

Abstract
Connexins (Cxs) are a family of vertebrate proteins constituents of gap junction channels (GJCs) that connect the cytoplasm of adjacent cells by the end-to-end docking of two Cx hemichannels. The intercellular transfer through GJCs occurs by passive diffusion allowing the exchange of water, ions, and small molecules. Despite the broad interest to understand, at the molecular level, the functional state of Cx-based channels, there are still many unanswered questions regarding structure-function relationships, perm-selectivity, and gating mechanisms. In particular, the ordering, structure, and dynamics of water inside Cx GJCs and hemichannels remains largely unexplored. In this work, we describe the identification and characterization of a believed novel water pocket—termed the IC pocket—located in-between the four transmembrane helices of each human Cx26 (hCx26) monomer at the intracellular (IC) side. Using molecular dynamics (MD) simulations to characterize hCx26 internal water structure and dynamics, six IC pockets were identified per hemichannel. A detailed characterization of the dynamics and ordering of water including conformational variability of residues forming the IC pockets, together with multiple sequence alignments, allowed us to propose a functional role for this cavity. An in vitro assessment of tracer uptake suggests that the IC pocket residue Arg-143 plays an essential role on the modulation of the hCx26 hemichannel permeability.
Introduction
The past decade has witnessed a growing interest toward the understanding of the role played by water inside pockets or cavities in proteins (1–5). The functional role of these cavities has been a matter of debate encompassing physicochemical, thermodynamic, and/or biophysical arguments (3,4,6). In particular, the hydration/dehydration of cavities has been related to activation/deactivation events on transmembrane proteins such as G protein-coupled receptors (7,8), ligand-binding processes (9–11), intermolecular recognition events (12–14), and even protein folding (13,14). Spontaneous nanoscale dewetting events have been first predicted in protein complex folding (15,16) and then associated to gating mechanism of ion channels (17–20). Despite this broad interest, the discovery, characterization, and analysis of protein cavities with or without water have been proven nontrivial even with the most sophisticated experimental techniques, such as x-ray crystallography, solution NMR, vibrational spectroscopy, and two-dimensional infrared spectroscopy in conjunction with hydrogen-deuterium exchange, among others (6,21–23). Meanwhile, during the past decade large-scale molecular dynamic (MD) simulations have been used as a complementary tool to observe, with atomistic detail, the structure and dynamics of water inside these protein cavities (24–28).
Connexins (Cxs) are a family of vertebrate proteins that constitute gap junction channels (GJCs). GJCs connect the cytoplasm of adjacent cells by the end-to-end docking of two connexin hemichannels producing a hydrophilic path between cells (29,30). The intercellular transfer through GJCs occurs by passive diffusion allowing the exchange of water, ions (e.g., Na+, K+, and Ca2+), small molecules such as peptides, metabolites, and signaling molecules (e.g., cAMP and inositol 1,4,5-trisphosphate) (29,31). Each hemichannel is formed by the oligomerization of six Cx subunits. Each Cx exhibits a characteristic topology of four transmembrane (TM) helices designated TM1–TM4, two extracellular loops termed E1 and E2, and one intracellular or cytoplasmic loop termed CL. Malfunction of GJCs or hemichannels is associated to pathologic conditions and genetic disorders that produce skin diseases, neurodegenerative and developmental diseases, cataracts, and most cases of hereditary deafness (see comprehensive reviews in previous studies (32,33)). In particular, mutations in the gene encoding the human Cx26 (hCx26) account for a large proportion of genetic deafness, leading either to nonsyndromic or syndromic disease. In syndromic deafness the hearing loss is associated with abnormal epidermal keratinisation (reviewed in (33)).
Despite the broad interest to further understand, at the molecular level, functional changes produced by mutations on Cx-based channels, there are still many unanswered questions regarding structure-function relationships, perm-selectivity, and gating mechanisms (34–37). In particular, the ordering, structure, and dynamics of water inside Cx GJCs and hemichannels remain largely unknown. Recently, the x-ray crystallographic structure of the hCx26 channel (38), solved at 3.5Å-resolution, has provided a structural basis for many other Cx channels also serving as a starting point to study structure-function relationships of these channels (39–43). One of many approaches to do so is by conducting systematic studies aimed to determine the presence and role played by structural water within proteins (8,13). However, the hCx26 crystallographic structure provided neither evidence on the presence of water molecules inside the main pore nor the existence of other relevant pockets that could be hydrated. Moreover, to our knowledge, neither theoretical nor experimental attempts to identify the presence and role of water inside Cx structures have been conducted so far.
To gain insights on the structure-function relationships coded into the molecular architecture of the hCx26 hemichannel, we conducted extensive MD simulations aimed to characterize the structure and dynamics of water molecules within this protein. By doing so, we have identified and characterized, to our knowledge, a novel water pocket, termed the IC pocket, which lays at the intracellular side of all hCx26 located in-between the four transmembrane helices of each monomer. Relying on these data and complemented with multiple sequence alignments, we elaborated on the functional role of the IC pocket: the position of Arg143 may regulate the IC pocket volume, modulating the dynamics of the N-terminal helix (NTH). To evaluate our functional hypothesis, a hemichannel permeability assessment was conducted through time-lapse imaging of ethidium (Eth) uptake, comparing hCx26Wt and mutants R143A, R143E, R143Q, and R143K. As a whole, our data suggest that the IC pocket and, furthermore, Arg-143 plays an essential role on the modulation of the hCx26 hemichannel permeability.
Materials and Methods
Modeling and simulation
The hCx26Wt hemichannel model was constructed using the coordinates from the Cx26 3.5Å x-ray structure (PDB ID:2ZW3) (38). Coordinates of Met1, the CL, the CT domain, and side chains for residues K15, S17, and S19 are not present in the x-ray structure. To obtain a complete Cx26-Wt hemichannel, all missing residues were modeled using MODELER (44) while keeping original x-ray coordinates fixed to model missing parts considering the hexameric symmetry of the hemichannel. Hydrogen atoms were assigned using the psf-gen module from VMD (24) and protonation states were assigned according to the pKa's predicted by PROPKA 3.0 (http://propka.ki.ku.dk) (45,46). Disulfide bonds identified in the crystal structure were created between C53–C180, C60–C174, and C64–C169 in each Cx subunit. The hCx26Wt hemichannel was then inserted into a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine lipid membrane considering the spatial arrangements of the protein with respect to the hydrocarbon core of the lipid bilayer, as obtained from the OPM database (47). A 150 × 150 × 120 Å box consisting of the protein, lipids, classic CHARMM TIP3 water molecules, and 150 mM KCl was generated using the membrane builder module of CHARMM-GUI (48). To respect the minimum image criteria, 15 Å at the z axis and 35 Å at both the x and y axis were defined as separation distance from the edge of the simulation cell. The CHARMM-22 (49) and the CHARMM-36 (50) force fields were used for protein and lipids, respectively. The final system was composed of 273.994 atoms, grouped by protein (22.344), water (18.0816), ions (350), and lipids (70.484). An initial system equilibration of 375 ps at 303.15 K was performed using the protocol developed by Woolf & Roux (51,52). Initial force constraints were gradually reduced to relax the system using NVT dynamics. The system was then equilibrated for 100 ns at 310 K using NPnAT dynamics with no constraints. Both methods were applied using the Langevin temperature control. Four independent MD simulations of 20 ns were performed by starting the simulation from different seeds, using NPnAT dynamics for data collecting and statistical analysis with set points of 1 atm and 310 K using the Nose–Hoover method and Langevin dynamics for temperature and piston fluctuation control, with a damping coefficient of 1 ps. All simulations were performed using NAMD2.9 (53). The PME method was used for full long-range electrostatics within a relative tolerance of 1 × 10−6. A cutoff distance of 12 Å was applied to real-space Ewald interactions, with a smooth switching function applied between 10 and 12 Å to account for Van der Waals (VdW) interactions. Multiple time steps were used with 2 fs for bonded interactions, 2 fs for short-range nonbonded interactions, and 4 fs for the full electrostatics evaluation using the r-RESPA method. The SHAKE algorithm was applied to constrain bond lengths to all hydrogen atoms. By plotting Cα-root-mean-squared (RMS) deviation and RMS fluctuation along the MD simulation, we assessed the structural equilibration reached by our hCx26Wt models, as seen in Figs. S1 and S2, respectively.
Sequence alignments and structural projection of the IC pocket conservation
To explore the relevance that each IC pocket residue may have on the hCx26 function, we evaluated the conservation level of these residues in between Cx subfamilies found in humans. To do so, we used as reference the work of Zardoya and Abascal (54). Using the BLOSUM64 scoring matrix, we generated a multiple sequence alignment of selected representatives of all human Cx subfamilies, Cxα, Cxβ, Cxγ, Cxδ, and Cxε, by using JalView (55). Resulting alignment was manually curated to fit the one presented by Abascal et al. (54). Finally, the curated alignment was used as input for the ConSurf server (56,57) to map the conservation of each IC pocket residue on the surface of a representative three-dimensional structure. The representative structure of the hCx26 was obtained by clustering using the Cα RMS deviation as metric, for every monomer from every frame, considering the four production runs. The central structure of the highest populated cluster was selected as the representative one (data not shown).
