

Abstract
Intragenic copy-number variants (CNVs) contribute to the allelic spectrum of both Mendelian and complex disorders. Although pathogenic deletions and duplications in SPAST (mutations in which cause autosomal-dominant spastic paraplegia 4 [SPG4]) have been described, their origins and molecular consequences remain obscure. We mapped breakpoint junctions of 54 SPAST CNVs at nucleotide resolution. Diverse combinations of exons are deleted or duplicated, highlighting the importance of particular exons for spastin function. Of the 54 CNVs, 38 (70%) appear to be mediated by an Alu-based mechanism, suggesting that the Alu-rich genomic architecture of SPAST renders this locus susceptible to various genome rearrangements. Analysis of breakpoint Alus further informs a model of Alu-mediated CNV formation characterized by small CNV size and potential involvement of mechanisms other than homologous recombination. Twelve deletions (22%) overlap part of SPAST and a portion of a nearby, directly oriented gene, predicting novel chimeric genes in these subjects’ genomes. cDNA from a subject with a SPAST final exon deletion contained multiple SPAST:SLC30A6 fusion transcripts, indicating that SPAST CNVs can have transcriptional effects beyond the gene itself. SLC30A6 has been implicated in Alzheimer disease, so these fusion gene data could explain a report of spastic paraplegia and dementia cosegregating in a family with deletion of the final exon of SPAST. Our findings provide evidence that the Alu genomic architecture of SPAST predisposes to diverse CNV alleles with distinct transcriptional—and possibly phenotypic—consequences. Moreover, we provide further mechanistic insights into Alu-mediated copy-number change that are extendable to other loci.
Introduction
DNA copy-number variants (CNVs) deleting or duplicating parts of genes contribute to exon shuffling and gene evolution, thus playing an important role in both Mendelian and common/complex traits.1, 2, 3, 4 CNVs in SPAST (2p22.3 [MIM 604277]) cause 8%–41% of autosomal-dominant spastic paraplegia 4 (SPG4 [MIM 182601]),5, 6, 7 the predominant form of hereditary spastic paraplegia (HSP).8, 9 Heterozygous CNVs deleting (n > 70 cases) and duplicating (n = 2 cases) various SPAST exons or the entire SPAST gene have been described.10, 11 However, it is not known how these CNVs form, whether and/or why SPAST is particularly susceptible to these rearrangements, and what the consequences of SPAST CNVs, either molecular or phenotypic, are in comparison to other types of mutations in the same gene.
Multiple molecular mechanisms have been described or proposed to generate small CNVs12, 13 commensurate with the genic/exonic scale of many or most rearrangements described in SPAST.14 Insights into the mechanism(s) of SPAST CNV formation, however, are largely absent because of methodological limitations: CNVs in SPAST are usually studied with multiplex ligation-dependent probe amplification (MLPA),6 a method that reveals the copy number of each of the 17 SPAST exons but leaves the exact location, sequence, and characteristics of CNV boundaries (i.e., breakpoint junctions) unknown.
We previously ascertained the nucleotide sequence of breakpoint junctions for three SPAST CNVs deleting the final exon of the gene.14 Although the size and genomic position of each deletion differed, all deletion breakpoints localized to pairs of directly oriented Alu elements, a repetitive DNA element of ∼300 base pairs (bp) present in >106 copies in the human genome.15 This Alu-Alu-mediated rearrangement phenomenon has been reported in studies of small, germline CNVs at other disease loci,16, 17, 18, 19, 20 somatic rearrangements found in malignancies,20, 21 and rearrangements arising during human evolution.22, 23, 24 Such variation has generally been attributed to Alu-Alu recombination, a proposed molecular mechanism in which homologous recombination (HR) utilizes nonallelic Alu pairs as substrates to generate CNVs.20, 25 Intriguingly, the CNVs in our small cohort exhibited features potentially inconsistent with formation by ectopic homologous recombination: one CNV was potentially complex, and breakpoint Alu pairs shared <90% identity. The remaining four SPAST CNVs with known breakpoints do not localize to Alus.11, 26, 27, 28 Therefore, we sought to further investigate the mechanism(s) generating SPAST CNVs, including the way in which the Alu-rich architecture of SPAST14 might render this gene or particular exons within it susceptible to CNV mutations.
SPG4, the clinical phenotype associated with SPAST mutation, is predominately characterized by progressive lower-limb weakness and spasticity caused by degeneration of upper motor neurons in the corticospinal tract.29 Occasional reports of additional phenotypes suggestive of CNS pathology exist,30, 31, 32 and subtle cognitive impairment has been suggested to be a moderately penetrant feature of the disease (reviewed by Dürr et al. in GeneReviews). Furthermore, severity and age of onset can vary. No consistent genotype-phenotype correlation exists for alleles in SPAST.33 Most disease-causing variants have been thought to represent loss-of-function alleles. However, we previously hypothesized that a subset of SPAST CNVs—those overlapping the 3′ end of the gene—might also affect expression of SLC30A6 (MIM 611148), the nearest gene to SPAST, located 8 kb downstream.14 SLC30A6 encodes a zinc transporter, ZnT6. Although SLC30A6 variants have not been found in association with any disease, zinc transporter (including ZnT6) dynamics are altered in the brains of some individuals with Alzheimer disease.34, 35 Thus, we investigated the possibility of SPAST CNVs dysregulating SLC30A6 expression, particularly given reports of individuals with comorbid spastic paraplegia and dementia.31, 36
We studied SPAST CNVs in an additional 51 subjects and now report diverse CNVs for which Alu-mediated formation predominates. These CNVs rearrange a variety of exons including 24 (47%) affecting the final (3′) exon of the gene and potentially forming fusion transcripts with or altering expression of SLC30A6 and other adjacent genes. In cultured lymphoblastoid cells from one subject with a SPAST exon 17 deletion, we identify multiple SPAST:SLC30A6 fusion transcripts. Finally, we propose alternative models to homologous recombination for Alu-mediated CNV formation based upon the observed breakpoint features of CNVs in SPAST; these are extendable to other loci, potentially enabling genome-wide predictions of Alu-mediated genome instability.
Subjects and Methods
Subjects
Subjects consisted primarily of individuals with a clinical diagnosis of spastic paraplegia and a previous finding of a SPAST CNV identified during clinical laboratory screening or a family-based research study (Table S1 available online). This study was approved by the Institutional Review Board for Human Subjects Research of Baylor College of Medicine (H-28128, H-25466, H-9170, H-29697). Remaining DNA samples were deidentified for our analyses. Subjects are further described in Tables 1 and S1. Owing to the anonymized nature of sample acquisitions, phenotypic details beyond a clinical diagnosis of spastic paraplegia are unknown in most cases. Samples from Athena Diagnostics were intentionally enriched for exon 17 CNVs. All other sources contributed all available CNVs in their collections.
Table 1.
Subjects and SPAST CNVs
| Subject | Del versus Dup | SPAST Exon(s) Affected (of 17) | Other Genes Deleted or Duplicated | CNV Size (kb) | Spastic Paraplegia | Origin | Novel Rearrangementa | Notes |
|---|---|---|---|---|---|---|---|---|
| A1 | del | 17 | – | 5.5 | y | USA | – | |
| A2 | del | 17 | – | 14.4 | y | USA | – | complex |
| A3 | del | 17 | SLC30A6 ex 1–5 (of 15) | 24.6 | y | USA | – | |
| A4 | del | 17 (partial) | – | 2.7 | y | USA | y | |
| A5 | del | 17 | SLC30A6 ex 1–2 (of 15) | 18.4 | y | USA | – | |
| A6 | del | 17 | SLC30A6 ex 1–2 (of 15) | 25.7 | y | USA | – | |
| A8 | del | 17 (partial) | – | 2.7 | y | USA | y | |
| A9 | del | 17 (partial) | – | 2.7 | y | USA | y | |
| A10 | dup | 13–15 | – | 6.2 | y | USA | y | tandem, directly oriented duplication |
| BAB 3166 | del | 13 | – | 1.3 | n (at age 16) | USA | NA | reported in Boone et al.26 |
| BAB 5112 | del | 5–17 | SLC30A6, NLRC4, YIPF4, BIRC6, MIR558, MIR4765, and TTC27 ex 1–5 (of 20) | 559.3 | y | USA | y | |
| BAB 3200 | del | 1–7 | DPY30 ex 1–3 (of 5) | 87.2 | y | USA | NA | complex; reported in de Ligt et al.27 |
| BAB 3327 | del | 17 | – | 5.5 | y | USA | – | |
| SPAST_1 | del | 17 (partial) | – | 2.7 | ?b | USA | y | |
| SPAST_3 | dup | 2–17 | SLC30A6, NLRC4, YIPF4, and BIRC6 ex 1–46 (of 74) | 432.2 | ?b | USA | y | tandem, directly oriented duplication |
| Spain 1 | del | 5–16 | – | 43.4 | y | Spain | y | |
| Spain 2 | del | 13–17 | SLC30A6, NLRC4, YIPF4, MIR558, and BIRC6 ex 1–66 (of 74) | 455.0 | y | Spain | y | |
| Spain 3 | del | 3–17 | SLC30A6 ex 1–5 (of 15) | 95.7 | y | Spain | – | |
| Spain 4 | del | 8–17 | SLC30A6 ex 1–5 (of 15) | 60.7 | y | Spain | – | |
| Spain 5 | del | 1 | – | 12.6 | y | Spain | – | |
| Spain 6 | del | 2–17 | SLC30A6 and NLRC4 ex 5–9 (of 9) | 170.1 | y | Spain | – | |
| Spain 7 | del | 10–16 | – | 16.0 | y | Spain | – | |
| Spain 8 | del | 2–7 | – | 48.9 | y | Spain | y | |
| Spain 9 | del | 6–7 | – | 5.0 | y | Spain | – | |
| JKF B | del | 16 | – | 2.0 | y | USA | – | |
| JKF C | dup | 9 | – | 6.7 | y | USA | y | tandem, directly oriented duplication |
| Ruhr 3 | del | 2–9 | – | 65.4 | y | Germany | – | |
| Ruhr 4 | del | 1 | – | 20.5 | y | Germany | – | |
| Ruhr 5 | del | 2–9 | – | 65.4 | y | Germany | – | |
| Ruhr 6 | del | 5–6 | – | 8.3 | y | Germany | – | |
| Ruhr 7 | del | 17 (partial) | – | 5.1 | y | Germany | y | |
| Ruhr 9 | del | 1 | – | 4.0 | y | Germany | – | |
| Ruhr 10 | del | 1 | – | 4.0 | y | Germany | – | |
| Ruhr 11 | del | 2–7 | – | 33.5 | y | Germany | y | |
| Ruhr 12 | del | 1 | – | 16.3 | y | Germany | – | |
| Ruhr 13 | del | 8–17 | SLC30A6 ex 1 (of 15) | 44.8 | y | Germany | – | |
| Ruhr 14 | del | 16–17 | SLC30A6 ex 1–4 (of 15) | 31.7 | y | Germany | – | |
| Ruhr 15 | del | 2–12 | – | 72.9 | y | Germany | y | |
| Ruhr 16 | del | 5 | – | 1.9 | y | Germany | – | |
| Ruhr 17 | del | 9–17 (partial) | – | 27.5 | y | Germany | y | |
| Ruhr 18 | del | 8–17 | SLC30A6 ex 1 (of 15) | 44.8 | y | Germany | – | |
| Ruhr 21 | del | 10–17 (partial) | – | 20.9 | y | Germany | y | |
| Ruhr 23 | del | 17 | – | 17.6 | y | Germany | – | |
| Ruhr 26 | del | 13–16 | – | 7.3c | y | Germany | – | |
| Ruhr 27 | del | 17 | SLC30A6 ex 1–2 (of 15) | 24.1 | y | Germany | – | complex |
| Ruhr 28 | del | 1 (partial) | – | 16.5 | y | Germany | – | |
| Ruhr 29 | del | 8–17 | SLC30A6, NLRC4, YIPF4, BIRC6, MIR558, MIR4765, TTC27, LINC00486, LOC100271832, LTBP1, 5S_rRNA | 1,283.9 | y | Germany | – | |
| Ruhr 31 | del | 8–9 | – | 9.3 | y | Germany | – | |
| Ruhr 32 | del | 2–13 | – | 59.4c | y | Germany | y | |
| Park 1 | del | 5–7 | – | 11.7 | y | Korea | – | |
| Park 2 | del | 5 (partial)–7 | – | 8.1 | y | Korea | y | |
| Park 3 | del | 8–9 | – | 8.9 | y | Korea | – | |
| Park 4 | del | 4–17 | SLC30A6 ex 1–9 (of 15) | 100.5 | y | Korea | – | |
| A37 | del | 17 | – | 10.8 | y | USA | NA | reported in Boone et al.14 |
| A38 | del | 17 | – | 16.8 | y | USA | NA | complex; reported in Boone et al.14 |
| A39 | del | 17 | – | 5.5 | y | USA | NA | reported in Boone et al.14 |
| Iwanaga | del | 1 (partial) | – | 2.3 | y | Japan | NA | reported in Iwanaga et al.28 |
| Mitne-Neto | dup | 10–12 | – | 4.0 | y | Brazil | NA | reported in Mitne-Neto et al.11; tandem, directly oriented duplication |
| Miura | del | 1–4 | DPY30 ex 1–3 (of 5) | 69.8 | y | Japan | NA | reported in Miura et al.40 |
Abbreviations are as follows: ex, exon; y, yes; n, no; NA, not applicable; –, no or none.
