

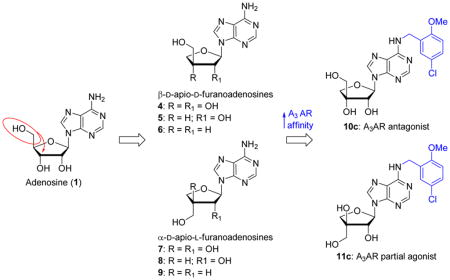
Abstract
Adenosine receptors (ARs) trigger signal transduction pathways inside the cell when activated by extracellular adenosine. Selective modulation of the A3AR subtype may be beneficial in controlling diseases such as colorectal cancer and rheumatoid arthritis. Here, we report the synthesis and evaluation of β-D-apio-D-furano- and α-D-apio-L-furanoadenosines and derivatives thereof. Introduction of a 2-methoxy-5-chlorobenzyl group at N6 of β-D-apio-D-furanoadenosine afforded an A3AR antagonist (10c, Ki = 0.98 μM), while a similar modification of an α-D-apio-L-furanoadenosine gave rise to a partial agonist (11c, Ki = 3.07 μM). The structural basis for this difference was examined by docking to an A3AR model; the antagonist lacked a crucial interaction with Thr94.
Keywords: G protein-coupled receptor, apionucleosides, Adenosine A3 receptor
INTRODUCTION
Transmembrane receptors coupled to G-proteins (heterotrimeric guanine nucleotide binding proteins) constitute a large receptor family, referred to as G protein-coupled receptors (GPCRs) or seven-transmembrane domain (7TM) receptors. Triggered by messenger molecules or signals outside the cell, GPCRs activate different signal transduction pathways inside the cell and, ultimately, cellular responses. The extracellular messengers range from photons to biogenic amines and other neurotransmitters to proteins. GPCRs are ubiquitous and involved in processes varying from directed chemotaxis[1] of small organisms (e.g. searching food for survival) to triggering apoptosis (programmed cell death) in large animals. They are the targets of almost 50% of marketed active pharmaceutical ingredients.[2]
GPCRs activated by extracellular adenosine are classified as adenosine receptors (ARs), which are divided in four different subtypes, i.e. A1, A2A, A2B and A3 ARs.[3,4] The amino acid sequence similarity between the human (h) A3AR and hA1, hA2A, hA2B AR is 54 %, 48 % and 44 %, respectively.[5] The ARs use different signaling pathways; the A1 AR and A3 AR are preferentially coupled to Gi-proteins, and upon activation lead to adenylate cyclase inhibition, while the A2A AR and A2B AR are preferentially coupled to Gs-proteins and lead to adenylate cyclase activation. In some cells (e.g., mast cells), the A2BAR is dually coupled to Gs and Gq and consequently also mobilizes calcium and activates phospholipase C and MAPK. [3,6]
Besides adenosine (1) itself, which is used clinically for the treatment of supraventricular tachycardia and in myocardial perfusion imaging,[7] only one AR-specific agent, the A2AAR agonist regedenoson, has so far been approved by the FDA. However, a relatively large group of AR ligands is currently under clinical evaluation.[8]
With the exception of compound 2, which was reported in the mid-1980s as being inactive at A1 and A2ARs,[9] and the recently reported carbocyclic analogue 3,[10] a weak A3AR agonist, 4′-hydroxymethyl transposed nucleosides have not been investigated as AR ligands.
This led us to employ a new and convenient method for the synthesis of apionucleosides from 1,2-O-isopropylidene-α-L-threose,[11] for the construction of suitably modified 9-(3-Chydroxymethyl-β-D-erythrofuranosyl)adenines (aka β-D-apio-D-furanoadenosines) (4-6) and 9-(3-C-hydroxymethyl-α-L-threofuranosyl)adenines (aka α-D-apio-L-furanoadenosines) (7-9) as potential A3AR modulators (Figure 1).
Figure 1.

Apio-type nucleosides previously tested for modulation of ARs (1-3) and target analogues of the present study (4-15).
On the one hand, we envisaged to substitute the N6 position of β-D-apio-D-furanoadenosine 4 with substituted benzyl groups known to enhance A3AR affinity;[12] on the other hand we planned to substitute the well-known ethylcarboxamide moiety in place of the 4′-CH2OH group.[13] We decided to introduce a benzyl moiety at the N6 position and an N-alkylcarboxamide moiety at the 5′ position because this combination was shown to be conducive to selectivity at the A3AR. For example, N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine and its 2-chloro analogue (Cl-IB-MECA) display Ki values at A3AR of ~1 nM and ≥50-fold selectivity for this subtype. [14] Subsequently, an N6-(3-chloro-5-methoxybenzyl) substitution was shown to be beneficial for A3AR selectivity,[12] and we incorporated this moiety in the current target compounds.
However, synthetic problems in introducing an ethylcarboxamide moiety motivated us to introduce a N-methyl or N-ethylcarbamoyloxymethyl group instead. Analogue modifications were carried out on α-D-apio-L-furanoadenosine 7.
RESULTS AND DISCUSSION
Chemistry
The β-D-apio-D-furanoadenosines 4-6 and the α-D-apio-L-furanoadenosines 7-9 were prepared by microwave assisted synthesis as described in ref. [11].
The L-threo analogue 7 was used as a model substrate for N6-derivatisation reactions. Treatment of 7 with the appropriate benzyl bromide first afforded the N1-benzyl derivative, which was isolated in the case of the 3-chlorobenzyl derivative 16 (Scheme 1). Upon prolonged heating with ammonia solution, the N1-benzyl intermediates were converted to the desired N6-benzylated compounds 11a-11c via Dimroth rearrangement.[14] Although in many cases this method suffered from low yields, it allowed gaining fast access to the target molecules. Using similar conditions β-D-apio-D-furanoadenosine 4 was benzylated at N6 affording the desired derivatives in low yield after purification by RP-HPLC or preparative TLC. In the case of 10c, using excess of the benzyl bromide led to degradation of the starting material and afforded bis-(2-methoxy-5-chlorobenzyl)adenine as observed by HRMS. By limiting the amount of this benzyl bromide to one equivalent, 10c could be obtained in 21% yield.
Scheme 1.

Synthesis of N6 substituted apioadenosines. Reagents and conditions: (a) (i) appropriate benzyl bromide, DMF, 50 °C, 48 h; (ii) 25% NH4OH, 50 °C, 48 h or 90 °C, 3 h, 20–56% over two steps; (b) (i) appropriate benzyl bromide, DMF, rt, 48 h; (ii) 25% NH4OH, 50 °C, 24 h, 8–21% over two steps.
Selective modification of the 5′-position requires a suitable protecting strategy. First attempts were made to per-silylate the hydroxyl groups of 7 (Scheme 2). Under mild conditions the tertiary hydroxyl group failed to react, while raising the temperature to 100 °C in DMF led to exclusive formation of the N6-imide 17. Since exploration of different solvents and scavenging bases did not allow the preparation of compound 18, the latter was obtained by treatment of 17 with ammonia in methanol.[15] Two attempts to selectively remove the TBDMS group from the primary hydroxyl of 18 resulted in complex reaction mixtures, from which isomers 20 and 21 proved inseparable by flash chromatography.[16]
Scheme 2.

Synthesis of sugar modified α-D-apio-L-furanoadenosines. Reagents and conditions: (a) TBDMSCl, imidazole, DMF, 100 °C, 3 days, 80%; (b) 7N NH3 in MeOH, rt, 18 h, 73%; (c) HF.pyridine, pyridine, THF, rt or TCA-H2O, THF, rt; (d) TBDMSCl, imidazole, DMF, rt, 18 h, 75–83%; (e) 22, TCA-H2O, THF, 0°C, 1 h, rt, 1 h, 87%; 23, HF.pyridine, pyridine, THF, 0°C, 1 h, rt, 1 h, 82%; (f) (i) CDI, THF, rt, 3h; (ii) EtNH2, rt, 16 h, 55–95%; (g) (i) RuCl3, NaIO4, AcCN-CCl4-H2O, rt, 7 h; (ii) CDI, THF, rt, 3 h; (iii) EtNH2, rt, 18 h, 9% over three steps; (h) NH4F, MeOH, 50 °C, 48 h, 70–94%.
The aforementioned problems led us to synthesize the bis-silylated products 22 and 23, from which the primary OH group could be selectively deprotected to provide 24 and 25 in excellent yields. Introduction of the desired 3′-carboxamide was tested on intermediate 25. Conversion of the primary hydroxyl group of 25 to the corresponding carboxylic acid via a TEMPO-BAIB oxidation gave the corresponding TEMPO ester (Entry 1, Table 1).[17] This may be due to the high catalyst loading in the reaction, since slow conversion of the aldehyde intermediate forced us to add extra amounts of reagents. Efforts to convert the TEMPO-ester to the ethylamide failed. Under conditions of Jones oxidation (CrO3 + dil. H2SO4) the starting material degraded. Oxidation using RuCl3-NaIO4,[18] followed by amide coupling, provided 27 and 28, albeit in very low yield. Removal of the TBDMS groups in 28 gave the desired 3′-ethylcarboxamide 15. Attempted ruthenium chloride oxidation of 24 resulted in decarboxylation and further oxidation to the ketone as the only product (Entry 2, Table 1). On the other hand, oxidation of 20 (as a mixture with 21) did not proceed beyond the aldehyde stage, thus weakening the prospects of synthesizing the corresponding alkylcarboxamide via the carboxylic acid route. Sequential treatment of 24 and 25 with carbonyldiimidazole and ethylamine afforded the 5′-O ethylcarbamate derivatives 26 and 27,[19] which were deprotected with NH4F in warm methanol to give the desired 5′-O-ethylcarbamate apionucleosides 13 and 14 in excellent yields.
Table 1.