Trajectory and water dynamics analyses
Trajectory analyses were conducted by in-house code and executing python scripts using the open-source software MDAnalysis; an object-oriented python library to analyze molecular dynamics trajectories (58).
Survival time of water molecules in the pocket
Local translational and rotational mobility of water molecules are related to the amount of time that a molecule is likely to remain in a given region (59). By measuring which fraction of molecules were in a given region at time t and remain there at a later time t = t+τ, the survival probability P(τ) can be computed. The decay rate of P(τ) depends on the size of the region and the mobility of molecules within it and is calculated as follows:
| (1) |
where N(t,τ) is the number of particles that remain in the selection at time t+τ, N(t) is the number of particles at time t, and T is the number of time steps contributing to P(τ).
Mean square displacement and diffusion rates
Diffusion coefficients are usually evaluated from the Einstein relation employing mean-squared displacement of a diffusing particle as = 2nDt, where brackets denote ensemble average, Δr(t) is the displacement after time lag t, D is the diffusion coefficient, and n is the dimensionality of the corresponding displacement metric Δr(t). However, when estimating the diffusivity in confined or in homogeneous fluids, the confining geometry and the time that particles spend in particular regions must be taken into account. In this case, the survival probability P(t) was included in the mean square displacement relation, as proposed by Liu and Berne (59), for the calculation of diffusion coefficients inside confined regions. Thus, in our case, = 6DP(t) t. Diffusion coefficients were estimated from MSD data in the 5 to 20 ps time interval. Data was fitted to a cubic polynomial and the local diffusion coefficients were related to the coefficient of the linear term. Errors were calculated as the difference between the slope of the MSD average curve and the curves produced by the deviation when considering the average MSD obtained from the four independent simulations. Diffusion coefficient errors were estimated by measuring the difference between the slope of the average function MSD/P(t) and the slope of the average function translated to the standard deviation generating two curves; one in the middle and the other in top/bottom of the standard deviation bars.
Orientational relaxation of water molecules
To characterize the ordering of water molecules inside the pockets, the orientational relaxation of water molecules inside the pockets was measured (60). Two vectors were considered to describe the rotational freedom of water molecules inside the pocket: 1), ûOH, the unit vector along the molecular OH bond, and 2), ûD, the unit vector along the dipole moment of the water molecule. By computing the rate of decay of the time correlation of these vectors, a detailed picture of how freely water molecules can rotate inside the pocket can be obtained. Specifically, the relevant time correlation is as follows:
| (2) |
where P2(x) = (3x2 − 1)/2 is the second-order Legendre polynomial and û is a unit vector used to specify the orientation of water molecules (ûOH; ûD).
Hydrogen bond population relaxation
Hydrogen bonds were identified by applying a geometric criteria: a water pair is considered to be hydrogen bonded if the oxygen–oxygen distance is less than 3.5 Å and the O-H···O angle is greater than 120°, with all O-H···O angles classified as being between 0° and 180°. To measure the H-bond lifetime inside the IC pocket, two different approximations were considered: 1), the intermittent existence of an H-bond, and 2), the continuous existence of an H-bond. In both cases, H-bonds were considered between water molecules inside the IC pocket or between water and residues forming the pocket, following a similar procedure by Rapaport (61). To do so, the autocorrelation function can be calculated as follows:
| (3) |
where hij(t) = 1 if there is a H-bond between a pair ij of H-bond donor and acceptor at time t (intermittent), or during the time interval 0 - t (continuous), and hij(t) = 0 otherwise.
IC pocket definition
The IC pocket region was geometrically approximated by a sphere of radius 6 Å, centered at the geometric center of the amino acid residues comprising the pocket (see Fig. 2 and Table 1). A water molecule was considered inside the pocket if its position vector was located within this region. The average and standard errors were obtained from block-averaging. The standard error was estimated by extrapolation to a block of infinite size.
Figure 2.
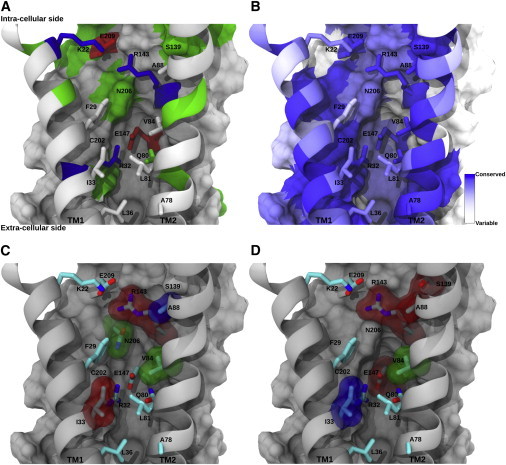
Conservation of residues in the IC pocket. (A) General overview of the IC pocket where constituent residues are rendered using licorice and color-coded by residue in equivalent orientation to Fig. 1E. Buried residues belonging to the IC pocket appear as colored surfaces. Transmembrane helices TM1 and TM2 are rendered using ribbons. (B) Residues belonging to the IC pocket, color-coded according to the ConSurf conservation score that appears at the bottom left. (C) IC pocket residues with associated mutations reported in the literature (see Table 2). The vdW representation is color-coded according to the functionality of the resulting mutant hCx26 hemichannel (red = diminished or null function; green = permeability and selectivity changes; blue = depending on the study may be classified red or green). (D) Mapping on the IC pocket of the amino acid residues reported by Skerret et al. (67). Residues surrounded by a transparent surface correspond to those that were mutated to cysteine showing relevant effects. Residues marked in red (R143, A88, S139, and E147) produced hemichannels with altered gating properties but normal conductance. Green-colored residues (V84) produced hemichannels with a diminished conductance. Blue-colored residues (I33) produced hemichannels with significant reduction in conductance. To see this figure in color, go online.
Table 1.
Residues forming the IC pocket classified by domain
| N-Ter | TM1 | TM2 | TM3 | TM4 | |||||
|---|---|---|---|---|---|---|---|---|---|
| 8 | THR | 22 | LYS | 80 | GLN | 133 | TRP | 202 | CYS |
| 11 | GLY | 25 | LEU | 84 | VAL | 136 | TYR | 205 | LEU |
| 12 | GLY | 26 | THR | 85 | SER | 139 | SER | 206 | ASN |
| 15 | LYS | 29 | PHE | 87 | PRO | 140 | ILE | 209 | GLU |
| 17 | SER | 32 | ARG | 88 | ALA | 143 | ARG | 213 | LEU |
| 33 | ILE | 91 | VAL | 144 | VAL | ||||
| 92 | ALA | 147 | GLU | ||||||
Assessment of hemichannel permeability
Hemichannel function was assessed through time-lapse imaging of ethidium (Eth) bromide (314 Dalton, +1) uptake. Briefly, cells plated on glass cover slips were bathed in ringer solution containing 1.8 mM Ca2+ and 50 μM Eth and tracer uptake was measured for 5 min. To induce hemichannel opening, the extracellular solution was removed and rinsed twice with Ca2+-Mg2+-free Hanks’ balanced salt solution (HBSS) (Invitrogen, Carlsbad, CA). Cells were then incubated in HBSS containing 50 μM Eth for 10 min, followed by the addition of hemichannel blocker La3+ (100 μM LaCl3; Sigma-Aldrich, St. Louis, MO) for another 5 min. Fluorescent images of different cells (defined as regions of interest) during nucleic acid stain uptake were obtained using a 40× objective in a Nikon (Chiyoda, Tokyo, Japan) TE-2000U inverted microscope. Capture was made using a Nikon DS-2WBc fast cooled monochromatic digital camera (8-bit) every 60 s (exposure time: 30 ms, gain: 0.5). Image analysis and quantification of fluorescence intensity were performed with Image J (http://rsbweb.nih.gov/ij/). Plasmid preparation, mutagenesis, and cell culture were performed as in (35). Finally, to normalize channel activity to expression levels, the constructs were inserted into pIRES vector (Clontech, Mountain View, CA) in which green fluorescent protein (GFP) and Cxs are components of the same bicistronic transcript of the pIRES vector.