Based on SPAST exon or combination of exons rearranged and ploidy (del versus dup). Rearrangement of part of an exon is considered to be distinct from rearrangement of the whole exon. Previously published CNVs derived from Loureiro et al.,5 Álvarez et al.,9 Boone et al.14,26 Sulek et al.,41 and Myers et al.42
Phenotype unknown.
CNV breakpoint could not be sequenced. CNV length is an average of the minimum and maximum CNV interval identified by aCGH.
Mapping CNV Breakpoints
DNA was extracted from whole blood using the Gentra Puregene Blood Kit (QIAGEN) or obtained from previously archived samples. A total of 50 CNVs in SPAST were confirmed and mapped using a custom comparative genomic hybridization (CGH) array targeted to SPAST and flanking regions (approximately one megabase 5′ and 3′). This Agilent 8 × 15 k probe oligonucleotide array (format = G4427A; design ID = 037541) was custom designed using Agilent eArray and contains approximately one probe per 200 base pairs (bp) throughout the targeted region. Array CGH procedures have been described.37 Hybridization controls were gender matched unless an individual’s gender was unknown, in which cases female DNA (HapMap individual NA15510) was used. Array image files were processed using Agilent Feature Extraction software (v.10) and CNVs called with Agilent Genomic Workbench software (v.7). One CNV (Ruhr 29) was mapped using a genome-wide Agilent 1M probe oligonucleotide CGH array (format = G4824A; design ID = 02159) because the CNV extended beyond the boundaries of our targeted array.
CNVs were further confirmed and mapped by long-range PCR with primers (Table S2) spanning their predicted breakpoints. PCR reagents and concentrations have been described previously.38 The thermal cycler was programmed as follows: 94°C × 1 min; 30 cycles of 94°C × 30 s followed by 68°C × 7 min; 72°C × 10 min. Breakpoint PCR products were treated with ExoStar (GE Healthcare) according to the manufacturer’s instructions or extracted from an agarose gel using the Zymoclean gel DNA recovery kit (Zymo Research), also according to the manufacturer’s instructions, then sequenced by Sanger di-deoxynucleotide sequencing (Lone Star Labs; SeqWright; Baylor College of Medicine Sequencing Core, Houston, TX, USA).
All genomic coordinates are based on the February 2009 genome build (GRCh37/hg19) unless otherwise specified. SPAST exon numbering is based on UCSC transcript variant uc002roc.3 (17 exons). SLC30A6 exon numbering is based on UCSC transcript variant uc002rof.2 (15 exons).
STR Analysis
Alleles at 15 short tandem repeats (STRs) and subject gender were assessed using the Applied Biosystems AmpFISTR Identifiler Direct PCR Amplification Kit (Life Technologies) according to the manufacturer’s instructions.
Haplotype Analysis
For each deletion found in multiple individuals, primer pairs (Table S2) were designed to amplify 5–10 kb flanking each side of the deletion. The deleted allele was selectively amplified or was isolated based on size by gel extraction as described above. Common SNPs (allele frequency ≥ 1% in dbSNP 138) within these amplicons were genotyped by Sanger sequencing using a custom set of sequencing primers (Table S2). To assess the frequency of the observed haplotypes in the population, we obtained 1,984 phased haplotypes from the 1000 Genomes Project.39
Cell Lines
Lymphoblasts were isolated from whole blood collected in acid citrate dextrose (ACD) tubes and transformed with Epstein-Barr virus to make lymphoblastoid cell lines (LCLs) by the Baylor College of Medicine Intellectual and Developmental Disabilities Research Center Tissue Culture Core. LCLs were cultured in GIBCO RPMI 1640 media with 20% FBS, then 10% FBS, and 1% antimycotic/antibiotic (all from Life Technologies) in 5% CO2.
RNA Extraction and cDNA Synthesis
Total RNA was extracted from confluent, transformed lymphoblastoid cells using TRIzol (Invitrogen). RNA was cleaned using the RNeasy kit (QIAGEN) and quantitated with a NanoDrop ND-1000 spectrophotometer (NanoDrop). Complementary DNA (cDNA) was synthesized using qScript cDNA SuperMix (Quanta Biosciences) according to the manufacturer’s instructions (input = 1 μg RNA). cDNA quality was assessed via gel electrophoresis (not shown). A mock cDNA synthesis reaction performed without the reverse transcriptase mixture served as a negative (“No RT”) control during cDNA PCR.
PCR and Sequencing of cDNA
cDNA (2 μl) was used as PCR substrate in a 25 μl reaction mix containing 0.5 μM of each primer (Table S2) and 1× Promega PCR Master Mix (Promega). Thermocycler conditions were as follows: 95°C × 2 min; 40 cycles of 95°C × 30 s, 57°C × 30 s, 72°C × 3 min; 72°C × 5 min. PCR products were sequenced by Sanger di-deoxynucleotide sequencing (Lone Star labs; Baylor College of Medicine Sequencing Core, Houston, TX, USA) using the above cDNA PCR primers.
Quantitative RT-PCR
Quantitative RT-PCR (qPCR) assessing the expression level of SLC30A6 was performed using the TaqMan assay (Life Technologies). The Hs01071782_m1 probe set was used for SLC30A6 (spans the boundary of exon 9 and exon 10). The Hs02758991_g1 probe set was used for GAPDH (control gene [MIM 138400]; probe set spans the boundary of exon 6 and exon 7). The PCR amplification step was performed with 1 μl cDNA utilizing TaqMan Fast Advanced Master Mix (Life Technologies) and the 7900HT Fast Real-Time PCR System (Applied Biosystems). Three technical replicates were performed for each sample (subject or control).
The comparative Ct method was used to generate the relative quantification (ΔCt) of the expression level of SLC30A6 over GAPDH in an individual. The distribution of the ΔCt for SLC30A6 was established by the control samples. The SLC30A6 ΔCt for the subject was compared to this distribution using the empirical cumulative distribution function.
Results
SPAST CNVs Exhibit Diverse Sizes, Genomic Positions, and Exons Rearranged
We analyzed 51 heterozygous CNVs in SPAST (48 deletions and 3 duplications), predominately identified in individuals with a diagnosis of spastic paraplegia (Table 1). CNVs were most commonly discovered by multiplex ligation-dependent probe amplification (MLPA; data not shown). MLPA indicates only the exon(s) deleted or duplicated by a CNV, not its boundaries (breakpoints), so we fine-mapped each rearrangement using array comparative genomic hybridization (aCGH). Array CGH confirmed each CNV (Figures 1 and S1) and determined the approximate size, extent, and genomic content of each rearranged interval, enabling junction-spanning (breakpoint) PCR and sequencing reactions to be performed. PCR amplified 50 of 51 (98%) CNV breakpoints (Figure S2), and sequencing delineated the breakpoint sequence and exact coordinates of 49 (98%) of these rearrangements (Figure S3; 46 deletions and 3 duplications).
Figure 1.

Targeted aCGH Confirms and Maps CNVs in SPAST
Array CGH log2 plots are aligned with models of SPAST and nearby genes. Very small CNVs are indicated by arrows. Coordinates refer to chromosome 2. No SPAST CNV was identified in either blood- or cell-culture-derived DNA from the control individual, BAB 3099.
A diversity of CNV alleles were identified (Tables 1 and S1, Figure 2A). To date, 40 different combinations of SPAST exons have been reported to be deleted in individuals with spastic paraplegia, and 2 duplicated;5, 9, 14, 40, 41 we now contribute 12 novel rearrangements of SPAST exons (Table 1), including 2 novel duplications.
Figure 2.