Products from oxidation reactions
| Entry | Reactant | Condition | Product | HRMS |
|---|---|---|---|---|
| 1 | 25 | TEMPO (0.7 eq), BAIB (4 eq), CH3CN-H2O, rt, 16h |
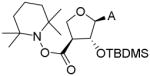
|
[M+H]+: 519.3119 |
| 2 | 24 | RuCl3 (0.7 eq), NaIO4 (4 eq), CH3CN-CCl4-H2O, rt, 7h |
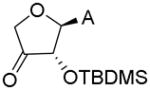
|
[M+H]+: 350.1656 [M+H3O]+: 368.1762 |
| 3 | 20 (+21) | RuCl3 (0.7 eq), NaIO4 (4 eq), CH3CN-CCl4-H2O, 50 °C, 2days |
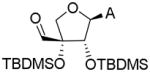
|
[M+H]+: 494.2609 [M+H3O]+: 512.2742 |
Given the problems encountered for the oxidation of α-D-apio-L-furanoadenosine (24), the corresponding D-furano epimer was only converted to carbamate 12 (Scheme 3). Silylation and desilylation of 4 rendered the 2′-O-monosilylated species 30, which upon treatment with carbonyldiimidazole and liq. methylamine gave 31. Deprotection of the TBDMS group furnished 5′-O-methylcarbamoyl-β-D-apio-D-furanoadenosine 12.
Scheme 3.
Synthesis of 5′-O-methylcarbamoyl-β-D-apio-D-furanoadenosine. Reagents and conditions: (a) TBDMSCl, imidazole, DMF, rt, 18 h, 74%; (b) TCA-H2O, THF, 0°C, 1 h, rt, 3 h, 35%; (c) (i) CDI, THF, rt, 3 h; (ii) MeNH2, rt, 16 h, 72%; (d) NH4F, MeOH, 50 °C, 48 h, 90%.
Pharmacological Evaluation
For the apio-type adenosine derivatives 4-16, we measured the binding affinities at the hA1, hA2A and hA3AR. The results are reported in Table 2. The ability of each of these adenosine derivatives to compete for radioligand binding at each of these hARs in cell membranes was evaluated at a fixed concentration of 10 μM. While most of the analogues inhibited binding to a negligible degree, it was feasible to determine full inhibition curves for compounds 10c and 11c at the A3AR (Figure 2). These two compounds display a modest degree of binding selectivity at the A3AR, but only with μM affinity for both. They were then examined in a functional assay,[12] i.e. inhibition of forskolin-stimulated cyclic AMP accumulation in an hA3AR-expressing CHO cell line. Compared to Cl-IB-MECA as reference full agonist, compound 11c was shown to be a partial agonist of the hA3AR, while compound 10c was inactive. The IC50 values for 11c and Cl-IB-MECA were 1560 ± 470 nM (50.9 ± 5.8% maximal efficacy) and 1.21 ± 0.35 nM (100% efficacy), respectively.
Table 2.
Percent inhibition of radioligand binding or binding affinities of apioadenosine derivatives at three hAR subtypes.a
| Compound | A1 AR, % inhibition | A2A AR, % inhibition | A3 AR, % inhibition or Ki (nM) |
|---|---|---|---|
| 4 | 6 ± 4% | 19±8% | 35 ± 4% |
| 5 | 6 ± 5% | 19±10% | 32 ± 2% |
| 6 | 10 ± 7% | 15±12% | 31 ± 2% |
| 7 | 0% b | 64% b | 10% |
| 8 | 74% b | 76% b | 7% |
| 9 | 0% b | ND | 7% |
| 10a | 10 ± 6% | 10±8% | 43 ± 5% |
| 10b | 27 ± 9% | 17±11% | 48 ± 6% |
| 10c | 12 ± 3% | 17±6% | 978 ± 175 |
| 11a | 19 ± 1% | 15 ± 5% | 44 ± 1% |
| 11b | 35 ± 5% | 22 ± 2% | 45 ± 1% |
| 11c | 13 ± 5% | 14 ± 5% | 3070 ± 750 |
| 12 | 12 ± 4% | 10±8% | 26 ± 6% |
| 13 | 9 ± 5% | 12 ± 4% | 6 ± 2% |
| 14 | 7 ± 2% | 2 ± 2% | 8 ± 1% |
| 15 | 8 ± 7% | 9 ± 5% | 10 ± 2% |
| 16 | 6 ± 5% | 12 ± 7% | 3 ± 2% |
Binding in membranes of CHO or HEK293 (A2AR only) cells stably expressing one of three hAR subtypes. Percent refers to inhibition of binding at 10 μM, unless noted, using agonists [3H]N6-[(R)-1-methyl-2-phenylethyl]adenosine, [3H]2-[p-(2-carboxyethyl)phenylethylamino]-5′-N-ethylcarboxamidoadenosine, or [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide, respectively. Values not in italics refer to the binding affinity at hA3ARs expressed as a Ki value. Values are mean ± S.E.M.
Percent refers to inhibition of binding at 50 μM. ND, not determined.
Figure 2.
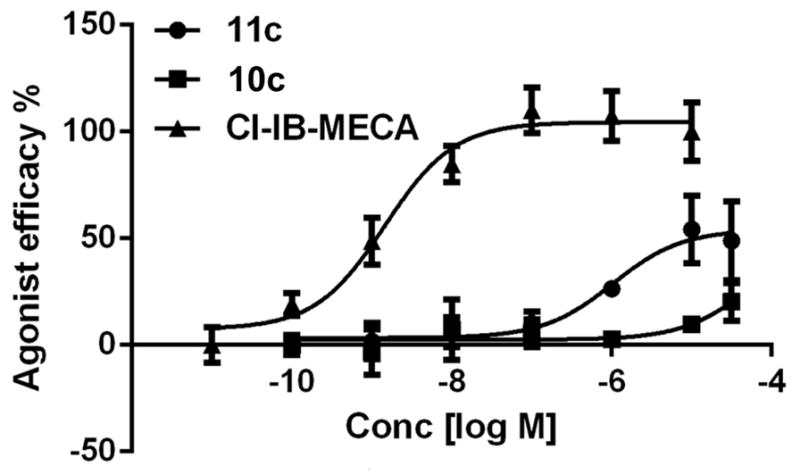
Effect of compounds 10c and 11c on forskolin-induced stimulation of cAMP production at the hA3AR expressed in CHO cells, compared to Cl-IB-MECA as reference full agonist (= 100%). Representative curves from 3 determinations are shown.
Docking studies performed at a hA3AR model
A hybrid homology model of the hA3AR based on an agonist-bound hA2AAR structure [20] (and modified with the position of TM2 based on the active state of the β2 adrenergic receptor) was built and used for docking of the nucleosides as described. [21],[22] The side chain of Asn250 (6.55, using Ballesteros-Weinstein notation [23]) in the hA3AR homology model strongly interacts with Cl-IB-MECA through two H-bonds involving the 6-amino group and the N7 atom of the adenine ring (Figure 3). Moreover, the adenine ring is anchored inside the binding site by a π–π stacking interaction with Phe168 (EL2) and strong hydrophobic contacts with Leu246 (6.51) and Ile268 (7.39). The N6-3-iodobenzyl ring is accommodated in a hydrophobic pocket delimited by TM5, TM6 and EL2 to form strong hydrophobic interactions with Val169 (EL2), Met174 (5.35) and Ile253 (6.58). Finally, the 3′- and 2′-hydroxyl groups of the ribose ring form H-bonds with Ser271 (7.42) and His272 (7.43), respectively, while the 5′-N-methyluronamido moiety forms a H-bond with Thr94 (3.36). These latter interactions involving the ribose moiety are particular to agonist binding, as shown by the comparison of the reported crystallographic structures of the hA2AAR in complex with agonists and antagonists, and are proposed to be important for the activation process in the AR family.
Figure 3.

Binding of A3AR full agonist Cl-IB-MECA
It has been previously shown that the 4′-truncation of nucleoside derivatives that are potent A3AR ligands leads to compounds with good affinity for the A3 receptor but with decreased efficacy, i.e. behaving as partial agonist or antagonist of this subtype. [24] Our docking of nucleoside analogues in the hA3AR homology model indicated that the absence of a hydroxymethyl or uronamide substituent at the 4′-position prevented these compounds from interacting with Thr94 (3.36). This residue is a conserved recognition point in agonist binding, and therefore the lack of this interaction was considered as a reason for the low efficacy profile of 4′-truncated nucleosides.
Both compounds 10c and 11c show docking poses at the hA3AR well superimposable to the binding mode of Cl-IB-MECA with conserved interactions stabilizing the adenine core and the N6 substituent. Differences can be observed in the interactions formed by the ribose moiety; in fact, both compounds form only two of the three H-bonds predicted for binding of the full agonist Cl-IB-MECA. In particular, compound 10c (Figure 4) forms two H-bonds with Ser271 (7.42) and His272 (7.43), but it cannot reach Thr94 in TM3, while compound 11c (Figure 5) forms H-bonds with Thr94 (3.36) and Ser271 (7.42) and not with His272 (7.43). Therefore, compound 10c show a binding mode similar to the one observed for other 4′-truncated nucleosides [22] and the missing interaction with Thr94 (3.36) is consistent with its lack of receptor activation, indicative of antagonist behavior. However, the ability of partial agonist 11c to bridge between TM3 and TM7 likely correlates with the ability to induce the conformational changes required for receptor activation, such as an inward movement of TM7.[3] Thus, the preferred modeled binding modes of 10c and 11c differ in the crucial interaction with Thr94, which seems to be associated with residual efficacy at the hA3AR in ribose-modified nucleosides.
Figure 4.

Binding of A3AR antagonist 10c
Figure 5.

Binding of A3AR partial agonist 11c
CONCLUSIONS
In summary, we have synthesized a small series of modified apioadenosines as potential A3AR ligands. The N6 substituted 9-(3-C-hydroxymethyl-β-D-erythrofuranosyl)adenines showed weak but selective binding affinity to the A3AR. A docking study allowed us to rationalize the lack of intrinsic efficacy of antagonist β-D-apio-D-furanoadenosine 10c and the partial agonist activity of α-D-apio-L-furanoadenosine 11c based on hydrogen bonding interaction with a key Thr residue. Synthetic problems in introducing a 3′-ethylcarboxamide moiety, motivated us to introduce a ((ethylcarbamoyl)oxy)methyl group instead. None of these 3′-altered derivatives showed highly potent binding affinity for the A3AR. Overall it may be concluded that substitution of an apiofuranose for a ribofuranose moiety is detrimental for binding to the ARs, but the addition of a favorable N6 substituent can partially compensate for this loss of binding affinity. Thus, structural changes in the sugar moiety, such as the configuration at the 3′ carbon, have major effects on the ability to activate the receptor, as well as binding recognition.