Results and Discussion
The IC pocket
While performing MD simulations to study water structure and dynamics within the hCx26 hemichannel, we identified, to our knowledge, six novel water pockets—one per monomer—that we termed IC pockets. Each IC pocket is located in-between the four transmembrane helices (TMHs) in the intracellular side of each hCx26 monomer (Fig. 1). Because of the low resolution and packing of the hCx26 crystal structure, the IC pocket remained unidentified until now. In this x-ray structure all pockets appear to be closed showing no co-crystallized waters (38). Interestingly, during the course of the equilibration run we observed that the four transmembrane helices of each monomer relax to form an IC pocket at its intracellular side. Structural analysis shows that each IC pocket is partly built by the N-terminal domain (NTH) of the corresponding monomer, as an accommodating space in-between the four TMHs. This rearrangement increases the channel diameter as compared with that provided by the crystal structure, in accordance to previous studies on hCx26 hemichannels (41,42). Importantly, the IC pocket is filled with water that can move from the cavity to the main pore, and vice versa, via a small aperture below NTH (Fig. 1 A to C), hydrating the interface between NTH and the rest of the monomer. In accordance, this hydration layer might play a role as a polar lubricant for NTH to be kept wet and away from hydrophobic collapse over TM1 and TM2. Therefore, we hypothesize that changes in water volume and/or flow between the IC pocket and the main pore may affect the dynamics of NTH, which might be relevant for the functional state of the hemichannel.
Figure 1.
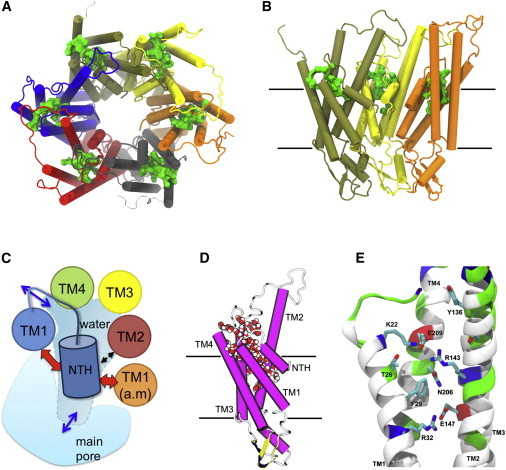
Overview of the IC pocket. (A) View from the intracellular side of the hCx26 hemichannel. The IC pocket location can be depicted by the presence of a green solid volume in every monomer. (B) View from inside the pore of the hCx26 hemichannel showing only three monomers for clarity. Protein is rendered in cartoon according to the secondary structure and colored by monomer. Water molecules inside each IC pocket are shown as a green solid volume (A) and (B). In (B) and (C), black solid lines depict the boundaries of the lipid membrane according to OPM database (see Methods). (C) Cartoon representation of one hCx26 monomer from a side view. Protein has been colored by secondary structure (magenta = α-helix; yellow = β-sheet; white = loop) and water molecules filling the IC pocket are shown in vdW representation colored by element (red = oxygen; white = hydrogen). (D) Schematic overview of the IC pocket location. Depicted cartoon represents a view from the intracellular side of one hCx26 monomer composing a hemichannel. Each circle represents a TM-helix. The orange circle represents TM1 from the clockwise adjacent monomer (a.m.). The blue cylinder represents NTH and the red and black arrows depict relevant interactions between NTH and residues from the same or the adjacent monomer. Blue arrows indicate the water entry and exit points. (E) Selected snapshot from the MD simulation depicting relevant residues forming the IC pocket. Protein is rendered as cartoon representation and colored by residue type (red = acidic; blue = basic; green = polar; white = nonpolar). Side chains are shown in licorice representation and colored by element (red = oxygen; blue = nitrogen; cyan = carbon). Hydrogen atoms and residues 1 to 14 (NTH) were omitted for clarity. To see this figure in color, go online.
Residues forming the IC pocket
By conducting a visual inspection of the IC pocket, we observed that it is mainly composed of small-sized noncharged polar and/or hydrophobic amino acids, rather than charged and/or bulky ones (Figs. 1 E and 2 A and Table 1). As it will be discussed later, two large residues located inside the pocket and facing water molecules, Phe-29 and Arg-143, escape this trend. The IC pocket interior is decorated with a mixture of polar and/or nonpolar amino acid residues, whereas charged amino acid residues forming salt-bridges that may act as embracing gates between the cavity and the exterior, compose both the IC and EC ends.
To consider the evolutive context, we proceeded to characterize the conservation of IC pocket residues between selected representatives of the Cx subfamilies by producing a multiple sequence alignment. The resulting alignment is presented in Fig. S3. As previously reported (54), an elevated conservation for a vast number of the IC pocket residues within and among the Cxs subfamilies is clearly visible (Fig. S3). In Fig. 2 B we present a mapping of the IC pocket conservation score onto a representative frame of the hCx26 monomer obtained from the MD production run. At first glance, it is clear that most amino acid residues range from moderately (light blue) to highly (blue) conserved. Moreover, most of the IC pocket residues are highly conserved within the beta subfamily and more loosely conserved among the Cx subfamilies (Fig. S3). Among the most highly conserved residues of the IC pocket, we found the two amino acid residues that form the EC or floor salt-bridge: Arg-32 (TM1) and Glu-147. When analyzing the salt-bridge formed by Lys-22 and Glu-209 located at the IC entry of the IC pocket, we realized that Glu-209 is highly conserved among Cx subfamilies being replaced by another negatively charged residue in ζ Cxs (Figs. 2 B and S3). As compared with Glu-209, Lys-22 is significantly less conserved but replaced by either a positively charged residue (Arg) or by a polar one (Gln). As a whole, the evidence suggest that the formation of a salt-bridge or an electrostatic interaction at both ends of the IC pocket could be relevant, either for a structural and/or a functional role on the hCx26 hemichannel. In agreement, these residues have been previously identified as structural constraints that reinforce hCx26 monomer stability (34,41).
In accordance to Figs. 2 B and S3, Gln-80 and Arg-143 are both conserved residues within the β subfamily. Gln-80 is highly conserved and only replaced by His in the Cx δ3 subfamily. Notably, Arg-143 is a highly conserved residue being replaced only by Lys in Cx α1, α4, β3, β5, and β7 subfamilies, supporting the relevance of its charge and size at this position.
Some mutations in the IC pocket are related with functional impairment of the hemichannel and hereditary deafness
The conservation pattern exhibited by several IC pocket residues suggests a possible functional role of this pocket. Therefore, we proceeded to compile reported mutations causing functional impairment of hCx26 hemichannels and GJC from the available literature. A comprehensive list of 16 mutations occurring at the IC pocket can be reviewed in Table S1. From them, nine mutations have been functionally characterized: T8M, I33T, V84L, A88S, A88S, R143W/Q, C202F, and N206S (33,62). As observed in Figs. S2 and 2 B and C, mutations on Ile-33, Val-84, and Arg-143, correspond to highly conserved residues, all of them facing water located within the IC pocket. According to functional studies, mutations such as I33T and R143W/Q diminish or abrogate the functional state of the channel (33). On the other hand, mutations such as V84L, A88S, T8M, and N206 change the permeability and selectivity of this channel (62). Moreover, mutations such as A88S and A88V produce aberrant hemichannels with increased permeability (63,64). Interestingly, some mutations that affect hemichannel function do not necessarily produce malfunctioning GJC and vice versa (65,66). Unfortunately, there is no actual study linking changes in hCx26 structural features with its functional state. However, important previous experiments should be mentioned. While conducting cysteine replacement accessibility assays on Cx32, Skerret et al. (2002) (67) showed that both mutations R143C and S139C induced a reverse gating phenotype. Despite the accessibility was only tested in the closed state of Cx32, no significant decrease in channel conductance was detected. This led to the suggestion that introducing Cys at R143 could produce a disulphide bond with C202 (C201 in Cx32), locking the channel in the closed position. Interestingly, during our simulations in which the hemichannel is in an open state, the distance between E147-Cβ and C202-Cβ was always >5Å for all monomers (data not shown). Interestingly, these authors also reported that mutation I33C presented no reactivity to MBB together with a reduction on the hemichannel conductance, a strong indicative of pore blocking by MBB implying direct exposure of the mutated residue to the pore. Notably, as it can be observed in Fig. 2 D), residues that did not react with MBB (A88, S138, R143, and E147) but affect gating and permeability of the channel when mutated to Cys, are actually located inside the IC pocket. This location could explain the lack of reactivity to MBB, a molecule that is too big to enter the IC pocket. Importantly for our findings, Cys replacement of these residues produced function disruption in Cx32 hemichannels.
Water dynamics slows down in the IC pocket
During our MD simulations, we found that the IC pocket becomes quickly hydrated from the early stage of equilibration remaining hydrated afterward for the rest of the production time with no noticeable dewetting events. The extent of hydration within this pocket was quantified by measuring the number of water molecules present along simulation time in each IC pocket (Figs. 3 A, S4, and S5 and Table 2). On average, 12 ± 3 water molecules were found to occupy each IC pocket, albeit the water capacity seems rather flexible in every pocket varying from 5 to 21, according to the minimum and maximum number of water molecules, respectively (Fig. 3 A). This variability implies differences in water volume between pockets that could be produced by local changes in rotamer states of residues lining the IC pocket. These sets of changes could collectively modulate the conformation of the pocket, indicating that its formation might depend on the internal dynamics of water within itself. To further explore this modulation, we characterized water individual behavior within the IC pocket along the simulation.