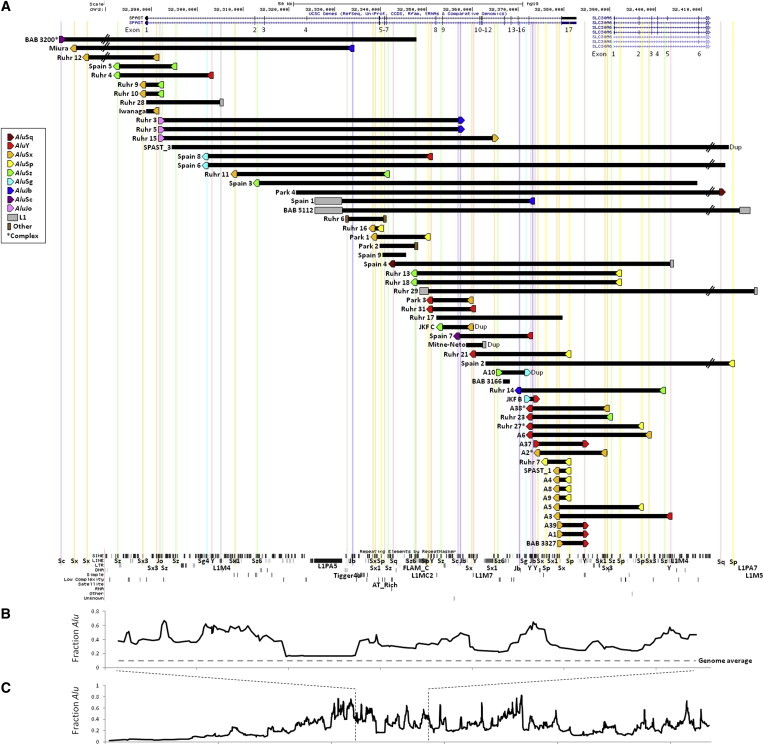
CNVs in SPAST Rearrange Diverse Exons and Have Breakpoints Predominately Localizing to Pairs of Alus in Direct Orientation
(A) CNVs are plotted by exact coordinates obtained by sequencing (black bars) and include both duplications (denoted “Dup”) and deletions (all others). Repetitive elements at CNV breakpoints are indicated by colored shapes and aligned with the RepeatMasker track (bottom, via UCSC Genome Browser) by colored lines. The genomic orientation of Alus is denoted by a right-facing (+) or left-facing (−) pentagon. Of 59 breakpoints, 39 (66%) localize to directly oriented Alu pairs. Note that BAB 3166 was identified in a young individual with no current spastic paraplegia26 and that the phenotypes of SPAST_1 and SPAST_3 are unknown; thus, these three CNVs might or might not be pathogenic. ∗Complex CNV (these CNVs are plotted as simple CNVs with only the first and final breakpoint repeat indicated). The breakpoints of Ruhr 26 and Ruhr 32 were not found, so these CNVs are not included.
(B) A moving average of Alu density (calculated in overlapping blocks of ten Alu elements) is aligned with (A), demonstrating that SPAST is overall an Alu-rich gene with “neighborhoods” of even higher Alu density. Alu data were downloaded from RepeatMasker.
(C) Zoomed-out plot of Alu density over a ∼1.1 Mb section of chromosome 2.
Of the CNVs we report, 36 (71%) occur in a single subject, and five groups of individuals had identical (shared regions of breakpoint microhomology) SPAST CNVs: Ruhr 9 and Ruhr 10 (del exon 1); Ruhr 3 and Ruhr 5 (del exons 2–9); Ruhr 13 and Ruhr 18 (del exons 8–17 and exon 1 of SLC30A6); A4, A8, A9, and SPAST_1 (partial del exon 17); and A1 and BAB 3327 (del exon 17; also identical to A39 from Boone et al.14). Identical CNVs in multiple subjects suggests either recurrent CNV formation or that individuals within a group are related or are the same individual. To investigate these possibilities, we analyzed 15 short tandem repeat alleles (STRs) and gender in these individuals (Table S3). No subjects sharing an identical CNV were monozygotic twins or the same individual, and in only one case (A1 and BAB 3327) were two such individuals consistent with a parent-child pair. To identify whether identical CNVs are observed on the same haplotype (i.e., potentially identical by descent) or on different haplotypes (i.e., identical by state, suggesting recurrent events), common SNPs flanking the CNVs were genotyped to establish a local haplotype. No flanking SNPs differ among individuals sharing the same CNV, thus failing to provide specific evidence for recurrence (Figure S6). For CNVs shared between Ruhr 13 and Ruhr 18, the haplotype surrounding the CNV is uncommon (5.1%), making recurrent events particularly unlikely. In other groups, the CNVs occurred on the most common haplotype.
We combined data from the 49 CNV breakpoint junctions delineated above with data from five SPAST CNVs we reported previously14, 26, 27 and three additional published SPAST CNVs,11, 28, 40 totaling 57 rearrangements and representing every SPAST CNV >30 bp for which exact breakpoint coordinates have been identified (Tables 1 and S1; Figure 2A). These CNVs range from 1,340 bp to 1,283,930 bp in size, with a mean of 73.1 kb, a median of 16.5 kb, and a distribution skewed toward smaller rearrangements (Figure 3). Thus, the majority of SPAST CNVs are of modest size when compared with the gene itself (94 kb).
Figure 3.
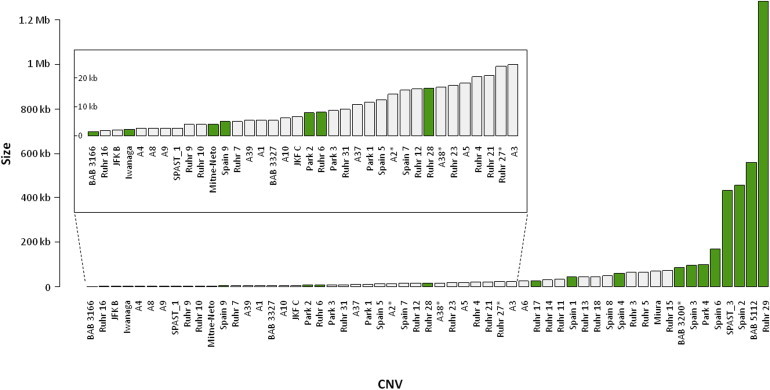
CNV Size: Skew toward Small Rearrangements and Association with Breakpoint Repeat Architecture
SPAST CNVs, plotted in ascending size order. Most SPAST CNVs are small (median size = 16.5 kb). CNVs flanked by Alu pairs (gray) are significantly smaller than remaining CNVs (green) (Kruskal-Wallis rank sum test, p = 0.00856; identical CNVs in multiple individuals [e.g., Ruhr 3 and 5] are collapsed to single entries, and CNVs published by other authors [which may be biased toward small, non-Alu-Alu-mediated events] are removed). ∗Complex CNV. BAB 5112 is LINE-LINE mediated. CNVs <25 kb are magnified in the inset to show detail. The size of complex CNVs is rounded to the nearest 0.1 kb.
All known SPAST duplications (Table 1, Figure 2) are tandem and directly oriented. We previously noted that SPAST duplications are significantly underrepresented when compared with deletions among subjects with SPG4,14 a trend that continues in the data presented here (Table 1). The pathogenic duplications in subjects A10 (exons 13–15) and Mitne-Neto (exons 10–12) are both expected to shift the reading frame; interestingly, the pathogenic duplication in JKF C (exon 9) is predicted to be in-frame.
CNV Breakpoints Predominately Localize to Pairs of Alu Elements in Direct Orientation
CNV boundaries, or breakpoints, occasionally preferentially localize to repetitive genomic elements, which can play a role in mediating CNV formation.13, 18 Repetitive elements present at the breakpoints of SPAST CNVs are shown in Figure 2 and listed in Table 2. Of 57 sequenced SPAST CNV breakpoints, 39 (68%) localize to directly oriented pairs of Alu repeats. Such CNVs demonstrate breakpoint “signatures” consistent with Alu-mediated CNV formation (i.e., the rearranged genome contains a complete hybrid Alu, each “half” of which is derived from one of the two breakpoint Alus that flanked the deleted or duplicated region prior to its loss or gain, respectively;22 Figure S3).
Table 2.
Repeats Present at CNV Breakpointsa
| Subject | Left Breakpoint Repeat | Right Breakpoint Repeat | Microhomology (bp) | % Identity of Repeatsb |
|---|---|---|---|---|
| A1 | AluSxc | AluY | 26 | 81.7 |
| A2c | AluSx | AluSx1 | complex | complex |
| A3 | AluSx1c | AluY | 7 | 86.9 |
| A4 | AluSx1c | AluSp | 15 | 84.8 |
| A5 | AluSx1c | AluSp | 9 | 88.6 |
| A6 | AluY | AluSx3c | 21 | 84.6 |
| A8 | AluSx1c | AluSp | 15 | 84.8 |
| A9 | AluSx1c | AluSp | 15 | 84.8 |
| A10 | AluSz6 | AluSg | 31 | 80.1 |
| BAB 3166 | – | – | 6 | – |
| BAB 5112 | L1PA5 | L1PA7 | 21 | 90.6 |
| BAB 3200 | AluSc | – | complex | – |
| BAB 3327 | AluSxc | AluY | 26 | 81.7 |
| SPAST_1 | AluSx1c | AluSp | 15 | 84.8 |
| SPAST_3 | – | – | 4 | – |
| Spain 1 | L1PA5 | AluJb | 4 | – |
| Spain 2 | – | AluSp | 2 | – |
| Spain 3 | AluSz6 | – | 2 | – |
| Spain 4 | AluSq | L1M4 | 8 | – |
| Spain 5 | AluSz | AluSz | 26 | 80.6 |
| Spain 6 | AluSg4 | – | 1 | – |
| Spain 7 | AluSc | AluY | 15 | 84.6 |
| Spain 8 | AluSg4 | AluYc | 9 | 83.7 |
| Spain 9 | – | – | 6 | – |
| JKF B | AluSg | AluY | 14 | 82.1 |
| JKF C | AluSz | AluSxc | 35 | 82.8 |
| Ruhr 3 | AluJo | AluJb | 19 | 77.6 |
| Ruhr 4 | AluSz | AluY | 25 | 75.8 |
| Ruhr 5 | AluJo | AluJb | 19 | 77.6 |
| Ruhr 6 | Tigger4a | AT_rich | 1 bp ins | – |
| Ruhr 7 | AluSp | AluSp | 8 | 87.9 |
| Ruhr 9 | AluSx3 | AluSzc | 5 | 82.7 |
| Ruhr 10 | AluSx3 | AluSzc | 5 | 82.7 |
| Ruhr 11 | AluSx1 | AluSz | 38 | 82.7 |
| Ruhr 12 | AluSx | AluSx3 | 9 | 77.4 |
| Ruhr 13 | AluSz6 | AluSp | 24 | 84.4 |
| Ruhr 14 | AluJb | AluSzc | 18 | 77.6 |
| Ruhr 15 | AluJo | AluY | 22 | 79.9 |
| Ruhr 16 | AluSx | AluSp | 20 | 79.1 |
| Ruhr 17 | – | – | 3 | – |
| Ruhr 18 | AluSz6 | AluSp | 24 | 84.4 |
| Ruhr 21 | AluY | AluSp | 33 | 83.2 |
| Ruhr 23 | AluY | AluSz | 11 | 85.6 |
| Ruhr 26d | NA | NA | NA | NA |
| Ruhr 27 | AluY | AluSp | complex | complex |
| Ruhr 28 | – | L1M4 | 4 | – |
| Ruhr 29 | L1MC2 | L1M5 | 0 | – |
| Ruhr 31 | AluYc | AluY | 5 | 90.7 |
| Ruhr 32d | NA | NA | NA | NA |
| Park 1 | AluSx1 | AluSp | 7 | 80.7 |
| Park 2 | – | FLAM_C | 3 | – |
| Park 3 | AluYc | AluSxc | 8 | 81.1 |
| Park 4 | – | AluSq | 4 | – |
| A37 | AluY | AluY | 8 | 89.4 |
| A38 | AluY | AluSx3 | complex | complex |
| A39 | AluSxc | AluY | 26 | 81.7 |
| Iwanaga | – | AluSx3 | 1 | – |
| Mitne-Neto | – | L1M7 | 0 | – |
| Miura | AluSx1 | AluJb | 9 | 77.1 |
Left, telomeric; right, centromeric; –, not applicable or no repeat present.