EXPERIMENTAL SECTION
Chemical Synthesis
All reagents were from standard commercial sources and of analytic grade. Dry solvents were obtained directly from commercial sources and stored on molecular sieves. Moisture sensitive reactions were carried out under argon atmosphere. Precoated Merck silica gel F254 plates were used for TLC, and spots were examined under ultraviolet light at 254 nm and further visualized by sulphuric acid-anisaldehyde spray. Column chromatography was performed on silica gel (200–400 mesh, 60 Å, Biosolve, Valkenswaard, The Netherlands). RP-HPLC was performed using Waters XBridge OBD™ Prep C18 5μm column @ 17.5 mL/min flow rate. NMR spectra were determined using a Varian Mercury 300 MHz spectrometer. Chemical shifts are given in ppm (δ) relative to the residual solvent signals or TMS as internal standard. Exact mass measurements were performed on a Waters LCT Premier XETM Time of flight (TOF) mass spectrometer equipped with a standard electrospray ionization (ESI) and modular LockSpray TM interface. Samples were infused in a CH3CN/water (1:1) mixture at 10 μL/min. NMR signals of sugar protons and carbons are indicated with a prime, and signals of base protons and carbons are given without a prime.
1′-[N1-(3-Chlorobenzyl)-adenin-9-yl]-α-D-apio-L-furanose bromide (16)
To a solution of α-D-apio-L-furanoadenosine 7 (25 mg, 0.094 mmol) in anh. DMF (1 mL) was added 3-chlorobenzyl bromide (50 μL, 0.374 mmol), and the mixture was stirred at 50 °C for 48 h. The solvent was evaporated under reduced pressure, and the residue was purified first by column chromatography (8–12% MeOH in CH2Cl2) and then with preparative thin layer chromatography (15% MeOH in CH2Cl2, Rf: 0.2) to afford the title compound 16 as a white solid (16 mg, 44%). 1H NMR (300 MHz, CD3OD) δ ppm 3.75 (d, J = 11.7 Hz, 1H, 5′-H), 3.85 (d, J = 11.7 Hz, 1H, 5′-H), 4.18 (d, J = 9.7 Hz, 1H, 4′-H), 4.25 (d, J = 9.4 Hz, 1H, 4′-H), 4.42 ( d, J = 0.9 Hz,1H, 2′-H), 5.63 (s, 2H, PhCH2), 6.15 (d, J = 1.5 Hz, 1H, 1′-H), 7.14 – 7.24 (m, 1H, Ph-H), 7.33 - 7.46 (m, 3H, Ph-H’s), 8.55 (s, 1H, 8-H), 8.68 (s, 1H, 2-H). 13C NMR (75 MHz, CD3OD) δ ppm 53.40 (PhCH2), 63.43 (5′-C), 78.14 (4′-C), 81.97 (2′-C), 82.77 (3′-C), 94.30 (1′-C), 120.91 (5-C), 126.28, 128.22 (Ph 2,6-Cs), 130.06, 131.98 (Ph 3,4-Cs), 136.26 (Ph 5-C), 136.52 (Ph 1-C), 144.79 (8-C), 148.09 (4-C), 148.42 (2-C), 152.29 (6-C). ESI-HRMS for [C17H18ClN5O4 + H]+ calcd, 392.1126; found, 392.1139.
1′-[N6-(3-Chlorobenzyl)-adenin-9-yl]-α-D-apio-L-furanose (11a)
To a solution of α-D-apio-L-furanoadenosine 7 (25 mg, 0.094 mmol) in anh. DMF (1 mL) was added 3-chlorobenzyl bromide (50μL, 0.374 mmol), and the mixture was stirred at 50 °C for 48 h. Ammonium hydroxide (25%, 3.0 mL) was added and the mixture stirred at 50 °C for 24 h (alternatively for 11a, at 90 °C for 3h). The volatiles were evaporated under reduced pressure and the residue purified by column chromatography (3–6% MeOH in CH2Cl2) to afford desired product 11a (20 mg, 56%) as a white solid. 1H NMR (300 MHz, DMSO-d6) δ ppm 3.62 (d, J = 5.6 Hz, 2H, 5′-H), 3.99 (d, J = 9.1 Hz, 1H, 4′-H), 4.05 (d, J = 9.1 Hz, 1H, 4′-H), 4.40 (dd, J = 5.0, 2.9 Hz, 1H, 2′-H), 4.64 (t, J = 5.6 Hz, 1H, 5′-OH), 4.71 (br.s, 2H, PhCH2), 5.32 (s, 1H, 3′-OH), 5.86 (d, J = 5.3 Hz, 1H, 2′-OH), 5.92 (d, J = 2.9 Hz, 1H, 1′-H), 7.23 – 7.37 (m, 3H, Ph-H’s), 7.39 (s, 1H, Ph-H), 8.23 (s, 1H, 2-H), 8.35 (s, 1H, 8-H), 8.44 (br.s, 1H, NH). 13C NMR (75 MHz, CD3OD) δ ppm 44.68 (PhCH2), 63.74 (5′-C), 77.69 (4′-C), 82.33 (2′-C), 82.73 (3′-C), 94.27 (1′-C), 120.82 (5-C), 126.96, 128.34, 128.56, 131.16 (Ph 2,3,4,6-C’s), 135.50 (Ph 5-C), 141.44 (8-C), 143.08 (Ph 1-C), 149.50 (4-C), 153.79 (2-C), 156.08 (6-C). ESI-HRMS for [C17H18ClN5O4 + H]+ calcd, 392.1126; found, 392.1105.
1′-[N6-(3-Iodobenzyl)-adenin-9-yl]-α-D-apio-L-furanose (11b)
Following the protocol for the synthesis of compound 11a, the title compound 11b (33 mg, 36%) was obtained from α-D-apio-L-furanoadenosine 7 (50 mg, 0.19 mmol) as a white solid. 1H NMR (300 MHz, CD3OD) δ ppm 3.76 (d, J = 11.6 Hz, 1H, 5′-H), 3.84 (d, J = 11.6 Hz, 1H, 5′-H), 4.12 (d, J = 9.5 Hz, 1H, 4′-H), 4.18 (d, J = 9.5 Hz, 1H, 4′-H), 4.37 (d, J = 1.5 Hz, 1H, 2′-H), 4.78 (br.s, 2H, PhCH2), 6.02 (d, J = 1.8 Hz, 1H, 1′-H), 7.07 (t, J = 7.8 Hz, 1H, Ph 5-H), 7.37 (d, J = 7.7 Hz, 1H, Ph 2-H), 7.58 (d, J = 7.9 Hz, 1H, Ph 4-H), 7.74 (s, 1H, Ph 6-H), 8.26 (s, 1H, 2-H), 8.29 (s, 1H, 8-H). 13C NMR (75 MHz, CD3OD) δ ppm 44.45 (PhCH2), 63.60 (5′-C), 77.54 (4′-C), 82.19 (2′-C), 82.59 (3′-C), 94.13 (1′-C), 94.96 (Ph 5-C), 120.68 (5-C), 127.84(Ph 2-C), 131.37 (Ph 3-C), 137.33 (Ph 4-C), 137.51 (Ph 6-C), 141.30 (8-C), 143.11 (Ph 1-C), 149.46 (4-C), 153.66 (2-C), 155.91 (6-C). ESI-HRMS for [C17H18IN5O4 + H]+ calcd, 484.0482; found, 484.0470.
1′-[N6-(2-Methoxy-5-chlorobenzyl)adenin-9-yl]-α-D-apio-L-furanose (11c)
Following the protocol used for the synthesis of compound 11a, α-D-apio-L-furanoadenosine 7 (50 mg, 0.19 mmol) was converted to the title compound 11c (15 mg, 20%), which was obtained as a white solid. 1H NMR (300 MHz, CD3OD) δ ppm 3.75 (d, J = 11.4 Hz, 1H, 5′-H), 3.84 (d, J = 11.7 Hz, 1H, 5′-H), 3.87 (s, 3H, Ph-OCH3), 4.12 (d, J = 9.3 Hz, 1H, 4′-H), 4.18 (d, J = 9.9 Hz, 1H, 4′-H), 4.37 (d, J = 1.2 Hz, 1H, 2′-H), 4.77 (br.s, 2H, PhCH2), 6.02 (d, J = 1.5 Hz, 1H, 1′-H), 6.94 (d, J = 8.7 Hz, 1H, Ph 5-H), 7.21 (dd, J = 8.7, 2.7 Hz, 1H, Ph 4-H), 7.25 (d, J = 2.4 Hz, 1H, Ph 6-H), 8.26 (s, 1H, 2-H), 8.29 (s, 1H, 8-H). 13C NMR (75 MHz, CD3OD) δ ppm 40.30 (PhCH2), 56.27 (PhOCH3), 63.60 (5′-C), 77.56 (4′-C), 82.17 (3′-C), 82.60 (2′-C), 94.15 (1′-H), 112.83 (Ph 3-C), 120.66 (5-C), 126.30 (Ph 5-C), 129.07 (Ph 6-C), 129.09 (Ph 4-C), 130.00 (Ph 1-C), 141.24 (8-C), 149.20 (4-C), 153.68 (2-C), 155.99 (6-C), 157.57 (Ph 2-C). ESI-HRMS for [C18H20ClN5O5 + H]+ calcd, 422.1231; found, 422.1236.
1′-[N6-(3-Chlorobenzyl)-adenin-9-yl]-β-D-apio-D-furanose (10a)
Following the protocol used for the synthesis of compound 11a, β-D-apio-D-furanoadenosine 4 (100 mg, 0.38 mmol) was converted to the title compound, which was first purified by silica-gel column chromatography and then by RP-HPLC (H2O/AcCN, 90/10→70/30 in 18 min, Rt = 12.5 min) to obtain 10a as white solid (16 mg, 11%). 1H NMR (300 MHz, DMSO-d6) δ ppm 3.46 (q, J = 11.0 Hz, 2H, 5′-H), 3.77 (d, J = 9.3 Hz, 1H, 4′-H), 4.32 (d, J = 9.1 Hz, 1H, 4′-H), 4.72 (br. s, 2H, PhCH2), 4.82 (d, J = 7.5 Hz, 1H, 2′-H), 4.87 (br.s, 1H, 3′-OH), 5.43 (br.s, 1H, 2′-OH), 5.90 (d, J = 7.7 Hz, 1H, 1′-H), 7.23 – 7.43 (m, 4H, Ph-H’s), 8.23 (s, 1H, 2-H), 8.38 (s, 1H, 8-H), 8.44 (br.s, 1H, NH). 13C NMR (75 MHz, DMSO-d6) δ ppm 42.41 (PhCH2), 62.38 (5′-C), 73.42 (2′-C), 74.59 (4′-C), 78.28 (3′-C), 87.81 (1′-C), 119.70 (5-C) 125.80, 126.58, 126.88, 130.14, 132.87 (Ph-C’s), 140.21 (8-C), 142.78 (Ph-C), 149.18 (4-C), 152.56 (2-C), 154.33 (6-C). ESI-HRMS for [C17H18ClN5O4 + H]+ calcd, 392.1120; found, 392.1127.