Figure 3.

Water dynamics inside the IC pocket of each hCx26 monomer. (A) Water occupancy depicted as a box plot showing the number of water molecules inside the IC pocket of each Cx26 monomer. Box boundaries represent the standard deviation from the average position marked by the small square. The central line on each box shows the median value. Whiskers represent percentile 5 (top) and 95 (bottom) and the stars denote maximum (top) and minimum (bottom) values. Data were collected from four independent MD simulations of 20 ns each (see Methods). (B) Survival probabilities P(t) for water molecules inside the IC pocket of each hCx26 monomer. Data was taken averaging per monomer for each independent MD simulation of 20 ns. Each P(t) was obtained by averaging for each of the time windows available at the given interval (from 1 to 50 ps). Error bars represent the standard deviation from the block-average depicted in solid and using monomer-colored lines. To see this figure in color, go online.
Table 2.
Water occupancy and water diffusion coefficients within the IC pocket
| Monomer | Water occupancy (number of water molecules) |
Diffusion coefficient |
||||
|---|---|---|---|---|---|---|
| 100 ns |
S1 |
S2 |
S3 |
S4 |
Average (S1–S4) |
|
| Average ± std. err. | Average ± std. err. | Average ± std. err. | Average ± std. err. | Average ± std. err. | D(A2/ps) ± err. | |
| M1 | 10 ± 0.2 | 10 ± 0.1 | 12 ± 0.2 | 10 ± 0.2 | 10 ± 0.2 | 0.015 ± 0.004 |
| M2 | 14 ± 0.4 | 15 ± 0.1 | 16 ± 0.3 | 16 ± 0.3 | 17 ± 0.2 | 0.052 ± 0.010 |
| M3 | 11 ± 0.5 | 10 ± 0.2 | 11 ± 0.2 | 9 ± 0.6 | 9 ± 0.2 | 0.020 ± 0.004 |
| M4 | 13 ± 0.3 | 14 ± 0.2 | 13 ± 0.3 | 14 ± 0.4 | 13 ± 0.2 | 0.034 ± 0.001 |
| M5 | 11 ± 0.3 | 12 ± 0.5 | 13 ± 0.3 | 13 ± 0.2 | 12 ± 0.4 | 0.032 ± 0.006 |
| M6 | 10 ± 0.5 | 12 ± 0.1 | 13 ± 0.1 | 12 ± 0.3 | 12 ± 0.3 | 0.035 ± 0.004 |
Because of the confinement, water in the IC pocket showed fairly different behavior than that of the bulk and therefore water should reside much longer into the pocket. Water residency was determined by calculating the survival probability P(t), which was obtained from autocorrelation function for the resident water molecules inside of each pocket. As seen in Fig. 3 B, survival probabilities slowly decay in all six IC pockets. The decay rates estimated from these P(t) curves range from 18 to 23 ps, indicating a much slower rate of exchange within the IC pocket compared with bulk water molecules (∼1.5 ps for bulk water). Interestingly, decay profiles of P(t) show differences between pockets (Fig. 3 B), reflecting a structural heterogeneity among them produced by an independent but concerted dynamics. To further explore this behavior, we calculated the diffusion coefficient (D) by deriving it from the mean square displacement (MSD/P(t)) over the confined waters (Table 2 and Fig. S6). Calculated D values suggested that water diffusion in the IC pocket is slowed down by an order of magnitude as compared with that of the bulk. Within the IC pocket, water diffuses with D = 0.028 ± 0.009 Å2/ps, whereas bulk water shows much faster diffusion (D = 0.27 Å2/ps) as measured for the same TIP3P water model at 300K. Consistently, each pocket shows noticeably different water dynamics providing a range of diffusion coefficients between 0.014 and 0.038 Å2/ps (Table 2). Moreover, water residence patterns appear to be different in every Cx26 pocket. This behavior could be expected because of the presence of polar amino acid residues (e.g., Ser, Thr, and Asn) together with charged amino acid residues (e.g., Arg, Lys, and Glu) lining the IC pocket to which water may produce stable electrostatic interactions. To test this hypothesis, we analyzed the orientational relaxation (OR) time of two vectors that allow the description of the rotational freedom of water molecules within the pocket: 1), ûOH the unit vector along the molecular OH bond, and 2), ûD the unit vector along the dipole moment of the water molecule. The OR time (C2,û (t)) of water molecules inside the IC pocket is shown in Fig. 4. A nonexponential decay slower than that of the bulk can be observed for both vectors. Interestingly, nonexponential behaviors for time correlation of both vectors has been reported for water in confined systems (68) and has been related to nongeometrical confinement effects, such as interaction between water molecules and the confining surface. Therefore, the slower decay of the water dipole OR correlation time (Fig. 4 A) with respect to the OH vector (Fig. 4 B) suggests that water molecules may adopt a preferential orientation, probably attributable to specific electrostatic interactions with amino acid residues lining the IC pocket.
Figure 4.

Orientational time correlation of water molecules within the IC pocket. (A) Correlation time C2,û(t) for ûdipole. (B) Correlation time C2,û(t) for ûOH. In both panels correlation time values obtained for bulk water are shown as a gray-dashed line. Data shown using solid lines correspond to the average obtained from each data point between the four independent 20 ns MD production runs. Error bars represent the standard deviation for every average point. To see this figure in color, go online.
Structural water molecules found in the IC pocket
As demonstrated by previous reports, long-lasting hydrogen bonds are indicatives of both ordered and structured water inside polar pockets, suggesting functional relevance of such water-protein interactions (6,69,70). Therefore, we computed the hydrogen bond population relaxation (HBPR) (61) formed between: 1), water molecules inside the IC pocket, 2), water molecules and residues that comprise the IC pockets, 3), water and Arg-143, and 4), water and Asn-206. Both the continuous and intermittent HBPR where considered, as it can be seen in Fig. S7. As expected, hydrogen bond networks between water molecules inside the IC pockets decay slower than that of bulk water, consistent with both the slower diffusion and longer residence time, as we previously discussed. On the other hand, an even slower rate of decay is detected between water molecules and specific amino acid residues comprising the IC pocket, denoting a strong interaction between water and the confining surface. In particular, a very slow decay rate of the HBs formed by Arg-143 and Asn-206 with water molecules was found, especially for the intermittent case (Fig. S7). These slow decay rates indicate that water molecules within the IC pocket behave as structural water and may play a functional role (68), as it will be discussed later.
Interestingly, HBPR decay inside the IC pocket suggests that charged residues, in particular Arg-143 and Asn-206, can directly influence water dynamics within the pocket. Thus, the IC pocket provides a favorable environment for polar/charged residues to make stable hydrogen bonds with water under nano-confinement (68,71,72) suggesting that water molecules within this pocket may have preferential orientation. Indeed, Arg-143 and Asn-206 acting as hydrogen donors will attract oxygen atoms from water molecules allowing two OH groups to spin around a relatively fixed dipole axis of the water molecule. Consequently, as expected, the ûdipole vector is to decay slower than that of the ûOH vector, as can be seen in Fig. 4.
Interplay between rotamer states of Arg-143 and Phe-29 with structural water in the IC pocket
Considering that water molecules are involved in stable hydrogen bonds with Arg-143, we hypothesize that the structural water inside the IC pocket might have played a significant role in influencing the rotamer states of residues protruding to the IC pocket.
Fig. S8 summarizes different states and conformations adopted by Arg-143 and Phe-29 by computing χ angle plots in all six Cx26 monomers during our simulations. The probability distribution of distances between interacting residues of the IC pocket can be reviewed in Fig. 5 A–C. Fig. S9 presents the average distance between the side chains of interacting residues within the IC pocket, as a function of simulation time for each monomer. A visual summary of these observations is provided in Fig. 5 D. Notably, Arg-143 may adopt three preferential states when paying attention to interacting distances (Fig. S9) and dihedral angles (Fig. S8).
Figure 5.