All Alu-Alu and LINE-LINE pairs are in matching genomic orientation (both negative or both positive) (Table S1, Figure 2A), with the exception of Ruhr 29.
Number of identical bases divided by smaller repeat length. Comparison by Needleman-Wunsch Global Alignment algorithm.
Repeat contains the consensus 13-nt sequence associated with meiotic recombination hotspots, some NAHR breakpoint regions, and bound in vitro by PRDM9.42, 43, 44 The Alu from which the inserted sequence in the complex A2 rearrangement derives contains this sequence.
Breakpoint unable to be sequenced (Ruhr 26) or failed breakpoint PCR (Ruhr 32).
Breakpoint Alu pairs exhibit variable sequence identity (range = 75.8%–90.7%), possess differing numbers of bases of microhomology at the hybrid junction (range = 5–38 bp), and comprise distinct Alu families (Table 2). Of the 39 breakpoint Alu pairings, 33 (85%) consist of Alus from different families. Several Alus are present at the breakpoints of multiple CNVs. These data are consistent with analyses of multiple Alu-mediated CNVs at additional disease loci (Table 4). The repeat present at a breakpoint of the greatest number of different CNVs is an AluY (Spain 7, A38, Ruhr 23, Ruhr 27, A6), as observed previously.16 Breakpoint crossover/microhomology locations occupy various positions within the Alu substrate pairs (Figure S4).
Table 4.
Properties of Alu-Mediated CNVs Derived from Large Studies of CNVs at Specific Loci
| Study | Gene | Syndrome | CNVs Studied (Unique) | Dups | Alu-Mediated CNVs | Average Breakpoint Alu Pair % Identity (Range) | Average Breakpoint Microhomology (Range) | Complex Alu-Mediated CNVs | Alus in Pair from Same Family | CNV size Avg. (Range) | Some Repeats Used in More than One CNV? |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Present | SPAST | Spastic paraplegia 4 | 57 (44) | 4 | 39/57 (68%) | 82.7% (75.8%–90.7%) | 17 bp (5–38 bp) | 3/39 (8%) | 6/39 (15%) | 19.0 kb (1.9–73 kb) | yes |
| Frank et al.16 | VHL (MIM 608537) | Von Hippel-Lindau syndrome | 33 (32) | 0 | 29/33 (88%) | not reported | 16 bp (2–45 bp) | 2/29 (7%) | 15/29 (52%) | 38.2 kb (0.8–249 kb) | yes |
| Kuiper et al.68 | EPCAM (TACSTD1 [MIM 185535]; upstream of MSH2 [MIM 609309]) | Lynch syndrome | 45 (19) | NAa | 45/45 (100%) | 81% (75%–91%) | 15 bp (3–32; one 0 bp) | 2/19 (11%) | 5/19 (26%) | 11.1 kbb (2.4–23.8 kb) | yes |
Only deletions were studied.
Deletions affecting MSH2 were specifically excluded, limiting the potential size of CNVs.
The eight largest CNVs are not Alu mediated (Figure 3). To determine statistically whether CNV size correlates with the presence or absence of Alus at CNV breakpoints, we collapsed CNVs found in multiple individuals (e.g., Ruhr 3 and 5) into single entries and removed the three SPAST CNVs described by other authors, which may be biased toward small, non-Alu-mediated events because such CNVs are more readily mapped and sequenced. Among this data set, CNVs flanked by Alu pairs are significantly smaller than non-Alu-mediated CNVs (Figure 3; Kruskal-Wallis rank sum test, p = 0.00856).
Three CNVs with proximal and distal breakpoints in Alus are potentially complex (A38, A2, and Ruhr 27). A38 has been described14 and is consistent with formation by two or three template switches during DNA replication or by NAHR and a novel, familial Alu polymorphism. The origin of A2 is consistent with two template switches or a small (11 bp) insertion. The origin of Ruhr 27 is consistent with two template switches, two distinct NAHR events, or an NAHR event and a novel, familial Alu polymorphism. It is unknown whether any of these CNVs is de novo, and parental DNA is unavailable.
In addition to the 39 CNVs in our cohort with pairs of Alus at breakpoints, other signatures are present in 32% of junctional sequences. These include 13 instances of microhomology not localizing to repeat pairs, two apparently blunt end “clean breaks,” one LINE-LINE recombination (BAB 5112), one 1 bp insertion, and one potentially complex CNV (BAB 3200) (Figure S3).
Some Breakpoint Alus Contain a 13-nt Consensus Motif Enriched at Meiotic Recombination Hotspots
A 13-nucleotide motif (consensus = CCNCCNTNNCCNC) has been delineated and found in proximity to and in association with >40% of meiotic recombination crossover hotspots, at NAHR crossover hotspots in low-copy repeats flanking some genomic disorder regions, and is bound in vitro by PRDM9, a protein with a role in meiotic homologous recombination.42, 43, 44 Alus are enriched for this motif. We analyzed sequence within or flanking SPAST (chr2: 32,280,000–32,412,000) and found 24 of these motifs, 23 of which are in or overlap Alus (Figure 4B, Table 3). This motif is present in one breakpoint Alu of ten Alu-mediated CNVs, at both breakpoint Alus of one CNV (Park 3), and in the Alu from which inserted breakpoint sequence is derived in one apparently complex rearrangement (A2) (Figure 4).
Figure 4.
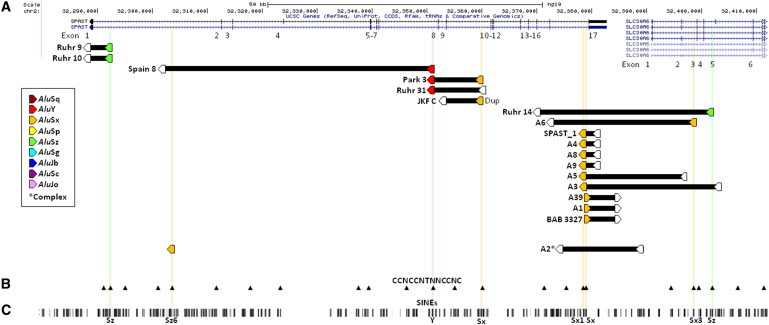
The Consensus 13-mer, CCNCCNTNNCCNC, Associated with Meiotic Recombination/NAHR Hotspots Is Present at Breakpoint Alus of Some Alu-Mediated CNVs
(A) CNVs (black bars) displayed with breakpoint Alus (pentagons; filled and intersected by a colored line if they contain the consensus 13-mer [CCNCCNTNNCCNC] associated with meiotic recombination/ NAHR hotspots42, 43). This motif is present at a single breakpoint Alu of ten Alu-mediated CNVs, at both breakpoint Alus of one CNV (Park 3), and in the Alu from which inserted breakpoint sequence is derived in one apparently complex rearrangement (A2). Abbreviation: Dup, duplication.
(B) All instances of the 13-mer (black arrowheads).
(C) SINE elements (RepeatMasker track via UCSC Genome Browser).
Table 3.
13-mer CCNCCNTNNCCNC Associated with Meiotic Recombination/NAHR Hotspots within and Flanking SPASTa
| Coordinate | Orientation | Within Alu? | Alu Used in CNV Breakpoint? |
|---|---|---|---|
| chr2: 32,290,968 | negative | – | – |
| chr2: 32,292,376 | positive | AluSz | Ruhr 9, Ruhr 10 |
| chr2: 32,294,873 | positive | AluY | – |
| chr2: 32,300,868 | positive | AluSz | – |
| chr2: 32,303,490 | positive | AluSz6 | A2 (complex) |
| chr2: 32,311,592 | negative | AluSx | – |
| chr2: 32,317,837 | negative | AluSx | – |
| chr2: 32,323,295 | negative | AluJr | – |
| chr2: 32,337,660 | positive | AluSg | – |
| chr2: 32,339,234 | negative | AluJo | – |
| chr2: 32,346,229 | negative | AluSx1 (overlaps) | – |
| chr2: 32,351,109 | positive | AluY | Park 3, Ruhr 31, Spain 8 |
| chr2: 32,355,085 | negative | AluJr (overlaps) | – |
| chr2: 32,360,010 | positive | AluSx | Park 3, JKF C |
| chr2: 32,371,388 | negative | AluJo | – |
| chr2: 32,375,404 | positive | AluSc | – |
| chr2: 32,378,630 | positive | AluSx1 | A3, A4, A5, A8, A9, SPAST_1 |
| chr2: 32,378,906 | negative | AluSx | A1, A39, BAB 3327 |
| chr2: 32,394,608 | negative | AluY | – |
| chr2: 32,398,678 | positive | AluSx3 | A6 |
| chr2: 32,399,507 | positive | AluJo | – |
| chr2: 32,402,087 | positive | AluSz | Ruhr 14 |
| chr2: 32,406,792 | negative | AluSx | – |
| chr2: 32,411,515 | negative | AluSx1 | – |
Chr2: 32,280,000–32,412,000.
Twenty-eight CNVs are Alu mediated, unique, and noncomplex. Of these CNVs’ 56 breakpoint Alus, 12 (21%) contain the 13-mer consensus motif. To investigate whether this represents an enrichment of the 13-mer motif in breakpoint Alus of Alu-mediated CNVs (and thus may be involved in their formation), we made the following comparisons to other groups of Alus. First, seven non-Alu-mediated CNVs have a single breakpoint in an Alu (Figure 2, Table 2); of these, none contain the 13-mer (0%); this paucity was not significant (p = 0.329, two-tailed Fisher exact test). Second, there are 188 Alus within and flanking SPAST (chr2: 32,280,000–32,412,000; Figure 4C); 23 of these contain the 13-mer (12%); this decrease is not significant (p = 0.126, two-tailed Fisher exact test).
Deletions Resulting in Chimeric Genes
Although 28 CNVs are purely intragenic, seven CNVs extend 5′ of SPAST, and for 22 CNVs a breakpoint maps beyond the 3′ end of the gene; of these CNVs overlapping SPAST, 17 affect additional genes (Figure 2A, Table 1). Two deletions extending beyond the 5′ end of SPAST (BAB 3200 and Miura) have telomeric breakpoints in another gene, DPY30 (MIM 612032). DPY30 is in reverse genomic orientation with SPAST; thus, a chimeric SPAST-DPY30 gene is less likely to result from these deletions. The effect of heterozygous loss of function of DPY30 is unclear, as an association between DPY30 and any human disease has not been described, although its ortholog appears to be developmentally important in C. elegans (reviewed by Miura et al.40).