1′-[N6-(3-Iodobenzyl)-adenin-9-yl]-β-D-apio-D-furanose (10b)
Following the protocol used for the synthesis of compound 11a, β-D-apio-D-furanoadenosine 4 (100 mg, 0.38 mmol) was converted to the title compound 10b, which was first purified by silica-gel column chromatography and then isolated as a white solid following trituration with methanol (15 mg, 8%).1H NMR (300 MHz, DMSO-d6) δ ppm 3.37 – 3.57 (m, 2H, 5′-CH2), 3.76 (d, J = 9.2 Hz, 1H, 4′-H), 4.32 (d, J = 9.2 Hz, 1H, 4′-H), 4.67 (br. s, 2H, PhCH2), 4.76 – 4.82 (m, 1H, 2′-H), 4.84 (s, 1H, 3′-OH), 4.90 (t, J = 5.5 Hz, 1H, 5′-OH), 5.40 (d, J = 6.9 Hz, 1H, 2′-H), 5.89 (d, J = 7.5 Hz, 1H, 1′-H), 7.11(t, J = 7.6 Hz, 1H, Ph 5-H), 7.36 (d, J = 7.6 Hz, 1H, Ph-H) 7.58 (d, J = 7.8 Hz, 1H, Ph-H) 7.72 (s, 1H, Ph 2-H), 8.22 (s, 1H, 2-H), 8.38 (s, 1H, 8-H), 8.41 (br.s, 1H, NH). 13C NMR (75 MHz, DMSO-d6) δ ppm 42.24 (PhCH2), 62.37 (5′-C), 73.39 (2′-C), 74.57 (4′-C), 78.26 (3′-C), 87.80 (1′-C), 94.71 (Ph-C), 119.72 (5-C), 126.59, 130.47, 135.32, 135.68 (Ph-C’s), 140.17 (8-C) 142.88 (Ph-C), 149.13 (4-C), 152.54 (2-C), 154.31 (6-C). ESI-HRMS for [C17H18IN5O4 + H]+ calcd, 484.0476; found, 484.0490.
1′-[N6-(2-Methoxy-5-chlorobenzyl)-adenin-9-yl]-β-D-apio-D-furanose (10c)
To a solution of β-D-apio-D-furanoadenosine 4 (60 mg, 0.23 mmol) in anh. DMF (2 mL) was added 2-methoxy-5-chlorobenzyl bromide (58 mg, 0.25 mmol), and the mixture was stirred at rt for 24 h. Solvent was evaporated in vacuo. The residue was treated with ammonium hydroxide (25%, 10 mL) and the mixture stirred at 55 °C for 24 h. The volatiles were evaporated under reduced pressure and the residue purified by silica-gel column chromatography (3–6% MeOH in CH2Cl2). The isolated residue was further purified by preparative thin layer chromatography to afford desired product 10c (20 mg, 21%) as a homogeneous white solid. 1H NMR (300 MHz, CD3OD) δ ppm 3.59 – 3.74 (q, J = 11.4 Hz, 2H, 5′-H), 3.87 (s, 3H, PhOCH3), 3.98 (d, J = 9.6 Hz, 1H, 4′-H), 4.49 (d, J = 9.6 Hz, 1H, 4′-H), 4.77 (br.s, 2H, PhCH2), 4.85 (d, J = 7.1 Hz, 1H, 2′-H), 6.01 (d, J = 7.1 Hz, 1H, 1′-H), 6.93 (d, J = 8.7 Hz, 1H, Ph 5-H), 7.20 (dd, J = 8.7, 2.7 Hz, 1H, Ph 4-H), 7.25 (d, J = 2.5 Hz, 1H, Ph 2-H), 8.25 (s, 2H, 2-H & 8-H). 13C NMR (75 MHz, CD3OD) δ ppm 40.25 (PhCH2), 56.25 (PhOCH3), 64.42 (5′-C), 75.99 (2′-C), 76.42 (4′-C), 79.85 (3′-C), 90.47 (1′-C), 112.79 (Ph-C) 120.99 (5-C), 126.27, 129.03, 129.05, 129.96 (Ph-C’s), 141.13 (8-C), 150.25 (4-C), 153.97 (2-C), 156.01 (6-C), 157.52 (Ph-C). ESI-HRMS for [C18H20ClN5O5 + H]+ calcd, 422.1226; found, 422.1235.
1′-[N6-(N,N-Dimethylformamidine)-adenin-9-yl]-2′,3′,5′-O-tris(tert-butyldimethylsilyl)-α-D-apio-L-furanose (17)
To a solution of α-D-apio-L-furanoadenosine 7 (150 mg, 0.56 mmol) in anh. DMF (5 mL) was added imidazole (458 mg, 6.73 mmol) and TBDMSCl (845 mg, 5.6 mmol). The mixture was heated to 100 °C for 3 days and cooled, and a saturated aqueous NH4Cl solution was added. The product was extracted with EtOAc (3 × 20 mL), and the combined organic layers were dried over anh. Na2SO4. The residue obtained after evaporation of the organic layers was subjected to column chromatography (15–30% EtOAc in hexanes) to afford 17 (300 mg, 80 %) as a colorless glassy solid. 1H NMR (300 MHz, CDCl3) δ ppm −0.38 (s, 3H, SiCH3), −0.07 (s, 3H, SiCH3), 0.00 (s, 3H, SiCH3), 0.11 (s, 3H, SiCH3), 0.14 (s, 3H, SiCH3), 0.15 (s, 3H, SiCH3), 0.18 (s, 3H, SiCH3), 0.78 (s, 9H, C(CH3)3), 0.93 (s, 9H, C(CH3)3), 0.96 (s, 9H, C(CH3)3), 3.21 (s, 3H, NCH3), 3.27 (s, 3H, NCH3), 3.71 (d, J = 10.5 Hz, 1H, 4′-H), 3.95 (d, J = 10.5 Hz, 1H, 4′-H), 4.25 (d, J = 8.5 Hz, 1H, 5′-H), 4.34 (d, J = 8.8 Hz, 1H, 5′-H), 5.16 (d, J = 6.2 Hz, 1H, 2′-H), 5.75 (d, J = 6.2 Hz, 1H, 1′-H), 7.94 (s, 1H, 8-H), 8.54 (s, 1H, 2-H), 8.97 (s, 1H, N6CH). ESI-HRMS for [C31H60N6O4Si3 + H]+ calcd, 665.4062; found, 665.4075.
1′-(Adenin-9-yl)-2′,3′,5′-O-tri(tert-butyldimethylsilyl)-α-D-apio-L-furanose (18)
The N6-dimethyformamidine derivative 17 (300 mg, 0.45 mmol) was dissolved in a 7N solution of ammonia in MeOH and stirred at rt for 18 h. The mixture was evaporated and the residue purified by column chromatography (20–40% EtOAc in hexanes) to afford 18 (200 mg, 73%) as a white foam. 1H NMR (300 MHz, CDCl3) δ ppm −0.34 (s, 3H, SiCH3), −0.04 (s, 3H, SiCH3), 0.01 (s, 3H, SiCH3), 0.09 (s, 3H, SiCH3), 0.14 (s, 3H, SiCH3), 0.15 (s, 3H, SiCH3), 0.17 (s, 3H, SiCH3), 0.79 (s, 9H, C(CH3)3), 0.92 (s, 9H, C(CH3)3), 0.97 (s, 9H, C(CH3)3), 3.71 (d, J = 10.5 Hz, 1H, 4′-H), 3.96 (d, J = 10.8 Hz, 1H, 4′-H), 4.25 (d, J = 8.8 Hz, 1H, 5′-H), 4.32 (d, J = 8.5 Hz, 1H, 5′-H), 5.13 (d, J = 6.2 Hz, 1H, 2′-H), 5.61 (s, 2H, NH2), 5.73 (d, J = 6.2 Hz, 1H, 1′-H), 7.87 (s, 1H, 8-H) 8.35 (s, 1H, 2-H). 13C NMR (75 MHz, CDCl3) δ ppm −5.29, −5.27, −5.19, −4.65, −2.85, −2.70 (SiCH3), 17.97, 18.41, 18.75 (C(CH3)3), 25.66, 26.04, 26.19 (C(CH3)3), 64.94 (5′-C), 74.45 (4′-C), 83.16 (2′ & 3′-C), 90.42 (1′-C), 120.57 (5-C), 140.12 (8-C), 150.07 (4-C), 153.22 (2-C), 155.55 (6-C). ESI-HRMS for [C28H55N5O4Si3 + H]+ calcd, 610.3640; found, 610.3651.
Method A
A solution of compound 18 (180 mg, 0.3 mmol) in anh. THF (4.25 mL) was placed in a Teflon flask under an inert atmosphere and cooled to 0 °C. In a separate polypropylene flask, anh. THF (1.25 mL) and anh. pyridine (0.5 mL) were placed under inert atmosphere and the mixture cooled to 0 °C. A solution of 70% HF in pyridine (0.5 mL) was added dropwise to the latter mixture. 1.7 mL of the resulting chilled THF-pyridine-HF.pyridine (2.5:1:1) mixture was added dropwise to the solution of 18, and the mixture was stirred at 0 °C for 1 h. The ice-bath was removed and reaction continued at rt for 24h. The reaction mixture was poured to an ice-cold solution of NaHCO3 (1.5 g in 8 mL) with vigorous stirring. The product was extracted with EtOAc (3 × 20 mL), washed with brine and dried over anh. Na2SO4. The residue obtained after evaporation was subjected to flash column chromatography (2–5% MeOH in CH2Cl2) to obtain following compounds as white foams: Starting material 18 (70 mg, 39%), compound 19 (33 mg, 29%) and a mixture of compounds 20 and 21 (~2:1, 37 mg, 25%).