Arg-143 may adopt three main conformations on the IC pocket. (A–C) Probability distribution of distances between Arg-143:Cζ and Glu-147:C∂; Arg-143:Cζ and Glu-209:C∂; and Arg-143:Cζ and center of mass of the phenyl ring of Phe-29, respectively, for each hCx26-monomer. (D) Panel shows representative conformations found for Arg-143 and Phe-29 from all six monomers along the MD production run. All monomers were aligned against one monomer from the crystal structure to observe, in one IC pocket, all the conformations adopted by Arg-143 and Phe-29. Other residues from the IC pocket are represented as a solid surface and colored by residue type (blue = positive; red = negative; green = polar; white = hydrophobic). Residues Arg-143 and Phe-29 are shown in licorice representation colored by monomer (black = M1; red = M2; green = M3; blue = M4; cyan = M5; magenta = M6) and labeled in red. Hydrogen atoms are omitted for clarity. The viewpoint is taken from inside the main pore facing NTH and TM2 (red arrow in the inset). Residues 1 to 18 and TM2 were omitted for clarity and are shown with dashed lines in the inset. The figure is oriented so that the upper boundary points toward the intracellular side. To see this figure in color, go online.
The “Up State,” in which Arg-143 is closer to Glu-209 (distance ≤ 5Å) than to Glu-147, could be stabilized by an ionic interaction with Glu-209 (M1, M6). Interestingly, monomers in which Arg-143 is forming an ionic interaction with either Glu-209 (Up) (M1 and M6) or Glu-147 (the “Down State,” see below) (M2), the side chain of Arg-143 shows less freedom of movement in terms of dihedral angles, as seen in the χ3 versus χ4 in Fig. S8.
The “Middle State,” is the one that more closely resembles the crystal structure. In this state, distances between Arg-143 and Glu-209 and Arg-143 and Glu-147, are both near to 9Å (Fig. S9 A and B). Notoriously, hCx26 monomers in which Arg-143 appears to be in the “Middle State” show the lower distances between Arg-143 and Phe-29 (∼4.5Å), an indicative of a possible cation-π interaction between both residues. Moreover, monomer M4, the clearest representative of this state, shows Phe-29 sandwiched between both Arg-32 and Arg-143 (data not shown). Interestingly, in monomers exhibiting the “Middle State,” Arg-143 shows a higher degree of freedom of movement in particular with respect to their guanidinium group reinforcing the notion that Arg-143 may not be involved in any stable ionic interaction.
The “Down State,” in which Arg-143 is closer to Glu-147 and therefore may form an ionic interaction with this residue, as for monomer 2 (Fig. S9) is also reflected by the observation of a stable trans conformation for χ3 (Fig. S8) along with a moderate freedom of movement of its guanidinium group. This might be possible because of the formation of a stable ionic interaction between both residues requires the χ3 angle of Arg-143 preferentially in transorientation.
On the other hand, Phe-29 also shows conformational variability among monomers as can be observed in Figs. 6, S8, and S9. It is clear that the distance between Arg-143 and Phe-29 depends on the conformation/orientation of both residues. Thus, the orientation variability of Arg-143 may not only be influenced by the surrounding Glu residues but also by the position of Phe-29 and vice versa. Accordingly, changes in the conformation and orientation of Arg-143 and/or Phe-29 may affect the water dynamics inside the IC pocket. Supporting this notion, mutation R143Q is associated with dominant high-frequency hearing loss (63,73,74) in GJCs. In addition, mutation R143W has been associated with recessive nonsyndromic sensorineural deafness (75–77). No mutational information is available to date for Phe-29. The conservation of the positive charged nature of the residue at position 143 (Figs. 2 B and S3) and the fact that replacing Arg for a neutral-polar residue such as Gln has a deleterious effect over the Cx26 function (63,73,74), suggests that the interplay between both residues may regulate water dynamics inside the IC pocket.
Figure 6.

Relation between dihedral angles χ3 of Arg-143 and χ1 of Phe-29 with water occupancy within the IC pocket. Water occupancy was defined as the number of water molecules inside the IC pocket by monomer in the hemichannel (see Methods). (A) Scatter plot of the dihedral angle χ3 (Cβ-Cγ- Cδ-Nε) of Arg-143 versus water occupancy. (B) Scatter plot of the dihedral angle χ1 (N-Cα -Cβ-Cγ) of Phe-29 versus water occupancy. Data gathered from the MD production run and color-coded by monomer (black = M1; red = M2; green = M3; blue = M4; cyan = M5; magenta = M6). To see this figure in color, go online.
An interplay between NTH and the IC pocket
As discussed previously, water molecules within the IC pocket form a layer that hydrates the interface between the NTH and the rest of the monomer. Therefore, variations in the volume and/or water flow from the pocket to the main pore should have an effect over the NTH position/orientation in each monomer. As suggested by previous studies, the NTH could be a key part of the fast-gating mechanism, one of the voltage-gating/sensing mechanisms proposed for Cxs (37,78,79). Consistent with this model, NTH conformational changes could produce a plug that reduces or blocks the channel pore as result of activation of the voltage sensor located in this helix. Moreover, available evidence suggests that the nature of the charged or polar residues located on the NTH domain could determine the gating polarity of the channel (79–82). Interestingly, the role of the N-terminal helix as a plug has been further supported by the cryo-EM of the M34A mutant where a pore-blocking density plug can be observed (83,84).
To obtain insights on structural changes produced on the NTH during our MD simulations, we measured the distance from the center of mass of NTH to the protein center in the z axis per monomer, and averaged over the six monomers and presented as a probability distribution, as it can be seen in Fig. S10 A and B. Two main populations of distances can be observed; the highest populated is the one where NTH is around 4Å, and the less populated is the one where NTH is between 6 to 7Å. Interestingly, the less-populated state is the one more similar to the crystal structure. Measurements of the angle formed between the principal axis of NTH and the z axis (Fig. S10 C and D) show that despite the two marked populations on the position of the NHT, no clear angular asymmetries emerge during our MD simulations, as compared with the crystal structure. This evidence suggests that each NTH acquires an equilibrium position and remains fluctuating around it during the rest of the simulation time, showing minor asymmetries as have been experimentally suggested (84).
To further explore the dynamics of the NTH, we decided to characterize the relationship between dihedral angles χ3 of Arg-143 and χ1 of Phe-29 with water occupancy within the IC pocket (Fig. 6). Water occupancy is higher when dihedral angle χ3 of Arg-143 is between 140° and 220°, corresponding to the “Down State” of R143 in the M2 monomer. On the other hand, when dihedral angle χ3 of Arg-143 is between 20° and 90°, and between 250° and 330°, similar water occupancies can be detected at the IC pocket. Dihedral angle χ1 of Phe-29 can be preferentially found in two positions. When dihedral angle χ1 of Phe-29 is between 160° and 240°, as in monomers M2, M5, and M6, a slightly higher water occupancy can be observed (Fig. 6 B), in comparison with that of angle χ1 between 250° and 320°.
Is the water volume of the IC pocket linked to hemichannel function on hCx26?
Data gathered in this work, together with previous observations, leads us to hypothesize about a functional role of the IC pocket linking it with a putative control of the hemichannel permeability. In particular, the interplay between IC pocket water volume and the position of both residues Arg-143 and Phe-29 may result in a volume-controlled gating mechanism of the hCx26 hemichannels. In accordance, changes on the orientation of Arg-143, for instance because of transmembrane voltage, may have a direct influence over structural water inside the IC pocket. Thus, the orientation of these residues and the volume of the IC pocket could be controlled by an external stimulus. Hence, the modulation of both the IC pocket volume and water dynamics could have a direct effect over the NTH orientation and/or freedom of movement, producing a direct outcome over its function as both a sensor of an external stimulus, and a channel gate. To better comprehend the proposed role of the IC pocket, it is important to mention that the involvement of the NTH as a plug or gate that moves to close the channel has received important criticism (37,83), in particular with regard to the energy needed for the process of reposition of the NTH domain to the open conformation. As proposed, the voltage-modulation of the volume of water inside the IC pocket could reduce significantly the energy requirements for the reposition of the NTH to the open conformation.
Arg-143 plays an essential role on hCx26 hemichannel permeability
To evaluate the existence of a structural and functional relationship between the IC pocket, Arg-143, and the hCx26 function, we conducted hemichannel functional assessments through time-lapse imaging of the Etd uptake (Fig. 7).
Figure 7.

Arg-143 plays an essential role on Cx26 hemichannels activity. To test the contribution of Arg-143 on the hemichannel activity, we replaced this residue by different charge/polarity residues through site-directed mutagenesis. The functional state of the hemichannels was assessed using the widely used Eth uptake assay. The Eth plotted curve represents the fluorescent intensity observed 5 min after incubation in solution free of Ca2+ and Mg2+. For all panels: WT Cx26 (white), Cx26R143A (red), Cx26143K (yellow), Cx26R143E (blue), and Cx26R143Q (green). (A) Time course of Eth uptake in a Ca2+-free solution to promote hemichannel opening. HeLa parental cells with MOCK transfection are depicted in gray. (B) Rate of Eth uptake extracted from the slopes of the curves showed in (A). (C) Relationship between the levels of Eth uptake and connexin level expression as a function of the GFP fluorescence intensity, under divalent cation-free solution. To see this figure in color, go online.