Thirteen deletions (and one duplication) that extend beyond the 3′ end of SPAST have centromeric breakpoints in other genes (SLC30A6 = 10 deletions; NLRC4 [MIM 606831] and TTC27 = 1 deletion each; BIRC6 [MIM 605638] = 1 deletion and 1 duplication) (Figure 5). SLC30A6, BIRC6, and TTC27 are in direct orientation with SPAST; thus, the 12 deletions of the 3′ end of SPAST and the 5′ end of these genes form chimeric genes in these subjects’ genomes (Figure 5A). Multiple transcript isoforms have been identified for SLC30A6, BIRC6, and TTC27; thus, multiple fusion transcripts incorporating 5′ SPAST exons and 3′ exons of these genes are possible in subjects with such deletions (Figure 5B). Eight of twelve deletions have at least one predicted in-frame transcript. The SPAST_3 duplication is not anticipated to disrupt SPAST and thus is not likely to convey pathogenic changes to the spastin protein (consistent with this subject’s unknown status for spastic paraplegia). However, it creates an additional BIRC6:SPAST chimeric gene, the predicted fusion transcript of which is in-frame (Figure 5C). RNA was not available from any of these deletion or duplication subjects to examine experimentally for fusion transcripts.
Figure 5.
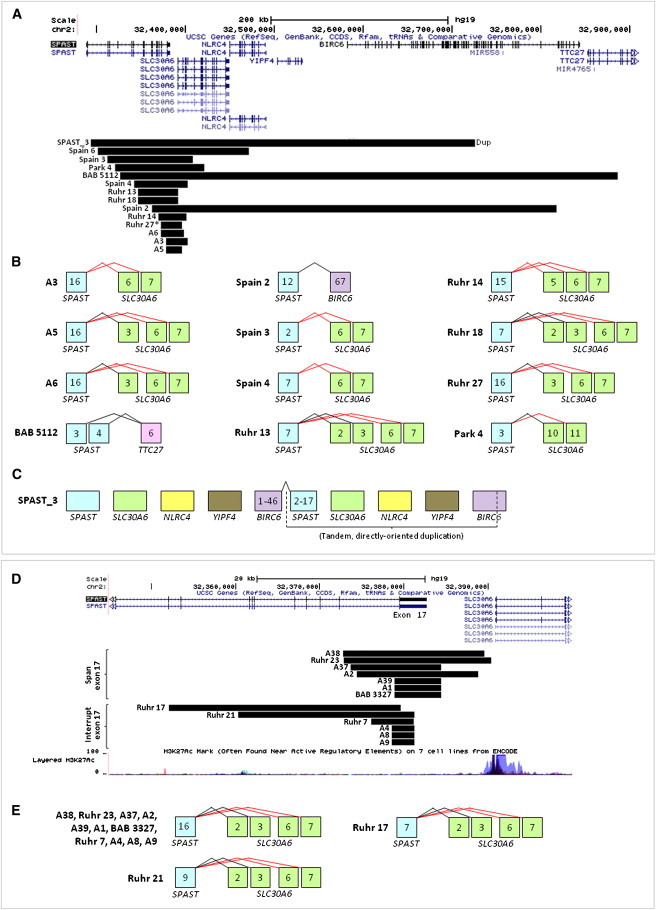
SPAST CNVs Extending into 3′ Genes Create Chimeric Genes, and Deletions Involving the Final Exon of SPAST Might Create SPAST:SLC30A6 Fusion Transcripts or Alter SLC30A6 Expression
(A–C) Deletions extending into 3′ genes.
(A) Fourteen CNVs (black bars) extend into genes 3′ of SPAST (13 deletions, 1 duplication). Three of the downstream genes (SLC30A6, BIRC6, TTC27) are in direct orientation with SPAST; thus, SPAST deletions intersecting these 3′ genes potentially yield chimeric genes.
(B) Potential fusion transcripts resulting from the deletions shown in (A) for which the 3′ intersected gene is directly oriented with SPAST. Potential fusion transcripts are based only on known splice isoforms of the 3′ genes and include all SPAST exons present, although other splice variants are possible. Predicted in-frame splicing is shown in black, out-of-frame in red. The deletion intersecting NLRC4 (Spain 6) is not shown, because this gene is oriented oppositely to SPAST.
(C) Potential fusion transcript resulting from the duplication in subject SPAST_3. This tandem, directly oriented duplication results in duplication of three genes (SLC30A6, NLRC4, YIPF4) and a potential in-frame transcript joining exons 1–46 of BIRC6 with exons 2–17 of SPAST; the normal, diploid copy number of full-length SPAST and BIRC6 are left intact.
(D and E) Deletions involving the final exon of SPAST.
(D) Deletions with 3′ breakpoints in the intergenic space between SPAST and SLC30A6 (top, “span exon 17”) or within the final exon of SPAST (exon 17) (bottom, “interrupt exon 17”). The former group of deletions may alter SLC30A6 expression by eliminating upstream SLC30A6 regulatory sequence (potential regulatory sequences shown by H3K27Ac track from UCSC), and both groups of CNVs may result in fusion transcripts with SLC30A6 by eliminating splicing to SPAST exon 17.
(E) Potential fusion transcripts resulting from the deletions shown in (D). Potential fusion transcripts are based only on known splice isoforms of SLC30A6 and include all SPAST exons present. Black, predicted in-frame; red, predicted out of frame.
None of the genes potentially involved in a fusion transcript (SLC30A6, BIRC6, and TTC27), nor any of the other genes affected by deletions spanning beyond the 3′ end of SPAST (NLRC4, YIPF4, MIR558, MIR4765, LINC00486, LOC100271832, LTBP1 [MIM 150390], and 5S_rRNA), is associated with Mendelian disease. Thus, the potential consequences to gene regulation or function of CNVs affecting these loci are unknown.
Deletion of SPAST Exon 17 Can Generate Fusion Transcripts with SLC30A6
Because SLC30A6 is the nearest gene downstream of SPAST, SPAST deletions with 3′ breakpoints in the 8 kb region between the two genes could potentially affect one or more SLC30A6 regulatory regions and/or result in splicing between the last remaining SPAST exon and SLC30A6. Seven such SPAST deletions exist among our data set, their 3′ breakpoints mapping within 6.5 kb to <1 kb of SLC30A6 (Figure 5). An additional seven deletions have 3′ breakpoints within the final exon (exon 17) of SPAST (Figure 5).
Nervous system RNA was not available from these subjects; however, a lymphoblastoid cell line from one subject with a complete exon 17 deletion (BAB 3327) was obtained. cDNA was prepared from this cell line and from a healthy control individual, BAB 3099;45 both blood-derived and lymphoblastoid-cell-line-derived DNA from the control individual is free of SPAST CNVs as determined by aCGH (Figure 1). Two PCRs of cDNA were performed, one with primers closely flanking the deletion (forward primer in exon 16 of SPAST and reverse primer in SLC30A6 exon 2) and one with primers flanking a larger area (forward primer in SPAST exon 12 and reverse primer in SLC30A6 exon 7, the first constitutive exon of this gene) (Figure 6). A few faint fusion transcript bands were present in the control individual. However, abundant, robust fusion transcript bands were present in the SPAST deletion CNV subject (BAB 3327) (Figures 6A–6C). Sequencing of all PCR products confirmed splicing from SPAST to SLC30A6 (Figure 6E), further indicating that deletion of SPAST exon 17 can alter the expression/transcript structure of a downstream gene.
Figure 6.
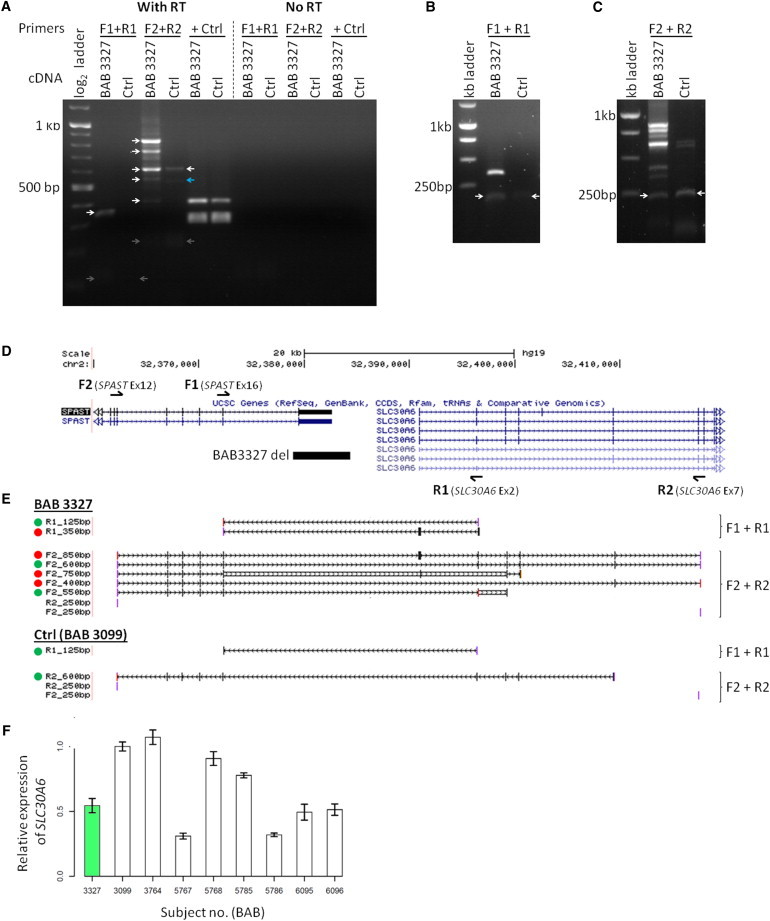
A Deletion of SPAST Exon 17 Yields Multiple SPAST:SLC30A6 Fusion Transcripts
(A–D) PCR primers (arrows above and below gene models in D) were designed to detect SPAST:SLC30A6 fusion transcripts in cDNA from subject BAB 3327, who has a deletion encompassing SPAST exon 17 (black bar in D). One primer set (F1 and R1) closely flanks the deletion, and the other (F2 and R2) spans a greater distance to account for alternative splicing known to exist in SLC30A6 (exon 7 is the first constitutive exon in this gene). PCR of cDNA (A) reveals multiple transcripts in BAB 3327 not present in the control individual (BAB 3099). The majority of bands (white arrows) were extracted and sequenced. One extracted band (blue arrow) failed sequencing. Four bands (gray arrows) required a second preparation/electrophoresis to be visualized, extracted, and sequenced (B and C, white arrows).
(E) Sequencing of PCR products in (A)–(C) demonstrates a variety of SPAST:SLC30A6 fusion transcript isoforms in BAB 3327, only some of which are present in the control individual (BAB 3099). Dots indicate predicted in-frame (green dots) or out-of-frame (red dots) transcripts. Reading frame is unclear for transcripts with no dot. PCR products were sequenced in both forward (F) and reverse (R) genomic orientation, although only one orientation may be shown.