Method B
To a cooled (0 °C) solution of compound 18 (70 mg, 0.12 mmol) in anh. THF (1 mL) was added a solution of trichloroacetic acid (507 mg, 3.1 mmol) in water (0.27 mL), and the mixture was stirred for 20 min at 0 °C, and for 24 h at rt. The reaction mixture was cooled and neutralized with a cold sat. NaHCO3 solution. The products were extracted with EtOAc (3 × 30 mL), the combined organic phase was dried over anh. Na2SO4, filtered, and evaporated under reduced pressure. The residue was purified by column chromatography to afford following compounds: Starting material 18 (20 mg, 29%), compound 19 (10 mg, 23%) and a mixture of compounds 20 and 21 (~2:1, 17 mg, 30%).
1′-(Adenin-9-yl)-3′-O-(tert-butyldimethylsilyl)-α-D-apio-L-furanose (19)
1H NMR (300 MHz, DMSO-d6) δ ppm 0.08 (s, 6H, (SiCH3)2), 0.90 (s, 9H, C(CH3)3), 3.74 (d, J = 10.5 Hz, 1H, 4′-H), 3.84 (d, J = 10.3 Hz, 1H, 4′-H), 3.99 (d, J = 9.1 Hz, 1H, 5′-H), 4.06 (d, J = 8.8 Hz, 1H, 5′-H), 4.41 (dd, J = 5.3, 3.22 Hz, 1H, 2′-H), 5.32 (s, 1H, 5′-OH), 5.88 (d, J = 5.6 Hz, 1H, 2′-OH), 5.90 (d, J = 3.2 Hz, 1H, 1′-H), 7.26 (br. s, 2H, NH2), 8.15 (s, 1H, 2-H), 8.30 (s, 1H, 8-H). 1H NMR (75 MHz, DMSO-d6) δ ppm −5.48, −5.37 (SiCH3), 18.05 (C(CH3)3), 25.79 (C(CH3)3), 64.25 (5′-C), 75.07 (4′-C), 79.93 (2′-C), 80.13 (3′-C), 90.57 (1′-C), 118.76 (5-C), 139.62 (8-C), 148.99 (4-C), 152.39 (2-C), 155.90 (6-C). ESI-HRMS for [C16H27N5O4Si +H]+ calcd, 382.1911; found, 382.1903.
1′-(Adenin-9-yl)-2′,3′-O-di(tert-butyldimethylsilyl)-α-D-apio-L-furanose (20)
1H NMR (300 MHz, DMSO-d6) δ ppm −0.18, −0.05, 0.08 (s, 4×3H, SiCH3), 0.78, 0.90 (s, 2×9H, C(CH3)3), 3.67 (d, J = 10.3 Hz, 1H, 5′-H), 3.82 (d, J = 10.3 Hz, 1H, 5′-H), 4.01 (d, J = 8.8 Hz, 1H, 4′-H), 4.11 (d, J = 8.8 Hz, 1H, 4′-H), 4.68 (d, J = 4.4 Hz, 1H, 2′-H), 5.33 (s, 1H, 5′-OH), 5.89 (d, J = 4.4 Hz, 1H, 1′-H), 7.27 (br. s., 2H, NH2), 8.15 (s, 1H, 2-H), 8.29 (s, 1H, 8-H). ESI-HRMS for [C22H41N5O4Si2 + H]+ calcd, 496.2775; found, 496.2775.
1′-(Adenin-9-yl)-3′,5′-O-di(tert-butyldimethylsilyl)-α-D-apio-L-furanose (21)
1H NMR (300 MHz, DMSO-d6) δ ppm 0.10, 0.10, 0.12, 0.13 (s, 4×3H, SiCH3), 0.87, 0.92 (s, 2×6H, C(CH3)3), 3.75 (d, J = 10.8 Hz, 1H, 5′-H), 3.87 (d, J = 11.1 Hz, 1H, 5′-H), 4.06 (d, J = 8.2 Hz, 1H, 4′-H), 4.19 (d, J = 8.2 Hz, 1H, 4′-H), 5.00 (t, J = 5.1 Hz, 1H, 2′-H), 5.76 (d, J = 5.9 Hz, 1H, 1′-H), 5.84 (d, J = 5.0 Hz, 1H, 2′-OH), 7.27 (br. s., 2H, NH2), 8.12 (s, 1H, 2-H), 8.24 (s, 1H, 8-H). ESIHRMS for [C22H41N5O4Si2 + H]+ calcd, 496.2775; found, 496.2775.
1′-(Adenin-9-yl)-2′,5′-O-di(tert-butyldimethylsilyl)-α-D-apio-L-furanose (22)
To a solution of α-D-apio-L-furanoadenosine 7 (150 mg, 0.56 mmol) in anh. DMF (2 mL) was added imidazole (155 mg, 2.28 mmol), followed by TBDMSCl (253 mg, 1.68 mmol) and the mixture was stirred at rt for 18 h. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layer was dried over anh. Na2SO4, filtered and evaporated. The residue was purified by column chromatography using 3–5% MeOH in CH2Cl2 to afford 22 (230 mg, 83%) as a white foam. 1H NMR (300 MHz, CDCl3) δ ppm 0.08 (s, 3H, SiCH3), 0.10 (s, 6H, SiCH3), 0.12 (s, 3H, SiCH3), 0.91 (s, 18H, C(CH3)3), 3.75 (d, J = 10.0 Hz, 1H, 4′-H), 3.92 (d, J = 10.3 Hz, 1H, 4′-H), 4.08 (d, J = 9.1 Hz, 1H, 5′-H), 4.21 (d, J = 9.4 Hz, 1H, 5′-H), 4.45 (d, J = 1.5 Hz, 1H, 2′-H), 5.80 (brs, 2H, NH), 5.95 (d, J = 1.8 Hz, 1H, 1′-H), 8.13 (s, 1H, 8-H) 8.32 (s, 1H, 2-H). 13C NMR (75 MHz, CDCl3) δ ppm −5.51, −5.31, −5.22, −4.55 (SiCH3), 17.87, 18.37 (C(CH3)3), 25.66, 25.91 (C(CH3)3), 63.15 (4′-C), 76.68 (5′-C), 81.43 (3′-C), 81.53 (2′-C), 92.75 (1′-C), 119.81 (5-C), 140.04 (8-C), 149.27 (4-C), 152.68 (2-C), 155.31 (6-C). ESI-HRMS for [C22H41N5O4Si2 + H]+ calcd, 496.2775; found, 496.2783.
1′-(Adenin-9-yl)-2′,5′-O-di(tert-butyldimethylsilyl)-3′-deoxy-α-D-apio-L-furanose (23)
To a solution of L-furano-3′-deoxy-D-apioadenosine 8 (170 mg, 0.67 mmol) in anh. DMF (2 mL) was added imidazole (230 mg, 3.38 mmol), followed by TBDMSCl (305 mg, 2.03 mmol) and the mixture was stirred at rt for 18h. The reaction mixture was diluted with water (10 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layer was dried over anh. Na2SO4, filtered and evaporated. The residue was purified by column chromatography using 3–5% MeOH in CH2Cl2 to afford 23 (242 mg, 75%) as a white foam. 1H NMR (300 MHz, CDCl3) δ ppm 0.05 (s, 6H, Si(CH3)2), 0.07 (s, 3H, SiCH3), 0.11 (s, 3H, SiCH3), 0.88 (s, 9H, C(CH3)3), 0.90 (s, 9H, C(CH3)3), 2.56 (qt, J = 7.8, 5.7 Hz, 1H, 3′-H), 3.75 (dd, J = 10.0, 7.9 Hz, 1H, 5′-H), 3.87 (dd, J = 10.1, 6.0 Hz, 1H, 5′-H), 4.08 (t, J = 8.4 Hz, 1H, 4′-H), 4.45 (dd, J = 8.2, 7.6 Hz, 1H, 4′-H), 4.85 (dd, J = 5.0, 2.05 Hz, 1H, 2′-H), 5.55 (br.s, 2H, NH2), 5.90 (d, J = 2.1 Hz, 1H, 1′-H), 7.87 (s, 1H, 8-H), 8.35 (s, 1H, 2-H). 13C NMR (75 MHz, CDCl3) δ ppm −5.47, −5.39, −5.28, −4.70 (SiCH3), 17.99, 18.26 (C(CH3)3), 25.69, 25.87 (C(CH3)3), 44.77 (3′-C), 60.02 (5′-C), 71.91 (4′-C), 76.02 (2′-C), 92.47 (1′-C), 120.45 (5-C), 138.77 (8-C), 149.35 (4-C), 152.95 (2-C), 155.24 (6-C). ESI-HRMS for [C22H41N5O3Si2] calcd, 480.2826; found, 480.2828.
1′-(Adenin-9-yl)-2′-O-(tert-butyldimethylsilyl)-α-D-apio-L-furanose (24)
Following the desilylation method B (which was slightly modified in that the reaction mixture was stirred for 2h at rt) 22 (210 mg, 0.42 mmol) was converted to 24 (140 mg, 87%), which was obtained as a white foam. 1H NMR (300 MHz, DMSO-d6) δ ppm −0.17 (s, 3H, SiCH3), −0.04 (s, 3H, SiCH3), 0.79 (s, 9H, C(CH3)3), 3.55 (dd, J = 11.1, 5.0 Hz, 1H, 5′-H), 3.64 (dd, J = 11.1, 5.3 Hz, 1H, 5′-H), 4.02 (d, J = 8.8 Hz, 1H, 4′-H), 4.08 (d, J = 8.8 Hz, 1H, 4′-H), 4.66 (d, J = 4.1 Hz, 1H, 2′-H), 4.74 (t, J = 5.3 Hz, 1H, 5′-OH), 5.35 (s, 1H, 3′-OH), 5.87 (d, J = 4.1 Hz, 1H, 1′-H), 7.27 (brs, 2H, NH2), 8.15 (s, 1H, 2-H), 8.32 (s, 1H, 8-H). 13C NMR (75 MHz, DMSO-d6) δ ppm −5.30 (Si(CH3)2), 17.49 (C(CH3)3), 25.37 (C(CH3)3), 61.85 (5′-C), 74.59 (4′-C), 79.92 (3′-C) 81.13 (2′-C) 89.86 (1′-C), 118.84 (5-C), 139.70 (8-C), 149.00 (4-C), 152.42 (2-C), 155.90 (6-C). ESIHRMS for [C16H27N5O4Si +H]+ calcd, 382.1911; found, 382.1908.