The uptake of Eth during the La3+ free time course can be characterized by three different groups: the one with higher hemichannel activity composed by mutant CxR143A; the one with medium activity composed by WT Cx26, CxR143K, and CxR143Q; and the one with diminished activity composed by CxR143E and the MOCK transfection (Fig. 7 A and B). To discard that differences in hemichannel activity were related to connexin level expression, levels of Eth uptake were plotted versus GFP expression levels (see Materials and Methods). As shown, regression values (r2) exhibit a high correlation between expression levels of CxR143A (r2 = 0.7) and Eth uptake, which is consistent with the apparent higher hemichannel activity elicited by this mutation when compared with WT Cx26 (r2 = 0.5) (Fig. 7 C). Therefore, the augmented activity of CxR143A could be merely produced by the higher abundance of hCx26 expressed in the HeLa cells. On the other hand, reduced Eth uptake observed for mutant CxR143Q (Fig. 7 A and B) is likely produced by hemichannels with diminished activity as demonstrated by the low correlation (r2 = 0.2) observed at Fig. 7 C). Notably, the low activity of mutant CxR143E corresponds with a near to zero correlation (r2 = 0.01) between levels of Eth uptake and GFP fluorescence. When evaluating the hemichannel activity of mutant CxR143K a nonsignificant change in the activity v/s WT Cx26 can be observed in Fig. 7 A and B, exhibiting a low correlation value (r2 = 0.3) (Fig. 7 C).
By conducting these in vitro studies, we demonstrated that changing the net charge and nature of Arg-143 to an amino acid residue with opposite polarity (E), the hemichannel permeability in cells expressing these mutants dramatically changes, as compared with those expressing the WT Cx26. As expected, mutation R143K does not impair hemichannel permeability, which is consistent with the notion that both, Arg and Lys, are conserved amino acid residues and natural constituents of the IC pocket in α and β Cxs, respectively. Finally, we also found that mutation R143Q, which is related with syndromic deafness, produced hemichannels with diminished activity, as we previously described (33).
Despite our data points toward a functional role of both the IC pocket and, in particular, a critical role of Arg-143 on the hCx26 hemichannel activity, unanswered questions remain on the precise role played by Arg-143. Importantly, Arginine residues have been associated to perm-selectivity and voltage-sensitivity functions in other proteins. In voltage-gated ion channels an Arg rich region in helix S4 is a key part of the voltage sensing mechanism (85–87). In aquaporins, a highly conserved Arg residue is a fundamental part of the selectivity filter (88–90) and different conformations of this residue inside the pore, affect the permeability of these channels (91–93). Moreover, the orientation of this Arg residue in aquaporins may response to external voltage, as described in MD simulations (94).
Unfortunately, the identification of the amino acid residues comprising the voltage sensor in Cx based channels has proven to be elusive (95). Previous studies have proposed that net charges located outside the N-terminus of Cx32-based channels can produce asymmetric voltage profiles across the channel pore that may induce movement of the voltage sensor (79). These charges include: R22, E208, R22, R142, and E146 from Cx32, equivalent to K22, E209, R143, and E147 in hCx26 (Fig. S2). In accordance to Oh et al. (2004) (79), as these amino acid residues will not move in response to changes of an external voltage, they should not be considered to be part of the voltage sensor. In the lights of our findings, the IC pocket, and in particular Arg-143, both play an important role in the hCx26 hemichannel activity. This role may include the participation in voltage sensing and gating of these hemichannels. Further experiments are currently under execution by our group to test the role of Arg-143 as a putative voltage sensor of hCx26 hemichannels.
Conclusions
We have identified, to our knowledge, a novel water pocket, termed IC pocket, in the intracellular side of each monomer of the Cx26 hemichannel. This pocket is located within the TM-helix bundle allowing the passage of water from the IC side to the main pore via a small opening beneath the NTH.
We characterized in detail the dynamics, ordering, and orientation of water molecules inside these pockets, finding a clear nonbulk-like behavior mainly because of interactions between water molecules and the protein interface.
When studying the composition and dynamics of the amino acid residues comprising the IC pocket we found that Arg-143 and Phe-29 establish strong interactions with water molecules within the IC pocket. These structural water molecules also influence the side-chain orientations (rotamer states) of residues such as Arg-143 and Phe-29, or vice versa.
Our in vitro experiments demonstrate that Arg-143 plays an essential role on the hCx26 hemichannel activity.
These findings might provide new insights into the molecular mechanism on gating of hCx26 hemichannels as well as novel potential drug design strategies for Cx-related diseases.
Acknowledgments
We thank Payel Das, Mike Pitman, Bruce Berne, Matteo Castelli, and Binquan Luan for helpful discussions.
This work was partially supported by FONDECYT 1130652 (T.P.-A.), FONDECYT 1130855 (A.D.M.), PFB16 Fundación Ciencia para la Vida, ACT1107 (T.P.-A.), ACT1104 (A.D.M.), and ICM-Economía P09-022-F Centro Interdisciplinario de Neurociencias de Valparaíso (T.P.-A., A.D.M., and J.C.S.). We thank IBM Blue Gene supercomputer for computational resources. RZ acknowledges the support from IBM Blue Gene Science Program. This research was partially supported by the supercomputing infrastructure of the NLHPC (ECM-02). Powered@NLHPC.
Footnotes
Raul Araya-Secchi and Tomas Perez-Acle contributed equally to this work.
Raul Araya-Secchi’s present address is Chemistry and Biochemistry Department, The Ohio State University, Columbus, OH.
Contributor Information
Tomas Perez-Acle, Email: tomas@dlab.cl.
Ruhong Zhou, Email: ruhongz@us.ibm.com.
Supporting Material
References
- 1.Jensen L.H. The structure of water in protein crystals. Dev. Biol. Stand. 1992;74:53–61. [PubMed] [Google Scholar]
- 2.Beckstein O., Sansom M.S. Liquid-vapor oscillations of water in hydrophobic nanopores. Proc. Natl. Acad. Sci. USA. 2003;100:7063–7068. doi: 10.1073/pnas.1136844100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Damjanović A., Schlessman J.L., García-Moreno E B. Role of flexibility and polarity as determinants of the hydration of internal cavities and pockets in proteins. Biophys. J. 2007;93:2791–2804. doi: 10.1529/biophysj.107.104182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matthews B.W., Liu L. A review about nothing: Are apolar cavities in proteins really empty? Protein Sci. 2009;18:494–502. doi: 10.1002/pro.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yin H., Feng G., Rasaiah J.C. Water in the polar and nonpolar cavities of the protein interleukin-1β. J. Phys. Chem. B. 2010;114:16290–16297. doi: 10.1021/jp108731r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schlessman J.L., Abe C., García-Moreno E B. Crystallographic study of hydration of an internal cavity in engineered proteins with buried polar or ionizable groups. Biophys. J. 2008;94:3208–3216. doi: 10.1529/biophysj.107.122473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grossfield A., Pitman M.C., Gawrisch K. Internal hydration increases during activation of the G-protein-coupled receptor rhodopsin. J. Mol. Biol. 2008;381:478–486. doi: 10.1016/j.jmb.2008.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pardo L., Deupi X., Campillo M. The role of internal water molecules in the structure and function of the rhodopsin family of G protein-coupled receptors. Chem. Bio. Chem. 2007;8:19–24. doi: 10.1002/cbic.200600429. [DOI] [PubMed] [Google Scholar]
- 9.González A., Perez-Acle T., Deupi X. Molecular basis of ligand dissociation in β-adrenergic receptors. PLoS ONE. 2011;6:e23815. doi: 10.1371/journal.pone.0023815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ladbury J.E. Just add water! The effect of water on the specificity of protein-ligand binding sites and its potential application to drug design. Chem. Biol. 1996;3:973–980. doi: 10.1016/s1074-5521(96)90164-7. [DOI] [PubMed] [Google Scholar]
- 11.Cooper A. Heat capacity effects in protein folding and ligand binding: a re-evaluation of the role of water in biomolecular thermodynamics. Biophys. Chem. 2005;115:89–97. doi: 10.1016/j.bpc.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 12.Li Z., Lazaridis T. Thermodynamics of buried water clusters at a protein-ligand binding interface. J. Phys. Chem. B. 2006;110:1464–1475. doi: 10.1021/jp056020a. [DOI] [PubMed] [Google Scholar]