(F) SLC30A6 qPCR. In lymphoblastoid cell lines, overall level of expression of SLC30A6 in BAB 3327 does not differ significantly from that of eight healthy controls (50th centile versus controls, empirical cumulative distribution function). Data plotted as mean ± standard deviation.
Effect of a SPAST Exon 17 Deletion on SLC30A6 Expression Level
The possibility exists that, by creating SPAST:SLC30A6 fusion transcripts or disrupting SLC30A6 upstream regulatory sequences, the SPAST exon 17 deletion in BAB 3327 may affect SLC30A6 expression level.46 qPCR indicated that the level of expression of SLC30A6 in a BAB3327 lymphoblastoid cell line does deviate significantly from that of eight pooled control lymphoblastoid cell lines (50th centile versus controls, empirical cumulative distribution function) (Figure 6).
Discussion
SPAST CNVs: Diverse Sizes, Boundaries, and Exons Rearranged
We delineated breakpoint junctions for 49 SPAST CNVs and document extensive diversity of disease-associated rearrangements in this gene, even for CNVs affecting a given exon. By increasing the number of published, disease-causing SPAST CNVs with known breakpoints from 7 to 54, we provide a resource for evaluating the pathogenicity of CNVs in SPAST identified during the course of genomic testing. Predicted in-frame deletions of exons 4, 8, and 9 in individuals with SPG4 suggest that these exons are indispensable, and a pathogenic, predicted in-frame duplication of exon 9 (24 amino acids) reinforces the sensitivity of the spastin AAA (ATPase Associated with various cellular Activities) domain to mutation47 (Figure S5). No SPAST exons have been demonstrated to be dispensable. However, exon 2 or 3 deletions have not been reported in persons with SPG4; thus, these in-frame exons may be candidates for exon skipping48 for individuals with pathogenic point mutations in them.
The CNV allele size versus frequency spectrum is skewed toward rearrangements of modest size relative to SPAST. A skewing toward small CNVs is reminiscent of the overall distribution of CNVs in personal genomes.49 It may also reflect the mechanism(s) of formation of SPAST CNVs, discussed below.
Alu Elements Destabilize SPAST, Leading to CNVs with Unique Properties
We found that more than two-thirds of SPAST CNVs have breakpoints in pairs of directly oriented Alus. Thus, Alu-mediated CNV formation is the predominant, but not exclusive, mechanism generating rearrangements in SPAST. The observed predominance of Alu-mediated CNVs suggests that the Alu-rich genomic architecture of SPAST (36% of the gene with segments of even higher Alu density;14 Figure 2B) renders this locus susceptible to a variety of genome rearrangements and may explain why an appreciable percentage of mutations causing SPG4 are CNVs.
Alu-mediated CNVs in our cohort share properties with Alu-mediated CNVs at other disease loci, including the presence of breakpoint microhomology, rearrangement between different Alu family members, and small size (Table 4). We found that Alu-mediated CNVs in SPAST are smaller than non-Alu-mediated CNVs (which include the eight largest rearrangements in our cohort). This could reflect a “boundary” created by a zone of low Alu density near SPAST. Alternatively, it suggests a potential size preference or limit as a general feature of Alu-mediated CNV formation. Human-specific Alu-mediated CNVs22 are even smaller (median 468 bp, mean 806 bp; range 101–7,255 bp) than Alu-mediated CNVs in disease loci, potentially a contributing factor to their becoming fixed in a species.
Proposed Mechanisms Generating Alu-Mediated CNVs
The hypothesized mechanism for Alu-Alu-mediated CNV formation has been homologous recombination between nonallelic Alus, i.e., Alu-Alu recombination.20, 50 The SPAST CNVs we report are generally consistent with this model: pairs of Alus are directly oriented and render a complete, hybrid Alu in the recombined genome; complex rearrangements are infrequent.43 However, the short length (<300 bp) and low percent identity (<91%) shared by Alus within each pair may be insufficient to meet the presumed requirements of ectopic homologous recombination derived from observational studies of NAHR at LCRs.51, 52 Furthermore, the three complex Alu-mediated CNVs are inconsistent with formation by NAHR unless a familial Alu polymorphism existed prior to the rearrangement. Some of the Alus at breakpoints of Alu-mediated CNVs contained the 13-nucleotide consensus motif enriched at meiotic recombination hotspots and at some LCRs involved in NAHR.42, 43 Although intriguing, any enrichment of this consensus motif in breakpoint Alus of Alu-mediated CNVs was not significant when compared to either Alus present at one breakpoint of non-Alu-mediated CNVs or to all Alus within and near SPAST. Finally, an analysis of previous studies at other loci (Table 4) demonstrates that moderate identity breakpoint Alu pairs and occasional complex events are general features of Alu-mediated CNVs. These findings may implicate an alternative non-homologous-recombination mechanism for Alu-mediated CNV formation.14
One alternative rearrangement mechanism may be microhomology-mediated DNA repair,53 for example microhomology-mediated end joining (MMEJ) or fork stalling and template switching (FoSTeS)/microhomology-mediated break-induced replication (MMBIR). These mechanisms are consistent with the microhomology present at the breakpoints of Alu-mediated CNVs among our and others’ data (Table 4). MMEJ may explain the predominance of deletions over duplications. FoSTeS/MMBIR may explain the occasional apparently complex Alu-mediated CNV.14
Another alternative mechanism may be single-strand annealing (SSA).54, 55 This double-strand break (DSB) repair pathway involves the annealing of directly oriented repeats flanking the DSB after they are made single-stranded by 5′ to 3′ end resection. The removal of the 3′ flaps and filling of gaps by DNA synthesis then results in a simple, small deletion. In a study of translocation mechanisms, Elliott et al.56 induced double-stranded breaks adjacent to a human Alu element on each of two nonhomologous chromosomes in cultured mouse cells; when the Alu elements were identical, SSA was the predominant mechanism generating translocations between these chromosomes. However, when the Alus were only 80% identical, the frequency of SSA diminished such that NHEJ became the predominant translocation-generating mechanism. Similarly, in yeast, a decrease of identity between two 205 bp repeats flanking a DSB site from 100% to 97% decreased the rate of SSA by 6-fold.57
Another potential contributing mechanism could be homeologous recombination, reduced-frequency recombination between substrates that are somewhat but not perfectly identical.58, 59, 60 This model is consistent with the low percent identity (75.8%–90.7%) shared by breakpoint Alu pairs in our study.61 In addition, in 33 of 39 (85%) Alu-Alu events in this study, the substrate pairs were from different Alu families. Whereas NAHR generates recurrent CNVs with high formation rates during meiosis,62 our data and others’ (Table 4) demonstrate that Alu-mediated CNVs are mostly or entirely nonrecurrent; thus, the formation rate of any particular rearrangement is low, again consistent with homeologous recombination.63 Although the probability of any one Alu-mediated CNV occurring is miniscule, with more than one million Alus in the genome, the cumulative ability of Alus to act as a recombination substrate and generate genomic rearrangements may be substantial.
SPAST deletions predominate over duplications. This suggests that, if formed by recombination, these events are most often intrachromatidal. Because Alus in close linear proximity along the same chromatid are generally expected to be in close three-dimensional proximity,64 and because three-dimensional proximity has been suggested to be important for generation of rearrangements,65 then perhaps small size is a general feature of Alu-mediated CNVs that is facilitated by chromosomal conformation itself. Long-range interactions, chromatin compartments,64 tethering of Alus, and genome reorganization during the cell cycle66 could also shape the spatial relationships of Alus and thus their probability of acting as rearrangement substrates. If these CNVs are formed by replicative template switching, small rearrangement size would be consistent with the known tendency of this process to occur more frequently over short distances.
Deletions Can Result in Chimeric Genes and Fusion Transcripts
Almost half of the CNVs we report extend beyond the boundaries of SPAST, many of which interrupt other genes. Although none of these other genes is known to be associated with Mendelian disease, their disruption could have phenotypic consequences. Moreover, 12 deletions extending 3′ of SPAST form chimeric genes that may yield fusion transcripts, also of unknown phenotypic consequence. Of note are the deletions forming chimeric genes between SPAST and SLC30A6 (solute carrier family 30, member 6) encoding a zinc transporter, ZnT6. Zinc and zinc transporter (including ZnT6) dynamics have been described to be altered in the brains of humans with Alzheimer disease and rodent models of dementia.34, 35, 67 Thus, the possibility of SPAST CNVs dysregulating SLC30A6 expression could potentially inform the clinical observation of comorbid spastic paraplegia and dementia.31 Neither RNA nor cells were available from any subjects with combined SPG4 and dementia.
Seven CNVs have 3′ breakpoints within the intergenic region between SPAST and SLC30A6, and an additional seven have 3′ breakpoints within the final exon of SPAST. The former group could affect SLC30A6 expression and all could potentially form fusion transcripts. We investigated this possibility experimentally using RNA from an available nonneuronal cell type (lymphoblastoid cell line) from one subject with a deletion encompassing the final exon of SPAST (exon 17). A few low-abundance fusion transcripts were present in a control individual, the significance of which is unknown. However, abundant and additional fusion transcripts were present in the SPAST deletion subject, indicating that a deletion of only an exon of SPAST can affect gene expression beyond the gene itself. The phenotypic consequence of this, if any, requires additional investigation.
The expression level of SLC30A6 in lymphoblastoid cell line RNA from this individual does not deviate significantly from that of eight control lymphoblastoid cell lines. Whether or not SLC30A6 expression is affected in this individual in neurons is unknown.
Summary
Molecular characterization of novel SPAST CNV alleles contributes to knowledge of pathogenic variants in this gene and provides insights into the importance of particular exons for spastin function. Our data suggest that the Alu-rich genomic architecture of SPAST predisposes to CNVs in this gene, contributing appreciably to its mutability and to SPG4 disease and potentially by a mechanism other than homologous recombination. Furthermore, these data allow the generation of hypotheses concerning Alus as a genome-destabilizing entity. We provide evidence that Alu-mediated rearrangements shape gene and genome structure and that the CNVs that are generated affect both SPAST and adjacent genes and extragenic regions. Thus, such CNVs can affect gene expression beyond SPAST itself, with potential phenotypic consequences.