1′-(Adenin-9-yl)-2′-O-(tert-butyldimethylsilyl)-3′-deoxy-α-D-apio-L-furanose (25)
Following the desilylation method A (which was slightly modified in that the reaction mixture was stirred for 1h at rt) starting material 23 (240 mg, 0.5 mmol) was converted to 25 (150 mg, 82%), obtained as a white foam. 1H NMR (300 MHz, CDCl3) δ ppm 0.08 (s, 3H, SiCH3), 0.11 (s, 3H, SiCH3), 0.91 (s, 9H, C(CH3)3), 2.14 (br. s., 1H, 5′-OH), 2.66 (tq, J = 7.9, 5.7 Hz, 1H, 3′-H), 3.89 (d, J = 5.6 Hz, 2H, 5′-H), 4.19 (t, J = 8.4 Hz, 1H, 4′-H), 4.46 (dd, J = 8.5, 7.9 Hz, 1H, 4′-H), 5.07 (dd, J = 5.7, 2.5 Hz, 1H, 2′-H), 5.56 (br. s., 2H, NH2), 5.89 (d, J = 2.4 Hz, 1H, 1′-H), 7.87 (s, 1H, 8-H), 8.35 (s, 1H, 2-H). 13C NMR (75 MHz, CDCl3) δ ppm −5.22, −4.72 (SiCH3), 17.94 (C(CH3)3), 25.70 (C(CH3)3), 43.69 (3′-C), 59.79 (5′-C), 70.79 (4′-C), 77.09 (2′-C), 92.38 (1′-C), 120.45 (5-C), 138.92 (8-C), 149.38 (4-C), 153.04 (2-C), 155.29 (6-C). ESI-HRMS for [C16H27N5O3Si + H]+ calcd, 366.1961; found, 366.1956.
1′-(Adenin-9-yl)-2′-O-(tert-butyldimethylsilyl)-5′-O-(N-ethylcarbamoyl)-α-D-apio-L-furanose (26)
To a solution of compound 24 (30 mg, 0.078 mmol) in anh. THF (2 mL) was added CDI (23 mg, 0.156 mmol) and the mixture was stirred at rt for 3 h. Cold ethylamine (0.3 mL) was added to the reaction mixture and stirring continued at rt for 16 h. Volatiles were evaporated, and the residue was suspended in CH2Cl2 and washed with sat. NH4Cl, water and brine. The organic layer was dried over anh. Na2SO4, filtered, volatiles evaporated. The residue was purified by column chromatography (1–3% MeOH in CH2Cl2) to afford the title compound 26 (17 mg, 52%) as a white foam. 1H NMR (300 MHz, CDCl3) δ ppm 0.01 (s, 3H, SiCH3), 0.06 (s, 3H, SiCH3), 0.91 (s, 9H, C(CH3)3), 1.14 (t, J = 7.2 Hz, 3H, NCH2CH3), 3.24 (qd, J = 7.3, 1.2 Hz, 2H, NCH2CH3) 4.06 (d, J = 9.4 Hz, 1H, 4′-H), 4.17 (d, J = 9.4 Hz, 1H, 4′-H), 4.32 (d, J = 11.4 Hz, 1H, 5′-H), 4.41 (d, J = 11.4 Hz, 1H, 5′-H), 4.56 (s, 1H, 2′-H), 4.84 (t, J = 5.1 Hz, 1H, CONH), 5.75 (s, 1H, 1′-H), 5.83 (br. s., 2H, NH2), 7.95 (s, 1H, 8-H), 8.32 (s, 1H, 2-H). 13C NMR (75 MHz, CDCl3) δ ppm −5.22 (SiCH3), −4.44 (SiCH3), 15.17 (NCH2CH3), 17.81 (C(CH3)3), 25.61 (C(CH3)3), 35.99 (NCH2CH3), 65.30 (5′-C), 76.41 (4′-C), 80.48 (3′-C), 82.81 (1′-C), 94.41 (1′-C), 120.43 (5-C), 140.41 (8-C), 148.43 (4-C), 152.50 (2-C), 155.70 (6-C), 156.44 (CO). ESI-HRMS for [C19H32N6O5Si + H]+ calcd, 453.2282; found, 453.2220.
1′-(Adenin-9-yl)-2′-O-(tert-butyldimethylsilyl)-3′-deoxy-5′-O-(N-ethylcarbamoyl)-α-D-apio-L-furanose (27)
Following the same protocol as described for 26, 30 mg (0.082 mmol) of 25 was converted to 27 (35 mg, 98%), obtained as a white foam. 1H NMR (300 MHz, CDCl3) δ ppm 0.12 (s, 3H, SiCH3), 0.18 (s, 3H, SiCH3), 0.92 (s, 9H, C(CH3)3), 1.12 (t, J = 7.2 Hz, 3H, NCH2CH3) 2.61 – 2.80 (m, 1H, 3′-H), 3.20 (quin, J = 6.6 Hz, 2H, NCH2CH3), 4.05 (t, J = 8.8 Hz, 1H, 4′-H), 4.14 – 4.34 (m, 2H, 5′-H), 4.44 (t, J = 8.1 Hz, 1H, 4′-H), 4.62 (t, J = 4.8 Hz, 1H, NH), 4.87 (d, J = 4.1 Hz, 1H, 2′-H), 5.65 (br.s, 2H, NH2), 5.87 – 5.96 ( d, J = 1.2 Hz, 1H, 1′-H), 7.87 (s, 1H, 8-H), 8.35 (s, 1H, 2-H). 13C NMR (75 MHz, CDCl3) δ ppm −5.29, −4.59 (Si(CH3)2), 15.25 ( NCH2CH3), 18.00 (C(CH3)3), 25.72 (C(CH3)3), 35.89 (NCH2CH3), 41.75 (3′-C), 61.03 (5′-C), 71.26 (4′-C), 75.99 (2′-C), 92.67 (1′-C), 120.44 (5-C), 138.51 (8-C), 149.24 (4-C), 153.00 (2-C), 155.30 (6-C), 155.97 (CO). ESI-HRMS for [C19H32N6O4Si] calcd, 437.2333; found, 437.2338.
1′-(Adenin-9-yl)-2′-O-(tert-butyldimethylsilyl)-3′-deoxy-5′-(N-ethyl)-carboxamido-α-D-apio-L-furanose (28)
To a suspension of compound 25 (30 mg, 0.082 mmol) in a 1:1:1.5 mixture of acetonitrile-CCl4-water (2 mL) was added NaIO4 (75 mg, 0.33 mmol), followed by RuCl3 (12 mg). The reaction mixture was stirred at rt for 7h and the solvents evaporated under vacuum. The residue was suspended in MeOH (5 mL), subjected to centrifugation and the clear supernatant liquid was decanted and evaporated. The residue was dried under high vacuum, and suspended in anh. THF (1.5 mL). To this suspension was added DMAP (23 mg, 0.19 mmol) and CDI (27 mg, 0.17 mmol), and the mixture was stirred at rt for 2–3 h. Cold ethylamine (0.3 mL) was added and stirring was continued at rt for 18h, after which volatiles in the reaction mixture were evaporated and the residue purified by column chromatography. The products were further purified by preparative thin layer chromatography (10% MeOH in CH2Cl2) to afford the title compound 28 (Rf: 0.4, 3 mg, 9%), as well as the carbamate 27 (Rf: 0.55, 3 mg, 8%). 1H NMR (300 MHz, CDCl3) δ ppm −0.01 (s, 3H, SiCH3), 0.06 (s, 3H, SiCH3), 0.87 (s, 9H, C(CH3)3), 1.18 (t, J = 7.3 Hz, 3H, NCH2CH3), 3.14 – 3.23 (m, 1H, 3′-H), 3.27 – 3.44 (m, 2H, NCH2CH3), 4.51 (dd, J = 8.8, 6.7 Hz, 1H, 4′-H), 4.57 (dd, J = 8.6, 7.2 Hz, 1H, 4′-H), 5.33 (dd, J = 5.9, 3.2 Hz, 1H, 2′-H), 5.64 (br. s., 2H, 6-NH2), 5.91 (d, J = 3.5 Hz, 1H, 1′-H), 6.09 (t, J = 4.5 Hz, 1H, 5′-NH), 7.86 (s, 1H, 8-H), 8.35 (s, 1H, 2-H). ESI-HRMS for [C18H30N6O3Si + H]+ calcd, 407.2227; found, 407.2222.
1′-(Adenin-9-yl)-3′-deoxy-5′-(N-ethyl)-carboxamido-α-D-apio-L-furanose (15)
To a solution of compound 28 (3 mg, 0.007 mmol) in MeOH (0.5 mL) in a polypropylene vessel was added NH4F (6 mg, 0.14 mmol), and the reaction mixture was stirred at 50 °C for 48 h. After addition of CH2Cl2 (1.5 mL), the reaction mixture was filtered through a celite pad. The filtrate was concentrated and the residue purified by column chromatography (5–7 % MeOH in CH2Cl2) to afford the title compound 15 (1.5 mg, 70 %) as a white solid after lyophilization. 1H NMR (300 MHz, DMSO-d6) δ ppm 1.02 (t, J = 7.2 Hz, 3H, NCH2CH3), 3.04 – 3.15 (m, 2H, NCH2CH3), 4.23 (t, J = 8.2 Hz, 1H, 4′-H), 4.35 (t, J = 8.1 Hz, 1H, 4′-H), 4.77 (br. s., 1H, 2′-H), 5.88 (d, J = 3.5 Hz, 1H, 2′-OH), 5.95 (d, J = 2.6 Hz, 1H, 1′-H), 7.26 (s, 2H, 6-NH2), 7.89 (t, J = 5.4 Hz, 1H, 5′-NH), 8.15 (s, 1H, 2-H), 8.23 (s, 1H, 8-H). 13C NMR (75 MHz, DMSO-d6) δ ppm 14.64 ( NCH2CH3), 33.43 ( NCH2CH3), 47.11 (3′-C), 69.12 (4′-C), 75.17 (2′-C), 90.38 (1′-C), 119.19 (5-C), 138.83 (8-C), 148.91 (4-C), 152.56 (2-C), 156.01 (6-C), 168.19 (CO). ESI-HRMS [C12H16N6O3 + H]+ calcd, 293.1362; found, 293.1360.