- 13.Imai T. Roles of water in protein structure and function studied by molecular liquid theory. Frontiers Biosci. 2009;14:1387–1402. doi: 10.2741/3314. [DOI] [PubMed] [Google Scholar]
- 14.Levy Y., Onuchic J.N. Water mediation in protein folding and molecular recognition. Annu. Rev. Biophys. Biomol. Struct. 2006;35:389–415. doi: 10.1146/annurev.biophys.35.040405.102134. [DOI] [PubMed] [Google Scholar]
- 15.Liu P., Huang X., Berne B.J. Observation of a dewetting transition in the collapse of the melittin tetramer. Nature. 2005;437:159–162. doi: 10.1038/nature03926. [DOI] [PubMed] [Google Scholar]
- 16.Zhou R., Huang X., Berne B.J. Hydrophobic collapse in multidomain protein folding. Science. 2004;305:1605–1609. doi: 10.1126/science.1101176. [DOI] [PubMed] [Google Scholar]
- 17.Jensen M.O., Borhani D.W., Shaw D.E. Principles of conduction and hydrophobic gating in K+ channels. Proc. Natl. Acad. Sci. USA. 2010;107:5833–5838. doi: 10.1073/pnas.0911691107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu F., Hummer G. Drying transition in the hydrophobic gate of the GLIC channel blocks ion conduction. Biophys. J. 2012;103:219–227. doi: 10.1016/j.bpj.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beckstein O., Biggin P.C., Sansom M.S.P. A hydrophobic gating mechanism for nanopores. J. Phys. Chem. B. 2001;105:12902–12905. [Google Scholar]
- 20.Birkner J.P., Poolman B., Koçer A. Hydrophobic gating of mechanosensitive channel of large conductance evidenced by single-subunit resolution. Proc. Natl. Acad. Sci. USA. 2012;109:12944–12949. doi: 10.1073/pnas.1205270109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matthews B.W., Morton A.G., Dahlquist F.W. Use of NMR to detect water within nonpolar protein cavities. Science. 1995;270:1847–1849. doi: 10.1126/science.270.5243.1847. [DOI] [PubMed] [Google Scholar]
- 22.DeFlores L.P., Tokmakoff A. Water penetration into protein secondary structure revealed by hydrogen-deuterium exchange two-dimensional infrared spectroscopy. J. Am. Chem. Soc. 2006;128:16520–16521. doi: 10.1021/ja067723o. [DOI] [PubMed] [Google Scholar]
- 23.Skinner J.L., Pieniazek P.A., Gruenbaum S.M. Vibrational spectroscopy of water at interfaces. Acc. Chem. Res. 2012;45:93–100. doi: 10.1021/ar200122a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hua L., Huang X., Berne B.J. Dynamics of water confined in the interdomain region of a multidomain protein. J. Phys. Chem. B. 2006;110:3704–3711. doi: 10.1021/jp055399y. [DOI] [PubMed] [Google Scholar]
- 25.Hua L., Huang X., Berne B.J. Nanoscale dewetting transition in protein complex folding. J. Phys. Chem. B. 2007;111:9069–9077. doi: 10.1021/jp0704923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Z., Shi B., Zhou R. Dewetting transitions in the self-assembly of two amyloidogenic β-sheets and the importance of matching surfaces. J. Phys. Chem. B. 2011;115:11137–11144. doi: 10.1021/jp2046454. [DOI] [PubMed] [Google Scholar]
- 27.Das P., Kapoor D., Matthews C.R. Interplay between drying and stability of a TIM barrel protein: a combined simulation-experimental study. J. Am. Chem. Soc. 2013;135:1882–1890. doi: 10.1021/ja310544t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bakowies D., van Gunsteren W.F. Simulations of apo and holo-fatty acid binding protein: structure and dynamics of protein, ligand and internal water. J. Mol. Biol. 2002;315:713–736. doi: 10.1006/jmbi.2001.5202. [DOI] [PubMed] [Google Scholar]
- 29.Kumar N.M., Gilula N.B. The gap junction communication channel. Cell. 1996;84:381–388. doi: 10.1016/s0092-8674(00)81282-9. [DOI] [PubMed] [Google Scholar]
- 30.Yeager M., Harris A.L. Gap junction channel structure in the early 21st century: facts and fantasies. Curr. Opin. Cell Biol. 2007;19:521–528. doi: 10.1016/j.ceb.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beblo D.A., Veenstra R.D. Monovalent cation permeation through the connexin40 gap junction channel. Cs, Rb, K, Na, Li, TEA, TMA, TBA, and effects of anions Br, Cl, F, acetate, aspartate, glutamate, and NO3. J. Gen. Physiol. 1997;109:509–522. doi: 10.1085/jgp.109.4.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willecke K., Eiberger J., Söhl G. Structural and functional diversity of connexin genes in the mouse and human genome. Biol. Chem. 2002;383:725–737. doi: 10.1515/BC.2002.076. [DOI] [PubMed] [Google Scholar]
- 33.Martinez A.D., Acuna R., Figueroa V., Maripillan J., Nicholson B. Gap-junction channels dysfunction in deafness and hearing loss. Antioxid. Redox Signal. 2009;11:309–322. doi: 10.1089/ars.2008.2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maeda S., Tsukihara T. Structure of the gap junction channel and its implications for its biological functions. Cell. Mol. Life Sci. 2011;68:1115–1129. doi: 10.1007/s00018-010-0551-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jara O., Acuña R., Martínez A.D. Critical role of the first transmembrane domain of Cx26 in regulating oligomerization and function. Mol. Biol. Cell. 2012;23:3299–3311. doi: 10.1091/mbc.E11-12-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bukauskas F.F., Verselis V.K. Gap junction channel gating. Biochim. Biophys. Acta. 2004;1662:42–60. doi: 10.1016/j.bbamem.2004.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bargiello T.A., Tang Q., Kwon T. Voltage-dependent conformational changes in connexin channels. Biochim. Biophys. Acta. 2012;1818:1807–1822. doi: 10.1016/j.bbamem.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maeda S., Nakagawa S., Tsukihara T. Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature. 2009;458:597–602. doi: 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]
- 39.Bicego M., Beltramello M., Mammano F. Pathogenetic role of the deafness-related M34T mutation of Cx26. Hum. Mol. Genet. 2006;15:2569–2587. doi: 10.1093/hmg/ddl184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pantano S., Zonta F., Mammano F. A fully atomistic model of the Cx32 connexon. PLoS ONE. 2008;3:e2614. doi: 10.1371/journal.pone.0002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kwon T., Harris A.L., Bargiello T.A. Molecular dynamics simulations of the Cx26 hemichannel: evaluation of structural models with Brownian dynamics. J. Gen. Physiol. 2011;138:475–493. doi: 10.1085/jgp.201110679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kwon T., Roux B., Bargiello T.A. Molecular dynamics simulations of the Cx26 hemichannel: insights into voltage-dependent loop-gating. Biophys. J. 2012;102:1341–1351. doi: 10.1016/j.bpj.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zonta F., Polles G., Mammano F. Permeation pathway of homomeric connexin 26 and connexin 30 channels investigated by molecular dynamics. J. Biomol. Struct. Dyn. 2012;29:985–998. doi: 10.1080/073911012010525027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sali A. Comparative protein modeling by satisfaction of spatial restraints. Mol. Med. Today. 1995;1:270–277. doi: 10.1016/s1357-4310(95)91170-7. [DOI] [PubMed] [Google Scholar]
- 45.Olsson M.H.M., Søndergaard C.R., Jensen J.H. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 46.Bas D.C., Rogers D.M., Jensen J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins. 2008;73:765–783. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- 47.Lomize M.A., Lomize A.L., Mosberg H.I. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 48.Jo S., Lim J.B., Im W. CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009;97:50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mackerell A.D., Jr., Feig M., Brooks C.L., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 50.Klauda J.B., Venable R.M., Pastor R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Woolf T.B., Roux B. Molecular dynamics simulation of the gramicidin channel in a phospholipid bilayer. Proc. Natl. Acad. Sci. USA. 1994;91:11631–11635. doi: 10.1073/pnas.91.24.11631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Woolf T.B., Roux B. Structure, energetics, and dynamics of lipid-protein interactions: A molecular dynamics study of the gramicidin A channel in a DMPC bilayer. Proteins. 1996;24:92–114. doi: 10.1002/(SICI)1097-0134(199601)24:1<92::AID-PROT7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 53.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abascal F., Zardoya R. Evolutionary analyses of gap junction protein families. Biochim. Biophys. Acta. 2013;1828:4–14. doi: 10.1016/j.bbamem.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 55.Troshin P.V., Procter J.B., Barton G.J. Java bioinformatics analysis web services for multiple sequence alignment—JABAWS:MSA. Bioinformatics. 2011;27:2001–2002. doi: 10.1093/bioinformatics/btr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ashkenazy H., Erez E., Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38:W529–W533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Armon A., Graur D., Ben-Tal N. ConSurf: an algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J. Mol. Biol. 2001;307:447–463. doi: 10.1006/jmbi.2000.4474. [DOI] [PubMed] [Google Scholar]