Acknowledgments
The authors thank the Spastic Paraplegia Foundation, Sandy Wilcock, and Amy Breman for assistance with subject/sample recruitment. We thank Gladys Zapata and Paula Patricia Hernandez for transforming lymphoblasts and Chad Shaw, Claudia Fonseca, and Avinash Dharmadhikari for technical advice. We are grateful to the Spanish Familial Spastic Paraplegia Association (AEPEF) and the European Hereditary Spastic Paraplegia Federation (Euro-HSP) for their support and collaboration. P.M.B. and I.M.C. are fellows of the Baylor College of Medicine Medical Scientist Training Program (T32GM007330). P.M.B. is supported by grants from the Wintermann Foundation and the Baylor Research Advocates for Student Scientists. I.M.C. was supported by a fellowship from the National Institute of Neurological Disorders and Stroke (F31 NS083159). C.R.B. is an HHMI Fellow of the Damon Runyon Cancer Research Foundation (DRG 2155-13). This work was supported in part by the US National Institute of Neurological Disorders and Stroke (RO1NS058529 to J.R.L.), the US National Human Genome Research Institute/National Heart, Blood and Lung Institute (U54HG006542), and the Spanish Fondo de Investigaciones Sanitarias (PS09/01830). J.K.F. is supported by the Spastic Paraplegia Foundation, the U.S. National Institute of Neurological Disorders and Stroke (RO1NS0679700), the U.S. Department of Veterans Affairs (Merit Review 5I01CX000344), the Geriatric Research Education and Clinical Center AAVAMC, and the Paul and Lois Katzman Family. J.R.L. holds stock ownership in 23andMe, Inc. and Ion Torrent Systems, Inc., and is a coinventor on multiple United States and European patents related to molecular diagnostics. M.J.S. is a shareholder of Genomic Consulting, a company related to the application of genomics in medicine. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular genetic testing offered in the Medical Genetics Laboratories. I.D.K. and S.D.B. are employees of Quest Diagnostics.
Published: July 24, 2014
Footnotes
Supplemental Data include Supplemental Methods, six figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.ajhg.2014.06.014.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Agilent eArray, https://earray.chem.agilent.com
GeneReviews, Dürr, A., Tallaksen, C., and Depienne, C. (2012). Spastic paraplegia 4, http://www.ncbi.nlm.nih.gov/books/NBK1122
International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
RepeatMasker, http://www.repeatmasker.org
UCSC Genome Browser, http://genome.ucsc.edu
Supplemental Data
References
- 1.Wiszniewski W., Hunter J.V., Hanchard N.A., Willer J.R., Shaw C., Tian Q., Illner A., Wang X., Cheung S.W., Patel A. TM4SF20 ancestral deletion and susceptibility to a pediatric disorder of early language delay and cerebral white matter hyperintensities. Am. J. Hum. Genet. 2013;93:197–210. doi: 10.1016/j.ajhg.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okamoto Y., Goksungur M.T., Pehlivan D., Beck C.R., Gonzaga-Jauregui C., Muzny D.M., Atik M.M., Carvalho C.M.B., Matur Z., Bayraktar S. Exonic duplication CNV of NDRG1 associated with autosomal-recessive HMSN-Lom/CMT4D. Genet. Med. 2014;16:386–394. doi: 10.1038/gim.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benito-Sanz S., Royo J.L., Barroso E., Paumard-Hernández B., Barreda-Bonis A.C., Liu P., Gracía R., Lupski J.R., Campos-Barros Á., Gómez-Skarmeta J.L., Heath K.E. Identification of the first recurrent PAR1 deletion in Léri-Weill dyschondrosteosis and idiopathic short stature reveals the presence of a novel SHOX enhancer. J. Med. Genet. 2012;49:442–450. doi: 10.1136/jmedgenet-2011-100678. [DOI] [PubMed] [Google Scholar]
- 4.Hjeij R., Lindstrand A., Francis R., Zariwala M.A., Liu X., Li Y., Damerla R., Dougherty G.W., Abouhamed M., Olbrich H. ARMC4 mutations cause primary ciliary dyskinesia with randomization of left/right body asymmetry. Am. J. Hum. Genet. 2013;93:357–367. doi: 10.1016/j.ajhg.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Loureiro J.L., Brandão E., Ruano L., Brandão A.F., Lopes A.M., Thieleke-Matos C., Miller-Fleming L., Cruz V.T., Barbosa M., Silveira I. Autosomal dominant spastic paraplegias: a review of 89 families resulting from a portuguese survey. JAMA Neurol. 2013;70:481–487. doi: 10.1001/jamaneurol.2013.1956. [DOI] [PubMed] [Google Scholar]
- 6.Depienne C., Fedirko E., Forlani S., Cazeneuve C., Ribaï P., Feki I., Tallaksen C., Nguyen K., Stankoff B., Ruberg M. Exon deletions of SPG4 are a frequent cause of hereditary spastic paraplegia. J. Med. Genet. 2007;44:281–284. doi: 10.1136/jmg.2006.046425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vandebona H., Kerr N.P., Liang C., Sue C.M. SPAST mutations in Australian patients with hereditary spastic paraplegia. Intern. Med. J. 2012;42:1342–1347. doi: 10.1111/j.1445-5994.2012.02941.x. [DOI] [PubMed] [Google Scholar]
- 8.McDermott C.J., Burness C.E., Kirby J., Cox L.E., Rao D.G., Hewamadduma C., Sharrack B., Hadjivassiliou M., Chinnery P.F., Dalton A., Shaw P.J., UK and Irish HSP Consortium Clinical features of hereditary spastic paraplegia due to spastin mutation. Neurology. 2006;67:45–51. doi: 10.1212/01.wnl.0000223315.62404.00. [DOI] [PubMed] [Google Scholar]
- 9.Álvarez V., Sánchez-Ferrero E., Beetz C., Díaz M., Alonso B., Corao A.I., Gámez J., Esteban J., Gonzalo J.F., Pascual-Pascual S.I., Group for the Study of the Genetics of Spastic Paraplegia Mutational spectrum of the SPG4 (SPAST) and SPG3A (ATL1) genes in Spanish patients with hereditary spastic paraplegia. BMC Neurol. 2010;10:89. doi: 10.1186/1471-2377-10-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sauter S., Miterski B., Klimpe S., Bönsch D., Schöls L., Visbeck A., Papke T., Hopf H.C., Engel W., Deufel T. Mutation analysis of the spastin gene (SPG4) in patients in Germany with autosomal dominant hereditary spastic paraplegia. Hum. Mutat. 2002;20:127–132. doi: 10.1002/humu.10105. [DOI] [PubMed] [Google Scholar]
- 11.Mitne-Neto M., Kok F., Beetz C., Pessoa A., Bueno C., Graciani Z., Martyn M., Monteiro C.B.M., Mitne G., Hubert P. A multi-exonic SPG4 duplication underlies sex-dependent penetrance of hereditary spastic paraplegia in a large Brazilian pedigree. Eur. J. Hum. Genet. 2007;15:1276–1279. doi: 10.1038/sj.ejhg.5201924. [DOI] [PubMed] [Google Scholar]
- 12.Zhang F., Gu W., Hurles M.E., Lupski J.R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genomics Hum. Genet. 2009;10:451–481. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu W., Zhang F., Lupski J.R. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1:4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boone P.M., Liu P., Zhang F., Carvalho C.M.B., Towne C.F., Batish S.D., Lupski J.R. Alu-specific microhomology-mediated deletion of the final exon of SPAST in three unrelated subjects with hereditary spastic paraplegia. Genet. Med. 2011;13:582–592. doi: 10.1097/GIM.0b013e3182106775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deininger P. Alu elements: know the SINEs. Genome Biol. 2011;12:236. doi: 10.1186/gb-2011-12-12-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Franke G., Bausch B., Hoffmann M.M., Cybulla M., Wilhelm C., Kohlhase J., Scherer G., Neumann H.P.H. Alu-Alu recombination underlies the vast majority of large VHL germline deletions: Molecular characterization and genotype-phenotype correlations in VHL patients. Hum. Mutat. 2009;30:776–786. doi: 10.1002/humu.20948. [DOI] [PubMed] [Google Scholar]
- 17.Lehrman M.A., Schneider W.J., Südhof T.C., Brown M.S., Goldstein J.L., Russell D.W. Mutation in LDL receptor: Alu-Alu recombination deletes exons encoding transmembrane and cytoplasmic domains. Science. 1985;227:140–146. doi: 10.1126/science.3155573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Smith A.J., Walters R.G., Coin L.J.M., Steinfeld I., Yakhini Z., Sladek R., Froguel P., Blakemore A.I.F. Small deletion variants have stable breakpoints commonly associated with alu elements. PLoS ONE. 2008;3:e3104. doi: 10.1371/journal.pone.0003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mei D., Lewis R., Parrini E., Lazarou L.P., Marini C., Pilz D.T., Guerrini R. High frequency of genomic deletions—and a duplication—in the LIS1 gene in lissencephaly: implications for molecular diagnosis. J. Med. Genet. 2008;45:355–361. doi: 10.1136/jmg.2007.056507. [DOI] [PubMed] [Google Scholar]
- 20.Deininger P.L., Batzer M.A. Alu repeats and human disease. Mol. Genet. Metab. 1999;67:183–193. doi: 10.1006/mgme.1999.2864. [DOI] [PubMed] [Google Scholar]
- 21.Strout M.P., Marcucci G., Bloomfield C.D., Caligiuri M.A. The partial tandem duplication of ALL1 (MLL) is consistently generated by Alu-mediated homologous recombination in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA. 1998;95:2390–2395. doi: 10.1073/pnas.95.5.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sen S.K., Han K., Wang J., Lee J., Wang H., Callinan P.A., Dyer M., Cordaux R., Liang P., Batzer M.A. Human genomic deletions mediated by recombination between Alu elements. Am. J. Hum. Genet. 2006;79:41–53. doi: 10.1086/504600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Babcock M., Pavlicek A., Spiteri E., Kashork C.D., Ioshikhes I., Shaffer L.G., Jurka J., Morrow B.E. Shuffling of genes within low-copy repeats on 22q11 (LCR22) by Alu-mediated recombination events during evolution. Genome Res. 2003;13:2519–2532. doi: 10.1101/gr.1549503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szabó Z., Levi-Minzi S.A., Christiano A.M., Struminger C., Stoneking M., Batzer M.A., Boyd C.D. Sequential loss of two neighboring exons of the tropoelastin gene during primate evolution. J. Mol. Evol. 1999;49:664–671. doi: 10.1007/pl00006587. [DOI] [PubMed] [Google Scholar]
- 25.Batzer M.A., Deininger P.L. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- 26.Boone P.M., Soens Z.T., Campbell I.M., Stankiewicz P., Cheung S.W., Patel A., Beaudet A.L., Plon S.E., Shaw C.A., McGuire A.L., Lupski J.R. Incidental copy-number variants identified by routine genome testing in a clinical population. Genet. Med. 2013;15:45–54. doi: 10.1038/gim.2012.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Ligt J., Boone P.M., Pfundt R., Vissers L.E.L.M., Richmond T., Geoghegan J., O’Moore K., de Leeuw N., Shaw C., Brunner H.G. Detection of clinically relevant copy number variants with whole-exome sequencing. Hum. Mutat. 2013;34:1439–1448. doi: 10.1002/humu.22387. [DOI] [PubMed] [Google Scholar]