1′-(Adenin-9-yl)-5′-O-(N-ethylcarbamoyl)-α-D-apio-L-furanose (13)
Following the protocol used for the synthesis of 15, compound 26 (20 mg, 0.044 mmol) was converted to the title compound 13 (14 mg, 94%), obtained as a white solid. 1H NMR (300 MHz, CD3OD) δ ppm 1.11 (t, J = 7.2 Hz, 3H, NCH2CH3), 3.14 (q, J = 7.0 Hz, 2H, NCH2CH3), 4.13 (d, J = 9.4 Hz, 1H, 4′-H), 4.20 (d, J = 9.4 Hz, 1H, 4′-H), 4.26 ( d, J = 11.4 Hz, 1H, 5′-H), 4.33 ( d, J = 11.1 Hz, 1H, 5′-H), 4.37 (d, J = 1.2 Hz, 1H, 2′-H), 6.04 (d, J = 1.8 Hz, 1H, 1′-H), 8.21 (s, 1H, 2-H), 8.31 (s, 1H, 8-H). 13C NMR (75 MHz, CD3OD) δ ppm 15.33 (NCH2CH3), 36.70 (NCH2CH3), 66.37 (5′-C), 77.43 (4′-C), 81.34 (3′-C), 81.93 (2′-C), 93.89 (1′-C), 120.30 (5-C), 141.53 (8-C), 150.00 (4-C) 153.69 (2-C), 157.35 (6-C), 158.83 (CO). ESI-HRMS for [C13H18N6O5 + H]+ calcd, 339.1417; found, 339.1409.
1′-(Adenin-9-yl)-3′-deoxy-5′-O-(N-ethylcarbamoyl)-α-D-apio-L-furanose (14)
Following the protocol used for the synthesis of 15, compound 27 (35 mg, 0.08 mmol) was converted to the title compound 14 (23 mg, 90%), obtained as a white solid. 1H NMR (300 MHz, DMSO-d6) δ ppm 1.00 (t, J = 7.2 Hz, 3H, NCH2CH3), 2.73 – 2.87 (m, 1H, 3′-H), 2.93 – 3.05 (m, 2H, NCH2CH3), 3.85 (t, J = 8.4 Hz, 1H, 4′-H), 4.03 (dd, J = 10.6, 8.3 Hz, 1H, 5′-H), 4.23 (dd, J = 10.9, 6.2 Hz, 1H, 5′-H), 4.37 (t, J = 7.8 Hz, 1H, 4′-H), 4.66 (t, J = 3.6 Hz, 1H, 2′-H), 5.86 (d, J = 4.8 Hz, 1H, 2′-OH), 5.92 (d, J = 2.1 Hz, 1H, 1′-H), 7.10 (t, J = 5.4 Hz, 1H, NH), 7.27 (br.s, 2H, NH2), 8.15 (s, 1H, 2-H), 8.23 (s, 1H, 8-H). 13C NMR (75 MHz, DMSO-d6) δ ppm 15.01 ( NCH2CH3), 35.00 ( NCH2CH3), 41.19 (3′-C), 60.76 (5′-C), 70.59 (4′-C), 74.24 (2′-C), 91.29 (1′-C), 119.24 (5-C), 139.05 (8-C), 148.73 (4-C), 152.55 (2-C), 155.99 (CO), 156.02 (6-C). ESI-HRMS for [C13H18N6O4 + H]+ calcd, 323.1468; found, 323.1472.
1′-(Adenin-9-yl)-2′,5′-O-di(tert-butyldimethylsilyl)-β-D-apio-D-furanose (29)
Following the reaction protocol used for the synthesis of compound 22, β-D-apio-D-furano adenosine 4 (150 mg, 0.56 mmol) was converted to the title compound 29 (207 mg, 74%) as white foam. 1H NMR (300 MHz, CDCl3) δ ppm −0.47, −0.07, 0.10, 0.12 (s’s, 4 × 3H, SiCH3), 0.81, 0.97 (s’s, 9H, C(CH3)3), 3.01 (s, 1H, 3′-OH), 3.50 (d, J = 10.3 Hz, 1H, 5′-H), 3.59 (d, J = 10.3 Hz, 1H, 5′-H), 4.02 (d, J = 9.4 Hz, 1H, 4′-H), 4.50 (dd, J = 9.4, 1.03 Hz, 1H, 4′-H), 5.41 (d, J = 6.6 Hz, 1H, 2′-H), 5.65 (br.s, 2H, NH2), 5.89 (d, J = 6.8 Hz, 1H, 1′-H), 7.86 (s, 1H, 2-H), 8.34 (s, 1H, 8-H). 13C NMR (75 MHz, CDCl3) δ ppm −5.68, −5.47, −5.22, −5.02 (SiCH3), 17.76, 18.13 (C(CH3)3) 25.54, 25.73 (C(CH3)3), 62.13 (5′-C), 73.95 (2′-C), 74.11 (4′-C), 78.79 (3′-C), 89.89 (1′-C), 120.43 (5-C), 140.03 (8-C), 150.11 (4-C), 153.23 (2-C), 155.46 (6-C). ESI-HRMS for [C22H41N5O4Si2 + H]+ calcd, 496.2775; found, 496.2781.
1′-(Adenin-9-yl)-2′-O-(tert-butyldimethylsilyl)-β-D-apio-D-furanose (30)
Following the desilylation method B (which was slightly modified in that the reaction mixture was stirred for 3 h at rt), compound 29 (200 mg, 0.4 mmol) was converted to 30 (90 mg, 35%), which was obtained as a white solid. 1H NMR (300 MHz, CDCl3) δ ppm −0.33, −0.03 (s’s, 2 × 3H, SiCH3), 0.84 (s, 9H, C(CH3)3), 3.28 (s, 1H, 3′-OH), 3.78 (s, 2H, 5′-H), 4.04 (d, J = 9.7 Hz, 1H, 4′-H), 4.45 (d, J = 9.7 Hz, 1H, 4′-H), 5.25 (d, J = 5.3 Hz, 1H, 2′-H), 5.71 (br.s, 2H, NH2), 5.78 (d, J = 5.3 Hz, 1H, 1′-H), 7.87 (s, 1H, 8-H), 8.36 (s, 1H, 2-H). 13C NMR (75 MHz, CDCl3) δ ppm −4.91, 0.15 (SiCH3), 17.96 (C(CH3)3), 25.69 (C(CH3)3), 65.64 (5′-C), 75.56 (2′-C), 75.74 (4′-C), 78.33 (3′-C), 91.67 (1′-C), 120.72 (5-C), 140.57 (8-C), 149.76 (4-C), 153.35 (2-C), 155.78 (6-C). ESI-HRMS for [C16H27N5O4Si + H]+ calcd, 382.1911; found, 382.1910.
1′-(Adenin-9-yl)-2′-O-(tert-butyldimethylsilyl)-5′-O-(N-methylcarbamoyl)-β-D-apio-Dfuranose (31)
Following the same protocol as described for 26 (which was slightly modified in that the liq. methylamine was reacted instead of liq. ethylamine), 85 mg (0.22 mmol) of 30 was converted to 31 (70 mg, 72%), obtained as a white foam. 1H NMR (300 MHz, CDCl3) δ ppm −0.42, −0.05 (s’s, 3H, SiCH3), 0.83 (s, 9H, C(CH3)3), 2.85 (d, J = 4.9 Hz, 3H, NCH3), 3.26 (br.s, 1H, 3′-OH), 4.09 (d, J = 9.8 Hz, 1H, 4′-H), 4.18 (d, J = 11.4 Hz, 1H, 5′-H), 4.28 (d, J = 11.4 Hz, 1H, 5′-H), 4.46 (d, J = 9.8 Hz, 1H, 4′-H), 4.90 (br.s, 1H, NH), 5.32 (d, J = 6.1 Hz, 1H, 2′-H), 5.81 (d, J = 6.1 Hz, 1H, 1′-H), 5.89 (br.m, 2H, NH2), 7.86 (s, 1H, 8-H), 8.35 (s, 1H, 2-H). 13C NMR (75 MHz, CDCl3) δ ppm −5.14, −5.09 (SiCH3), 17.76 (C(CH3)3), 25.52 (C(CH3)3), 27.71 (NCH3), 65.43 (5′-C), 74.99 (2′-C), 75.13 (4′-C), 90.60 (1′-C), 120.60 (5-C), 140.49 (8-C), 149.86 (4-C), 153.17 (2-C), 155.65 (6-C), 156.60 (C=O). ESI-HRMS for [C18H30N6O5Si + H]+ calcd, 439.2125; found, 439.2128.
1′-(Adenin-9-yl)-5′-O-(N-methylcarbamoyl)-β-D-apio-D-furanose (12)
Following the protocol used for the synthesis of 15, compound 31 (60 mg, 0.13 mmol) was converted to the title compound 12 (40 mg, 90%), obtained as a white solid. 1H NMR (300 MHz, CD3OD) δ ppm 2.73 (s, 3H, NCH3), 3.99 (d, J = 9.6 Hz, 1H, 4′-H), 4.15 – 4.31 (q, J = 11.2 Hz, 2H, 5′-H), 4.46 (d, J = 9.8 Hz, 1H, 4′-H), 4.91 (d, J = 7.1 Hz, 1H, 2′-H), 5.99 (d, J = 7.1 Hz, 1H, 1′-H), 8.21 (s, 1H, 2-H), 8.28 (s, 1H, 8-H). 13C NMR (75 MHz, CD3OD) δ ppm 27.53 (NCH3), 66.69 (5′-C), 76.15 (2′-C), 76.43 (4′-C), 78.55 (3′-C), 90.40 (1′-C), 120.72 (5-C), 141.79 (8-C), 150.86 (4-C), 153.92 (2-C), 157.33 (6-C), 159.21 (C=O). ESI-HRMS for [C12H16N6O5 + H]+ calcd, 325.1255; found, 325.1260.