- 58.Michaud-Agrawal N., Denning E.J., Beckstein O. MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011;32:2319–2327. doi: 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu P., Harder E., Berne B.J. On the calculation of diffusion coefficients in confined fluids and interfaces with an application to the liquid−vapor interface of water. J. Phys. Chem. B. 2004;108:6595–6602. [Google Scholar]
- 60.Yeh Y.-l., Mou C.-Y. Orientational relaxation dynamics of liquid water studied by molecular dynamics simulation. J. Phys. Chem. B. 1999;103:3699–3705. [Google Scholar]
- 61.Rapaport D.C. Hydrogen bonds in water. Mol. Phys. 1983;50:1151–1162. [Google Scholar]
- 62.Xu J., Nicholson B.J. The role of connexins in ear and skin physiology—functional insights from disease-associated mutations. Biochim. Biophys. Acta. 2013;1828:167–178. doi: 10.1016/j.bbamem.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang J., Scherer S.S., Yum S.W. Dominant Cx26 mutants associated with hearing loss have dominant-negative effects on wild type Cx26. Mol. Cell. Neurosci. 2011;47:71–78. doi: 10.1016/j.mcn.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mhaske P.V., Levit N.A., White T.W. The human Cx26-D50A and Cx26-A88V mutations causing keratitis-ichthyosis-deafness syndrome display increased hemichannel activity. Am. J. Physiol. Cell Physiol. 2013;304:C1150–C1158. doi: 10.1152/ajpcell.00374.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beyer E.C., Lipkind G.M., Berthoud V.M. Structural organization of intercellular channels II. Amino terminal domain of the connexins: sequence, functional roles, and structure. Biochim. Biophys. Acta. 2012;1818:1823–1830. doi: 10.1016/j.bbamem.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sosinsky G.E., Nicholson B.J. Structural organization of gap junction channels. Biochim. Biophys. Acta. 2005;1711:99–125. doi: 10.1016/j.bbamem.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 67.Skerrett I.M., Aronowitz J., Nicholson B.J. Identification of amino acid residues lining the pore of a gap junction channel. J. Cell Biol. 2002;159:349–360. doi: 10.1083/jcb.200207060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Milischuk A.A., Ladanyi B.M. Structure and dynamics of water confined in silica nanopores. J. Chem. Phys. 2011;135:174709. doi: 10.1063/1.3657408. [DOI] [PubMed] [Google Scholar]
- 69.Yu H., Rick S.W. Free energy, entropy, and enthalpy of a water molecule in various protein environments. J. Phys. Chem. B. 2010;114:11552–11560. doi: 10.1021/jp104209w. [DOI] [PubMed] [Google Scholar]
- 70.Williams M.A., Goodfellow J.M., Thornton J.M. Buried waters and internal cavities in monomeric proteins. Protein Sci. 1994;3:1224–1235. doi: 10.1002/pro.5560030808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kumar P., Han S., Stanley H.E. Anomalies of water and hydrogen bond dynamics in hydrophobic nanoconfinement. J. Phys. Condens. Matter. 2009;21:504108. doi: 10.1088/0953-8984/21/50/504108. [DOI] [PubMed] [Google Scholar]
- 72.Park S., Moilanen D.E., Fayer M.D. Water dynamics—the effects of ions and nanoconfinement. J. Phys. Chem. B. 2008;112:5279–5290. doi: 10.1021/jp7121856. [DOI] [PubMed] [Google Scholar]
- 73.Löffler J., Nekahm D., Janecke A.R. Sensorineural hearing loss and the incidence of Cx26 mutations in Austria. Eur. J. Hum. Genet. 2001;9:226–230. doi: 10.1038/sj.ejhg.5200607. [DOI] [PubMed] [Google Scholar]
- 74.Yum S.W., Zhang J., Scherer S.S. Dominant connexin26 mutants associated with human hearing loss have trans-dominant effects on connexin30. Neurobiol. Dis. 2010;38:226–236. doi: 10.1016/j.nbd.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Park H.J., Hahn S.H., Kim H.N. Connexin26 mutations associated with nonsyndromic hearing loss. Laryngoscope. 2000;110:1535–1538. doi: 10.1097/00005537-200009000-00023. [DOI] [PubMed] [Google Scholar]
- 76.Wang H.L., Chang W.T., Huang P.C. Functional analysis of connexin-26 mutants associated with hereditary recessive deafness. J. Neurochem. 2003;84:735–742. doi: 10.1046/j.1471-4159.2003.01555.x. [DOI] [PubMed] [Google Scholar]
- 77.Mani R.S., Ganapathy A., Anand A. Functional consequences of novel connexin 26 mutations associated with hereditary hearing loss. Eur. J. Hum. Genet. 2009;17:502–509. doi: 10.1038/ejhg.2008.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kronengold J., Srinivas M., Verselis V.K. The N-terminal half of the connexin protein contains the core elements of the pore and voltage gates. J. Membr. Biol. 2012;245:453–463. doi: 10.1007/s00232-012-9457-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Oh S., Rivkin S., Bargiello T.A. Determinants of gating polarity of a connexin 32 hemichannel. Biophys. J. 2004;87:912–928. doi: 10.1529/biophysj.103.038448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Purnick P.E., Oh S., Bargiello T.A. Reversal of the gating polarity of gap junctions by negative charge substitutions in the N-terminus of connexin 32. Biophys. J. 2000;79:2403–2415. doi: 10.1016/S0006-3495(00)76485-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oh S., Abrams C.K., Bargiello T.A. Stoichiometry of transjunctional voltage-gating polarity reversal by a negative charge substitution in the amino terminus of a connexin32 chimera. J. Gen. Physiol. 2000;116:13–31. doi: 10.1085/jgp.116.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Verselis V.K., Ginter C.S., Bargiello T.A. Opposite voltage gating polarities of two closely related connexins. Nature. 1994;368:348–351. doi: 10.1038/368348a0. [DOI] [PubMed] [Google Scholar]
- 83.Oshima A., Tani K., Sosinsky G.E. Three-dimensional structure of a human connexin26 gap junction channel reveals a plug in the vestibule. Proc. Natl. Acad. Sci. USA. 2007;104:10034–10039. doi: 10.1073/pnas.0703704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Oshima A., Tani K., Fujiyoshi Y. Asymmetric configurations and N-terminal rearrangements in connexin26 gap junction channels. J. Mol. Biol. 2011;405:724–735. doi: 10.1016/j.jmb.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pathak M., Kurtz L., Isacoff E. The cooperative voltage sensor motion that gates a potassium channel. J. Gen. Physiol. 2005;125:57–69. doi: 10.1085/jgp.200409197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jogini V., Roux B. Dynamics of the Kv1.2 voltage-gated K+ channel in a membrane environment. Biophys. J. 2007;93:3070–3082. doi: 10.1529/biophysj.107.112540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schwaiger C.S., Börjesson S.I., Lindahl E. The free energy barrier for arginine gating charge translation is altered by mutations in the voltage sensor domain. PLoS ONE. 2012;7:e45880. doi: 10.1371/journal.pone.0045880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Beitz E., Wu B., Zeuthen T. Point mutations in the aromatic/arginine region in aquaporin 1 allow passage of urea, glycerol, ammonia, and protons. Proc. Natl. Acad. Sci. USA. 2006;103:269–274. doi: 10.1073/pnas.0507225103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hub J.S., Grubmuller H., de Groot B.L. Aquaporins. Springer; Berlin: 2009. Dynamics and energetics of permeation through aquaporins. What do we learn from molecular dynamics simulations? pp. 57–76. [DOI] [PubMed] [Google Scholar]
- 90.Araya-Secchi R., Garate J.A., Perez-Acle T. Molecular dynamics study of the archaeal aquaporin AqpM. BMC Genomics. 2011;12(Suppl 4):S8. doi: 10.1186/1471-2164-12-S4-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jensen M.O., Mouritsen O.G. Single-channel water permeabilities of Escherichia coli aquaporins AqpZ and GlpF. Biophys. J. 2006;90:2270–2284. doi: 10.1529/biophysj.105.073965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hedfalk K., Törnroth-Horsefield S., Neutze R. Aquaporin gating. Curr. Opin. Struct. Biol. 2006;16:447–456. doi: 10.1016/j.sbi.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 93.Jiang J., Daniels B.V., Fu D. Crystal structure of AqpZ tetramer reveals two distinct Arg-189 conformations associated with water permeation through the narrowest constriction of the water-conducting channel. J. Biol. Chem. 2006;281:454–460. doi: 10.1074/jbc.M508926200. [DOI] [PubMed] [Google Scholar]
- 94.Hub J.S., Aponte-Santamaría C., de Groot B.L. Voltage-regulated water flux through aquaporin channels in silico. Biophys. J. 2010;99:L97–L99. doi: 10.1016/j.bpj.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ek-Vitorin J.F., Burt J.M. Structural basis for the selective permeability of channels made of communicating junction proteins. Biochim. Biophys. Acta. 2013;1828:51–68. doi: 10.1016/j.bbamem.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.