- 28.Iwanaga H., Tsujino A., Shirabe S., Eguchi H., Fukushima N., Niikawa N., Yoshiura K., Eguchi K. Large deletion involving the 5′-UTR in the spastin gene caused mild phenotype of autosomal dominant hereditary spastic paraplegia. Am. J. Med. Genet. A. 2005;133A:13–17. doi: 10.1002/ajmg.a.30510. [DOI] [PubMed] [Google Scholar]
- 29.Salinas S., Proukakis C., Crosby A., Warner T.T. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7:1127–1138. doi: 10.1016/S1474-4422(08)70258-8. [DOI] [PubMed] [Google Scholar]
- 30.Nielsen J.E., Johnsen B., Koefoed P., Scheuer K.H., Grønbech-Jensen M., Law I., Krabbe K., Nørremølle A., Eiberg H., Søndergård H. Hereditary spastic paraplegia with cerebellar ataxia: a complex phenotype associated with a new SPG4 gene mutation. Eur. J. Neurol. 2004;11:817–824. doi: 10.1111/j.1468-1331.2004.00888.x. [DOI] [PubMed] [Google Scholar]
- 31.Murphy S., Gorman G., Beetz C., Byrne P., Dytko M., McMonagle P., Kinsella K., Farrell M., Hutchinson M. Dementia in SPG4 hereditary spastic paraplegia: clinical, genetic, and neuropathologic evidence. Neurology. 2009;73:378–384. doi: 10.1212/WNL.0b013e3181b04c6c. [DOI] [PubMed] [Google Scholar]
- 32.Ribaï P., Depienne C., Fedirko E., Jothy A.-C., Viveweger C., Hahn-Barma V., Brice A., Durr A. Mental deficiency in three families with SPG4 spastic paraplegia. Eur. J. Hum. Genet. 2008;16:97–104. doi: 10.1038/sj.ejhg.5201922. [DOI] [PubMed] [Google Scholar]
- 33.Erichsen A.K., Inderhaug E., Mattingsdal M., Eiklid K., Tallaksen C.M.E. Seven novel mutations and four exon deletions in a collection of Norwegian patients with SPG4 hereditary spastic paraplegia. Eur. J. Neurol. 2007;14:809–814. doi: 10.1111/j.1468-1331.2007.01861.x. [DOI] [PubMed] [Google Scholar]
- 34.Smith J.L., Xiong S., Markesbery W.R., Lovell M.A. Altered expression of zinc transporters-4 and -6 in mild cognitive impairment, early and late Alzheimer’s disease brain. Neuroscience. 2006;140:879–888. doi: 10.1016/j.neuroscience.2006.02.049. [DOI] [PubMed] [Google Scholar]
- 35.Lovell M.A., Smith J.L., Markesbery W.R. Elevated zinc transporter-6 in mild cognitive impairment, Alzheimer disease, and pick disease. J. Neuropathol. Exp. Neurol. 2006;65:489–498. doi: 10.1097/01.jnen.0000229237.98124.91. [DOI] [PubMed] [Google Scholar]
- 36.White K.D., Ince P.G., Lusher M., Lindsey J., Cookson M., Bashir R., Shaw P.J., Bushby K.M. Clinical and pathologic findings in hereditary spastic paraparesis with spastin mutation. Neurology. 2000;55:89–94. doi: 10.1212/wnl.55.1.89. [DOI] [PubMed] [Google Scholar]
- 37.Gonzaga-Jauregui C., Zhang F., Towne C.F., Batish S.D., Lupski J.R. GJB1/Connexin 32 whole gene deletions in patients with X-linked Charcot-Marie-Tooth disease. Neurogenetics. 2010;11:465–470. doi: 10.1007/s10048-010-0247-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boone P.M., Bacino C.A., Shaw C.A., Eng P.A., Hixson P.M., Pursley A.N., Kang S.-H.L., Yang Y., Wiszniewska J., Nowakowska B.A. Detection of clinically relevant exonic copy-number changes by array CGH. Hum. Mutat. 2010;31:1326–1342. doi: 10.1002/humu.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miura S., Shibata H., Kida H., Noda K., Toyama T., Iwasaki N., Iwaki A., Ayabe M., Aizawa H., Taniwaki T., Fukumaki Y. Partial SPAST and DPY30 deletions in a Japanese spastic paraplegia type 4 family. Neurogenetics. 2011;12:25–31. doi: 10.1007/s10048-010-0260-7. [DOI] [PubMed] [Google Scholar]
- 41.Sulek A., Elert E., Rajkiewicz M., Zdzienicka E., Stepniak I., Krysa W., Zaremba J. Screening for the hereditary spastic paraplaegias SPG4 and SPG3A with the multiplex ligation-dependent probe amplification technique in a large population of affected individuals. Neurol. Sci. 2013;34:239–242. doi: 10.1007/s10072-011-0899-3. [DOI] [PubMed] [Google Scholar]
- 42.Myers S., Freeman C., Auton A., Donnelly P., McVean G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat. Genet. 2008;40:1124–1129. doi: 10.1038/ng.213. [DOI] [PubMed] [Google Scholar]
- 43.Sasaki M., Lange J., Keeney S. Genome destabilization by homologous recombination in the germ line. Nat. Rev. Mol. Cell Biol. 2010;11:182–195. doi: 10.1038/nrm2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baudat F., Buard J., Grey C., Fledel-Alon A., Ober C., Przeworski M., Coop G., de Massy B. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science. 2010;327:836–840. doi: 10.1126/science.1183439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu P., Erez A., Nagamani S.C.S., Dhar S.U., Kołodziejska K.E., Dharmadhikari A.V., Cooper M.L., Wiszniewska J., Zhang F., Withers M.A. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell. 2011;146:889–903. doi: 10.1016/j.cell.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ligtenberg M.J.L., Kuiper R.P., Chan T.L., Goossens M., Hebeda K.M., Voorendt M., Lee T.Y.H., Bodmer D., Hoenselaar E., Hendriks-Cornelissen S.J.B. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3′ exons of TACSTD1. Nat. Genet. 2009;41:112–117. doi: 10.1038/ng.283. [DOI] [PubMed] [Google Scholar]
- 47.Shoukier M., Neesen J., Sauter S.M., Argyriou L., Doerwald N., Pantakani D.V.K., Mannan A.U. Expansion of mutation spectrum, determination of mutation cluster regions and predictive structural classification of SPAST mutations in hereditary spastic paraplegia. Eur. J. Hum. Genet. 2009;17:187–194. doi: 10.1038/ejhg.2008.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aartsma-Rus A. Overview on DMD exon skipping. Methods Mol. Biol. 2012;867:97–116. doi: 10.1007/978-1-61779-767-5_7. [DOI] [PubMed] [Google Scholar]
- 49.Wheeler D.A., Srinivasan M., Egholm M., Shen Y., Chen L., McGuire A., He W., Chen Y.-J., Makhijani V., Roth G.T. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 50.Lehrman M.A., Russell D.W., Goldstein J.L., Brown M.S. Alu-Alu recombination deletes splice acceptor sites and produces secreted low density lipoprotein receptor in a subject with familial hypercholesterolemia. J. Biol. Chem. 1987;262:3354–3361. [PubMed] [Google Scholar]
- 51.Reiter L.T., Hastings P.J., Nelis E., De Jonghe P., Van Broeckhoven C., Lupski J.R. Human meiotic recombination products revealed by sequencing a hotspot for homologous strand exchange in multiple HNPP deletion patients. Am. J. Hum. Genet. 1998;62:1023–1033. doi: 10.1086/301827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Metzenberg A.B., Wurzer G., Huisman T.H.J., Smithies O. Homology requirements for unequal crossing over in humans. Genetics. 1991;128:143–161. doi: 10.1093/genetics/128.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ottaviani D., LeCain M., Sheer D. The role of microhomology in genomic structural variation. Trends Genet. 2014;30:85–94. doi: 10.1016/j.tig.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 54.Haber J.E. Transpositions and translocations induced by site-specific double-strand breaks in budding yeast. DNA Repair (Amst.) 2006;5:998–1009. doi: 10.1016/j.dnarep.2006.05.025. [DOI] [PubMed] [Google Scholar]
- 55.Pâques F., Haber J.E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elliott B., Richardson C., Jasin M. Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol. Cell. 2005;17:885–894. doi: 10.1016/j.molcel.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 57.Sugawara N., Goldfarb T., Studamire B., Alani E., Haber J.E. Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc. Natl. Acad. Sci. USA. 2004;101:9315–9320. doi: 10.1073/pnas.0305749101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mézard C., Pompon D., Nicolas A. Recombination between similar but not identical DNA sequences during yeast transformation occurs within short stretches of identity. Cell. 1992;70:659–670. doi: 10.1016/0092-8674(92)90434-e. [DOI] [PubMed] [Google Scholar]
- 59.Yang D., Waldman A.S. Fine-resolution analysis of products of intrachromosomal homeologous recombination in mammalian cells. Mol. Cell. Biol. 1997;17:3614–3628. doi: 10.1128/mcb.17.7.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bailis A.M., Rothstein R. A defect in mismatch repair in Saccharomyces cerevisiae stimulates ectopic recombination between homeologous genes by an excision repair dependent process. Genetics. 1990;126:535–547. doi: 10.1093/genetics/126.3.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rossetti L.C., Goodeve A., Larripa I.B., De Brasi C.D. Homeologous recombination between AluSx-sequences as a cause of hemophilia. Hum. Mutat. 2004;24:440. doi: 10.1002/humu.9288. [DOI] [PubMed] [Google Scholar]
- 62.Dittwald P., Gambin T., Szafranski P., Li J., Amato S., Divon M.Y., Rodríguez Rojas L.X., Elton L.E., Scott D.A., Schaaf C.P. NAHR-mediated copy-number variants in a clinical population: mechanistic insights into both genomic disorders and Mendelizing traits. Genome Res. 2013;23:1395–1409. doi: 10.1101/gr.152454.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Waldman A.S., Liskay R.M. Differential effects of base-pair mismatch on intrachromosomal versus extrachromosomal recombination in mouse cells. Proc. Natl. Acad. Sci. USA. 1987;84:5340–5344. doi: 10.1073/pnas.84.15.5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fudenberg G., Getz G., Meyerson M., Mirny L.A. High order chromatin architecture shapes the landscape of chromosomal alterations in cancer. Nat. Biotechnol. 2011;29:1109–1113. doi: 10.1038/nbt.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Naumova N., Imakaev M., Fudenberg G., Zhan Y., Lajoie B.R., Mirny L.A., Dekker J. Organization of the mitotic chromosome. Science. 2013;342:948–953. doi: 10.1126/science.1236083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bush A.I. The metal theory of Alzheimer’s disease. J. Alzheimers Dis. 2013;33(Suppl 1):S277–S281. doi: 10.3233/JAD-2012-129011. [DOI] [PubMed] [Google Scholar]
- 68.Kuiper R.P., Vissers L.E.L.M., Venkatachalam R., Bodmer D., Hoenselaar E., Goossens M., Haufe A., Kamping E., Niessen R.C., Hogervorst F.B.L. Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Hum. Mutat. 2011;32:407–414. doi: 10.1002/humu.21446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.