Pharmacological assay procedures
Cyclic AMP accumulation assay
Intracellular cAMP levels were measured with a competitive protein binding method.[25] Chinese hamster ovary (CHO) cells that expressed the recombinant hA3AR were harvested by trypsinization. After centrifugation and resuspension in medium, cells were plated in 24-well plates in 0.5 mL medium. After 24 h, the medium was removed, and cells were washed three times with 1 mL DMEM, containing 50 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES), pH 7.4. Cells were then treated with agonists and/or test compounds in the presence of rolipram (10 μM) and adenosine deaminase (3 units/mL). Forskolin (10 μM) was added to the medium after 45 min. After the addition of forskolin, the incubation was continued an additional 15 min. The reaction was terminated upon removal of the supernatant, and cells were lysed upon the addition of 200 μL of 0.1 M ice-cold HCl. The cell lysate was resuspended and stored at −20°C. For determination of cAMP production, protein kinase A (PKA) was incubated with [3H]cAMP (2 nM) in K2HPO4/EDTA buffer (K2HPO4, 150 mM; EDTA, 10 mM), 20 μL of the cell lysate, and 30 μL 0.1 M HCl or 50 μL of cAMP solution (0–16 pmol/200 μL for standard curve). Bound radioactivity was separated by rapid filtration through Whatman GF/C filters and washed once with cold buffer. Bound radioactivity was measured by liquid scintillation spectrometry.
Binding assays
Binding assays were performed using CHO cells stably expressing the recombinant hA1AR or hA3AR or HEK-293 cells stably expressing the hA2AAR.[24] Binding and functional parameters were calculated using Prism 5.0 software (GraphPAD, San Diego, CA, USA). IC50 values obtained from competition curves were converted to Ki values using the Cheng-Prusoff equation.[26] Data were expressed as mean ± standard error of the mean.
Molecular Modeling Methods
A homology model of the hA3AR was built based on a hybrid structural template using the homology modeling tool implemented in the MOE suite,[27] as previously reported.[21,28] In particular, an agonist-bound structure of the closely related hA2AAR (PDB code 3QAK)[20] was used as a template for most of the hA3AR structure except for the extracellular terminus of TM2 (residues from Val63 to Ser73) and EL1 (residues from Leu74 to Tyr81). The extracellular portion of TM2 was based on the X-ray structure of the hβ2 adrenergic receptor in complex with the Gs protein (PDB code 3SN6).[29] Molecular docking of apio-adenosine derivatives (prepared for docking using the build panel and the LigPrep panel implemented in the Schrödinger suite)[30] at the hA3AR hybrid model was performed by means of the Glide[31] module of the Schrödinger suite. A Glide Grid was centered on the centroid of some key residues of the binding pocket of adenosine receptors, namely Phe (EL2), Asn (6.55), Trp (6.48) and His (7.43). The Glide Grid was built using an inner box (ligand diameter midpoint box) of 10 Å × 10 Å × 10 Å and an outer box that extended 20 Å in each direction from the inner one. Docking of compounds was performed in the rigid binding site using the XP (extra precision) procedure. The top scoring docking poses for each ligand were subjected to visual inspection and analysis of protein-ligand interactions to select the proposed binding conformations.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases.
Abbreviations
- AR
adenosine receptor
- BAIB
bis-acetoxyiodobenzene
- cAMP
adenosine 3′,5′-cyclic phosphate
- CDI
carbonyldiimidazole
- Cl-IB-MECA
2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine
- CHO
Chinese hamster ovary
- DMF
N,N-dimethylformamide
- DMEM
Dulbecco’s modified Eagle’s medium
- EDTA
ethylenediaminetetraacetic acid
- GPCR
G protein-coupled receptor
- HEK
human embryonic kidney
- I-AB-MECA
N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyl-uronamide
- NECA
5′-N-ethylcarboxamidoadenosine
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HRMS
high resolution mass spectroscopy
- NMR
nuclear magnetic resonance
- R-PIA
N6-R-phenylisopropyladenosine
- TBDMS
tert-butyldimethylsilyl
- TCA
trichloroacetic acid
- TEMPO
(2,2,6,6-tetramethylpyperidin-1yl)-oxyl
- THF
tetrahydrofuran
- TLC
thin layer chromatography
- TM
transmembrane helical domain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Adler L, Tso WW. Science. 1974;184:1292–1294. doi: 10.1126/science.184.4143.1292. [DOI] [PubMed] [Google Scholar]
- 2.Bleicher KH, Böhm HT, Müller K, Alanine AI. Nat Rev Drug Discov. 2003;2:369–378. doi: 10.1038/nrd1086. [DOI] [PubMed] [Google Scholar]
- 3.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Müller CE. Curr Top Med Chem. 2003;3:445–462. doi: 10.2174/1568026033392174. [DOI] [PubMed] [Google Scholar]; (b) Fishman P, Bar-Yehuda S. Curr Top Med Chem. 2003;3:463–469. doi: 10.2174/1568026033392147. [DOI] [PubMed] [Google Scholar]
- 5.(a) Murrison EM, Goodson SJ, Edbrooke MR, Harris CA. FEBS Lett. 1996;384:243–246. doi: 10.1016/0014-5793(96)00324-9. [DOI] [PubMed] [Google Scholar]; (b) Fredholm BB, Arslan G, Halldner L, Kull B, Schulte G, Wasserman W. Naunyn-Schmiedeberg’s Arch Pharmacol. 2000;362:364–374. doi: 10.1007/s002100000313. [DOI] [PubMed] [Google Scholar]
- 6.Klinger M, Freissmuth M, Nanoff C. Cell Signal. 2002;14:99–108. doi: 10.1016/s0898-6568(01)00235-2. [DOI] [PubMed] [Google Scholar]
- 7.Poulsen SA, Quinn RJ. Bioorg Med Chem. 1998;6:619–641. doi: 10.1016/s0968-0896(98)00038-8. [DOI] [PubMed] [Google Scholar]
- 8.Jacobson KA, Balasubramanian R, Deflorian F, Gao ZG. Purinerg Signal. 2012;8:419–436. doi: 10.1007/s11302-012-9294-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor MD, Moos WH, Hamilton HW, Szotek DS, Patt WC, Badger EW, Bristol JA, Bruns RF, Heffner TG, Mertz TE. J Med Chem. 1986;29:346–353. doi: 10.1021/jm00153a008. [DOI] [PubMed] [Google Scholar]
- 10.Gao ZG, Jeong LS, Moon HR, Kim HO, Choi WJ, Shin DH, Elhalem E, Comin MJ, Melman N, Mamedova L, Gross AS, Rodriguez JB, Jacobson KA. Biochem Pharmacol. 2004;67:893–901. doi: 10.1016/j.bcp.2003.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toti KS, Derudas M, Pertusati F, Sinnaeve D, Van den Broeck F, Margamuljana L, Martins JC, Herdewijn P, Balzarini J, McGuigan C, Van Calenbergh S. J Org Chem. 2014 doi: 10.1021/jo500659e. [DOI] [PubMed] [Google Scholar]
- 12.Ohno M, Gao ZG, Rompaey PV, Tchilibon S, Kim SK, Harris BA, Gross AS, Duong HT, Van Calenbergh S, Jacobson KA. Bioorg Med Chem. 2004;12:2995–3007. doi: 10.1016/j.bmc.2004.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Galen PJM, Van Bergen AH, Gallo-Rodriguez C, Melman N, Olah ME, IJzerman AP, Stiles GL, Jacobson KA. Mol Pharmacol. 1994;45:1101–1111. [PMC free article] [PubMed] [Google Scholar]
- 14.Gallo-Rodriguez C, Ji X, Melman N, Siegman BD, Sanders LS, Orlina J, Fischer B, Pu Q, Olah ME. J Med Chem. 1994;37:636–646. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ju J, Kim DH, Bi L, Meng Q, Bai X, Li Z, Li X, Marma MS, Shi S, Wu L, Edwards JR, Romu A, Turro NJ. Proc Nat Acad Sci USA. 2006;103:19635–19640. doi: 10.1073/pnas.0609513103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Evano G, Schaus JV, Panek JS. Org Lett. 2004;6:525–528. doi: 10.1021/ol036284k. [DOI] [PubMed] [Google Scholar]; (b) Pradere U, Amblard F, Coats SJ, Schinazi RF. Org Lett. 2012;14:4426–4429. doi: 10.1021/ol301937v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Epp JB, Widlanski TS. J Org Chem. 1999;64:293–295. doi: 10.1021/jo981316g. [DOI] [PubMed] [Google Scholar]
- 18.Debnath J, Dasgupta S, Pathak T. Chem Eur J. 2012;18:1618–1627. doi: 10.1002/chem.201102816. [DOI] [PubMed] [Google Scholar]
- 19.Shao Y, Ding H, Tang W, Lou L, Hu L. Bioorg Med Chem. 2007;15:5061–5075. doi: 10.1016/j.bmc.2007.05.045. [DOI] [PubMed] [Google Scholar]
- 20.Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tosh DK, Deflorian F, Phan K, Gao ZG, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. J Med Chem. 2012;55:4847–4860. doi: 10.1021/jm300396n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tosh DK, Paoletta S, Phan K, Gao ZG, Jacobson KA. ACS Med Chem Lett. 2012;3:596–601. doi: 10.1021/ml300107e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ballesteros JA, Weinstein H. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 24.Melman A, Wang B, Joshi BV, Gao ZG, de Castro S, Heller CL, Kim SK, Jeong LS, Jacobson KA. Bioorg Med Chem. 2008;16:8546–8556. doi: 10.1016/j.bmc.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nordstedt C, Fredholm BB. Anal Biochem. 1990;189:231–234. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- 26.Cheng Y-C, Prusoff WH. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 27.Molecular Operating Environment (MOE), version 2012.10. Chemical Computing Group Inc; 1255 University St., Suite 1600, Montreal, QC, H3B 3×3 (Canada): [Google Scholar]
- 28.Paoletta S, Tosh DK, Finley A, Gizewski E, Moss SM, Gao ZG, Auchampach JA, Salvemini D, Jacobson KA. J Med Chem. 2013;56:5949–5963. doi: 10.1021/jm4007966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah STA, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schrödinger Suite. Schrödinger, LLC; New York, NY: 2012. [Google Scholar]
- 31.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shaw DE, Shelley M, Perry JK, Francis P, Shenkin PS. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.