and localization of the P protein oligomerization domain. J Gen Virol. 2004;85(Pt 6):1643–1653. doi: 10.1099/vir.0.79830-0. [DOI] [PubMed] [Google Scholar]
- 50.Oliveira AP, et al. Human respiratory syncytial virus N, P and M protein interactions in HEK-293T cells. Virus Res. 2013;177(1):108–112. doi: 10.1016/j.virusres.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 51.Marie JC, et al. Cell surface delivery of the measles virus nucleoprotein: A viral strategy to induce immunosuppression. J Virol. 2004;78(21):11952–11961. doi: 10.1128/JVI.78.21.11952-11961.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ravanel K, et al. Measles virus nucleocapsid protein binds to FcgammaRII and inhibits human B cell antibody production. J Exp Med. 1997;186(2):269–278. doi: 10.1084/jem.186.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bangham CR, et al. Human and murine cytotoxic T cells specific to respiratory syncytial virus recognize the viral nucleoprotein (N), but not the major glycoprotein (G), expressed by vaccinia virus recombinants. J Immunol. 1986;137(12):3973–3977. [PubMed] [Google Scholar]
- 54.Cherrie AH, Anderson K, Wertz GW, Openshaw PJ. Human cytotoxic T cells stimulated by antigen on dendritic cells recognize the N, SH, F, M, 22K, and 1b proteins of respiratory syncytial virus. J Virol. 1992;66(4):2102–2110. doi: 10.1128/jvi.66.4.2102-2110.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Venter M, Rock M, Puren AJ, Tiemessen CT, Crowe JE., Jr Respiratory syncytial virus nucleoprotein-specific cytotoxic T-cell epitopes in a South African population of diverse HLA types are conserved in circulating field strains. J Virol. 2003;77(13):7319–7329. doi: 10.1128/JVI.77.13.7319-7329.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McDermott DS, Knudson CJ, Varga SM. Determining the breadth of the respiratory syncytial virus-specific T cell response. J Virol. 2014;88(6):3135–3143. doi: 10.1128/JVI.02139-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lalvani A, et al. Rapid effector function in CD8+ memory T cells. J Exp Med. 1997;186(6):859–865. doi: 10.1084/jem.186.6.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gubser PM, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol. 2013;14(10):1064–1072. doi: 10.1038/ni.2687. [DOI] [PubMed] [Google Scholar]
- 59.Choudhuri K, et al. Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature. 2014;507(7490):118–123. doi: 10.1038/nature12951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hallak LK, Collins PL, Knudson W, Peeples ME. Iduronic acid-containing glycosaminoglycans on target cells are required for efficient respiratory syncytial virus infection. Virology. 2000;271(2):264–275. doi: 10.1006/viro.2000.0293. [DOI] [PubMed] [Google Scholar]
- 61.Gomez RS, et al. Respiratory syncytial virus detection in cells and clinical samples by using three new monoclonal antibodies. J Med Virol. 2014;86(7):1256–1266. doi: 10.1002/jmv.23807. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.